

Abstract
Learning processes mediating conditioning and extinction of contextual fear require activation of several key signaling pathways in the hippocampus. Principal hippocampal CA1 neurons respond to fear conditioning by a coordinated activation of multiple protein kinases and immediate early genes, such as cFos, enabling rapid and lasting consolidation of contextual fear memory. The extracellular signal-regulated kinase (Erk) additionally acts as a central mediator of fear extinction. It is not known however, whether these molecular events take place in overlapping or nonoverlapping neuronal populations. By using mouse models of conditioning and extinction of fear, we set out to determine the time course of cFos and Erk activity, their cellular overlap, and regulation by afferent cholinergic input from the medial septum. Analyses of cFos+ and pErk+ cells by immunofluorescence revealed predominant nuclear activation of either protein during conditioning and extinction of fear, respectively. Transgenic cFos-LacZ mice were further used to label in vivo Fos+ hippocampal cells during conditioning followed by pErk immunostaining after extinction. The results showed that these signaling molecules were activated in segregated populations of hippocampal principal neurons. Furthermore, immunotoxin-induced lesions of medial septal neurons, providing cholinergic input into the hippocampus, selectively abolished Erk activation and extinction of fear without affecting cFos responses and conditioning. These results demonstrate that extinction mechanisms based on Erk signaling involve a specific population of CA1 principal neurons distinctively regulated by afferent cholinergic input from the medial septum.
Introduction
Extinction of contextual fear takes place when aversive events associated with a particular environment consistently stop occurring. This process is essential for the cessation of ongoing fear responses rooted in past stressful experiences. Significant theoretical and experimental effort has been undertaken to elucidate the mechanisms underlying extinction to advance strategies for treatment of anxious patients displaying persistent contextual fear in the absence of aversive reinforcement (Grillon, 2002). Presently, extinction is thought to reflect a learning process regulated among the basolateral amygdala, prefrontal cortex and hippocampus (Bouton, 2004; Maren and Quirk, 2004; Sotres-Bayon et al., 2004). Whereas the prefrontal cortex involves excitatory glutamatergic-mediated mechanisms to form new extinction memories (Milad and Quirk, 2002), and the amygdala recruits learning mechanisms involving both excitatory and inhibitory neurotransmission to regulate expression of fear (Falls et al., 1992; Chhatwal et al., 2005; Berlau and McGaugh, 2006; Herry et al., 2008), the mechanisms by which the hippocampus contributes to extinction are not known in detail.
Several processes mediated by the hippocampus have been implicated in extinction of contextual fear. Some of them, such as retrieval of the conditioning memory (Ouyang and Thomas, 2005) or its destabilization (Lee et al., 2008) involve modifications of the neuronal subsets involved in conditioning (Reijmers et al., 2007). New extinction learning, however, is likely to recruit distinctive cellular and molecular mechanisms.
At a molecular level, principal hippocampal CA1 neurons respond to fear conditioning by a coordinated activation of multiple signaling pathways and immediate early genes, such as cFos, enabling rapid and lasting consolidation of contextual fear memory (Radulovic et al., 1998; Matsuo et al., 2008). Most of these pathways, involving protein kinase C (Tronson et al., 2008), cAMP-dependent protein kinase (Isiegas et al., 2006), and cyclin-dependent kinase 5 (Sananbenesi et al., 2007), show downregulated activity during extinction. The extracellular signal-regulated kinase (Erk), however, acting as a central mediator of fear extinction (Szapiro et al., 2003; Chen et al., 2005; Tronson et al., 2008; Ryu et al., 2008), shows robust and sustained somatonuclear phosphorylation (pErk) (Fischer et al., 2007).
The main objective of the present study was to determine the cellular overlap of cFos+ hippocampal neurons activated by fear conditioning and pErk+ neurons activated by extinction and establish their regulation by afferent hippocampal input. We demonstrated that Erk-dependent extinction mechanisms are fully distinguishable at a molecular, cellular and circuit level from mechanisms underlying conditioning of fear.
Materials and Methods
Animals.
Nine-week old C57BL/6 mice were obtained at 9 weeks of age. Frozen embryos of cFos-LacZ transgenic mice (Schilling et al., 1991) were re-derived in the Northwestern University transgenic core facility. Experiments were replicated in 3 backcrosses of cFos-LacZ mice in the C57BL/6 strain. The mice were individually housed in a satellite facility adjacent to the behavioral equipment. The facility was provided with a separate ventilation system (15 air exchanges per hour), a 12 h dark/light cycle (7:00 A.M.–7:00 P.M.), 40–50% humidity, and 20 ± 2°C temperature. All studies were approved by the Animal Care and Use Committee of Northwestern University in compliance with National Institutes of Health standards. The number of mice per group was 8–15.
Antibodies.
Single labeling was performed using rabbit polyclonal anti-cFos (Oncogene, 1:20,000), mouse monoclonal anti-di-phosphoErk (Sigma, 1:16,000) or goat polyclonal anti-AChE antibodies (1:200). Double labeling was performed using rabbit polyclonal anti-cFos (Oncogene, 1:5000), mouse monoclonal anti-di-phosphoErk (Sigma,1:4000), rabbit polyclonal anti-fluorescein (Genetex) reacting with fluorescein-di-β-d-galactopyranoside (FDG) (1:500) and rabbit polyclonal anti-GAD65,67 (Millipore Bioscience Research Reagents, 1:100). Immunotoxin, consisting of saporin (SAP) conjugated to a rabbit anti-mouse neurotrophin low affinity (p75) receptor antibody and unconjugated SAP were purchased from Advanced Targeting Systems.
Surgery and cannulation.
Double guided cannula (Plastic One) was placed into the dorsal hippocampus (vs bregma: anteroposterior, 1.5 mm; mediolateral, 1 mm; dorsoventral, 2 mm), lateral ventricles (vs bregma: anteroposterior, 0.5 mm; mediolateral, 1 mm; dorsoventral, 2 mm) or medial septum (vs bregma: anteroposterior, 0.4 mm; mediolateral, 0 mm; dorsoventral, 4 mm) as described earlier (Radulovic et al., 1999). The coordinates were selected for intrahippocampal (i.h.), intracerebroventricular (i.c.v.) and intraseptal (i.s.) injections. The gauge of the guide and injection cannulae was 26 and 28, respectively.
