

Abstract
Aims/hypothesis
Mitochondria frequently change their shapes by fusion and fission and these morphological dynamics play important roles in mitochondrial function and development as well as programmed cell death. The goal of this study is to investigate whether: (1) mitochondria in mouse coronary endothelial cells (MCECs) isolated from diabetic mice exhibit increased fragmentation; and (2) chronic treatment with a superoxide anion (O2 −) scavenger has a beneficial effect on mitochondrial fragmentation in MCECs.
Methods
MCECs were freshly isolated and lysed for protein measurement, or cultured to determine mitochondrial morphology and O2 − production. For the ex vivo hyperglycaemia experiments, human coronary endothelial cells were used.
Results
Elongated mitochondrial tubules were observed in MCECs isolated from control mice, whereas mitochondria in MCECs from diabetic mice exhibited augmented fragmentation. The level of optic atrophy 1 (OPA1) protein, which leads to mitochondrial fusion, was significantly decreased, while dynamin-related protein 1 (DRP1), which leads to mitochondrial fission, was significantly increased in MCECs from diabetic mice. Diabetic MCECs exhibited significantly higher O2 − concentrations in cytosol and mitochondria than control MCECs. Administration of the O2 − scavenger TEMPOL to diabetic mice for 4 weeks led to a significant decrease in mitochondrial fragmentation without altering the levels of OPA1 and DRP1 proteins in MCECs. High-glucose treatment for 24 h significantly induced mitochondrial fragmentation, which was restored by TEMPOL treatment. In addition, excess O2 − production, either in cytosol or in mitochondria, significantly increased mitochondrial fragmentation.
Conclusions/interpretation
These data suggest that lowering the O2 − concentration can restore the morphological change in mitochondria and may help improve mitochondrial function in diabetic MCECs.
Electronic supplementary material
The online version of this article (doi:10.1007/s00125-010-1770-4) contains supplementary material, which is available to authorised users.
Keywords: DRP1, Endothelium, Fission, Fusion, Mitochondrial dynamics, O2−, OPA1, Type 1 diabetes
Introduction
Diabetic cardiomyopathy, a prominent cardiovascular complication, has been recognised as a microvascular disease and a primary cause of morbidity and mortality in diabetic patients. The pathogenesis of diabetic cardiomyopathy involves coronary endothelial cell (EC) dysfunction, fibrosis, cardiac myocyte dysfunction and cardiac myocyte necrosis. We recently reported that coronary ECs from diabetic mice are dysfunctional in regulation of vascular tone and angiogenesis [1]. In addition, we and other investigators [2, 3] demonstrated that EC apoptosis is significantly increased in the diabetic heart; however, the molecular mechanism is not clear.
Recent work has highlighted the importance of mitochondrial dynamics in cells and animal physiology. Because mitochondria constantly fuse and divide, an imbalance of these two processes dramatically alters overall mitochondrial morphology [4]. It is now clear that mitochondrial dynamics play important roles in mitochondrial functions, including development, apoptosis and functional complementation of mitochondrial DNA (mtDNA) mutations by content mixing [5–11].
There are at least five proteins which regulate mitochondrial fusion and fission: optic atrophy 1 (OPA1), mitofusins 1 and 2 (MFN1, MFN2), dynamin-related protein 1 (DRP1) and fission 1 (FIS1). MFN1, MFN2 and OPA1 are essential for mitochondrial fusion and FIS1 and DRP1 are required for mitochondrial fission in mammals [7, 12]. MFN1 and MFN2 reside on the outer membrane with the N-terminal GTPase, where it is predicted to have a coil protruding into the cytosol [13], while OPA1 is an intermembrane-space protein [14]. These proteins work together to promote mitochondrial fusion [15]. DRP1 exists largely in a cytosolic pool, but a fraction localises to puncta on mitochondria [16]. FIS1 has a single transmembrane domain at the C-terminal end such that the bulk of the molecule is exposed to the cytosol [17]. It has been suggested that FIS1 recruits DRP1 from the cytosol to mitochondria for the fission reaction [17, 18]. Those proteins are associated with different kinds of disease, most of which are related to neuropathy [19–25]. Leinninger et al. previously reported that a change in the DRP1 protein level is caused by hyperglycaemia [26]. Other studies indicate a close relationship between MFN2 and type 2 diabetes and obesity [27, 28].
Mitochondria are a primary source of reactive oxygen species (ROS) during production of ATP by the complexes of the respiratory chain. While ROS production is occurring in mitochondria throughout life, mtDNA is more sensitive than genomic DNA to ROS-induced damage, as it is not protected by histones and its repair capabilities are limited [29]. There is increasing evidence showing the involvement of the superoxide anion (O2 −) in the pathogenesis of diabetes-associated vascular complications [30, 31]. High-glucose treatment of cultured endothelial cells induces overproduction of mitochondrial O2 − [32]. In vivo studies show overproduction of cytosolic O2 − in aortic endothelial cells in type 1 diabetes [33–35]. There is, however, no direct evidence demonstrating the change of mitochondrial O2 − concentration in vivo or the relationship between O2 − concentration and mitochondrial fission in ECs in diabetes.
In this study, we show increased mitochondrial fragmentation, decreased OPA1 and increased DRP1 protein production in mouse coronary endothelial cells (MCECs) isolated from diabetic mice. In addition, we found that morphological changes in mitochondria that occurred as a result of diabetes were restored by O2 − scavenger treatment without changing OPA1 and DRP1 protein levels.
Methods
Materials
M199, antibiotic reagents, dispase II, Mito Tracker Green FM, MitoSOX Red and dihydroethidium (DHE) were purchased from Invitrogen (Carlsbad, CA, USA). Anti-OPA1, anti-MNF1, anti-MNF2, anti-FIS1, anti-DRP1 and anti-actin antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-CD31 and endothelial cell growth supplement (ECGS) were obtained from BD Biosciences (San Jose, CA, USA). Collagenase II was purchased from Worthington Biochemical (Lakewood, NJ, USA). All other chemicals were from Sigma-Aldrich (St Louis, MO, USA).
