

Abstract
Purpose of review
The purpose of the present review is to describe new, innovative strategies of diagnosing and treating specific human cancers using a cadre of radiolabeled regulatory peptides.
Recent findings
Peptide receptor-targeted radionuclide therapy is a method of site-directed radiotherapy that specifically targets human cancers expressing a cognate receptor-subtype in very high numbers. Ideally, the procedure targets only the primary or metastatic disease and is minimally invasive, with little radiation damage to normal, collateral tissues. For treatment strategies of this type to be effective, it is critical to evaluate the toxicity of the treatment protocol, the radiation dosimetry of the therapeutic regimen, and the biological profile of the radiopharmaceutical, including biodistribution and pharmacokinetics of the drug. Site-directed molecular imaging procedures via γ-scintigraphy can address many of the critical issues associated with peptide receptor-targeted radionuclide therapy and it is, therefore, necessary to describe the effective balance between the clinical benefits and risks of this treatment strategy.
Summary
Continued development in the design or chemical structure of radiolabeled, biologically active peptides could do much to improve the targeting ability of these drugs, thereby creating new and innovative strategies for diagnosis or treatment of human cancers.
Keywords: molecular imaging, peptides, regulatory, therapy
Introduction
Molecular imaging is defined as the visualization, characterization, and measurement of biological processes at the molecular and cellular levels in humans and other living systems. Molecular imaging is a noninvasive procedure that involves clinical in-vivo administration of a biologically active, receptor-specific, targeting vector (i.e., peptides, small proteins, antibody fragments, or intact antibodies) conjugated to a radioligand, nanoparticle, and/or fluorescent/magnetic resonance imaging (MRI) probe followed by acquisition and quantification of signal by positron emission tomography (PET), single photon emission computed tomography (SPECT), MRI, fluorescence imaging, or ultrasound [1].
Radiolabeled, regulatory peptides continue to hold promise for early diagnosis or treatment of human disease [2•]. The interest in radiolabeled peptides for molecular imaging or peptide receptor-targeted radionuclide therapy (PRTRT) stems from their ability to be easily produced, rapid clearance from whole blood, ease of penetration into the tumor vascular endothelium, relatively low immunogenicity, and rapid excretion from the human body [1]. Furthermore, radiolabeled regulatory peptides can be chemically tuned to target cell-surface receptors that tend to be expressed in very high numbers on human cancer cells, offering the advantage of identifying and treating diseased human tissue with minimal collateral damage to neighboring, normal tissues [2•].
Peptide-based targeting vectors for molecular imaging or PRTRT (Fig. 1) are composed of a targeting vector (regulatory peptide), pharmacokinetic modifier (hydrophilic aliphatic or amino acid tethering molecule), bifunctional chelating agent (BFCA, a complexing agent capable of producing a kinetically-inert, in-vivo radiometal/ligand complex while linking it to the regulatory peptide), and radiometal [γ-emitting or positron-emitting (γ or β+, Table 1) diagnostic or β-emitting or α-emitting (β− or α, Table 2) therapeutic radionuclide] [3].
Figure 1.
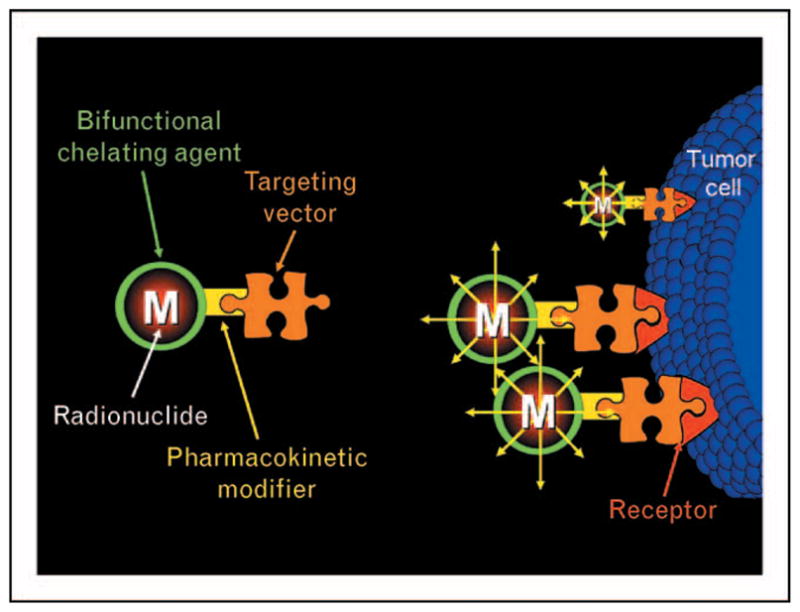
A representative outline of a peptide-based targeting vector for molecular imaging or therapy of specific human cancers
Table 1.