Fear conditioning and extinction.
Contextual fear conditioning was performed with an automated system (TSE Inc.) and consisted of a single exposure to context (3 min) followed by a footshock (2 s, 0.7 mA, constant current) as described previously (Radulovic et al., 1999). The extinction trials were performed at 24 h intervals and consisted of nonreinforced 3-min exposures to the context (Fischer et al., 2004, 2007). Context-dependent freezing was measured every 10th second over 3 min by two observers unaware of the experimental conditions and expressed as percentage of total number of observations.
Lesions of medial septal cholinergic neurons.
Unconjugated SAP or μ p75 SAP, serving to lesion cholinergic medial septal neurons, were injected at a single dose of 180 ng/0.2 μl in the medial septum over 2 min. The cannula position was determined for each mouse at end of experiments during histological examination of brain tissue and only data obtained from mice with correctly inserted cannula were analyzed.
Labeling of cFos-LacZ in vivo by FDG.
For studies aimed to determine the colocalization of neurons showing upregulation of cFos and pErk, all mice were initially habituated to the injection procedure by one daily intrahippocampal injection of artificial CSF in 10% DMSO (aCSF; 0.25 μl/site) for 4 consecutive days. The nontoxic precipitating fluorescent substrate of LacZ, FDG [10 mm in aCSF/10% DMSO (Nirenberg and Cepko, 1993), 0.25 μl/site], or vehicle was injected at a rate of 1 μl/2 min. Mice received either FDG alone or were trained and injected with FDG immediately thereafter. One-half of the latter group was left undisturbed for 5 consecutive days, whereas the other half was exposed to extinction trials. Extinction was performed by re-exposing the mice to the conditioning context for 3 min/d on 5 consecutive days. The number of FDG+, pErk+ or double positive neurons was determined by immunofluorescence.
Immunohistochemistry.
Brains were collected 1 h after training, or indicated extinction tests. The selected time points were previously shown to give maximal cFos and pErk signals after conditioning and extinction, respectively (Radulovic et al., 1998; Fischer et al., 2007). Mice were anesthetized with an intraperitoneal injection of 240 mg/kg Avertin 1 h after indicated trials and transcardially perfused with ice-cold 4% paraformaldehyde in phosphate buffer (pH 7.4, 150 ml/mouse). Brains were postfixed for 48 h in the same fixative and then immersed for 24 h each in 10%, 20%, and 30% sucrose in phosphate buffer. After the tissue was frozen by liquid nitrogen, 50 μm-thick coronal sections were used for performing free-floating immunohistochemistry with primary antibodies to cFos, pErk, fluorescein or GAD65,67. Biotinylated secondary antibody and ABC peroxidase complex (Vector) were used for signal amplification and DAB (Sigma), FITC (fluorescein isothiocyanate) or rhodamine (tyramide signal amplification, NEN Life Sciences) as visualizing substrates. Control procedures involved immunostaining without primary or secondary antibodies and isotype controls (e.g., Fig. S2C, available at www.jneurosci.org as supplemental material). For the characterization of FDG signals, selected brain sections were coverslipped with Vectashield (Vector) containing the nuclear counterstain 4′,6-diamidino-2-phenylindole (DAPI).
Quantification of immunostaining signals was performed as described previously (Fischer et al., 2007). Digital images were captured with a cooled color charge-coupled device camera (RTKE Diagnostic Instruments) and SPOT software for Macintosh. Image J was used for image processing. Cell counts from the dorsohippocampal CA1 subfield was performed using three consecutive dorsohippocampal sections per mouse (Brown et al., 1998). For double labeling, the separate FITC and rhodamine captures were digitally combined to produce composite images. Equal cutoff thresholds were applied to all captures to remove background autofluorescence. For each capture, cell counts were performed within 100 μm2 grids (∼7 grids/section) for three sections of each CA1. Counts of individual signals were first performed separately, followed by identification and counting of double positive cells on the composite images. An overlapping signal of FITC and rhodamine fluorescence in nuclei that were sharply in focus in a single focal plane was used as a criterion for nuclear colocalization of FDG and pErk (Patterson et al., 2001). The measures for each capture were averaged to give the number of pErk+, cFos+ and FDG+ nuclei and expressed per 0.1 mm2 area. Finally, the proportional number (percentage) of double positive neurons from total pErk immunopositive neurons was calculated. Representative images (4–5 per group) were captured using a Zeiss LSM5 Pascal confocal microscope. Between 7 and 12 z-slices were obtained for each channel which were then converted into a z-projection, using a maximum projection algorithm (MetaMorph, Molecular Devices); thus, there was no loss of signal from any slice when the z-stack is produced (Schrick et al., 2007).
Data analysis.
Statistically significant differences were determined by one- (molecular analyses, factor Group) or two-way ANOVA (behavioral analyses, Group × Test interactions) followed by Scheffe′s test for post hoc comparisons. The results are presented as mean ± SEM.
