

Abstract
Abnormalities of genomic methylation patterns are lethal or cause disease, but the cues that normally designate CpG dinucleotides for methylation are poorly understood. We have developed a new method of methylation profiling that has single-CpG resolution and can address the methylation status of repeated sequences. We have used this method to determine the methylation status of >275 million CpG sites in human and mouse DNA from breast and brain tissues. Methylation density at most sequences was found to increase linearly with CpG density and to fall sharply at very high CpG densities, but transposons remained densely methylated even at higher CpG densities. The presence of histone H2A.Z and histone H3 di- or trimethylated at lysine 4 correlated strongly with unmethylated DNA and occurred primarily at promoter regions. We conclude that methylation is the default state of most CpG dinucleotides in the mammalian genome and that a combination of local dinucleotide frequencies, the interaction of repeated sequences, and the presence or absence of histone variants or modifications shields a population of CpG sites (most of which are in and around promoters) from DNA methyltransferases that lack intrinsic sequence specificity.
The human genome contains ∼28 million CpG sites, about 60% of which are methylated at the 5 position of the cytosine. Methylation of relatively CpG-rich promoters causes strong transcriptional repression (Stein et al. 1982; Lorincz et al. 2002), and many experiments have demonstrated faithful inheritance of methylation patterns over many cell divisions in mammalian somatic cells (Wigler et al. 1981; Lorincz et al. 2002). This heritability means that genomic methylation patterns could have many biological functions, and many such functions have been proposed over the past 50 yr. However, much controversy as to the biological roles of genomic methylation patterns remains because of the lack of information about the genome-wide structure of methylation patterns. A further concern is the common use of cultured cells in methylation profiling studies; genomic methylation patterns are unstable in cultured cells, and promoters of tissue-specific genes that are methylated in cultured cells are usually unmethylated in both expressing and nonexpressing tissues (Jones et al. 1990).
Half of all CpG sites are contained in repetitive DNA (Rollins et al. 2006), but existing methods of methylation profiling are largely or completely unable to evaluate methylation at dispersed and tandem-repeated sequences. This is a severe shortcoming, as the methylation of such sequences can have strong effects on phenotype. Human ICF syndrome is caused by mutations in the DNMT3B gene that prevent methylation of specific classes of tandem-repeated sequences (Xu et al. 1999), while Fragile X syndrome is caused by de novo methylation provoked by expansion of a CGG repeat tract at the FMR1 locus (Sutcliffe et al. 1992). Transposon insertion alleles of mouse genes such as nonagouti (also known as agouti) (a) and Axin1 show highly variable penetrance and expressivity that are dependent on the methylation state of the transposon (Michaud et al. 1994; Rakyan et al. 2001).
We have developed a new method called Methyl-MAPS (methylation mapping analysis by paired-end sequencing) that can provide coverage of up to 82.4% of the CpG sites in the genome (Supplemental Fig. S1). This method probes methylation status at single-copy and repetitive elements, each of which represents ∼50% of the CpGs in the genome (Rollins et al. 2006). The method combines enzymatic fractionation of the genome into methylated and unmethylated compartments with deep sequencing to provide a comprehensive profile of genomic methylation patterns. A comparison of Methyl-MAPS to other techniques for methylation profiling shows that Methyl-MAPS provides high coverage of single-copy and repeated sequences at relatively low cost (Supplemental Table S3). We have applied Methyl-MAPS to determine the structure of genomic methylation patterns at both fine and gross levels and have found sequence contexts and specific chromatin marks that are tightly associated with methylation status.
Results and Discussion
The methylated compartment of the genome was isolated by digestion with five methylation-sensitive restriction endonucleases (RE), while the unmethylated compartment was isolated by limit digest with the methylation-dependent McrBC complex (Supplemental Fig. S1). Paired-end libraries were prepared, and 25 bases from both ends of each DNA molecule were determined by sequencing-by-ligation on Applied Biosystems SOLiD System DNA sequencers (see Methods). CpG methylation was then determined by analyzing which CpGs were resistant or sensitive to cleavage by McrBC or RE (Supplemental Fig. S1). The use of paired-end sequencing allows direct determination of the methylation status of interspersed repeated sequences, as in the majority of cases one or both end tags are anchored in unique sequence. A total of 15,154,064 unmethylated sequences and 21,393,168 methylated sequences from somatic DNA were mapped to unique locations in the genome (hg18, mm9), and mean coverage at sites cleavable by McrBC and RE was 7.1X (Supplemental Table S1). The methylation status of 152,693,954 CpG dinucleotides was determined in human breast DNA, 75,676,854 in human brain DNA, and 52,896,012 in mouse brain DNA, for a total of 281,266,820 CpG sites.
We validated the Methyl-MAPS results by comparison to bisulfite methylation analysis on the Illumina Infinium HumanMethylation 27 beadchip and found that Pearson's correlation coefficient for methylation data obtained via the two unrelated methods was 0.84 for breast 1 and 0.87 for breast 2. This is substantially greater than correlations obtained by pairwise comparison of other DNA methylation profiling methods (Irizarry et al. 2008). To further confirm that accurate methylation data were obtained by Methyl-MAPS, sequences known to be methylated on the female X chromosome and at imprinted loci were examined. Promoter-associated islands on female chr X were much more methylated than were promoter-associated islands on the male chr X, whereas islands on male and female autosomes were less methylated (Supplemental Fig. S2A). An analysis of all known differentially methylated regions (DMRs) at imprinted loci showed DMRs to be methylated at intermediate densities, as expected for sequences subject to allele-specific methylation (Supplemental Table S2; Supplemental Fig. S2B).