Metal-based radionuclides (γ and β+ ) for molecular imaging
| Radionuclide | Production/availability | Decay | Eγ/Eβ+ (keV) |
|---|---|---|---|
| Cu-64 | Cyclotron | EC, β−, and β+ | γ, 1345.8; β−, 578, β+, 651 |
| Ga-66 | Cyclotron | EC and β+ | γ, 1039.3 and 2752.2; β+, 4153 |
| Ga-67 | Cyclotron | EC | γ, 93.3, 184.6, and 300.2 |
| Ga-68 | Cyclotron | EC and β+ | γ, 1077.3; β+, 1899 |
| Y-86 | Cyclotron | EC and β+ | γ, 1076.7, 627.8, and 1153.1; β+, 1248 |
| Tc-99m | 99Mo/99mTc Generator | IT | γ, 142.7 |
| In-111Cycl | Cyclotron | EC | γ, 171.3 and 245.4 |
| Pb-203 | Cyclotron | Y | γ, 279.2 |
Table 2.
Metal-based radionuclides (α and β−) for peptide receptor-targeted radionuclide therapy
| Radionuclide | Production/availability | Decay | Eβ−/Eα(MeV) |
|---|---|---|---|
| Y-90 | 90Sr/90Y Generator | β− | β−, 2.27 |
| Rh-105 | Reactor | β− and γ | β−, 0.566, 0.248; γ, 0.3192 |
| Pm-149 | Reactor | β− and γ | β−, 1.072; γ, 0.286 |
| Sm-153 | Reactor | β− and γ | β−, 0.69, 0.64; γ, 0.1032, 0.0697 |
| Ho-166 | Reactor | β− and γ | β−, 1.855, 1.773; γ, 0.0806, 1.3794 |
| Lu-177 | Reactor | β− and γ | β−, 0.497; γ, 0.2084, 0.1129 |
| Re-188 | 188W/188Re Generator | β− and γ | β−, 2.118, 1.962; γ, 0.155 |
| Re-186 | Reactor | β− and γ | β−, 1.071, 0.933; γ, 0.1372 |
| At-211 | Accelerator | EC, α, and γ | γ, 0.687, 0.6696; α, 5.868 |
| Pb-212 | 224Ra/212Pb Generator | β− and γ | β−, 0.569, 0.335; γ, 0.3000, 0.2386 |
| Bi-212 | 224Ra/212Bi Generator | β−, γ, and α | β−, 2.251; γ, 0.7273; α, 6.051 |
| Bi-213 | 225Ac/213Bi Generator | β−, γ, and α | β−, 1.42, 1.02; γ, 0.4405, 0.3237; α, 5.87, 5.55 |
Herein we report recent investigations that describe advances and new targeting strategies using radiolabeled regulatory peptides for molecular imaging or PRTRT of specific human disease. Specifically, we attempt to highlight somatostatin, gastrin-releasing peptide, and melanocortin receptor-targeting peptides, and recent advances in radiolabeled, multimeric/multivalent regulatory peptides for targeting multiple cell-surface receptor subtypes.
Somatostatin receptor-targeting peptides
To date, radiolabeled analogues of somatostatin (SST) have been the gold standard for peptide-receptor imaging and therapeutic agents. For example, the clinical successes of octreoscan for visualizing primary and meta-static SST receptor-positive tumors have paved the way for exploration and radiolabeling of other biologically active regulatory peptides described herein. SST, a peptide hormone expressed in the central and peripheral nervous systems, exists naturally in two forms: SST-14 and SST-28. SST-14 and SST-28 have a cyclic structure due to the presence of a single disulfide bond. Five known G protein-coupled receptor SST subtypes exist, SSTR1–5. SSTR1–5 are expressed in very high numbers on tumors of neuroendocrine origin, with SSTR2 being the most prolific. Therefore, radiolabeled agents based upon SSTR-targeting peptides are viable candidates for site-directed molecular imaging or PRTRT of neuroendocrine receptor-expressing tumors [3].
SSTR-targeting regulatory peptides have proven their diagnostic and therapeutic efficacy in the clinic and are the benchmark of which all other radiolabeled regulatory peptides should be compared to. Until now, much of the focus for targeting the SSTR2 with radiolabeled SST derivatives has centered upon octreotide [D-Phe-Cys-Phe-D-Trp-Lys-Thr-Cys-Thr(ol)], [Tyr3] octreotide [D-Phe-Cys-Tyr-D-Trp-Lys-Thr-Cys-Thr(ol)], and [Tyr3]-octreotate [D-Phe-Cys-Tyr-D-Trp-Lys-Thr-Cys-Thr]. For many years, PRTRT of primary and metastatic tumors of neuroendocrine origin has focused upon DOTA (1,4,7,10-tetraazacyclotetradodecane −1,4,7,10-tetraacetic acid)-conjugated SSTR-targeting peptides radiolabeled with β−-emitting Y-90 or Lu-177 [3]. Miederer et al. [4], however, have recently evaluated α-emitting 225Ac-DOTATOC for its therapeutic efficacy in neuroendocrine tumors and compared it directly to [177Lu-DOTATOC]. Their studies have found that dose administrations up to 20 kBq had no significant toxicity in rodents. Furthermore, they determined that administration of 12–20 kBq reduced the growth rate of tumors and showed improved therapeutic efficacy over [177Lu-DOTATOC]. Kwekkeboom et al. [5] have evaluated the toxicity, efficacy, and survival of patients presenting with gastroenteropancreatic neuroendocrine tumors that were treated with [177Lu-DOTA-Tyr3]octreotate. Their studies showed that treatment of patients with [177Lu-DOTA-Tyr3]octreotate presented few adverse effects, including myelodysplastic syndrome and temporary, non-fatal, liver toxicity. Furthermore, they determined that tumor response and survival compared favorably to a limited number of alternative treatment strategies. Muros et al. [6] have recently evaluated the response and outcomes of five patients presenting with advanced neuroendocrine tumors upon being treated with either [90Y-DOTATOC] or [177Lu-DOTATATE]. In this clinical trial, they determined that the average survival was of approximately 28 months with a good-to-excellent quality of life response being observed in all patients. These recent studies and others have shown that PRTRT of neuroendocrine tumors via radiolabeled SSTR-targeting peptides is clearly a success and should be considered a model for future treatment strategies for tumors of this general type.