Results
Predominant nuclear activation of cFos after conditioning and pErk during extinction of fear in hippocampal neurons
Several studies have shown that retrieval of the contextual fear memory (Ouyang and Thomas, 2005) and temporary destabilization (Lee et al., 2008) or depotentiation (Kim et al., 2007) of its underlying circuits are needed for fear extinction. These processes involve, at least in part, the neuronal subset encoding the conditioning fear memory. Fear conditioning potently activates the c-fos promoter causing an increase of the cFos protein in hippocampal neurons during fear conditioning (Radulovic et al., 1998; Reijmers et al., 2007). We therefore first examined whether cFos and pErk are coactivated in hippocampal neurons in response to conditioning and extinction of fear. A naive group consisted of mice that were left undisturbed in their home cages throughout the experiments. Mice repeatedly exposed to context alone were not used in the present study because our previous analyses revealed that this treatment did not trigger cFos (Radulovic et al., 1998) or pErk responses (our unpublished observations). Brain sections were analyzed 1 h after training (T) or 1 h after the fifth extinction test (E5) (Fig. 1A). This time point was selected because when they are activated by conditioning or extinction of fear, both cFos and pErk show maximal hippocampal levels ∼1 h later (Atkins et al., 1998; Radulovic et al., 1998; Chen et al., 2005; Fischer et al., 2007). Thus, colocalization analyses are possible in this time frame. Immunohistochemical analyses of cFos and pErk revealed that the upregulation of these proteins represented transient and temporally dissociated molecular alterations the T and E5 groups (Fig. 1B,C). Furthermore, although detectable signals of both proteins were observed after E2, <5% of either cFos or pErk+ cells showed colocalization of both proteins. This lack of overlap was specific for the hippocampus when compared with the amygdala, in which 29 and 41% of cFos and pErk+ cells, respectively, were double positive (Fig. 2A–C). Two possibilities could account for the segregated cFos and pErk signals: (1) pErk was upregulated instead of cFos within cells previously involved in fear conditioning, suggesting that conditioning and extinction are processed by the same cells using different molecular mechanisms, or (2) pErk upregulation during extinction took place within a separate subset of hippocampal cells.
Figure 1.
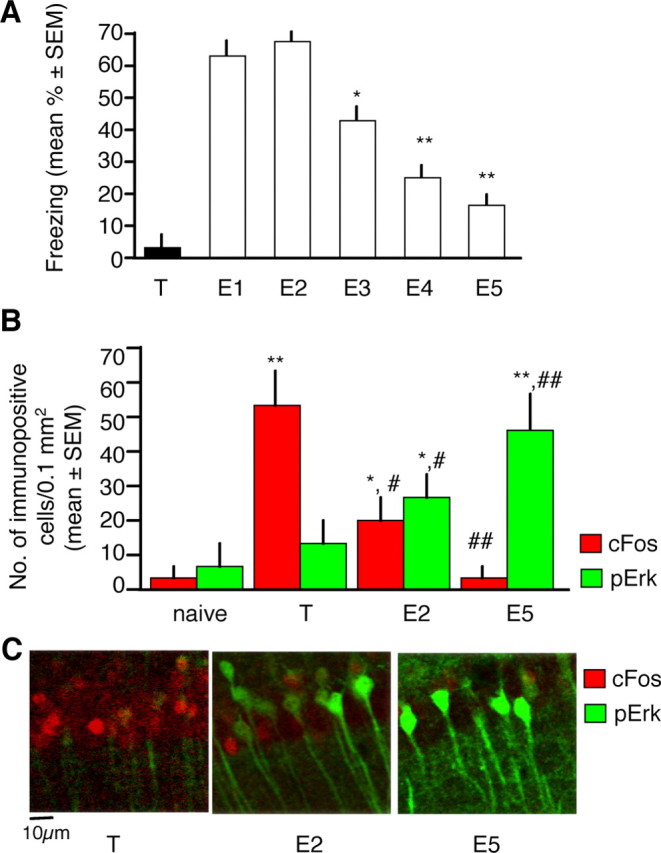
Activation of cFos and pErk within the nuclei of pyramidal CA1 cells after contextual fear conditioning and extinction. A, Freezing behavior is robustly induced by a single training (T) trial but progressively decreases with repeated extinction (E1–E5) trials (*p < 0.05; **p < 0.01 vs E1). n = 10; error bars = mean ± SEM. B, Within CA1 cells, nuclear cFos was maximal after training and declined during extinction. Significant increase of nuclear pErk, however, was observed only during extinction (*p < 0.05; **p < 0.001 vs naive; #p < 0.05; ##p < 0.01 vs T). n = 32 (4 per group); error bars = mean ± SEM. C, Representative micrographs demonstrating cFos and pErk signals 1 h after training (T, right) or after the second extinction test 2 (E2, middle) and the fifth extinction test (E5).
Figure 2.

Lack of cFos/pErk colocalization in the hippocampus but not the amygdala. Colocalization of cFos and pErk was determined 1 h after E2. A, Areas selected for analyses are outlined. B, cFos and pErk+ cells did not colocalize in the hippocampal CA1 subfield, whereas colabeling was observed in the basolateral amygdala (BLA) as revealed by 30 ± 3% of pErk+ neurons; 43 ± 8 cFos+ neurons showing overlap (*p < 0.05; **p < 0.001 vs naive). The naive group is presented as a dashed line. C, Representative micrographs.