The methylation status of the BIK (BCL2 interactor and killer) gene in DNA from normal human breast tissue is shown in Figure 1A. The pattern of methylation of this gene is typical in that the CpG-rich promoter and flanking sequences are unmethylated, whereas the bulk of the gene is methylated. Methyl-MAPS can be used to directly measure the methylation of repetitive sequences, as shown in Figure 1B. The SVA retrotransposon in this repeat-rich genomic region is densely covered by methylated fragments, which is typical of both dispersed and repeated sequences in the mammalian genome.
Figure 1.

High-resolution genome-wide methylation profiling and genome-wide DNA methylation trends. (A) Browser view of Methyl-MAPS data from the genomic region spanning the BIK gene. Individual mapped sequence reads are shown in the top raw data tracks. Red sequences were resistant to methylation-sensitive restriction endonucleases (RE) and are therefore methylated. Blue sequences were resistant to the methylation-dependent McrBC complex and are unmethylated. Tick marks in both tracks along the top of the figure and within each sequence indicate locations of individual RE and McrBC recognition sequences. Methylation data is also presented in a concise view, where each CpG is assigned a methylation score from the ratio of methylated to total (unmethylated and methylated) sequences covering each CpG site. The bulk of the BIK gene is methylated, while the CpG-rich promoter is unmethylated. (B) Methylation of the SVA retrotransposon in a repeat-rich region of chr 19. While the CpG density is comparable to that of the CpG island of the BIK gene shown in A, the SVA retrotransposon is densely methylated.
Analysis of the observed methylation patterns revealed a significant relationship between CpG density and methylation density (Fig. 2). Previously, it has been established that regions of high CpG density in promoters are largely unmethylated (Weber et al. 2008); however, only promoter regions were studied, and the relationship between CpG density and methylation at nonpromoter sequences and in repeats has not been elucidated. In single-copy DNA, the fraction of methylated CpGs was found to increase with CpG density up to a density of 0.025 (one out of 40 nucleotides [nt] is a C in a CpG dinucleotide), where 70% of the CpG sequences were methylated (Fig. 2A,C). Over 90% of the CpGs found in single-copy sequence are found at these lower CpG densities with higher levels of methylation. At higher CpG densities, methylation density fell off sharply, and methylation of unique sequences was lowest at very CpG-rich promoters. A similar pattern was seen for repeated sequences (Fig. 2B); for these sequences, methylation increased up to a CpG density of 0.07 (one out of ∼15 nt is a C in a CpG dinucleotide), where 80% of CpGs were methylated; thus, these repeated sequences, which are largely composed of transposable elements, continue to be methylated at very high CpG densities. Methylation in repeated sequences was low only in CpG containing simple sequence repeats of 2–6 nt. These methylation patterns were very similar in human breast and brain DNA (Supplemental Fig. S3) and in mouse brain DNA (Supplemental Fig. S4), and indicate that these trends are fundamental features of the methylation program in mammalian somatic tissues. The unmethylated, CpG-dense compartment was found to be populated by two very different sequence types: single-copy promoter-associated CpG islands (Fig. 2C) and simple sequence repeats of 2–6 nt (Fig. 2B). The overall trend of increasing CpG methylation with increasing CpG density at low- to mid-CpG densities with very low levels of CpG methylation at high CpG densities was observed across all sequence classes (Fig. 3D; Supplemental Fig. S7).
Figure 2.

Relationship of CpG density and methylation for repeated and unique sequences. (A) CpG methylation is plotted as a function of CpG density for four distinct genomic compartments (single copy, retrotransposons, simple repeats, and other repeats). Approximately 50% of the CpGs in the genome are contained in both repeats (B) and unique sequences (C). Each curve is divided into four CpG density regions; the CpG composition of each is shown in the bar charts on the right. The large majority of CpGs are contained in region 1 in both plots (A, 96%; B, 81.9%). (B) The majority of low-CpG density CpGs are contained in SINE and LINE elements, while the highly unmethylated high-density CpGs are primarily found in simple repeats. (C) The majority of low-CpG density CpGs are contained in intergenic and intronic unique sequences, while the highly unmethylated high-density CpGs are primarily found in promoter-associated regions.
Figure 3.

Relationship between CpG methylation at Alu retrotransposons and proximity to methylated and unmethylated promoters. Alu methylation is plotted as a function of distance from the TSS (A) and to the 3′ splice site of the first exon (B) of methylated (red) and unmethylated (blue) first exons. When near unmethylated first exons, Alu elements are also unmethylated. Alu methylation correlates with first-exon methylation when Alus are within ∼1 kb of the TSS or 3′ edge of the first exon. (C) Negative selection of Alu elements near unmethylated promoters. The ratio of the fraction of methylated promoters with Alus near the first exon to the fraction of unmethylated promoters with Alus near the first exon is plotted as a function of the distance to the TSS. This suggests that unmethylated Alus near promoters are deleterious and are lost from the population by selection. (D) The methylation status of the three major classes of Alu retrotransposons. Note that AluY (the only active Alu in the human genome) remains heavily methylated, even at very high CpG densities.
Figure 4B shows DNA methylation and CpG distributions averaged across 16,181 RefSeq genes. As expected from the presence of CpG islands at most promoters, the density of CpG dinucleotides was very high in first exons, with the high CpG density extending well 5′ and 3′ of the first exon. These CpGs were undermethylated, with the density of unmethylated CpGs reaching a maximum at the transcription start site (TSS). CpG-poor promoter regions are partially methylated (Supplemental Figs. S5, S7), but the methylation density is likely to be too low to enforce transcriptional silencing (Kass et al. 1997; Weber et al. 2008). Figure 4C shows that methylation status across multiple sequence compartments is very similar between unrelated individuals.