Until recently, clinical evaluation in human patients has been limited to SSTR-targeting agonist ligands. Agonist ligands elicit a biological response from the cell and are effectively endocytosed and internalized upon binding to the receptor. Antagonist ligands, on the other hand, are not internalized and have previously been deemed inappropriate for targeted molecular imaging and PRTRT. Recent studies with SSTR-targeting antagonist ligands, however, have indicated that the preferable use of agonist ligands for in-vivo molecular imaging and PRTRT clearly bears reconsideration [1]. Anderson and coworkers have recently reported synthesis of [CB-TE2A-sst2-ANT] (Fig. 2) that has been radiolabeled with Cu-64. This new conjugate is a potentially useful SSTR antagonist for PET imaging of SSTR-positive tumors. In this study, they determined that [64Cu-CB-TE2A-sst2-ANT] showed higher tumor-to-blood and tumor-to-muscle ratios as compared with [64Cu-CB-TE2A-Y3-TATE], an SSTR2-positive agonist. MicroPET/CT imaging of male Lewis rats bearing AR42J tumors showed high-quality diagnostic images with excellent tumor-to-background contrast at 4 h postintravenous injection [7•]. In addition, Cescato et al. [8] recently reported a series of new DOTA-conjugated SSTR2 antagonists that exhibit antagonist-like behavior in vitro. Pertaining to this original work, Reubi and Maecke [2•] have recently indicated that a clinical trial using a radiolabeled DOTA-conjugated SSTR2 antagonist is currently underway.
Figure 2.

The chemical structure of [CB-TE2A-sst2-ANT]-targeting vector
Gastrin-releasing peptide receptors
Gastrin-releasing peptide (GRP) receptors are present in high concentrations in human cancers such as prostate, breast, pancreatic, and small cell lung carcinoma [9••]. Mammalian bombesin receptors are G protein-coupled, seven-transmembrane receptors having the capacity to be endocytosed upon binding by an effective agonist ligand. There are three known mammalian bombesin receptor subtypes: BB1 (neuromedin B receptors, NMBR), BB2 (gastrin-releasing peptide receptors, GRPRs), and BB3 (BRS-3) [10]. To date, design and development of radio-labeled agents for the mammalian bombesin receptors have focused on the highly prolific BB2 receptor subtype targeted by bombesin peptide (BBN) or BBN-like analogues. BBN is the 14 amino acid amphibian peptide analogue of the 27 amino acid mammalian regulatory peptide GRP. BBN and GRP share a homologous 7 amino acid amidated C-terminus, [-WAVGHLM-NH2], which is essential for high-affinity receptor binding to GRPR. The ability of GRPR agonists to be rapidly internalized coupled with a high incidence of GRPR expression on various neoplasias has been a driving force for design and development of new diagnostic and therapeutic agents targeting GRPR-positive tumors [11•].
[BBN(7–14)NH2] agonist has been the primary focus of many research groups. 64Cu-labeled [BBN(7–14)NH2] radiopharmaceuticals have been of interest due to the ideal nuclear characteristics of Cu-64, making them useful for in-vivo molecular imaging and PRTRT. Hoffman and Smith [12•] and Prasanphanich et al. [13•] have radiolabeled [NO2A-8-Aoc-BBN(7–14)NH2] conjugate (Fig. 3) with Cu-64 that has resulted in tumor uptake of 3.59 ± 0.70 %ID/g at 1 h postintravenous injection in PC-3 tumor-bearing severe combined immunodeficient (SCID) mice and 2.27 ± 0.08 %ID/g at 1 h postintravenous injection in T-47D tumor-bearing SCID mice. High-quality, high-contrast microPET images were obtained in both mouse models. Another successful [BBN(7–14)NH2] conjugate is [177Lu-AMBA], developed at Bracco for diagnostic imaging and treatment of meta-static disease. Biodistribution studies on PC-3 tumor-bearing mice showed that [177Lu-AMBA] had very low kidney uptake (2.95 and 0.91 %ID/g, 1 and 24 h postintravenous injection, respectively) and high tumor accumulation (6.35 and 3.39 %ID/g, 1 and 24 h postintravenous injection, respectively) [14]. Currently, [177Lu-AMBA] is undergoing phase I clinical trials as a systemic radiotherapy for hormone refractory prostate cancer [11•].
Figure 3.