Animal preparation
All investigations conformed to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH Publication No. 85-23, Revised 1985). This study was conducted in accordance with the guidelines established by the Institutional Animal Care and Use Committee at the University of California, San Diego. Male NIH Swiss mice, 6 weeks of age, were purchased from Harlan Sprague Dawley (Indianapolis, IN, USA) and mice in the diabetic group received a single injection of streptozotocin (160 mg/kg, dissolved in citrate buffer, i.v.). All data were obtained from mice 10 weeks after injection. Administration of 4-hydroxy-2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPOL, an O2 − scavenger) was started at 6 weeks after streptozotocin injection and continued for 4 weeks (1 mmol/l in drinking water). Plasma glucose levels were 8.1 ± 1.0 mmol/l in control mice, 40.2 ± 4.5 mmol/l in diabetic mice and 35.6 ± 3.5 mmol/l in TEMPOL-treated diabetic mice.
Isolation of mouse coronary vascular endothelial cells
MCECs were isolated as described previously [1]. Briefly, dissected heart tissues were minced and incubated with M199 containing 1 mg/ml collagenase II and 0.6 U/ml dispase II, for 1 h at 37°C. The digested material was filtered through sterile 40 μm nylon mesh and washed in 2% (vol./vol.) FCS–M199. Subsequently, the cells were incubated with Dynabeads (Invitrogen) which were prepared as follows: beads coated with sheep anti-rat IgG were incubated with purified rat anti-mouse CD31 monoclonal antibody (1 μg/ml) at 4°C overnight and then washed with PBS containing 0.1% (wt/vol.) BSA and 2 mmol/l EDTA. The cell suspension was incubated with beads for 1 h at 4°C and then beads attached to endothelial cells were captured by the Dynal magnet (Invitrogen).
Quantification of mitochondrial morphology in living cells
Isolated MCECs were cultured in M199 (containing 5 mmol/l glucose) supplemented with 20% (vol./vol.) FCS, 100 μg/ml ECGS, 100 U/ml penicillin, 100 μg/ml streptomycin, 50 mg/l d-valine and 16 U/ml heparin. Cells were plated on glass chamber slides coated with 1% (wt/vol.) gelatine. After 3 days of recovery from isolation, mitochondrial morphology was assessed. Cells were stained with 100 nmol/l Mito Tracker Green FM for 30 min to visualise mitochondrial morphology and then washed three times with media. Images were captured with a DeltaVision deconvolution microscope system (Applied Precision, Issaquah, WA, USA) located at the University of California, San Diego Cancer Center microscope facility. Using a ×60 (numerical aperture 1.4) lens, images of ∼50 serial optical sections, spaced by 0.15 μm, were acquired. The data sets were deconvolved and optical sections merged to produce three-dimensional pictures using SoftWorx software (Applied Precision) on a Silicon Graphics Octane workstation [36] and the volume of individual mitochondria was measured by SoftWRx 2.50 software (www.api.com/pdfs/lifescience/AppNote-3Dmodeling.pdf). The morphological changes were assessed with mitochondrial volume (μm3) as a variable. The volume of individual mitochondria (total mitochondria number was 500–2,000 per cell) was measured and the average individual mitochondrial volume per cell was calculated. The average individual mitochondrial volume from each cell was used to calculate the mean and SEM (6–15 cells per experiment).
Cytosolic and mitochondrial O2− measurement in ECs
O2 − was detected by using the fluorescent probe DHE (for cytosol O2 − detection) or MitoSOX Red (for mitochondrial O2 − detection). Cells were preloaded with 50 μmol/l DHE or 5 μmol/l MitoSOX Red for 30 min before capturing images. Cytosolic DHE exhibits blue fluorescence, but once it is oxidised by O2 − to ethidium bromide (EB), it intercalates within the cell’s DNA, staining its nucleus a fluorescent red. The index of cytosolic O2 − concentration is described as a ratio of EB and DHE. MitoSOX Red is cell permeant and is rapidly and selectively targeted to the mitochondria. Once in the mitochondria, MitoSOX Red is oxidised by O2 − and exhibits red fluorescence. A value of red fluorescence intensity in the mitochondria was measured and mitochondrial O2 − concentration is described as a normalised value by fluorescence intensity in control cells.
Western blot analysis
After isolation of MCECs, cells were lysed and centrifuged at 16,000 g for 10 min at 4°C. Supernatant fractions were used as protein samples. Samples were separated on SDS-polyacrylamide gels and transferred to nitrocellulose membranes. Blots were then incubated with a primary antibody (against: OPA1, MFN1, MFN2, FIS1 or DRP1 [1:500]; or actin [1:4,000]) followed by incubation with a horseradish-peroxidase-conjugated secondary antibody. The immunoblots were detected with Western Lightning ECL Detection Reagent (Perkin Elmer LAS, Norton, OH, USA). Band intensity was normalised to actin controls and expressed in arbitrary units.
Determination of protein oxidation in the hearts
Cell extracts were freshly prepared from whole hearts by homogenisation in 500 μl lysate buffer. Protein carbonyl content was measured according to the manufacturer’s protocol (OxiBlot Protein Oxidation Detection Kit, Chemicon International, Temecula, CA, USA) [37]. Briefly, protein samples (15 μg/lane) were derivatised with dinitrophenyl hydrazine, fractionated by 12% SDS-PAGE and transferred to nitrocellulose membranes. The derivatised proteins were sequentially reacted with rabbit anti-dinitrophenyl antibody and horseradish peroxidase-conjugated goat anti-rabbit IgG and were visualised by chemiluminescence.