Figure 4.

(A,B) CpG distributions and methylation patterns in human genes. m5CpG and CpG densities are shown in relation to enhancers (A), TSS, exon splice sites, stop codons, and poly(A) sites (B). Note spikes in CpG and m5CpG densities at the 5′ and 3′ ends of exons and internal to the stop codon in the last exon. (C) Comparison of methylation patterns in normal breast tissue from two individuals. Methylation status of each CpG with high coverage is computed for each sample. The frequency of such points is then plotted as a function of the methylation score for each sample. Heat map indicates frequency. Values in the left corner are unmethylated in both samples. Values in the right corner are methylated in both samples. Values along the horizontal are equivalently methylated in each sample. Some sequence classes have a wide range of methylation states, such as intronic and intergenic single-copy sequences and LINEs, LTRs, and DNA transposons. Other classes such as SINEs, exons, simple repeats, and promoters are polarized.
CpG islands near TSSs are unmethylated (Supplemental Fig. S6); however, only ∼40% of computationally annotated CpG islands are located near TSSs. Our analysis of length and methylation density showed that non-TSS islands were much more likely to be methylated and were much shorter than were TSS islands (Supplemental Fig. S6). Both tendencies were less pronounced for intergenic CpG islands, some of which may be associated with novel TSSs for genes that encode unknown transcripts.
Within coding regions of genes, we also found unanticipated patterns of methylation at the borders of exons. We observed an increase in the density of CpG sites at the 5′ and 3′ ends of internal exons (Majewski and Ott 2002), and these CpG sites were relatively highly methylated. As can be seen in Figure 4C, the sequence compartments in which the fraction of unmethylated CpG sites is lowest are SINE (largely Alu) transposons and internal exons of cellular genes. The presence of densely methylated coding exons was surprising, as 5-methylcytosine (m5C) is a premutagenic modified base that leads to C → T mutations at a rate 18-fold higher than the average of all other point mutations (Kondrashov 2003). The high methylation and CpG densities at exon ends could increase the efficiency of splice-site selection via recruitment of MECP2, which has been reported to bind to m5C and has been reported to be required for accurate pre-mRNA splicing (Young et al. 2005); however, the increase in CpG density and methylation density is also apparent just 5′ of the stop codon, which is not associated with a splice site.
There is considerable interest in the relationship between DNA methylation and histone modifications. Large databases that describe the distribution of histone modifications and chromatin proteins over the genome have been derived by chromatin immunoprecipitation (Barski et al. 2007; Mikkelsen et al. 2007) or DNase I cleavage (Boyle et al. 2008), followed by deep sequencing. We used these data to test for correlations of histone variants and bound chromatin proteins with patterns of DNA methylation. H3K36 methylation, H3K27 methylation, H3K79 methylation, and H3K9 di-and trimethylation showed no strong correlation with DNA methylation (Fig. 5A,B). In contrast, di- and trimethylation of lysine 4 of histone H3 (H3K4) showed a strong negative correlation with DNA methylation. While previously it was shown that H3K4Me2 was associated with unmethylated promoters (Weber et al. 2008), it is interesting that little correlation was found with H3K4Me1, and the strength of the correlation increased with the level of modification at H3K4. This is consistent with the finding that de novo methylation is targeted to DNA sequences associated with histones that are unmethylated at H3K4 via a domain in the methylation regulator DNMT3L that specifically recognizes unmethylated H3K4 (Ooi et al. 2007).
Figure 5.

Relationship between DNA methylation, histone modification, chromatin proteins, and nucleosome positioning. (A) m5CpG and CpG densities, ChIP-seq scores, and DNase hypersensitivity scores are plotted relative to promoter TSSs for 16,181 RefSeq genes. (B) CpG methylation plotted as a function of histone modifications, chromatin factors, and RNA polymerase II occupancy. Note the strong negative relationship between DNA methylation and density of H3K4me3 and H2A.Z and the lack of a strong association between DNA methylation status and most histone modifications.
The binding of the H2A.Z histone variant correlates inversely with DNA methylation (Fig. 5), demonstrating that these two marks may be mutually exclusive in mammals, as was found recently in plants (Zilberman et al. 2008). Binding of CTCF also correlated globally with unmethylated DNA, in agreement with previous reports that CTCF binds to unmethylated DMRs at specific loci (Bell and Felsenfeld 2000; Hark et al. 2000). DNA methylation patterns have been shown to be subject to somatic inheritance in mammals, whereas there is little evidence for the mitotic inheritance of histone marks and histone variants. DNA methylation in mammals is dominant over histone methylation, as shown by the faithful inheritance of DNA methylation patterns on DNA introduced into cells (Wigler et al. 1981; Lorincz et al. 2002). As shown in Supplemental Figure 8, analysis of CpG loss rates across primates indicates that the genomic methylation patterns observed in somatic cells are similar to those of male germ cells.
Comparison of our data to that from H1 ES cells (Lister et al. 2009) show that at the time of establishment, DNA methylation patterns at lower CpG densities begin as highly methylated (Supplemental Fig. S9). Over time, in differentiated cells some methylation is lost in these CpG-poor regions. The data in Figures 2 and 4 suggest that the domains around CpG-dense promoters may be inherently refractory to DNA methylation. To test this hypothesis we examined the methylation state of Alu elements that fall into promoter domains near first exons. Alu elements are normally highly methylated (Fig. 3D), but Alu elements located within ∼1000 bp of unmethylated first exons tend to be unmethylated (Fig. 3A,B). This supports the hypothesis that single-copy CpG-rich regions are shielded from the DNA methylation machinery. Interestingly, Alu elements are also depleted from these unmethylated domains (Fig. 3C), which suggests that Alu elements that insert into these unmethylated regions reduce host fitness and are lost from the population by selection.