The chemical structure of [NO2A-8-Aoc-BBN(7–14)NH2]-targeting vector
Modification of [BBN(7–14)NH2] to improve receptor-targeting ability is also of high priority. For example, Schweinsberg et al. [15] have synthesized [99mTc-(CO)3-Ala(NTG)-βAla-βAla-BBN(7–14)NH2], where Leu13 and Met14 have been replaced by Cha and Nle, respectively. This peptide modification has been reported to increase metabolic stability in human plasma and PC-3 cells. The use of the polar glycated linker had beneficial pharmacokinetic effects by significantly reducing liver accumulation (0.6 vs. 2.4 %ID/g, 1.5 h postintravenous injection), increasing PC-3 tumor uptake (3.6 %ID/g, 1.5 h), and improving tumor-to-background ratios in vivo (17-fold at 1.5 h postintravenous injection).
The use of BBN antagonists has begun the latest trend in GRPR targeting with BBN. Cescato et al. [9••] have explored the use of the antagonist, [N4-[D-Phe6,Leu-NHEt13,des-Met14]bombesin(6–14)] (demobesin 1). Their results showed comparable in-vitro binding to that of the agonists, [BBN(1–14)NH2] and [N4-[Pro1,Tyr4,N-le14]bombesin] (demobesin 4), in GRPR-transfected HEK293 cells, PC-3 cells, and human prostate cancer specimens. In PC-3 cells at 4 and 24 h, [99mTc-demobesin 1] uptake was four-fold and two-fold better than that of [99mTc-demobesin 4]. In fact, when compared in PC-3 tumors, [99mTc-demobesin 1] labels GRPRs more intensely and longer than any other GRPR agonist [2•]. Reubi and colleagues [2•,9••] suggest that GRPR antagonists might be the future in bombesin radiopharmaceuticals.
Melanocortin receptor-targeting peptides
α-Melanocyte stimulating hormone (α-MSH) is a trideca-peptide of the general structure [Ac-S1YSMEH-FRWGKPV13-NH2] that regulates skin pigmentation in most vertebrates [16•,17]. Biological activity of α-MSH in vivo is mediated by virtue of interactions with the G protein-coupled melanocortin 1 receptor (MC1R), one of five known subtypes of melanocortin receptors. α-MSH receptors have been identified to be present on melanoma cell lines and on human melanoma tissue samples, catalyzing efforts to identify radiolabeled α-MSH peptide analogues for targeted imaging and therapy of melanocortin receptor-expressing tumors [16•,17–20].
Recent studies for specific targeting of the MC1R have been reported predominantly by Quinn and colleagues [16•,17–20]. Much of their original work has focused on the cyclic regulatory peptide CCSMH, [[Cys3,4,10, D-Phe7,Arg11]αMSH3–13]. The presence of a nonradioactive rhenium metal center coordinated to sulfhydryl functional groups on the cysteine residues affords cyclization of the peptide, making it highly resistant to chemical and proteolytic degradation without compromising the biological integrity of the molecule [17,18].
Miao et al. [16•] have previously described a successful PRTRT study involving the α-emitting conjugate [212Pb-DOTA-Re(Arg11)CCMSH], wherein they were able to cure 45% of B16/F1 murine melanoma-bearing C57 mice in a 120-day study. Recent studies by this group report the ‘matched pair’ [203Pb-DOTA-Re(Arg11) CCMSH] analogue. ‘Matched pairs’ offer the unique opportunity to use information derived from routine patient diagnostic SPECT or PET studies to determine the degree of receptor density on primary and metastatic tissues prior to the administration of the corresponding therapeutic analogue. In this way, treatment is only administered to patients previously demonstrating expression of the target receptor. Furthermore, the diagnostic radiopharmaceutical can be invaluable in prescreening receptor-positive patients for therapy with respect to drug pharmacokinetics, receptor density, and patient dosimetry, potentially reducing or eliminating unsuccessful radiotherapeutic regimens. In this study, they reported very high tumor accumulation (12.00 ± 3.20 % ID/g, 1 h postintravenous injection) and retention (3.43 ± 1.12 % ID/g, 48 h postintravenous injection) of radioactivity in B16/F1 melanoma-bearing C57 mice. The SPECT resolution of Pb-203 was approximately 1.6 mm and very similar to Tc-99m. High-quality, high- contrast microSPECT diagnostic images were obtained in B16/F1 melanoma-bearing C57 mice, demonstrating the effectiveness of [203Pb-DOTA-Re(Arg11)CCMSH] to be used as a ‘matched pair’ radiopharmaceutical for [212Pb-DOTA-Re(Arg11)CCMSH] [16•].
Cutler et al. have reported the microwave synthesis of [68Ga-DOTA-Re(Arg11)CCMSH]. Ga-68 is an ideal radio-nuclide for identification of human tumors or metastatic disease via clinical PET. In this study, they reported the microwave synthesis of high-specific activity [68Ga-DOTA-Re(Arg11)CCMSH]. The new conjugate displayed very high-tumor accumulation (6.27 ± 1.60 % ID/g, 1 h postintravenous injection) and retention (5.85 ± 1.32 % ID/g, 4 h postintravenous injection) of radioactivity in tumor and produced high-resolution, high-contrast micro-PET images in B16/F1 melanoma-bearing C57 mice [17].