Measurement of plasma 8-iso-PGF2α level
To assess oxidant stress in the plasma, 8-iso-PGF2α level in the plasma was measured using the Isoprostane Oxidative Stress Assay Kit B (Enzo Life Sciences International, Plymouth Meeting, PA, USA). The kit uses a polyclonal antibody to 8-iso-PGF2α to bind, in a competitive manner, the 8-iso-PGF2α in the sample or an alkaline phosphatase molecule which has 8-iso-PGF2α covalently attached to it. Plasma samples were collected and stored at −20°C before use. To hydrolyse the ester bond, 25 μl of 10 mol/l NaOH was added to 100 μl of plasma. The samples were heated at 45°C for 2 h, and 25 μl of concentrated HCl (12.1 mol/l) was added to neutralise the samples. The samples were then centrifuged for 5 min at 20,817 g and the clear supernatant fraction was used in the assay. After a simultaneous incubation at room temperature, the excess reagents were washed away and substrate was added. After a short incubation time the enzyme reaction was stopped and the yellow colour generated was read on a microplate reader at 405 nm. The intensity of the bound yellow colour is inversely proportional to the concentration of 8-iso-PGF2α in either standards or samples. The measured optical density was used to calculate the concentration of 8-iso-PGF2α.
Ex vivo high glucose treatment in ECs
To test the effect of high glucose ex vivo, we used human coronary artery endothelial cells (HCECs) purchased from Cell Applications (San Diego, CA, USA). HCECs were used only for the experiments with results shown in Figs 6, 7 and 8, but other experiments were carried out with freshly isolated MCECs. For high-glucose treatment (HG), 20 mmol/l glucose was added to the media (the final glucose concentration was 25 mmol/l). In a control group of cells, equimolar mannitol was added to exclude the potential effect of changes in osmolarity (normal glucose [NG]: glucose concentration, 5 mmol/l). After 24 h, ECs were used for the measurement of O2 − production, mitochondrial fission, and OPA1 protein production. In another group of cells, TEMPOL (10 μmol/l) was added to the media 1 h before application of high glucose.
Fig. 6.

High-glucose treatment over 24 h increases mitochondrial fragmentation and ROS generation in cytosol and mitochondria, whereas scavenging O2 − significantly increases mitochondrial tubular formation without changing OPA1 and DRP1 protein levels. High glucose (25 mmol/l) or normal glucose (5 mmol/l glucose + 20 mmol/l mannitol) was added in the culture media for 24 h. TEMPOL was added 1 h before high glucose application. a Representative images showing mitochondrial morphology in ECs cultured in NG, HG and HG+TEMPOL. Bar = 5 μm. b Summarised data for mitochondrial volume (μm3) in ECs cultured in NG (n = 11 cells), HG (n = 10 cells) and HG+TEMPOL (HG+T, n = 10 cells). Data are mean ± SEM. *p < 0.05 vs NG. † p < 0.05 vs HG. c Western blot showing OPA1, DRP1 and actin protein levels in ECs cultured in NG, HG and HG+T for 24 h. d, e Summarised data for (d) OPA1 and (e) DRP1 protein levels (normalised by actin level) in ECs cultured in NG, HG and HG + T. Data are mean ± SEM. For each group, n = 2 samples. f, h Representative images showing (f) cytosolic O2 − production and (h) mitochondrial O2 − production in ECs cultured in NG and HG. g, i Summarised data showing the arbitrary unit of (g) cytosolic O2 − (ratio of EB/DHE; NG = 6 cells, HG = 8 cells) and (i) mitochondrial O2 − (MitoSOX intensity normalised by NG; NG n = 12 cells, HG n = 8 cells). Data are mean ± SEM. *p < 0.05 vs NG
Fig. 7.

O2 − regulates mitochondrial morphology. Cells were incubated with compounds that generate O2 − in cytosol (10 μmol/l DETA), or in mitochondria (100 μmol/l menadione) for 1 h at room temperature and then stained with Mito Tracker Green to visualise mitochondrial morphology. a Representative images showing mitochondrial morphology in HCECs with or without DETA. Bar = 5 μm. b Summarised data for mitochondrial volume (μm3) in ECs. Data are mean ± SEM, n = 6 cells in each group. * p < 0.05 vs without DETA (−DETA). c Western blot showing OPA1, DRP1 and actin protein levels in HCECs with or without DETA. d Representative images showing mitochondrial morphology in HCECs with or without menadione. Scale bar, 5 μm. e Summarised data of mitochondrial volume (μm3) in ECs without menadione (−mena, n = 12 cells) and with menadione (+mena, n=6 cells). Data are mean ± SEM. *p < 0.05 vs without menadione. Excess O2 − generation in cytosol or mitochondria significantly increased mitochondrial fragmentation without changing OPA1 and DRP1 protein levels
Fig. 8.

Downregulation of DRP1 protein level decreases high-glucose-treatment-induced mitochondrial fragmentation in HCECs. Drp1–shRNA (shDrp1) or control–shRNA (shCont) plasmids (1 μg) were transfected into HCECs by electroporation. Two days after the transfection, cells were treated with high glucose (25 mmol/l) or normal glucose (5 mmol/l glucose + 20 mmol/l mannitol) for 24 h. a Representative images showing mitochondrial morphology in ECs with control–shRNA in NG (NG w/shCont), with control–shRNA in HG (HG w/shCont), and with Drp1–shRNA in HG (HG w/shDrp1). Scale bar, 5 μm. b Summarised data for mitochondrial volume (μm3). Data are mean ± SEM, n = 8 cells in each group. *p < 0.05 vs NG w/shCont. c Western blot showing DRP1 and actin protein levels on day 3 after Drp1 or control shRNA transfection. Drp1–shRNA transfection successfully decreased DRP1 protein concentration
Drp1–small hairpin (sh)RNA transfection in HCEC
Downregulation of DRP1 in HCECs was achieved using the Drp1 (also known as Dmn1l)–shRNA plasmid (Santa Cruz Biotechnology) and the Amaxa Nucleofector technology (Amaxa, Gaithersburg, MD, USA) [38]. Specific protein knockdown was verified with western blotting. A high level of glucose was added to the media on day 2 after the transfection.
Statistical analysis
Values are expressed as mean ± SEM. In the case of mitochondrial volume, the volume of an individual mitochondrion (total mitochondria number was 500–2,000 per cell) was measured and the average individual mitochondrial volume per cell was calculated. The average individual mitochondrial volume from each cell was used to calculate the mean and SEM (6–15 cells per experiment). Bonferroni tests for multiple statistical comparisons and Student’s t test for unpaired samples were carried out to identify significant differences. Differences were considered to be statistically significant when p < 0.05.