DNA methylation has long been believed to regulate gene expression via programmed removal of DNA methylation from promoters by passive or active methylation to allow lineage-specific gene expression. Arguments against this model have been raised (Walsh and Bestor 1999), and it has recently been reported that the gain of DNA methylation at promoters in cells differentiating in vitro is much more prevalent than is a loss of promoter methylation (Mohn et al. 2008). It has recently been shown that the patterns of histone modification and histone variants at promoters are only weakly related to the level of expression of genes, while chromatin modifications at enhancers are strongly associated with cell-type-specific gene expression (Heintzman et al. 2009). We examined the CpG and methylation density of 27,065 enhancers identified by Heintzmann and colleagues and found that enhancers are characterized by very low levels of CpG and DNA methylation (Fig. 4A). This indicates that enhancer methylation is unlikely to be involved in cell-type-specific gene expression. The lack of cell-type-specific methylation at either enhancers or promoters indicates that DNA methylation is likely to have a negligible or very small role in development, and that the methylation changes seen at some low-CpG promoters are likely to be a result of transcriptional activation rather than a cause.
Our genome-wide data reveals features of methylation patterns that were not apparent in previous experiments that covered small fractions of the genome (Eckhardt et al. 2006; Meissner et al. 2008; Mohn et al. 2008; Weber et al. 2008) or have known biases with respect to CpG density (Supplemental Table S3; Down et al. 2008). The likelihood of methylation of a CpG dinucleotide depends in part on the local sequence environment: High CpG density increases the probability that a CpG will be methylated up to a limit, after which very high CpG densities repel DNA methylation. This trend includes exonic CpGs, which tend to be methylated. Other factors that have been implicated in shaping genomic methylation patterns include the piRNA pathway, which targets classes of transposons for de novo methylation specifically in male germ cells (Carmell et al. 2007) and the binding of transcription factors, such as SP1 to methylated target sites, which can induce demethylation of local CpG sites in dividing mammalian cells (Matsuo et al. 1998; Lin et al. 2000).
Data shown here indicate that methylation is the default state of nucleosomal DNA and could explain how genomic methylation patterns are established and maintained by DNA methyltransferases whose sequence specificity is limited to the CpG dinucleotide. The heritability of genomic methylation patterns clearly shows that once established, DNA methylation is dominant over chromatin modifications. Sequences such as imprinting control regions, CpG islands of the inactive X chromosome, and some transposons and retroviruses are methylated as a result of poorly understood pathways that direct de novo methylation specifically to these sequences. The data indicate that the bulk of the genome is methylated as the default state, and unmethylated regions are protected from a promiscuous DNA methylating system by a combination of very high CpG densities and histone modifications and variants (di- and trimethylated H3K4 and H2A.Z) that repel DNA methyltransferase complexes.
Methods
Isolation of genomic DNA
Human breast DNA was prepared from post-surgical specimens of normal breast tissue adjacent to breast cancer tissue. Human brain DNA was collected from gray matter from the left dorsolateral prefrontal cortex. Mouse brain (strain 129 SvEv, from Taconic) DNA was collected from the left hemisphere with the cerebellar tissue removed.
DNA was prepared from tissues by pulverizing tissue under liquid nitrogen and digesting overnight at 55°C (10 mM NaCl, 10 mM Tris at pH 8, 25 mM EDTA at pH 8, 0.5% SDS, 100 ng/mL proteinase K). DNA was purified by phenol-chloroform extraction and RNase A treatment, followed by ethanol precipitation. DNA was stored in 10 mM Tris, 1 mM EDTA (pH 8) at −20°C. High molecular weight quality of DNA was confirmed by agarose gel electrophoresis and the DNA was quantified on a Qubit fluorometer (Invitrogen).
Enzymatic fractionation by methylation state
Unmethylated and methylated compartments were obtained by limit digestions of 10–15 μg of DNA with McrBC or five tetranucleotide methylation-sensitive restriction endonucleases (referred to as RE), respectively (New England BioLabs). Two rounds of McrBC digestion were performed to ensure sufficient digestion (1× NEB Buffer 2, 2× GTP, 1× BSA, 10 U of McrBC per microgram starting DNA at 37°C for 4–6 h). Each round of digestion was followed by phenol-choloroform extraction (phenol:chloroform:isoamyl alcohol [25:24:1] at pH 8) and ethanol precipitation (add 1 mg/mL glycogen [0.01 vol], 3 M sodium acetate at pH 5.3 [0.1 vol], and ethanol [2 vol]; incubate at −20°C for 15 min; centrifuge at 20,800g at 4°C for 15 min; wash with 600 μL of cold 70% ethanol, and centrifuge at 20,800g at 4°C for 5 min; air dry and resuspended in 1× TE [10 mM Tris, 1 mM EDTA at pH 8]). Phenol-choloroform extraction and ethanol precipitation is further referred to as PCP. Following McrBC digestion, excess GTP was removed by purification with a Sephadex G50 column (GE Healthcare). RE digestion was conducted in three consecutive rounds. Digestion was conducted with 10 U of restriction enzyme per microgram of starting DNA with HpaII and HpyCH4IV (1× NEB Buffer 1, 37°C, 4–6 h), AciI and HhaI (1× NEB Buffer 3, 1× BSA, 37°C, 4–6 h), and BstUI (1× NEB Buffer 2, 60°C, 2–3 h). Each round of digestion was followed by PCP.