Wie et al. [18] have recently described the preparation of [CHX-A″-Re(Arg11)CCMSH] (Fig. 4) and radiosynthesis of corresponding In-111, Y-86, and Ga-68 conjugates. CHX-A″ is a poly(aminocarboxylate) chelating ligand for lanthanide or lanthanide-like radioelements based upon DTPA (diethylaminetriaminepentaacetic acid). CHX-A″ can be radiolabled using milder conditions and under shorter incubation periods as compared with DOTA. In this study, the new conjugates displayed high tumor uptake and retention in B16/F1 melanoma-bearing C57 mice with 4.17 ± 0.94 (In-111), 4.68 ± 1.02 (Y-86), and 2.68 ± 0.69 (Ga-68) %ID/g present in tumor at 4 h postintravenous injection, respectively. While high-resolution, high-contrast microPET images were obtained in rodent models, tumor accumulation was moderated by low-specific activity radiolabeled peptide.
Figure 4.

The chemical structure of [CHX-A-Re(Arg11)CCMSH]-targeting vector
New and improved radiolabeling and targeting strategies for the MC1R on melanoma are clearly warranted. Alternative to CCSMH-targeting vector, Miao et al. [19] and Guo et al. [20] have recently reported lactam bridge-cyclized α-MSH peptide analogues for diagnostic molecular imaging of melanoma. In these studies, they reported high tumor uptake and retention of radioactivity in either B16/F1 or B16/F10 melanoma-bearing C57 mice. Although high-resolution, high-contrast microPET images of primary and metastatic disease were obtained for the new conjugates, the results do not appear to be altogether superior to the metal-cyclized [Re(Arg11) CCMSH] regulatory peptide analogues. Additionally, Raposinho et al. [21,22] have used a pyrazolyl ligand framework to stabilize the low-valent, fac-[99mTc(CO)3]+ metal center for potential SPECT imaging of MC1R-positive melanoma tumors. Lastly, Bapst et al. [23] have evaluated glycosylated DOTA-α-MSH analogues radiolabeled with In-111. They have determined that high-tumor uptake and low-renal retention of conjugate in B16/F1 melanoma-bearing B6D2F1 female mice clearly brings impetus for design and development of new radiolabeled regulatory peptides for targeted diagnosis and PRTRT of MC1R-positive human tumors.
Arginine–glycine–aspartic acid multimeric receptor-targeting peptides
Angiogenesis is the physiological process for growth of new blood vessels from existing ones and is a regulatory mechanism for transition of tumor tissue from a dormant to a malignant or metastatic state [24]. Integrins are cell-surface transmembrane glycoproteins existing as αβ heterodimers. αvβ3 and αvβ5-integrin subtypes are expressed on the endothelial cells of tumor neovasculature during angiogenesis and form the basis of investigations for molecular imaging and PRTRT of angiogenesis and tumor formation in vivo. The αvβ3-integrin is known to be expressed in very high numbers in many tumor cell types, including lung carcinomas, osteosarcomas, breast cancer, and glioblastomas [25]. Radiolabeled peptides containing the amino acid sequence [Arg-Gly-Asp] (RGD) are nonregulatory peptides that have been used extensively to target αvβ3 receptors upregulated on tumor cells and neovasculature, therefore providing a molecular vehicle for early detection of rapidly growing tumors and metastatic disease [25].
The clinical utility of monomeric radiolabeled peptides can be limited by a number of factors, including receptor density, binding affinity, and pharmacokinetics. For example, high-quality, high tumor-to-background PET or SPECT images require a high degree of receptor expression on tumor cells as compared with normal, collateral tissue. The number of effective receptors may differ significantly during tumor development resulting in significantly reduced uptake and retention of targeting vector, limiting the quality of the diagnostic image. Also, the binding affinity of monomeric peptides can be relatively low as compared with multimeric-targeting vectors [26•]. Cyclic RGD-containing peptide analogues are not considered to be a member of the regulatory peptide family [2•]. Nonetheless, radiolabeled dimeric or tetrameric cyclic RGD peptides have shown significant improvements to the αvβ3-integrin-binding domain as compared with radiolabeled monomeric RGD-targeting vectors and their use does bear some discussion. For example, Liu et al. [27] have shown the Ga-68 NOTA (1,4,7-triazacyclononane-1,4,7-triacetic acid) labeled dimer, [E[c(RGDyK)]2], and tetramer, [E{E[c(RGDyK)]2}2], to have improved tumor uptake in U87MG glioblastoma xenografts when compared with the Ga-68 NOTA labeled monomer, [c(RGDyK)]. Radio-labeled monomeric regulatory peptides may also be limited by pharmacokinetic considerations. For example, clearance properties and excretion rates of targeting vector may limit the diagnostic imaging utility of a specific monomeric probe [26•]. For these reasons, multi-meric or multivalent regulatory peptide probes have recently become a new avenue for diagnostic molecular imaging and PRTRT of tumors expressing either singly or multitargetable receptors [24,25,26•,27–29].