Upregulation of cFos and pErk in segregated hippocampal principal CA1 neurons
To delineate between these possibilities, we tagged the hippocampal neurons of mice expressing a cFos-LacZ fusion protein driven by the c-fos promoter (Schilling et al., 1991). The fusion protein, responding to exogenous stimuli in a manner similar to that of endogenous cFos (Smeyne et al., 1992), was visualized with FDG, a nontoxic precipitating substrate of LacZ allowing for stable in vivo labeling of activated neurons (Nirenberg and Cepko, 1993). Indirect immunofluorescence was used to enhance the signal intensity. Untreated cFos-LacZ mice acquired and extinguished conditioned fear in a manner similar to that of their wild-type littermates and C57BL/6 mice (F(2,27) = 0.45, p = 0.98) (Fig. S1A, available at www.jneurosci.org as supplemental material), and these behaviors remained unaltered after treatment with FDG or vehicle (F(3,28) = 0.76, p = 1.02) (Fig. S1B, available at www.jneurosci.org as supplemental material). Furthermore, extinction of fear was also Erk-dependent in cFos-LacZ mice, as it was reported earlier for the C57BL/6 strain, as revealed by significant persistence of fear after inhibition of the Erk up-stream activator mitogen-activated and extracellular signal regulated kinase by U0126 (F(1,14) = 8.23, p < 0.05) (Fig. S1C, available at www.jneurosci.org as supplemental material). The possibility that genetically introduced cFos-LacZ protein might involve somewhat smaller cell population than endogenous cFos (Smeyne et al., 1992) or interferes with Erk activation was examined by comparing these molecular responses between cFos-LacZ, wild-type and C57BL/6 mice. We did not observe significant differences in the number of cFos (F(3,16) = 1.88, p = 1.92) and pErk (F(2,12) = 1.25, p = 1.13) signals determined after conditioning and E5, respectively, between these mouse strains (Fig. S1D,E, available at www.jneurosci.org as supplemental material). The FDG signals (Fig. S1D, available at www.jneurosci.org as supplemental material) were not significantly reduced when compared with cFos signals in cFos-LAcZ mice despite showing a tendency toward a decrease. Possibly, earlier studies showed a greater difference because LacZ activity was compared with general fos-like immunoreactivity rather than specific cFos signals. The observed labeling pattern thus suggested that the cFos-LacZ mouse model would be appropriate for the analyses of FDG+ and pERK+ cells. We subsequently performed i.h. injections of FDG alone or in conjunction with training and extinction (Fig. 3A). Whereas injection of FDG alone did not cause an increase of immunofluorescent signals above baseline (Fig. 3B, top), injection of FDG after fear conditioning resulted in a significant increase of FDG+ signals in the CA1 hippocampal subfield, F(2,38) = 43.93, p < 0.001, that persisted over 5 consecutive days with or without exposure to extinction trials (Fig. 3B, middle and bottom). The FDG puncta were smaller than typical cFos nuclear labeling, probably because the substrate becomes diluted by diffusion through the brain tissue. Accordingly, a control experiment using i.c.v. injection of FDG revealed signals of large nuclear size (similar to cFos) in the lateral septal area adjacent to the ventricles, followed by a size reduction within the hipocampal tissue (Fig. 3C). FDG injections did not cause a lesion within the dorsal hippocampus, as revealed by cresyl violet staining (Fig. 3D). The FDG+ signals were specific (Fig. 3E) and nuclear, as revealed by their colocalization with cFos (Fig. 3F) and the nuclear counterstain DAPI (Fig. 3G). Because in 98% of the counted nuclei (from a total of 500 CA1 neurons randomly picked from sections of different experiments) we detected only one FDG punctum/nucleus, each punctum was counted as one cell. The somatonuclear pERK immunostaining, upregulated in CA1 cells of the extinction group (F(2,38) = 87.53, p < 0.001) (Fig. 4A,B; Fig. S2A, available at www.jneurosci.org as supplemental material) showed less then 10% overlap with FDG. Namely, the number of FDG+/pErk+ neurons was not significantly elevated above baseline levels, F(2,38) = 0.757, p = 0.47 (Fig. 4A–C), as determined by both conventional and confocal microscopy. This is unlikely the result of pErk localization in interneurons, because pErk, as it is well known for cFos, was predominantly increased in excitatory, principal CA1 neurons (Fig. 4D). We also examined whether the absence of pErk signals was caused by inhibitory effects of cFos-LacZ or FDG on Erk activation within individual cells. In a separate group of mice, we injected FDG i.c.v. to obtain FDG signals in the cortical areas adjacent to the hippocampus. Contrary to the hippocampus, FDG+ and pErk+ signals in the parietal cortex exhibited 42% overlap (7–9 double positive cells from 20 pErk+ cells/0.1 mm2) in this brain area (Fig. 4E; Fig. S2B, available at www.jneurosci.org as supplemental material). These findings indicated that cFos-LacZ did not interfere with Erk activity. Rather, segregated subsets of excitatory cFos+ and pErk+ CA1 neurons responded to fear conditioning and extinction, respectively. Thus, Erk activity triggered by nonreinforced trials most likely did not involve modifications of the conditioning fear memory but instead processed a new extinction memory.
Figure 3.

Long-term labeling of cFos-LacZ+ neurons activated by fear conditioning. A, Experimental design for cFos-LacZ labeling. Four aCSF injections were performed once a day for four consecutive days to habituate the cFos response to the injection procedure before FDG administration. Control mice were injected with FDG alone without prior training or subsequent contextual exposures. Mice of the training group were injected with FDG immediately after training and left undisturbed for 5 consecutive days. The extinction group consisted of mice exposed to training, FDG injection, and five extinction trials. B, FDG signals (green) were weak in the control group (top). Note strong FDG signals in the groups injected with FDG immediately after training with (middle panel) or without extinction (bottom panel). Background was subtracted using the same threshold for each section to eliminate interference of autofluorescence. C, Control section obtained from a mouse with FDG injected into the brain ventricles showing the effect of a diffusion gradient from the ventricles into the hippocampal tissue on the size of FDG+ signals. The size of the signals was smaller than was observed in vitro or after surface application (Nirenberg and Cepko, 1993), probably because of dilution after diffusing within the brain tissue. The size of FDG signals decreased from 10 μm (neurons close to the chorioid plexus) to 0.5–3 μm (100 μm laterally within the hippocampal tissue). D, Image of cresyl violet staining showing an intact CA1 area immediately dorsal to the injection site. E, Micrographs showing lack of cFos-LacZ+ labeling under control conditions when primary (left) or secondary (middle) antibody (Ab) was omitted or FDG was not injected (right). F, Micrographs showing nuclear colocalization of FDG and cFos signals. G, Using DAPI as a nuclear counterstain, we determined that most nuclei contained one FDG punctum. Red arrows, cFos+ cells; blue arrows, DAPI+ nuclei.
Figure 4.