Paired-end library preparation
Paired-end libraries were prepared from the methylated and unmethylated DNA compartments by following an adaptation of Applied Biosystems' SOLiD System Mate-paired Library Preparation Protocol. Adaptor sequences can be found in the manufacturer's instructions. DNA fragment ends were repaired using T4 DNA polymerase and T4 polynucleotide kinase (1× NEB PNK buffer, 0.1 mg/mL BSA, 0.4 mM ATP, 0.4 mM dNTPs, 50 U of enzyme) at 12°C for 15 min, then 25°C for an additional 15 min. Unincorporated dNTPs were removed with a Sephadex G50 column followed by PCP. Endogenous EcoP15I sites were methylated with EcoP15I (1× NEB Buffer 3, 1× BSA, 360 μM SAM, 10 U of enzyme per measured microgram of DNA). Successful EcoP15I methylation was confirmed by resistance to digestion by EcoP15I of a control EcoP15I-methylated sample performed in parallel with library samples. A 100 molar ratio excess of CAP adaptors (containing EcoP15I sites) was ligated onto the end-polished, EcoP15I-methylated DNA molecules (1× Invitrogen ligase buffer [5×], 30 U of Ambion T4 DNA ligase), followed by PCP.
Samples were run on a 1% TAE low-melt agarose gel at 45 V for 2.5 h. The gel was size fractionated at 800 bp to remove digested DNA and unligated CAP adaptors. DNA fragments were collected in several size fractions (McrBC: 0.8–2 kb, 2–5 kb, >5 kb; RE: 0.8–1.5 kb, 1.5–3 kb, 3–6 kb, >6 kb) and extracted using the GENECLEAN Glassmilk Spin Column Kit (MP Biomedicals). DNA from each size fractionation was circularized separately with a 3 molar excess of biotinylated internal adaptor (T30B) at 16°C overnight (1× Ambion ligation buffer and 30 U of Ambion T4 DNA ligase) under concentration conditions that promote 95% circularization efficiency of DNA molecules of each length range. The DNA concentration I was calculated as I (ng/uL) = [J/(95% intramolecular)] − J and 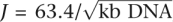 , where J is the Jacobson-Stockmayer factor, the effective local concentration of one end of the DNA molecule in the vicinity of the other (Collins and Weissman 1984). An additional 2.5 U of ligase was added 1 h after circularization commenced, and again 1 h before the reaction was terminated. Uncircularized DNA molecules were degraded with plasmid-safe DNAse treatment (62.5 μM ATP, 0.0625 U/μL ATP-dependent DNase) (Epicentre) and the enzyme was heat inactivated (70°C for 20 min). Following PCP, the four RE fractions were combined into <2-kb and >2-kb fractions. Excess nucleotides were removed using a G50 Sephadex column. Circularized DNA was digested with EcoP15I (10 U per 100 ng of circularized DNA) overnight at 37°C (1× NEB Buffer 3, 1× BSA, 2 mM ATP, 0.1 mM Sinefungin). Fresh ATP, BSA, and Sinefungin were added the following morning 1 h before termination of the reaction by heat inactivation. The digested DNA was end repaired by 5 U of Klenow (New England BioLabs) in the presence of 25 μM dNTPs at room temperature for 30 min, followed by heat inactivation at 65°C for 20 min. Sixty molar excess P1ds and P2ds linkers containing primer sequences for PCR were ligated onto the ends of the DNA molecules in 250 uM ATP and 20 U of T4 DNA ligase (Ambion).
, where J is the Jacobson-Stockmayer factor, the effective local concentration of one end of the DNA molecule in the vicinity of the other (Collins and Weissman 1984). An additional 2.5 U of ligase was added 1 h after circularization commenced, and again 1 h before the reaction was terminated. Uncircularized DNA molecules were degraded with plasmid-safe DNAse treatment (62.5 μM ATP, 0.0625 U/μL ATP-dependent DNase) (Epicentre) and the enzyme was heat inactivated (70°C for 20 min). Following PCP, the four RE fractions were combined into <2-kb and >2-kb fractions. Excess nucleotides were removed using a G50 Sephadex column. Circularized DNA was digested with EcoP15I (10 U per 100 ng of circularized DNA) overnight at 37°C (1× NEB Buffer 3, 1× BSA, 2 mM ATP, 0.1 mM Sinefungin). Fresh ATP, BSA, and Sinefungin were added the following morning 1 h before termination of the reaction by heat inactivation. The digested DNA was end repaired by 5 U of Klenow (New England BioLabs) in the presence of 25 μM dNTPs at room temperature for 30 min, followed by heat inactivation at 65°C for 20 min. Sixty molar excess P1ds and P2ds linkers containing primer sequences for PCR were ligated onto the ends of the DNA molecules in 250 uM ATP and 20 U of T4 DNA ligase (Ambion).
M280 Strepavidin beads (Invitrogen) were prewashed with once 1× BBW buffer (2% Tween 20, 2% Triton X-100, 10 mM EDTA), 1× BSA, and 1× BW buffer (10 mM Tris-HCl, 1 M NaCl, 1 mM EDTA). DNA was bound through the biotin tag on the internal adaptor to 15 μL of prewashed M280 strepavidin beads (Invitrogen) for 15 min at room temperature in a final concentration of 1× BW buffer. Bound beads were washed once with 1× BBW buffer, twice with 1X BW buffer, and once with 1× NEB Buffer 2. The beads were resuspended in 1× NEB Buffer 2 with 500 μM dNTPs and 15 U of DNA polymerase I (New England BioLabs) and incubated at 16°C for 30 min. A test PCR at 20 cycles was done to confirm the presence of the expected 156-bp DNA product; cycles were titrated up and down as needed to produce sufficient product. A large-scale PCR was performed on the entire population of beads (95°C for 5 min, 18–22 cycles of 95°C for 15 sec, 62°C for 15 sec, 70°C for 1 min, followed by a final extension at 70°C for 5 min and storage at 4°C indefinitely). Each 50-μL reaction contained 1 uL of bead template, 1 μL of 50 μM L-PCR-P1 primer, 1 μL of 50 μM L-PCR-P2 primer, and 47 μL of Platinum PCR Supermix (Invitrogen). Reactions were combined and ethanol precipitated.