Results
Increased mitochondrial fragmentation in coronary ECs isolated from diabetic mice
Previous studies in human umbilical vein endothelial cells indicate that short-time high-glucose treatment (15 min) induces mitochondrial fragmentation [39]. To test the effect of hyperglycaemia in vivo, mitochondrial morphology was compared in primary cultured MCECs isolated from control and diabetic mice. As shown in Fig. 1, there are elongated mitochondrial tubules in ECs isolated from control mice, whereas ECs from diabetic mice exhibit augmented mitochondrial fragmentation.
Fig. 1.

Mouse coronary endothelial cells isolated from diabetic mice exhibit enhanced mitochondrial fragmentation. a Representative images showing mitochondrial morphology in primary cultured MCECs isolated from control and diabetic mice. There are elongated mitochondrial tubules in MCECs isolated from control mice and MCECs from diabetic mice exhibit augmented mitochondrial fragmentation. Scale bar, 5 μm. b Summarised data for individual mitochondrial volume (μm3). For each group, n = 10 cells. Data are mean ± SEM. *p < 0.05 vs control
Levels of mitochondrial fission- and fusion-related proteins
Three mitochondrial fusion-related proteins, OPA1, MFN1 and MFN2, and two mitochondrial fission-related proteins, FIS1 and DRP1, were tested in MCECs (Fig. 2). OPA1 protein level was significantly decreased in MCECs freshly isolated from diabetic mice compared with control mice (control, 1.00 ± 0.10; diabetic, 0.52 ± 0.03; p < 0.05, n = 4 samples for each group). There was no difference in protein levels of MNF2 between control and diabetic mice (control, 1.00 ± 0.09; diabetic, 0.88 ± 0.06; p = 0.38, n = 2 samples for each group), while MFN1 exhibited a non-significant trend to increase in diabetic mice (control, 1.00 ± 0.07; diabetic, 1.54 ± 0.48; p = 0.38, n = 2 samples for each group). FIS1 protein level was unchanged in diabetic MCECs (control, 1.00 ± 0.28; diabetic, 0.99 ± 0.18; p = 0.99, n = 2 samples for each group), whereas DRP1 protein level was significantly higher in MCECs from diabetic than from control mice (control, 1.03 ± 0.29; diabetic, 2.40 ± 0.21; p < 0.05, n = 4 samples for each group). These data suggest that of these fusion- and fission-related proteins, OPA1 and DRP1 might play the most influential role related to the mitochondrial morphological change seen in MCECs from diabetic mice.
Fig. 2.

Change in protein levels related to mitochondrial fission or fusion in diabetic MCECs. Western blots showing OPA1, MFN1 and MFN2 (mitochondrial fusion-related proteins), and FIS1 and DRP1 (mitochondrial fission-related proteins). Actin was used as a loading control. OPA1 is significantly decreased in MCECs isolated from diabetic mice compared with MCECs from control mice (control, 1.00 ± 0.10: diabetic, 0.52 ± 0.03; p < 0.05, n = 4 samples for each group), whereas DRP1 is significantly increased in MCECs from diabetic mice (control, 1.03 ± 0.29; diabetic, 2.40 ± 0.21; p < 0.05, n = 4 samples for each group)
Increased cytosolic and mitochondrial O2− production in MCECs in diabetic mice
It has been shown that O2 − production is augmented in aortic ECs in diabetes [33–35]. We tested whether MCECs from diabetic mice also have increased O2 − production. As shown in Fig. 3a, b, cytosolic O2 − concentration was significantly higher in MCECs isolated from diabetic mice than in MCECs from control mice. In addition, mitochondrial O2 − production in MCECs was significantly increased in diabetic mice compared with control mice (Fig. 3c, d).
Fig. 3.

MCECs from diabetic mice exhibit increased cytosolic and mitochondrial O2 − production. a, c Representative images showing (a) cytosolic O2 − production and (c) mitochondrial O2 − production in primary cultured MCECs from control (C) and diabetic (D) mice. b, d Arbitrary units of (b) cytosolic O2 − (ratio of EB/DHE; control n = 15 cells, diabetic n = 15 cells) and (d) mitochondrial O2 − (MitoSOX intensity normalised by control; control n = 10 cells, diabetic n = 6 cells). Data are mean ± SEM. *p < 0.05 vs control
Augmented oxidative stress was decreased by chronic O2− scavenger administration in diabetic mice
The O2 − scavenger TEMPOL was administrated to diabetic mice for 4 weeks in drinking water. As shown in Fig. 4a, several proteins in the heart were oxidised. TEMPOL treatment markedly decreased the oxidised level of these proteins (Fig. 4b). The oxidative stress marker 8-iso-PGF2α was also increased in the plasma of diabetic mice and TEMPOL treatment significantly decreased the level of 8-iso-PGF2α (Fig. 4c). These data suggest that 4 week TEMPOL treatment beneficially decreases the oxidative stress in diabetes and may subsequently decrease the damage induced by augmented oxidative stress systemically.
Fig. 4.