Much of the work of Liu et al. [25,26•,28] has focused on multimeric regulatory peptide probes. Their focus has centered on RGD/BBN multimeric peptide conjugates for integrin/GRPR dual receptor imaging, as many GRPR-positive tumors are also αvβ3-integrin-positive. Most recent work of Liu et al. [26•,28] has focused on PC-3 tumor uptake in nude mice with Ga-68 or Cu-64 radio-labeled [NOTA-RGD-BBN(7–14)NH2] (Fig. 5), wherein the RGD and BBN-targeting motifs are linked by a glutamic acid. Their studies showed [68Ga-NOTA-RGD-BBN(7–14)NH2] and [68Ga-NOTA-BBN(7–14) NH2] having comparable uptake at all time-points in xeno-grafted PC-3 tumors [26•]. [68Ga-NOTA-RGD] monomer, on the other hand, showed much lower accumulation. At 2 h postintravenous injection, PC-3 tumor uptake was shown to be approximately 4.0, 3.2, and 0.80 %ID/g for [68Ga-NOTA-RGD-BBN(7–14)NH2], [68Ga-NOTA-BBN(7–14)NH2], and [68Ga-NOTA-RGD], respectively. Clearance of the peptide heterodimer was primarily via the renal/urinary excretion pathway (about 5.0 %ID/g, 2 h post-intravenous injection). Furthermore, uptake and retention of conjugate in hepatic tissue for [68Ga-NOTA-RGD-BBN(7–14)NH2] was significantly improved as compared with [68Ga-NOTA-BBN(7–14)NH2] [26•]. Some degree of improvement in retention of tracer was seen by radiolabeling these conjugates with Cu-64 radionuclide. For example, at 20 h postintravenous injection, PC-3 tumor retention was 2.04 ± 0.35, 0.44 ± 0.39, and 0.55 ± 0.32 %ID/g for [64Cu-NOTA-RGD-BBN(7–14)NH2], [64Cu-NOTA-BBN(7–14)NH2], and [64Cu-NOTA-RGD], respectively. Once again, clearance of the peptide 64Cu-heterodimer was primarily through the kidneys (1.87 ± 0.41 %ID/g, 20 h postintravenous injection). This new conjugate also exhibited lower liver retention (0.98 ± 0.40 %ID/g, 20 h post-intravenous injection) as compared with BBN (4.20 ± 0.53 %ID/g, 20 h postintravenous injection) and RGD (1.67 ± 0.69 %ID/g, 20 h postintravenous injection) monomeric conjugate. All tracers cleared quickly from the blood, with less than 0.75 %ID/g remaining at 30 min postintra-venous injection [28].
Figure 5.

The chemical structure of [NOTA-BBN-RGD] heterodimer-targeting vector
Another multimeric regulatory peptide probe currently being evaluated is RGD/α-MSH. Yang et al. [29] have developed [99mTc-RGD-Lys-(Arg11)CCMSH] for dual imaging of integrin and MC1R-expressing tumors. Bio-distribution studies on B16/F1 melanoma tumor-bearing C57 mice have shown high accumulation (14.83 ± 2.94 %ID/g, 2 h postintravenous injection) and prolonged retention (7.59 ± 2.04 %ID/g, 24 h postintravenous injection) of conjugate in tumor tissue. These values are 1.1 and 2.3 times higher than those observed with [99mTc-(Arg11)CCMSH]. Although tumor uptake was favorable, it was overshadowed by the massive kidney uptake (67.12 ± 8.79 %ID/g, 2 h postintravenous injection) and retention (40.26 ± 10.83 %ID/g, 24 h post-intravenous injection). At 4 h postintravenous injection, the renal accumulation of [99mTc-RGD-Lys-(Arg11) CCMSH] was approximately 12.5 times the renal uptake of [99mTc-(Arg11)CCMSH]. They suggest that the positive charge on the lysine residue [between the RGD and (Arg11)CCMSH moiety] contributed to electrostatic interaction between the positively charged peptide and the negatively charged tubule cells, resulting in the higher renal uptake [29].
Conclusion
For more than a decade, the field of nuclear medicine has been investigating the usage of radiolabeled regulatory peptides for molecular targeting of receptors highly expressed on specific human tumors. The clinical successes of octreoscan have paved the way for exploration and radiolabeling of other biologically active regulatory peptides and some of them have been described herein. Other cancer types that express regulatory peptide receptors in very high numbers include neuropeptide-Y (NPY) receptors in breast tumors, glioblastomas, and sarcomas; glucagon-like-peptide-1 (GLP-1) receptors in insulinomas; cholecystokinin-2 (CCK2) receptors in medullary thyroid cancers; and neurotensin receptors in pancreatic cancers [2•]. Continued design and development of multi-meric or multivalent peptides capable of targeting multiple receptor subtypes highly expressed on human cancers could do much to improve image resolution and contrast, all but eliminating the high false-positive rates and nontarget uptake that oftentimes limits some of the clinically approved radiopharmaceuticals from widespread usage for diagnosis of malignant tissues. Furthermore, the ability to target specific receptors highly expressed on the surfaces of human cancer cells creates the opportunity to use PRTRT as a highly selective treatment strategy for tumor targeting or as a mechanism for this treatment strategy to complement traditional, clinically useful chemotherapeutic regimens of treatment.
Acknowledgments
The present review is the result of work supported with resources and the use of facilities at the Radiopharmaceutical Sciences Institute within the Harry S. Truman Memorial Veterans’ Hospital, Columbia, MO 65201 and the University of Missouri School of Medicine, Columbia, MO 65211, USA. This work was funded in part by The United States Department of Veterans’ Affairs VA Merit Award and the National Institutes of Health (5T32EB004822).