A 6% DNA retardation PAGE gel (Invitrogen) was prerun for 5 min at 115 V. Using Ficoll loading dye, the samples and a 25-bp ladder (Invitrogen) were run on a 6% DNA retardation PAGE gel in 1× TBE at 115 V for 45 min. To visualize the bands, the gel was stained in 1:6000 ethidium bromide (10 mg/mL) in 1× TBE for 5 min and destained twice in water. The 156-bp library band was dissected from the gel and shredded. DNA was extracted from the shredded gel in 200 μL of PAGE elution buffer (1:5 7.5 M ammonium acetate in 1× TE) at room temperature for 20 min and 250 μL PAGE elution buffer at 37°C for 40 min. The gel and buffer were separated on a 0.45 micron filter spin column. The DNA was ethanol precipitated. The resuspended DNA was purified with a MinElute Reaction Cleanup Kit (Qiagen) and eluted in 20 μL of 10 mM Tris and an additional 20 μL of 10 mM Tris was added to bring the final library volume to 40 μL. DNA quantity was assessed on a Qubit fluorometer (Invitrogen) and quality and quantity on a Bioanalyzer DNA 1000 LabChip (Agilent).
DNA sequencing
Individual library fragments were amplified on 1-μm beads using emulsion-PCR according to the Applied Biosystems SOLiD System emulsion PCR standard protocol. Samples were subjected to paired-end sequencing on an Applied Biosystems SOLiD System DNA sequencer. Twenty-five base-pair sequences were obtained for both R2 and F2 tags on each bead. Mouse and Human brain samples were sequenced using the same protocol at Agencourt Biosciences.
Tag mapping
Initial tag mappings were performed with the Applied Biosystems SOLiD System software analysis package. Paired tags were each individually mapped in color space, allowing up to two mismatches in each 25-bp tag to the human hg18 sequence obtained from the UCSC Genome Browser (http://genome.ucsc.edu). Up to 10 hits per chromosome were recorded for each tag. A mate pair was reported if a single uniquely placed pair could be made from the hits of each tag that met the constraints of order, orientation and distance (0–15 kb). If no mate pair could be made, then mate-pair rescue was attempted in which each hit was used as an anchor and the region where an appropriate mate should be found was scanned while allowing a total of four mismatches in both tags. A rescued mate pair was reported if a single uniquely placed pair could be made.
Data filtering and CpG analysis
A custom Perl script was written to parse the output files from the Applied Biosystems SOLiD System and filter sequences that did not have at least one restriction site (McrBC or RE, respectively) at the fragment ends. Since methylated (RE) and unmethylated (McrBC) compartments are sampled independently, it is important to find the correct ratio of RE:McrBC fragments that represents the “true” distribution that would be obtained from a random sampling of the genome. This ratio can be determined numerically since, if you fix the total number of McrBC + RE fragments, then using the ratio that matches the underlying “true” distribution should yield the maximum physical coverage. This ratio was estimated by finding the ratio of McrBC and RE fragments that maximized coverage on a subset of chromosomes (chromosomes 16–21). All McrBC and RE fragments were then overlapped with an indexed list of CpGs in hg18. The number of unmethylated observances at each CpG, nu, was set equal to the number of RE fragments that terminated at that CpG plus the number of McrBC fragments containing that CpG in its interior (>50 bp from the end). The number of methylated observances, nm, at a particular CpG was calculated as the sum of all RE fragments to which a particular CpG was interior. The coverage of a CpG at position i is then given as C(i) = nm(i) + nu(i) and the methylation score is calculated as m(i) = nm(i)/C(i).
CpG island, RepeatMasker, RefSeq gene data, and other genomic annotation information was downloaded from the UCSC Genome Browser website (http://genome.ucsc.edu). The average methylation score of a genomic element e is calculated as
where N(e) is the total number of CpGs in that element and where C(i) ≥ 7 and each CpG at position i are both McrBC and RE sites. All annotation and methylation data, indexed by the CpG site was then stored in a MySQL database that could be used directly for calculations.
Model gene analysis
A total of 16,841 RefSeq genes with annotated coding sequence start and end points and greater than two exons were considered for this analysis. Analysis of CpG density (ρCpG) was performed as in Majewski and Ott (2002). meCpG density (ρmeCpG) at position x relative to the end of an exon was estimated as
where
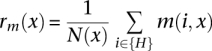 |
and H = {all CpG that are McrBC and RE sites and C(i) ≥ 7}.
CpG island analysis
Each island was annotated according to its genomic location (promoter, exon, intron, intergenic) in hierarchical fashion. Promoter islands were defined as islands that occur within 1 kb of a gene TSS. Exon and intron islands must overlap exons and introns, respectively. All islands not labeled as promoter, exon, or intron were labeled as intergenic. CpG island lengths for islands of each class could then be calculated directly. Average methylation scores, 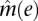 , were then calculated for island CpGs with coverage C(i) ≥ 7 and which were both McrBC and RE sites that had a particular annotation and a particular CpG density. Scores were similarly computed for promoter-associated islands on chr X and autosomes.
, were then calculated for island CpGs with coverage C(i) ≥ 7 and which were both McrBC and RE sites that had a particular annotation and a particular CpG density. Scores were similarly computed for promoter-associated islands on chr X and autosomes.