Conditioning and extinction of fear activate segregated cFos+ and pErk+ cell subsets. A, Confocal images reveal stably labeled cFos-LacZ (FDG+) cells in mice exposed to training or training followed by extinction, but not in untrained mice injected with FDG alone. cFos-LacZ and pErk were upregulated (**p < 0.001 vs FDG control) in nonoverlapping cell subsets after conditioning and extinction, respectively, as revealed by lack of FDG and pErk colabeling. n = 41 (13–15 per group); error bars = mean ± SEM. The naive group is presented as a dashed line. B, Representative micrograph showing very low overlap between pErk (red, occasionally white because of intense fluorescence) and FDG (green) nuclear signals (only one indicated cell in the field is pErk+/FDG+). C, Orthogonal projections along the FDG-positive cell. The projections are marked with an arrow along the YZ (ii–iv) and XZ (v–vii) planes. The channels for FDG (green; ii and v), pERK (red; iii and vi) and pERK + FDG (overlay; iv–vii) are presented. Arrows indicate FDG-positive cell nuclei. D, Confocal images excluding pErk+ labeling of CA1 interneurons. Micrographs obtained during confocal microscopy of a representative section labeled for pErk (red) and GAD65,67 (green) after extinction were overlaid revealing lack of colocalization. E, Confocal images demonstrating colocalization of FDG and pErk signals in the parietal cortex. Dashed squares outlined cells or puncta for colocalization analysis; green stars, FDG+ cells; yellow stars, pErk+/FDG+ cells; yellow arrow, overlays of pErk+ fibers and FGD puncta (these signals were not counted as nuclear colocalization).
Given that in some conditioning paradigms one-trial training is insufficient to trigger ceiling behavioral and molecular responses (Navarro et al., 2000), a possibility remained that the used experimental approach did not allow for optimal codetection of cFos and pErk (each showing upregulation in 9–15% of CA1 principal cells). By increasing the number of training trials or shock intensity, we did not see further enhancement of conditioned freezing (Fig. S3A,B, available at www.jneurosci.org as supplemental material). Notably, the use of a 1.5 mA shock resulted in slower extinction, indicating a stronger fear memory, however this effect was accompanied by reduced pErk and unaltered cFos responses of CA1 neurons (Fig. S3C, available at www.jneurosci.org as supplemental material). So far, we were not able to identify conditioning/extinction conditions causing increased levels of both cFos and pErk thus enhancing the odds of their codetection. Nevertheless, the consistent lack of overlap in the one-trial conditioning paradigm is strongly suggestive of disparate cell populations upregulating cFos and pErk in the hippocampus as opposed to control brain areas.
Selective regulation of pErk+ neurons by afferent cholinergic hippocampal input from the medial septum
We next studied the regulation of pErk and cFos responses by hippocampal input. Two key inputs from the entorhinal cortex and medial septum (Wheal and Miller, 1980) are thought to provide the hippocampus with signals on actual and predicted events, respectively (Buhusi and Schmajuk, 1996; Gray and McNaughton, 2000; Vinogradova, 2001). Because the latter signals have been selectively implicated in extinction of conditioned responses (Gray and McNaughton, 2000), we hypothesized that elimination of hippocampal cholinergic input from the medial septum would be critical for pErk upregulation and fear extinction. To test this hypothesis, we performed cholinergic lesions of the medial septum. The toxin SAP, conjugated to a rabbit anti-mouse antibody recognizing the low affinity p75 neurotrophin receptor expressed on cholinergic neurons was injected into the medial septum (Fig. 5A,B). The injection caused a significant loss of cholinergic markers within the medial septum and cholinergic denervation of the hippocampus (Fig. 5C,G), as determined at the end of the experiment for each mouse. Twenty days after injection of toxin or immunotoxin, mice with septohippocampal cholinergic lesions did not exhibit alterations of activity (t18 = 1.32, p = 0.83) shock response (t18 = 1.44, p = 0.79) or freezing behavior during training (Fig. 5D,E). The mice acquired contextual fear normally but did not show fear extinction even after multiple nonreinforced trials (Fig. 5E) when compared with control mice injected with unconjugated SAP (Group × Extinction interaction: F(6,84) = 3.693, p < 0.01), alone or training followed by extinction. Notably, the lesions did not affect cFos responses triggered by fear conditioning, t4 = 0.43, p = 0.65, but significantly attenuated pErk responses, t12 = 10.966, p < 0.01, triggered by extinction (Fig. 4F,G). On the basis of these findings, the septal cholinergic input was identified an important regulator of the hippocampal pErk+ cell subset and fear extinction.
Figure 5.

Lesions of medial septal cholinergic neurons impair contextual fear extinction and Erk signaling while leaving fear conditioning and cFos responses intact. A, Experimental design describing the timing of surgery, toxin injection, training and extinction. B, Localization of injection sites of SAP and μ p75-SAP in the medial septum. C, Immunostaining for AChE (top) in the medial septum of SAP and μ p75-SAP-injected mice. Sections were collected immediately posterior to the injection site. The same sections counterstained with cresyl violet (bottom) do not reveal gross cell loss in the medial septum. LS, lateral septum; MS, medial septum. D, Mice with μ p75-SAP-induced cholinergic lesions did not show alterations of exploratory activity or shock responses during training. E, Permanent immunotoxic lesion of cholinergic cells of the medial septum initiated 20 d before training (n = 7 per group) preserved fear conditioning but impaired fear extinction (**p < 0.01). F, Anatomical level of the dorsal hippocampus (left) used for quantification of the number of cFos+ (middle) and pErk+ (right) CA1 neurons after training and extinction, respectively, in sections of mice injected with SAP- and μ p75-SAP into the medial septum. Note specific impairment of the pErk response (***p < 0.01). Error bars = mean ± SEM. G, Effects of septally injected μ p75-SAP on AChE (n = 10 per group), pErk (n = 7 per group), and cFos immunoreactivity (n = 3 per group) in the hippocampus. μ p75-SAP produced hippocampal cholinergic denervation (left) resulting in intact responses of cFos+ cells after training (middle) but impaired responses of pErk+ cells after extinction (right).
Discussion
By using stable visualization of cFos-LacZ+ cells, we established that hippocampal principal neurons responding to fear conditioning (cFos+) and extinction (pErk+) represented nonoverlapping cell subsets. Together with data demonstrating selective regulation of pErk+ neurons by afferent cholinergic input, we isolated and extinction-specific mechanism involving distinctive molecular, cellular and circuit regulation when compared with cFos-dependent conditioning mechanisms. Although the data do not rule out the possibility that pErk mechnisms may exhibit cellular overlap with cFos-independent mechanisms, the observed dissociation from cFos is significant given its strong causal association to fear conditioning (Fleischmann et al., 2003).