References and recommended reading
Papers of particular interest, published within the annual period of review, have been highlighted as:
• of special interest
•• of outstanding interest
Additional references related to this topic can also be found in the Current World Literature section in this issue (p. 103).
- 1.Jong MD, Breeman WAP, Kwekkeboom DJ, et al. Tumor imaging and therapy using radiolabeled somatostatin analogues. Acc Chem Res. 2009;42:873–880. doi: 10.1021/ar800188e. [DOI] [PubMed] [Google Scholar]
- 2•.Reubi JC, Maecke HR. Peptide-based probes for cancer imaging. J Nucl Med. 2008;49:1735–1738. doi: 10.2967/jnumed.108.053041. A review of regulatory peptide probes currently used for cancer imaging. [DOI] [PubMed] [Google Scholar]
- 3.Khan IU, Beck-Sickinger AG. Targeted tumor diagnosis and therapy with peptide hormones as radiopharmaceuticals. Anticancer Agents Med Chem. 2008;8:186–199. doi: 10.2174/187152008783497046. [DOI] [PubMed] [Google Scholar]
- 4.Miederer M, Henriksen G, Alke A, et al. Preclinical evaluation of the α-particle generator nuclide 225Ac for somatostatin receptor radiotherapy of neuroendocrine tumors. Clin Cancer Res. 2008;14:3555–3561. doi: 10.1158/1078-0432.CCR-07-4647. [DOI] [PubMed] [Google Scholar]
- 5.Kwekkeboom DJ, de Herder WW, Kam BL, et al. Treatment with the radio-labeled somatostatin analog [177Lu-DOTA0, Tyr3]octreotate: toxicity, efficacy, and survival. J Clin Oncol. 2008;26:2124–2130. doi: 10.1200/JCO.2007.15.2553. [DOI] [PubMed] [Google Scholar]
- 6.Muros MA, Varsavsky M, Rozas PI, et al. Outcome of treating advanced neuroendocrine tumors with radiolabeled somatostatin analogues. Clin Transl Oncol. 2009;11:48–53. doi: 10.1007/s12094-009-0310-5. [DOI] [PubMed] [Google Scholar]
- 7•.Wadas TJ, Eiblmaier M, Zheleznyak A, et al. Preparation and biological evaluation of 64Cu-CB-TE2A-sst2-ANT, a somatostatin antagonist for PET imaging of somatostatin receptor-positive tumors. J Nucl Med. 2008;49:1819–1827. doi: 10.2967/jnumed.108.054502. The study evaluates 64Cu-CB-TE2A-sst2-ANT as a potential PET radiopharmaceutical for SSTR2-positive tumors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cescato R, Erchegyi J, Waser B, et al. Design and in vitro characterization of highly sst2-selective somatostatin antagonists suitable for radiotargeting. J Med Chem. 2008;51:4030–4037. doi: 10.1021/jm701618q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9••.Cescato R, Maina T, Nock B, et al. Bombesin receptor antagonists may be preferable to agonists for tumor targeting. J Nucl Med. 2008;49:318–326. doi: 10.2967/jnumed.107.045054. [99mTc-labeled] antagonistic demobesin 1 showed more pronounced uptake in PC-3 tumors than that of 99mTc-labeled agonistic demobesin 4, suggesting that the future of GRPR targeting lies in bombesin antagonists. [DOI] [PubMed] [Google Scholar]
- 10.Jensen RT, Battey JF, Spindel ER, et al. Mammalian bombesin receptors: nomenclature, distribution, pharmacology, signaling, and functions in normal and disease states. Pharmacol Rev. 2008;60:1–42. doi: 10.1124/pr.107.07108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11•.Thomas R, Chen J, Roudier MM, et al. In vitro binding evaluation of 177Lu AMBA, a novel 177Lu-labeled GRP-R agonist for systemic radiotherapy in human tissues. Clin Exp Metastasis. 2009;26:105–119. doi: 10.1007/s10585-008-9220-0. Pharmacological analysis of 177Lu-AMBA justifies its use for clinical application in GRP and NMB receptor-positive tumors. [DOI] [PubMed] [Google Scholar]
- 12•.Hoffman TJ, Smith CJ. True radiotracers: Cu-64 targeting vectors based upon bombesin peptide. Nucl Med Biol. 2009;36:579–585. doi: 10.1016/j.nucmedbio.2009.03.007. A review of the recent developments in Cu-64 radiochemistry and radiopharmaceutical design based upon bombesin. [DOI] [PubMed] [Google Scholar]
- 13•.Prasanphanich AF, Retzloff L, Lane SR, et al. In vitro and in vivo analysis of [64Cu-NO2A-8-Aoc-BBN(7–14)NH2]: a site-directed radiopharmaceutical for positron-emission tomography imaging of T-47D human breast cancer tumors. Nucl Med Biol. 2009;36:171–181. doi: 10.1016/j.nucmedbio.2008.11.005. Evaluation of 64Cu-bombesin radiopharmaceutical uptake in T-47D tumors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lantry LE, Cappelletti E, Maddalena ME, et al. 177Lu-AMBA: synthesis and characterization of a selective 177Lu-labeled GRP-R agonist for systemic radiotherapy of prostate cancer. J Nucl Med. 2006;47:1144–1152. [PubMed] [Google Scholar]