DMR analysis
Methylation scores were calculated for all CpGs in each well-curated differentially methylated region (see Supplemental Table 2 online) that are McrBC and RE sites with coverage C(i) ≥ 7.
Histone analysis
Chromatin immunoprecipitation with massively parallel sequencing (ChIP-seq) data was obtained directly from http://dir.nhlbi.nih.gov/papers/lmi/epigenomes/hgtcell.aspx. The number of fragments covering each CpG (assuming a 200-bp length, which is equal to the average fragment length) for each mark was then computed and normalized to the total number of sequences obtained. Methylation scores could then be computed as a function of the normalized ChIP-seq score.
Promoter analysis
Promoters were defined as the region 1-kb centered on the transcription start site (TSS) of RefSeq genes whose cdsStart and cdsStop were annotated as complete. Promoter distribution was calculated as a function of CpG observed/expected (O/E) calculated across the entire 1-kb region where
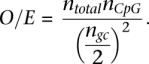 |
Promoter distribution was also calculated as a function of CpG density of the 1-kb region centered on the TSS. The average methylation score, , was then calculated for all CpGs in the 1-kb region centered on the TSS with the given CpG O/E or CpG density.
CpG annotation
Nonrepeat-associated CpGs were annotated as promoter, exon, intron, or intergenic. CpGs were assigned a single category given preference in the order of the above list. Exon and intron locations were based on complete RefSeq entries. Promoters regions were defined as 1-kb upstream of or downstream from the TSS. For humans, a list of TSSs was obtained using the SwitchGear TSS track (http://www.switchgeargenomics.com) on the UCSC Genome Browser. For mouse, a list of TSSs was obtained from the UCSC knownGene track. Repeat-associated CpGs were annotated from the RepeatMasker entries (http://www.repeatmasker.org) from the appropriate tracks on the UCSC Genome Browser. Average methylation scores, , were then calculated for CpGs that had a particular annotation and a particular CpG density.
CpGs associated with exons of complete RefSeq entries were classified as belonging to first, internal, and last exons. Average methylation scores, , were then calculated for CpGs that had a particular annotation and a particular CpG density.
Alu analysis
CpGs were annotated as belonging to AluS, AluY, or AluJ from the RepeatMasker annotations downloaded from UCSC's Genome Browser website. Average methylation scores, , were then calculated for CpGs that had a particular annotation and a particular CpG density as for other elements. A total of 16,181 well-annotated RefSeq genes were divided into CpG-rich (HCG) and CpG-poor (LCG) promoter classes using an observed/expected (O/E, calculated as above) cutoff of 0.35.
The average methylation score, , of Alu-annotated CpGs was computed as a function of their distance to annotated RefSeq genes with either methylated or unmethylated first exons. Exon methylation was determined if over 70% of the sites that were both McrBC and RE sites had coverage C(i) ≥ 5. Exons were deemed methylated if the average methylation score of these CpGs was greater than 0.7 and unmethylated if this score was less than 0.3. Alu average methylation scores are smoothed with a 200-bp sliding window.
To compute the fraction of Alus near methylated and unmethylated promoters, the Alu density as a distance to the TSS was calculated for each gene. Promoter methylation was determined if over 70% of the sites that were both McrBC and RE sites had coverage C(i) ≥ 5. Promoters were defined as the region 1 kb upstream of the TSS. Promoters were deemed methylated if the average methylation score of these CpGs was greater than 0.7 and unmethylated if this score was less than 0.3, and the ratio of fraction of methylated to the fraction of unmethylated promoters with nearby Alus was computed. This ratio was then smoothed with a 100-bp sliding window.
Estimate of the theoretical coverage of Methyl-MAPS
As an example to calculate the theoretical maximum coverage, assume the genome is 60% methylated. There are 28,163,863 CpGs in the human genome (hg18), implying that 16,898,318 are methylated and 11,265,545 are unmethylated. Of the methylated CpGs, ∼37% (6,252,378) will be RE sites; and of unmethylated CpGs, ∼76% (8,561,814) will be McrBC sites. Thus, in theory, 14,814,192 CpGs could be probed using this technique. In reality there is not a single copy of the genome present, and methylation patterns can be heterogeneous (see Fig. 1A); therefore, the number of CpG sites that can be probed will be higher. These numbers are highly dependent on the exact fraction of the genome that is methylated.
Acknowledgments
We thank J.J. Mann, A. Dwork, and P. Graham for providing the human brain tissue; J. Manning for providing informatics support for the tag mappings; and L. Zhang and B. Coleman for aid with the sequencing. We thank Reiner Schultz for the annotated list of DMRs. This work was funded by NHGRI (T.H.B.), NCI (T.H.B. and J.R.E.), NIMH (F.H.), NIDA (J.A.G.), DOD and NIMH (A.H.O.), and the Simons Foundation (J.A.G.).
Footnotes
[Supplemental material is available online at http://www.genome.org. The methylation data from this study have been submitted to the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo) under accession no. GSE21242.]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.101535.109.