Taking into consideration the established role of Erk signaling in both conditioning (Atkins et al., 1998) and extinction (Szapiro et al., 2003), it was surprising that pErk+ cells activated by extinction represented a segregated neuronal subset. Codetection of cFos/FDG and pErk might have been reduced because we used single trial fear conditioning, nevertheless this paradigm conditions were sufficient to trigger robust behavioral and molecular effects during both conditioning and extinction. We therefore hypothesize that the observed specificity is primarily based on the subcellular localization of pErk. Namely, whereas transient somatonuclear increase of pErk has been reported in some fear conditioning paradigms (Trifilieff et al., 2006; Sindreu et al., 2007), we and others have shown that hippocampal responses to synaptic potentiation (Winder et al., 1999), contextual aversive conditioning (Sananbenesi et al., 2002; Feld et al., 2005), or stress hormones (Kovalovsky et al., 2002) primarily require somatodendritic pErk. Extinction, however, triggers rapid, robust and sustained somatonuclear Erk activation (Fischer et al., 2007). Immunobolot studies further reveal that Erk phosphorylation triggered by contextual extinction is significantly faster and stronger when compared with fear conditioning, revealing maximal levels shortly before and during initial extinction while returning to baseline levels once extinction has been fully established (Fischer et al., 2007; Ryu et al., 2008). Because cellular and subcellular localization constraining Erk's proximity to interacting molecules is a critical determinant of its actions (Sweatt, 2004; Schrick et al., 2007), the unique contribution of this kinase to fear extinction versus conditioning may therefore rely on sustained nuclear activity in the absence of co-upregulation of cFos, cAMP response element binding protein (Tronson et al., 2008), Cdk5 (Sananbenesi et al., 2007), and possibly other molecules required for conditioning but not extinction of fear. The down-stream effects of nuclear pErk, likely to involve distinctive gene responses and structural modifications of the identified neuronal subset, remain to be elucidated.
The dissociation of the identified hippocampal mechanism underlying extinction versus conditioning of fear was additionally documented by specific regulation via medial septal cholinergic input. As has been previously reported for the immediate early gene Arc (Fletcher et al., 2007), cFos and somatodendritic pErk were unaffected by permanent immunotoxin-induced lesions of cholinergic afferents. The levels of somatonuclear pErk, however, were significantly reduced. Accordingly, mice acquired conditioned fear normally but showed resistance to extinction. These findings provide strong molecular and cellular support for the comparator theory (Buhusi and Schmajuk, 1996; Gray and McNaughton, 2000; Vinogradova, 2001) proposing that the hippocampal responses to mismatch between anticipated and delivered reinforcement provided by septal and cortical inputs, respectively, is critical for extinction of conditioned responses. Interestingly, the septal cholinergic input facilitates somatic but depresses dendritic field potentials of hippocampal pyramidal neurons (Rovira et al., 1982). Such effects could account for the sustained somatonuclear Erk activity during extinction when compared with conditioning of fear. The identification of the main cholinergic receptor subtypes contributing to hippocampal Erk activation will help to further develop specific tools serving to facilitate fear extinction.
Together, our findings are consistent with recent observations obtained by electrophysiological recordings identifying separate amygdalar excitatory neurons activated during conditioning and extinction of fear (Herry et al., 2008). However, whereas the activity of those cells has been predominantly linked to the expression fear, as revealed by high correlation with freezing behavior, the hippocampal formation is unlikely to be directly involved in fear regulation. Accordingly, the increase of pErk activity in the hippocampal CA1 neurons occurs in a specific time window shortly preceding the decline of freezing and lasting only until extinction has been established (Fischer et al., 2007; Ryu et al., 2008). These findings strongly suggest that pErk activation within a distinctive subset of principal cells initiates new extinction learning. Lack of pErk signals in interneurons additionally indicated that this learning process involves excitatory but not inhibitory neurotransmission. Based on the known connections of the hippocamal CA1 subfield, the identified hippocampal Erk+ neurons may provide input to the prefrontal cortex and basolateral amygdala thereby initiating broad neuronal alterations within the fear extinction circuitry.
Footnotes
This work was supported by National Institute of Mental Health Grant MH073669 and Dunbar Funds to J.R. We thank Dr. James I. Morgan (St. Joseph Hospital, Memphis, TN) for providing frozen embryos of cFos-LacZ mice, Warren Tourtelotte and the Northwestern University Transgenic Core Facility for deriving the cFos-LacZ mouse lines from frozen embryos, Dr. Eva Redei for reading and discussing this manuscript, and Dan Sylvester for assistance with the preparation of this manuscript.