Diabetes increases oxidative stress and administration of the O2
− scavenger, TEMPOL (T), restores the level of oxidative stress. TEMPOL was added in drinking water (1 mmol/l) for 4 weeks. a Representative image showing the pattern of oxidised proteins. Whole heart was used for this experiment to avoid unwelcome oxidation during EC isolation. Arrows indicate proteins where oxidative level was increased by diabetes and decreased by chronic TEMPOL treatment. C, control; D, diabetic; D+T, diabetic + TEMPOL. b Columns show the summarised data of the intensity of band 1. Actin was used as a loading control. Data are mean ± SEM. *p < 0.05 vs control; †
p < 0.05 vs diabetic. For each group, n = 2 samples. c Plasma levels of 8-iso-PGF2α, an oxidative stress marker, were increased in diabetic mice. TEMPOL treatment significantly decreased 8-iso-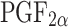 levels. Data are mean ± SEM. †
p < 0.05 vs diabetic. For each group, n = 7 samples
levels. Data are mean ± SEM. †
p < 0.05 vs diabetic. For each group, n = 7 samples
Administration of TEMPOL for 4 weeks restored the mitochondrial morphological changes in MCECs from diabetic mice
To examine whether increased O2 − production in MCECs regulates mitochondrial fragmentation in diabetes, TEMPOL was administrated to diabetic mice and then MCECs were isolated from control, diabetic and TEMPOL-treated diabetic mice. TEMPOL administration significantly decreased mitochondrial fragmentation in diabetic MCECs (Fig. 5a, b), but the levels of OPA1 and DRP1 proteins were not changed by TEMPOL administration (Fig. 5c–e). In addition, levels of other fusion–fission-related proteins were not affected by TEMPOL treatment (Electronic Supplementary Material [ESM] Fig. 1). This implies that restoration of mitochondrial morphology might be a direct effect of the decrease in O2 − concentration by TEMPOL administration, but one that is not mediated by the change in fusion–fission-related protein level.
Fig. 5.

Administration of the O2 − scavenger (TEMPOL, T) for 4 weeks restores mitochondrial morphological changes in diabetic MCECs without altering OPA1 and DRP1 protein levels, as detected by western blot. a Representative images showing mitochondrial morphology in primary cultured MCECs from control, diabetic and TEMPOL-treated diabetic mice. Bar = 5 μm. b Summarised data for mitochondrial volume (μm3) in MCECs from control (C, n = 15 cells), diabetic (D, n = 15 cells) and diabetic + TEMPOL (D+T, n = 11 cells). *p < 0.05 vs control, † p < 0.05 vs diabetic. c Western blot showing OPA1, DRP1 and actin in MCECs from control, diabetic and diabetic+TEMPOL mice. d, e Summarised data showing protein levels of (d) OPA1 and (e) DRP1 (normalised by actin level) in MCECs isolated from hearts. Data are mean ± SEM from three samples in each group. *p < 0.05 vs control. TEMPOL treatment significantly increased mitochondrial volume, but the decrease in OPA1 protein and the increase in DRP1 protein in diabetic MCECs are not restored by TEMPOL treatment
Mitochondrial fragmentation induced by 24 h high-glucose treatment was restored by TEMPOL treatment in HCECs
HCECs were exposed to NG (5 mmol/l glucose + 20 mmol/l mannitol), HG (25 mmol/l glucose) or TEMPOL+HG for 24 h and mitochondrial morphological changes were determined. Figure 6a, b demonstrates that TEMPOL treatment significantly increased the mitochondrial volume that was decreased by high-glucose treatment. In contrast to the result of hyperglycaemia in vivo, the effect of 24 h high-glucose levels on mitochondrial fragmentation ex vivo was not related to a change in OPA1 protein level. On the other hand, DRP1 was increased by high glucose and TEMPOL did not have an effect on DRP1 protein level ex vivo in line with the in vivo data (Fig. 6c–e). Other fusion–fission-related protein levels are shown in ESM Fig. 2. These data imply that increased DRP1 may contribute to excess mitochondrial fragmentation independent of increased O2 − production under hyperglycaemic conditions.
High-glucose treatment over 24 h significantly increased cytosolic and mitochondrial O2 − concentrations (Fig. 6f–i). To test whether excess O2 − production has a direct effect on mitochondrial fragmentation, we used two types of O2 − generator: one for cytosol (10 μmol/l DETA) and another for mitochondria (100 μmol/l menadione). Cells were incubated with O2 − generators for 1 h at room temperature and then stained with Mito Tracker Green to visualise mitochondrial morphology. As shown in Fig. 7, overproduction of O2 − in either cytosol or mitochondria caused significant mitochondrial fragmentation without changing OPA1 and DRP1 protein levels, suggesting that hyperglycaemia ex vivo might lead to mitochondrial fragmentation by the increase in O2 − production and DRP1 protein upregulation, but that they might then contribute to mitochondrial fragmentation separately. Levels of other fusion–fission-related proteins are shown in ESM Fig. 3.
Drp1–shRNA transfection induced mitochondrial elongation under high-glucose treatment
To test the effect of DRP1 downregulation on high-glucose-induced mitochondrial fission, Drp1–shRNA (1 μg) was transfected in HCECs by electroporation. Two days after transfection, cells were treated with high glucose or mannitol for 24 h. As shown in Fig. 8, decreased level of DRP1 protein led to increased mitochondrial elongation under high-glucose treatment, suggesting that high glucose causes, at least in part, mitochondrial fragmentation by increasing the DRP1 protein level.
Discussion
For the first time, a quantification of mitochondrial morphological changes in living cells, comparing control and diabetic cells, was performed. Mitochondria in MCECs isolated from control mice appear as elongated tubular structures with many branches. In contrast, MCECs from diabetic mice have more fragmented mitochondria with significantly smaller volume than mitochondria in control MCECs (Fig. 1). The physiological significance of the continual fusion and fission of mitochondria is still under debate. A possible function for fusion could be a rescue mechanism for damaged mitochondria involving exchange of mtDNA and/or mitochondrial protein [11]. The predominant gene responsible for autosomal-dominant optic atrophy has been identified as OPA1 [19, 20] and those patients exhibit significantly lower copy number of mitochondrial DNA molecules, which may result from decreased mitochondrial fusion [21]. A recent report demonstrated that Fis1 transfection in clone 9 cells led to increased cell apoptosis. A targeted null mutation of either Mfn1 or Mfn2 results in mid-gestational lethality [23]. Another study shows a naturally occurring neuropathy is associated with Mfn2 mutation [24]. In addition, MFN2 was identified as a suppressor of obesity [27, 28] and hypertension [25]. Taken together, these data suggest that increased mitochondrial fission disrupts the normal function of different kinds of cells and may cause organ failure and systemic disease. Figure 2 demonstrates that the fusion-related protein OPA1 level is significantly decreased and fission-related protein DRP1 is significantly increased in MCECs from diabetic mice compared with control mice, but other fusion- or fission-related protein levels do not change (MFN2 and FIS1) or change in a way which is contrary to our expectations (MFN1). The decrease in OPA1 and the increase in DRP1 protein level might be, at least in part, one of the causes of increased mitochondrial fragmentation in diabetic MCECs. As the increase in MFN1 protein is supposed to increase mitochondrial fusion, we hypothesise that in this cell type MFN1 is not a major player in the determination of mitochondrial morphology and further study is required to test this hypothesis.