References
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K 2007. High-resolution profiling of histone methylations in the human genome. Cell 129: 823–830 [DOI] [PubMed] [Google Scholar]
- Bell AC, Felsenfeld G 2000. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature 405: 482–488 [DOI] [PubMed] [Google Scholar]
- Boyle AP, Davis S, Shulha HP, Meltzer P, Margulies EH, Weng Z, Furey TS, Crawford GE 2008. High-resolution mapping and characterization of open chromatin across the genome. Cell 132: 311–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmell MA, Girard A, van de Kant HJ, Bourc'his D, Bestor TH, de Rooij DG, Hannon GJ 2007. MIWI2 is essential for spermatogenesis and repression of transposons in the mouse male germline. Dev Cell 12: 503–514 [DOI] [PubMed] [Google Scholar]
- Collins FS, Weissman SM 1984. Directional cloning of DNA fragments at a large distance from an initial probe: A circularization method. Proc Natl Acad Sci 81: 6812–6816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Down TA, Rakyan VK, Turner DJ, Flicek P, Li H, Kulesha E, Gräf S, Johnson N, Herrero J, Tomazou EM, et al. 2008. A Bayesian deconvolution strategy for immunoprecipitation-based DNA methylome analysis. Nat Biotechnol 26: 779–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckhardt F, Lewin J, Cortese R, Rakyan VK, Attwood J, Burger M, Burton J, Cox TV, Davies R, Down TA, et al. 2006. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat Genet 38: 1378–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hark AT, Schoenherr CJ, Katz DJ, Ingram RS, Levorse JM, Tilghman SM 2000. CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature 405: 486–489 [DOI] [PubMed] [Google Scholar]
- Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, et al. 2009. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 459: 108–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Ladd-Acosta C, Carvalho B, Wu H, Brandenburg SA 2008. Comprehensive high-throughput arrays for relative methylation (CHARM). Genome Res 18: 780–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PA, Wolkowicz MJ, Rideout WM 3rd, Gonzales FA, Marziasz CM, Coetzee GA, Topscott SJ 1990. De novo methylation of the MyoD1 CpG island during the establishment of immortal cell lines. Proc Natl Acad Sci 87: 6117–6121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kass SU, Landsberger N, Wolffe AP 1997. DNA methylation directs a time-dependent repression of transcription initiation. Curr Biol 7: 157–162 [DOI] [PubMed] [Google Scholar]
- Kondrashov AS 2003. Direct estimates of human per nucleotide mutation rates at 20 loci causing Mendelian diseases. Hum Mutat 21: 12–18 [DOI] [PubMed] [Google Scholar]
- Lin IG, Tomzynski TJ, Ou Q, Hsieh CL 2000. Modulation of DNA binding protein affinity directly affects target site demethylation. Mol Cell Biol 20: 2343–2349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, et al. 2009. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462: 315–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorincz MC, Schübeler D, Hutchinson SR, Dickerson DR, Groudine M 2002. DNA methylation density influences the stability of an epigenetic imprint and Dnmt3a/b-independent de novo methylation. Mol Cell Biol 22: 7572–7580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majewski J, Ott J 2002. Distribution and characterization of regulatory elements in the human genome. Genome Res 12: 1827–1833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo K, Silke J, Georgiev O, Marti P, Giovannini N, Rungger D 1998. An embryonic demethylation mechanism involving binding of transcription factors to replicating DNA. EMBO J 17: 1446–1453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, Zhang X, Bernstein BE, Nusbaum C, Jaffe DB, et al. 2008. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 454: 766–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaud EJ, van Vugt MJ, Bultman SJ, Sweet HO, Davisson MT, Woychick RP 1994. Differential expression of a new dominant agouti allele (Aiapy) is correlated with methylation state and is influenced by parental lineage. Genes Dev 8: 1463–1472 [DOI] [PubMed] [Google Scholar]
- Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, et al. 2007. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448: 553–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohn F, Weber M, Rebhan M, Roloff TC, Richter, Stadler MB, Bibel M, Schübler D 2008. Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol Cell 30: 755–766 [DOI] [PubMed] [Google Scholar]
- Ooi SK, Qiu C, Bernstein E, Li K, Jia D, Yang Z, Erdjument-Bromage H, Tempst P, Lin SP, Allis CD, et al. 2007. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 448: 714–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakyan VK, Preis J, Morgan HD, Whitelaw E 2001. The marks, mechanisms and memory of epigenetic states in mammals. Biochem J 15: 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollins RA, Haghighi F, Edwards JR, Das R, Zhang, Ju J, Bestor TH 2006. Large-scale structure of genomic methylation patterns. Genome Res 16: 157–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein R, Razin A, Cedar H 1982. In vitro methylation of the hamster adenine phosphoribosyltransferase gene inhibits its expression in mouse L cells. Proc Natl Acad Sci 79: 61–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutcliffe JS, Nelson DL, Zhang F, Pieretti M, Caskey CT, Saxe D, Warren ST 1992. DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum Mol Genet 1: 1397–1400 [DOI] [PubMed] [Google Scholar]
- Walsh CP, Bestor TH 1999. Cytosine methylation and mammalian development. Genes Dev 13: 26–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber M, Hellmann I, Stadler MB, Ramos L, Pääbo S, Rebhan M, Schübeler D 2008. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet 39: 457–466 [DOI] [PubMed] [Google Scholar]
- Wigler M, Levy D, Perucho M 1981. The somatic replication of DNA methylation. Cell 24: 33–38 [DOI] [PubMed] [Google Scholar]
- Xu GL, Bestor TH, Bourc'his D, Hsieh C-L, Tommerup N, Bugge M, Hulten M, Qu X, Russo JJ, Viegas-Pequignot E 1999. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature 402: 187–191 [DOI] [PubMed] [Google Scholar]
- Young JI, Hong EP, Castle JC, Crespo-Barreto J, Bowman AB, Rose MF, Kang D, Redman R, Johnson JM, Berget S, et al. 2005. Regulation of RNA splicing by the methylation-dependent transcriptional repressor methyl-CpG binding protein 2. Proc Natl Acad Sci 102: 17551–17556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilberman D, Coleman-Derr D, Ballinger T, Henikoff S 2008. Histone H2A.Z and DNA methylation are mutually antagonistic chromatin marks. Nature 456: 125–129 [DOI] [PMC free article] [PubMed] [Google Scholar]