References
- Atkins CM, Selcher JC, Petraitis JJ, Trzaskos JM, Sweatt JD. The MAPK cascade is required for mammalian associative learning. Nat Neurosci. 1998;1:602–609. doi: 10.1038/2836. [DOI] [PubMed] [Google Scholar]
- Berlau DJ, McGaugh JL. Enhancement of extinction memory consolidation: the role of the noradrenergic and GABAergic systems within the basolateral amygdala. Neurobiol Learn Mem. 2006;86:123–132. doi: 10.1016/j.nlm.2005.12.008. [DOI] [PubMed] [Google Scholar]
- Bouton ME. Context and behavioral processes in extinction. Learn Mem. 2004;11:485–494. doi: 10.1101/lm.78804. [DOI] [PubMed] [Google Scholar]
- Brown HE, Garcia MM, Harlan RE. A two focal plane method for digital quantification of nuclear immunoreactivity in large brain areas using NIH-image software. Brain Res Brain Res Protoc. 1998;2:264–272. doi: 10.1016/s1385-299x(98)00003-8. [DOI] [PubMed] [Google Scholar]
- Buhusi CV, Schmajuk NA. Attention, configuration, and hippocampal function. Hippocampus. 1996;6:621–642. doi: 10.1002/(SICI)1098-1063(1996)6:6<621::AID-HIPO6>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Chen X, Garelick MG, Wang H, Lil V, Athos J, Storm DR. PI3 kinase signaling is required for retrieval and extinction of contextual memory. Nat Neurosci. 2005;8:925–931. doi: 10.1038/nn1482. [DOI] [PubMed] [Google Scholar]
- Chhatwal JP, Myers KM, Ressler KJ, Davis M. Regulation of gephyrin and GABAA receptor binding within the amygdala after fear acquisition and extinction. J Neurosci. 2005;25:502–506. doi: 10.1523/JNEUROSCI.3301-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falls WA, Miserendino MJ, Davis M. Extinction of fear-potentiated startle: blockade by infusion of an NMDA antagonist into the amygdala. J Neurosci. 1992;12:854–863. doi: 10.1523/JNEUROSCI.12-03-00854.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feld M, Dimant B, Delorenzi A, Coso O, Romano A. Phosphorylation of extra-nuclear ERK/MAPK is required for long-term memory consolidation in the crab Chasmagnathus. Behav Brain Res. 2005;158:251–261. doi: 10.1016/j.bbr.2004.09.005. [DOI] [PubMed] [Google Scholar]
- Fischer A, Sananbenesi F, Schrick C, Spiess J, Radulovic J. Distinct roles of hippocampal de novo protein synthesis and actin rearrangement in extinction of contextual fear. J Neurosci. 2004;24:1962–1966. doi: 10.1523/JNEUROSCI.5112-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A, Radulovic M, Schrick C, Sananbenesi F, Godovac-Zimmermann J, Radulovic J. Hippocampal Mek/Erk signaling mediates extinction of contextual freezing behavior. Neurobiol Learn Mem. 2007;87:149–158. doi: 10.1016/j.nlm.2006.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleischmann A, Hvalby O, Jensen V, Strekalova T, Zacher C, Layer LE, Kvello A, Reschke M, Spanagel R, Sprengel R, Wagner EF, Gass P. Impaired long-term memory and NR2A-type NMDA receptor-dependent synaptic plasticity in mice lacking c-Fos in the CNS. J Neurosci. 2003;23:9116–9122. doi: 10.1523/JNEUROSCI.23-27-09116.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher BR, Baxter MG, Guzowski JF, Shapiro ML, Rapp PR. Selective cholinergic depletion of the hippocampus spares both behaviorally induced Arc transcription and spatial learning and memory. Hippocampus. 2007;17:227–234. doi: 10.1002/hipo.20261. [DOI] [PubMed] [Google Scholar]
- Gray J, McNaughton N. The neuropsychology of anxiety. Ed 2. Oxford: Oxford UP; 2000. [Google Scholar]
- Grillon C. Startle reactivity and anxiety disorders: aversive conditioning, context, and neurobiology. Biol Psychiatry. 2002;52:958–975. doi: 10.1016/s0006-3223(02)01665-7. [DOI] [PubMed] [Google Scholar]
- Herry C, Ciocchi S, Senn V, Demmou L, Müller C, Lüthi A. Switching on and off fear by distinct neuronal circuits. Nature. 2008;454:600–606. doi: 10.1038/nature07166. [DOI] [PubMed] [Google Scholar]
- Isiegas C, Park A, Kandel ER, Abel T, Lattal KM. Transgenic inhibition of neuronal protein kinase A activity facilitates fear extinction. J Neurosci. 2006;26:12700–12707. doi: 10.1523/JNEUROSCI.2743-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Lee S, Park K, Hong I, Song B, Son G, Park H, Kim WR, Park E, Choe HK, Kim H, Lee C, Sun W, Kim K, Shin KS, Choi S. Amygdala depotentiation and fear extinction. Proc Natl Acad Sci U S A. 2007;104:20955–20960. doi: 10.1073/pnas.0710548105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovalovsky D, Refojo D, Liberman AC, Hochbaum D, Pereda MP, Coso OA, Stalla GK, Holsboer F, Arzt E. Activation and induction of NUR77/NURR1 in corticotrophs by CRH/cAMP: involvement of calcium, protein kinase A, and MAPK pathways. Mol Endocrinol. 2002;16:1638–1651. doi: 10.1210/mend.16.7.0863. [DOI] [PubMed] [Google Scholar]
- Lee SH, Choi JH, Lee N, Lee HR, Kim JI, Yu NK, Choi SL, Lee SH, Kim H, Kaang BK. Synaptic protein degradation underlies destabilization of retrieved fear memory. Science. 2008;319:1253–1256. doi: 10.1126/science.1150541. [DOI] [PubMed] [Google Scholar]
- Maren S, Quirk GJ. Neuronal signalling of fear memory. Nat Rev Neurosci. 2004;5:844–852. doi: 10.1038/nrn1535. [DOI] [PubMed] [Google Scholar]
- Matsuo N, Reijmers L, Mayford M. Spine-type-specific recruitment of newly synthesized AMPA receptors with learning. Science. 2008;319:1104–1107. doi: 10.1126/science.1149967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milad MR, Quirk GJ. Neurons in medial prefrontal cortex signal memory for fear extinction. Nature. 2002;420:70–74. doi: 10.1038/nature01138. [DOI] [PubMed] [Google Scholar]
- Navarro M, Spray KJ, Cubero I, Thiele TE, Bernstein IL. cFos induction during conditioned taste aversion expression varies with aversion strength. Brain Res. 2000;887:450–453. doi: 10.1016/s0006-8993(00)03032-8. [DOI] [PubMed] [Google Scholar]