It has been documented that hyperglycaemia induces excess ROS production, including O2 −, in many cell types [31–35, 40–43] and local ROS formation is considered to be a major contributing factor to endothelial dysfunction, including endothelial cell apoptosis [42–44], abnormalities in cell cycling [44] and delayed replication [41]. Our data demonstrate that MCECs isolated from diabetic mice exhibit significantly higher production of cytosolic and mitochondrial O2 − (Fig. 3) and, interestingly, that chronic reduction of O2 − by TEMPOL administration increased mitochondrial volume to a level even above that of control MCECs (Fig. 5a,b) without changing OPA1 and DRP1 protein levels (Fig. 5c–e). This suggests that O2 − concentration might serve as a direct regulator of mitochondrial morphology. Our ex vivo data also support this hypothesis. As seen in Fig. 6, HCECs exposed to high glucose for 24 h show significantly decreased mitochondrial volume, which is reversed by treatment with the O2 − scavenger TEMPOL. This high-glucose-treatment-induced mitochondrial fragmentation was also decreased by Drp1–shRNA transfection (Fig. 8), implying that these morphological changes by 24 h-high-glucose exposure are not mediated by the fusion-related protein OPA1, but by DRP1 protein upregulation and/or by excess O2 − production in the cytosol and mitochondria. In addition, exogenous O2 − overproduction by DETA or menadione directly changed the mitochondrial morphology and increased fragmentation (Fig. 7). These data suggest that increased O2 − in ECs may potentially cause mitochondrial fragmentation in the diabetic heart.
There is increasing evidence to show that ROS can directly initiate mitochondrial fragmentation in different cell types [45–48]; the detailed mechanisms are, however, still unclear. It is known that O2 − can activate protein kinase C (PKC) in ECs [49, 50], although PKC activation can also stimulate O2 − generation. It is thus possible that morphological dynamics in mitochondria could be controlled by PKC activation initiated by O2 −. Our data show that treatment with 100 nmol/l phorbol 12-myristate 13-acetate (PMA, a PKC activator) significantly decreased mitochondrial volume (vehicle 0.01% (vol./vol.) DMSO, 0.24 ± 0.01 μm3; PMA, 0.15 ± 0.02 μm3; p < 0.05, n = 5 each group). Although these data imply that PKC directly regulates mitochondrial morphology, it doesn’t define the relationship between PKC and O2 −. It would be necessary to use a PKC inhibitor to identify whether O2 −-mediated mitochondrial fission is controlled by PKC activation in diabetic MCECs.
These data suggest that O2 − overproduction, a decrease in the fusion-related protein OPA1 and an increase in the fission-related protein DRP1 in MCECs in diabetic mice lead to mitochondrial fragmentation, and that treatment with an O2 − scavenger may help improve mitochondrial function by decreasing mitochondrial fragmentation.
Electronic supplementary material
Below is the link to the electronic supplementary material.
(PDF 157 kb)
(PDF 140 kb)
(PDF 71 kb)
Acknowledgements
This work was supported by the grants of HL66917 and MD00220 (W. H. Dillmann) and DK083506 (A. Makino) from the National Institutes of Health.
Duality of interest
The authors declare that there is no duality of interest associated with this study.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Abbreviations
- DETA
Diethyldithiocarbamic acid
- DHE
Dihydroethidium
- DRP1
Dynamin-related protein 1
- EB
Ethidium bromide
- EC
Endothelial cell
- ECGS
Endothelial cell growth supplement
- FIS1
Fission 1
- HCECs
Human coronary endothelial cells
- HG
High glucose
- MCECs
Mouse coronary endothelial cells
- MFN
Mitofusin
- mtDNA
Mitochondrial DNA
- NG
Normal glucose
- O2−
Superoxide anion
- OPA1
Optic atrophy 1
- PKC
Protein kinase C
- PMA
Phorbol 12-myristate 13-acetate
- ROS
Reactive oxygen species
- shRNA
Small hairpin RNA
- TEMPOL
4-Hydroxy-2,2,6,6-tetramethylpiperidine-1-oxyl
References
- 1.Makino A, Platoshyn O, Suarez J, Yuan JX, Dillmann WH. Downregulation of connexin40 is associated with coronary endothelial cell dysfunction in streptozotocin-induced diabetic mice. Am J Physiol Cell Physiol. 2008;295:C221–C230. doi: 10.1152/ajpcell.00433.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Camp TM, Tyagi SC, Senior RM, Hayden MR. Gelatinase B(MMP-9) an apoptotic factor in diabetic transgenic mice. Diabetologia. 2003;46:1438–1445. doi: 10.1007/s00125-003-1200-y. [DOI] [PubMed] [Google Scholar]
- 3.Yoon YS, Uchida S, Masuo O, et al. Progressive attenuation of myocardial vascular endothelial growth factor expression is a seminal event in diabetic cardiomyopathy: restoration of microvascular homeostasis and recovery of cardiac function in diabetic cardiomyopathy after replenishment of local vascular endothelial growth factor. Circulation. 2005;111:2073–2085. doi: 10.1161/01.CIR.0000162472.52990.36. [DOI] [PubMed] [Google Scholar]
- 4.Bereiter-Hahn J, Voth M. Dynamics of mitochondria in living cells: shape changes, dislocations, fusion, and fission of mitochondria. Microsc Res Tech. 1994;27:198–219. doi: 10.1002/jemt.1070270303. [DOI] [PubMed] [Google Scholar]