- 15.Schweinsberg C, Maes V, Brans L, et al. Novel glycated [99mTc(CO)3]-labeled bombesin analogues for improved targeting of gastrin-releasing peptide receptor-positive tumors. Bioconjugate Chem. 2008;19:2432–2439. doi: 10.1021/bc800319g. [DOI] [PubMed] [Google Scholar]
- 16•.Miao Y, Figueroa SD, Fisher DR, et al. 203Pb-labeled α-melanocyte-stimulating hormone peptide as an imaging probe for melanoma detection. J Nucl Med. 2008;49:823–829. doi: 10.2967/jnumed.107.048553. Development of a 203Pb-labeled α-MSH SPECT imaging agent for its potential use as a matched pair for α-therapy with 212Pb-labeled α-MSH. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cantorias MV, Figueroa SD, Quinn TP, et al. Development of high-specific-activity 68Ga-labeled DOTA-rhenium-cyclized α-MSH peptide analog to target MC1 receptors overexpressed by melanoma tumors. Nucl Med Biol. 2009;36:505–513. doi: 10.1016/j.nucmedbio.2009.01.017. [DOI] [PubMed] [Google Scholar]
- 18.Wie L, Zhang X, Gallazzi F, et al. Melanoma imaging using 111In-, 86Y- and 68Ga-labeled CHX-A″-Re(Arg11)CCMSH. Nucl Med Biol. 2009;36:345–354. doi: 10.1016/j.nucmedbio.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miao Y, Gallazzi F, Guo H, Quinn TP. 111In-labeled lactam bridge-cyclized α-melanocyte stimulating hormone peptide analogues for melanoma imaging. Bioconjugate Chem. 2008;19:539–547. doi: 10.1021/bc700317w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo H, Shenoy N, Gershman BM, et al. Metastatic melanoma imaging with an 111In-labeled lactam bridge-cyclized α-melanocyte-stimulating hormone peptide. Nucl Med Biol. 2009;36:267–276. doi: 10.1016/j.nucmedbio.2009.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Raposinho PD, Xavier C, Correia JDG, et al. Melanoma targeting with α-melanocyte stimulating hormone analogs labeled with fac-[99mTc(CO)3]+: effect of cyclization on tumor-seeking properties. J Biol Inorg Chem. 2008;13:449–459. doi: 10.1007/s00775-007-0338-3. [DOI] [PubMed] [Google Scholar]
- 22.Raposinho PD, Correia JDG, Alves S, et al. A 99mTc(CO)3-labeled pyrazolyl-α-melanocyte-stimulating hormone analog conjugate for melanoma targeting. Nucl Med Biol. 2008;35:91–99. doi: 10.1016/j.nucmedbio.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 23.Bapst JP, Calame M, Tanner H, Eberle AN. Glycosylated DOTA-α-melano-cyte-stimulating hormone analogues for melanoma targeting: influence of the site of glycosylation on in vivo biodistribution. Bioconjugate Chem. 2009;20:984–993. doi: 10.1021/bc900007u. [DOI] [PubMed] [Google Scholar]
- 24.Shi J, Kim YS, Zhai S, et al. Improving tumor uptake and pharmacokinetics of 64Cu-labeled cyclic RGD peptide dimers with Gly3 and PEG4 linkers. Bioconjugate Chem. 2009;20:750–759. doi: 10.1021/bc800455p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu Z, Yan Y, Chin FT, et al. Dual integrin and gastrin-releasing peptide receptor targeted tumor imaging using 18F-labeled PEGylated RGD-bombesin heterodimer 18F-FB-PEG3-Glu-RGD-BBN. J Med Chem. 2009;52:425–432. doi: 10.1021/jm801285t. [DOI] [PubMed] [Google Scholar]
- 26•.Liu Z, Niu G, Wang F, Chen X. 68Ga-labeled NOTA-RGD-BBN peptide for dual integrin and GRPR-targeted tumor imaging. Eur J Nucl Med Mol Imaging. 2009;36:1483–1494. doi: 10.1007/s00259-009-1123-z. A study of integrin/GRPR-targeting properties with an RGD–BBN heterodimeric peptide. [DOI] [PubMed] [Google Scholar]
- 27.Liu Z, Niu G, Shi J, et al. 68Ga-labeled cyclic RGD dimers with Gly3 and PEG4 linkers: promising agents for tumor integrin αvβ3 PET imaging. Eur J Nucl Med Mol Imaging. 2009;36:947–957. doi: 10.1007/s00259-008-1045-1. [DOI] [PubMed] [Google Scholar]
- 28.Liu Z, Li ZB, Cao Q, et al. Small-animal PET of tumors with 64Cu-labeled RGD-bombesin heterodimer. J Nucl Med. 2009;50:1168–1177. doi: 10.2967/jnumed.108.061739. [DOI] [PubMed] [Google Scholar]
- 29.Yang J, Guo H, Gallazzi F, et al. Evaluation of a novel Arg-Gly-Asp-conjugated α-melanocyte stimulating hormone hybrid peptide for potential melanoma therapy. Bioconjugate Chem. 2009;20:1634–1642. doi: 10.1021/bc9001954. [DOI] [PMC free article] [PubMed] [Google Scholar]