- Nirenberg S, Cepko C. Targeted ablation of diverse cell classes in the nervous system in vivo. J Neurosci. 1993;13:3238–3251. doi: 10.1523/JNEUROSCI.13-08-03238.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang M, Thomas SA. A requirement for memory retrieval during and after long-term extinction learning. Proc Natl Acad Sci U S A. 2005;102:9347–9352. doi: 10.1073/pnas.0502315102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson SL, Pittenger C, Morozov A, Martin KC, Scanlin H, Drake C, Kandel ER. Some forms of cAMP-mediated long-lasting potentiation are associated with release of BDNF and nuclear translocation of phospho-MAP kinase. Neuron. 2001;32:123–140. doi: 10.1016/s0896-6273(01)00443-3. [DOI] [PubMed] [Google Scholar]
- Radulovic J, Kammermeier J, Spiess J. Relationship between fos production and classical fear conditioning: effects of novelty, latent inhibition, and unconditioned stimulus preexposure. J Neurosci. 1998;18:7452–7461. doi: 10.1523/JNEUROSCI.18-18-07452.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radulovic J, Rühmann A, Liepold T, Spiess J. Modulation of learning and anxiety by corticotropin-releasing factor (CRF) and stress: differential roles of CRF receptors 1 and 2. J Neurosci. 1999;19:5016–5025. doi: 10.1523/JNEUROSCI.19-12-05016.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reijmers LG, Perkins BL, Matsuo N, Mayford M. Localization of a stable neural correlate of associative memory. Science. 2007;317:1230–1233. doi: 10.1126/science.1143839. [DOI] [PubMed] [Google Scholar]
- Rovira C, Cherubini E, Ben-Ari Y. Opposite actions of muscarinic and nicotinic agents on hippocampal dendritic negative fields recorded in rats. Neuropharmacology. 1982;21:933–936. doi: 10.1016/0028-3908(82)90087-9. [DOI] [PubMed] [Google Scholar]
- Ryu J, Futai K, Feliu M, Weinberg R, Sheng M. Constitutively active Rap2 transgenic mice display fewer dendritic spines, reduced extracellular signal-regulated kinase signaling, enhanced long-term depression, and impaired spatial learning and fear extinction. J Neurosci. 2008;28:8178–8188. doi: 10.1523/JNEUROSCI.1944-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sananbenesi F, Fischer A, Schrick C, Spiess J, Radulovic J. Phosphorylation of hippocampal Erk-1/2, Elk-1, and p90-Rsk-1 during contextual fear conditioning: interactions between Erk-1/2 and Elk-1. Mol Cell Neurosci. 2002;21:463–476. doi: 10.1006/mcne.2002.1188. [DOI] [PubMed] [Google Scholar]
- Sananbenesi F, Fischer A, Wang X, Schrick C, Neve R, Radulovic J, Tsai LH. A hippocampal Cdk5 pathway regulates extinction of contextual fear. Nat Neurosci. 2007;10:1012–1019. doi: 10.1038/nn1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling K, Luk D, Morgan JI, Curran T. Regulation of a fos-lacZ fusion gene: a paradigm for quantitative analysis of stimulus-transcription coupling. Proc Natl Acad Sci U S A. 1991;88:5665–5669. doi: 10.1073/pnas.88.13.5665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrick C, Fischer A, Srivastava DP, Tronson NC, Penzes P, Radulovic J. N-cadherin regulates cytoskeletally associated IQGAP1/ERK signaling and memory formation. Neuron. 2007;55:786–798. doi: 10.1016/j.neuron.2007.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sindreu CB, Scheiner ZS, Storm DR. Ca2+-stimulated adenylyl cyclases regulate ERK-dependent activation of MSK1 during fear conditioning. Neuron. 2007;53:79–89. doi: 10.1016/j.neuron.2006.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeyne RJ, Schilling K, Robertson L, Luk D, Oberdick J, Curran T, Morgan JI. fos-lacZ transgenic mice: mapping sites of gene induction in the central nervous system. Neuron. 1992;8:13–23. doi: 10.1016/0896-6273(92)90105-m. [DOI] [PubMed] [Google Scholar]
- Sotres-Bayon F, Bush DE, LeDoux JE. Emotional perseveration: an update on prefrontal-amygdala interactions in fear extinction. Learn Mem. 2004;11:525–535. doi: 10.1101/lm.79504. [DOI] [PubMed] [Google Scholar]
- Sweatt JD. Mitogen-activated protein kinases in synaptic plasticity and memory. Curr Opin Neurobiol. 2004;14:311–317. doi: 10.1016/j.conb.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Szapiro G, Vianna MR, McGaugh JL, Medina JH, Izquierdo I. The role of NMDA glutamate receptors, PKA, MAPK, and CAMKII in the hippocampus in extinction of conditioned fear. Hippocampus. 2003;13:53–58. doi: 10.1002/hipo.10043. [DOI] [PubMed] [Google Scholar]
- Trifilieff P, Herry C, Vanhoutte P, Caboche J, Desmedt A, Riedel G, Mons N, Micheau J. Foreground contextual fear memory consolidation requires two independent phases of hippocampal ERK/CREB activation. Learn Mem. 2006;13:349–358. doi: 10.1101/lm.80206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronson NC, Schrick C, Fischer A, Sananbenesi F, Pagès G, Pouysségur J, Radulovic J. Regulatory mechanisms of fear extinction and depression-like behavior. Neuropsychopharmacology. 2008;33:1570–1583. doi: 10.1038/sj.npp.1301550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinogradova OS. Hippocampus as comparator: role of the two input and two output systems of the hippocampus in selection and registration of information. Hippocampus. 2001;11:578–598. doi: 10.1002/hipo.1073. [DOI] [PubMed] [Google Scholar]
- Wheal HV, Miller JJ. Pharmacological identification of acetylcholine and glutamate excitatory systems in the dentate gyrus of the rat. Brain Res. 1980;182:145–155. doi: 10.1016/0006-8993(80)90837-9. [DOI] [PubMed] [Google Scholar]
- Winder DG, Martin KC, Muzzio IA, Rohrer D, Chruscinski A, Kobilka B, Kandel ER. ERK plays a regulatory role in induction of LTP by theta frequency stimulation and its modulation by beta-adrenergic receptors. Neuron. 1999;24:715–726. doi: 10.1016/s0896-6273(00)81124-1. [DOI] [PubMed] [Google Scholar]