- 5.Scott SV, Cassidy-Stone A, Meeusen SL, Nunnari J. Staying in aerobic shape: how the structural integrity of mitochondria and mitochondrial DNA is maintained. Curr Opin Cell Biol. 2003;15:482–488. doi: 10.1016/S0955-0674(03)00070-X. [DOI] [PubMed] [Google Scholar]
- 6.Heath-Engel HM, Shore GC. Mitochondrial membrane dynamics, cristae remodelling and apoptosis. Biochim Biophys Acta. 2006;1763:549–560. doi: 10.1016/j.bbamcr.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 7.Chan DC. Mitochondrial fusion and fission in mammals. Annu Rev Cell Dev Biol. 2006;22:79–99. doi: 10.1146/annurev.cellbio.22.010305.104638. [DOI] [PubMed] [Google Scholar]
- 8.Chen H, Chan DC. Emerging functions of mammalian mitochondrial fusion and fission. Hum Mol Genet. 2005;14:R283–R289. doi: 10.1093/hmg/ddi270. [DOI] [PubMed] [Google Scholar]
- 9.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakada K, Inoue K, Ono T, et al. Inter-mitochondrial complementation: mitochondria-specific system preventing mice from expression of disease phenotypes by mutant mtDNA. Nat Med. 2001;7:934–940. doi: 10.1038/90976. [DOI] [PubMed] [Google Scholar]
- 11.Ono T, Isobe K, Nakada K, Hayashi JI. Human cells are protected from mitochondrial dysfunction by complementation of DNA products in fused mitochondria. Nat Genet. 2001;28:272–275. doi: 10.1038/90116. [DOI] [PubMed] [Google Scholar]
- 12.Frazier AE, Kiu C, Stojanovski D, Hoogenraad NJ, Ryan MT. Mitochondrial morphology and distribution in mammalian cells. Biol Chem. 2006;387:1551–1558. doi: 10.1515/BC.2006.193. [DOI] [PubMed] [Google Scholar]
- 13.Rojo M, Legros F, Chateau D, Lombes A. Membrane topology and mitochondrial targeting of mitofusins, ubiquitous mammalian homologs of the transmembrane GTPase Fzo. J Cell Sci. 2002;115:1663–1674. doi: 10.1242/jcs.115.8.1663. [DOI] [PubMed] [Google Scholar]
- 14.Olichon A, Emorine LJ, Descoins E, et al. The human dynamin-related protein OPA1 is anchored to the mitochondrial inner membrane facing the inter-membrane space. FEBS Lett. 2002;523:171–176. doi: 10.1016/S0014-5793(02)02985-X. [DOI] [PubMed] [Google Scholar]
- 15.Cipolat S, Martins de Brito O, Dal Zilio B, Scorrano L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci USA. 2004;101:15927–15932. doi: 10.1073/pnas.0407043101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smirnova E, Griparic L, Shurland DL, van der Bliek AM. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell. 2001;12:2245–2256. doi: 10.1091/mbc.12.8.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mozdy AD, McCaffery JM, Shaw JM. Dnm1p GTPase-mediated mitochondrial fission is a multi-step process requiring the novel integral membrane component Fis1p. J Cell Biol. 2000;151:367–380. doi: 10.1083/jcb.151.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yoon Y, Krueger EW, Oswald BJ, McNiven MA. The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol Cell Biol. 2003;23:5409–5420. doi: 10.1128/MCB.23.15.5409-5420.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alexander C, Votruba M, Pesch UE, et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet. 2000;26:211–215. doi: 10.1038/79944. [DOI] [PubMed] [Google Scholar]
- 20.Delettre C, Lenaers G, Griffoin JM, et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet. 2000;26:207–210. doi: 10.1038/79936. [DOI] [PubMed] [Google Scholar]
- 21.Kim JY, Hwang JM, Ko H, Seong MW, Park BJ, Park SS. Mitochondrial DNA content is decreased in autosomal dominant optic atrophy. Neurology. 2005;64:966–972. doi: 10.1212/01.WNL.0000163768.58168.3A. [DOI] [PubMed] [Google Scholar]
- 22.Lodi R, Tonon C, Valentino ML, et al. Deficit of in vivo mitochondrial ATP production in OPA1-related dominant optic atrophy. Ann Neurol. 2004;56:719–723. doi: 10.1002/ana.20278. [DOI] [PubMed] [Google Scholar]
- 23.Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003;160:189–200. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zuchner S, Mersiyanova IV, Muglia M, et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot–Marie–Tooth neuropathy type 2A. Nat Genet. 2004;36:449–451. doi: 10.1038/ng1341. [DOI] [PubMed] [Google Scholar]
- 25.Chen KH, Guo X, Ma D, et al. Dysregulation of HSG triggers vascular proliferative disorders. Nat Cell Biol. 2004;6:872–883. doi: 10.1038/ncb1161. [DOI] [PubMed] [Google Scholar]
- 26.Leinninger GM, Backus C, Sastry AM, Yi YB, Wang CW, Feldman EL. Mitochondria in DRG neurons undergo hyperglycemic mediated injury through Bim, Bax and the fission protein Drp1. Neurobiol Dis. 2006;23:11–22. doi: 10.1016/j.nbd.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 27.Bach D, Naon D, Pich S, et al. Expression of Mfn2, the Charcot–Marie–Tooth neuropathy type 2A gene, in human skeletal muscle: effects of type 2 diabetes, obesity, weight loss, and the regulatory role of tumor necrosis factor alpha and interleukin-6. Diabetes. 2005;54:2685–2693. doi: 10.2337/diabetes.54.9.2685. [DOI] [PubMed] [Google Scholar]
- 28.Bach D, Pich S, Soriano FX, et al. Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism. A novel regulatory mechanism altered in obesity. J Biol Chem. 2003;278:17190–17197. doi: 10.1074/jbc.M212754200. [DOI] [PubMed] [Google Scholar]
- 29.Wei YH, Lee HC. Oxidative stress, mitochondrial DNA mutation, and impairment of antioxidant enzymes in aging. Exp Biol Med (Maywood) 2002;227:671–682. doi: 10.1177/153537020222700901. [DOI] [PubMed] [Google Scholar]
- 30.Giugliano D, Ceriello A, Paolisso G. Oxidative stress and diabetic vascular complications. Diab Care. 1996;19:257–267. doi: 10.2337/diacare.19.3.257. [DOI] [PubMed] [Google Scholar]
- 31.Hink U, Li H, Mollnau H, et al. Mechanisms underlying endothelial dysfunction in diabetes mellitus. Circ Res. 2001;88:E14–E22. doi: 10.1161/01.res.88.2.e14. [DOI] [PubMed] [Google Scholar]
- 32.Mukhopadhyay P, Rajesh M, Yoshihiro K, Hasko G, Pacher P. Simple quantitative detection of mitochondrial superoxide production in live cells. Biochem Biophys Res Commun. 2007;358:203–208. doi: 10.1016/j.bbrc.2007.04.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Turkseven S, Kruger A, Mingone CJ, et al. Antioxidant mechanism of heme oxygenase-1 involves an increase in superoxide dismutase and catalase in experimental diabetes. Am J Physiol Heart Circ Physiol. 2005;289:H701–H707. doi: 10.1152/ajpheart.00024.2005. [DOI] [PubMed] [Google Scholar]
- 34.Wendt MC, Daiber A, Kleschyov AL, et al. Differential effects of diabetes on the expression of the gp91phox homologues nox1 and nox4. Free Radic Biol Med. 2005;39:381–391. doi: 10.1016/j.freeradbiomed.2005.03.020. [DOI] [PubMed] [Google Scholar]
- 35.Alp NJ, Mussa S, Khoo J, et al. Tetrahydrobiopterin-dependent preservation of nitric oxide-mediated endothelial function in diabetes by targeted transgenic GTP-cyclohydrolase I overexpression. J Clin Invest. 2003;112:725–735. doi: 10.1172/JCI17786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jhunjhunwala S, van Zelm MC, Peak MM, et al. The 3D structure of the immunoglobulin heavy-chain locus: implications for long-range genomic interactions. Cell. 2008;133:265–279. doi: 10.1016/j.cell.2008.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cooksey RC, Jouihan HA, Ajioka RS, et al. Oxidative stress, beta-cell apoptosis, and decreased insulin secretory capacity in mouse models of hemochromatosis. Endocrinology. 2004;145:5305–5312. doi: 10.1210/en.2004-0392. [DOI] [PubMed] [Google Scholar]
- 38.Csiszar A, Ahmad M, Smith KE, et al. Bone morphogenetic protein-2 induces proinflammatory endothelial phenotype. Am J Pathol. 2006;168:629–638. doi: 10.2353/ajpath.2006.050284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu T, Robotham JL, Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc Natl Acad Sci USA. 2006;103:2653–2658. doi: 10.1073/pnas.0511154103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baynes JW. Role of oxidative stress in development of complications in diabetes. Diabetes. 1991;40:405–412. doi: 10.2337/diabetes.40.4.405. [DOI] [PubMed] [Google Scholar]
- 41.Zou MH, Shi C, Cohen RA. High glucose via peroxynitrite causes tyrosine nitration and inactivation of prostacyclin synthase that is associated with thromboxane/prostaglandin H(2) receptor-mediated apoptosis and adhesion molecule expression in cultured human aortic endothelial cells. Diabetes. 2002;51:198–203. doi: 10.2337/diabetes.51.1.198. [DOI] [PubMed] [Google Scholar]
- 42.Piconi L, Quagliaro L, Assaloni R, et al. Constant and intermittent high glucose enhances endothelial cell apoptosis through mitochondrial superoxide overproduction. Diabetes/Metab Res Rev. 2006;22:198–203. doi: 10.1002/dmrr.613. [DOI] [PubMed] [Google Scholar]
- 43.Nishikawa T, Edelstein D, Du XL, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404:787–790. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- 44.Abraham NG, Kushida T, McClung J, et al. Heme oxygenase-1 attenuates glucose-mediated cell growth arrest and apoptosis in human microvessel endothelial cells. Circ Res. 2003;93:507–514. doi: 10.1161/01.RES.0000091828.36599.34. [DOI] [PubMed] [Google Scholar]
- 45.Jou MJ. Pathophysiological and pharmacological implications of mitochondria-targeted reactive oxygen species generation in astrocytes. Adv Drug Deliv Rev. 2008;60:1512–1526. doi: 10.1016/j.addr.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 46.Jendrach M, Mai S, Pohl S, Voth M, Bereiter-Hahn J. Short- and long-term alterations of mitochondrial morphology, dynamics and mtDNA after transient oxidative stress. Mitochondrion. 2008;8:293–304. doi: 10.1016/j.mito.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 47.Knott AB, Perkins G, Schwarzenbacher R, Bossy-Wetzel E. Mitochondrial fragmentation in neurodegeneration. Nat Rev Neurosci. 2008;9:505–518. doi: 10.1038/nrn2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pletjushkina OY, Lyamzaev KG, Popova EN, et al. Effect of oxidative stress on dynamics of mitochondrial reticulum. Biochim Biophys Acta. 2006;1757:518–524. doi: 10.1016/j.bbabio.2006.03.018. [DOI] [PubMed] [Google Scholar]
- 49.Ali MH, Mungai PT, Schumacker PT. Stretch-induced phosphorylation of focal adhesion kinase in endothelial cells: role of mitochondrial oxidants. Am J Physiol Lung Cell Mol Physiol. 2006;291:L38–L45. doi: 10.1152/ajplung.00287.2004. [DOI] [PubMed] [Google Scholar]
- 50.Ogata N, Yamamoto H, Kugiyama K, Yasue H, Miyamoto E. Involvement of protein kinase C in superoxide anion-induced activation of nuclear factor-kappa B in human endothelial cells. Cardiovasc Res. 2000;45:513–521. doi: 10.1016/S0008-6363(99)00364-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 157 kb)
(PDF 140 kb)
(PDF 71 kb)