

Abstract
Several pro-angiogenic/pro-inflammatory factors involved in endometrial cancer are regulated by leptin but the signaling mechanisms responsible for these leptin induced actions are largely unknown. Here we report that in benign (primary and HES) and cancer endometrial epithelial cells, EEC (An3Ca, SK-UT2 and Ishikawa) leptin in a dose-dependent manner regulates vascular endothelial growth factor, (VEGF); interleukin-1 beta, (IL-1β); leukemia inhibitory factor, (LIF) and their respective receptors, VEGFR2, IL-1R tI and LIFR. Remarkably, leptin induces a greater increase in VEGF/VEGFR2 and LIF levels in cancer than in benign cells. However, IL-1β was only increased by leptin in benign primary-EEC. Cancer-EEC expressed higher levels of leptin receptor (full-length OB-Rb and short isoforms) in contrast to benign primary-EEC. Leptin-mediated activation of JAK2 (janus kinase 2) was upstream to the activation of PI-3K (phosphatidylinositol-3 kinase) and/or MAPK (mitogen activated protein kinase) signaling pathways. Leptin induction of cytokines/receptors generally involved JAK2 and MAPK activation but PI-3K phosphorylation was required for leptin increase of LIF, IL-1/IL-1R tI. Leptin-mediated activation of mTOR (mammalian target of Rapamycin), mainly linked to MAPK, played a central role in leptin regulation of all cytokines and receptors. These results suggest that leptin's effects are cell-specific and could confer a proliferative or cell survival advantage or possibly promote endometrial thickness. Leptin's effects on pro-angiogenic molecules were more evident in malignant versus benign cells and may imply that there is an underlying shift in leptin induced cell signaling pathways in endometrial cancer cells.
Keywords: leptin, pro-angiogenesis molecules, endometrial epithelial cells, endometrial cancer, leptin signaling pathways, VEGF, VEGFR2, LIF, LIFR, IL1, IL-1R
INTRODUCTION
Leptin actions are more often than not related to energy balance. However, leptin is also recognized for its contributions to reproduction, angiogenesis, proliferation and inflammation. Therefore it is not surprising that leptin's actions are now being linked to the development and pathogenesis of cancer and endometriosis (1-9).
The leptin sequence is highly conserved among mammalian species (10). In contrast, its receptor, OB-R has several isoforms (11) believed to be produced by alternative splicing. OB-Rb (full-length and functional isoform) and OB-Ra (a short isoform) are main isoforms found in diverse tissues (12), including the endometrium (13-15). Higher concentrations of OB-Ra to OB-Rb in endometrial cancer cells have been described (16) but the implication of this ratio is unclear.
Leptin binds to OB-Rb in an absolute specific way leading to the activation of any one of canonical signaling pathways (17) including JAK2/STAT3, PI-3K/AKT1 (18, 19) and MAPK/ERK1/2 (20). Although not as common, other, kinases such as p38 protein kinase (21) and jun n terminal kinase (JNK) (22), protein kinase C (PKC) and AMP-activated protein kinase (AMPK) (23) are activated in response to leptin in some cell types.
Endometrial epithelial cells (EEC) express OB-R and are targets for leptin actions (13-15, 24). Accumulating evidence support the idea that leptin signaling is essential for the acquisition of endometrial receptivity and successful embryo implantation (25, 26) but aberrant leptin actions have been found involved in pathological conditions (i.e., endometriosis and endometrial cancer) (1, 2, 8, 9, 16, 27-29).
Leptin's pleiotropic actions in the endometrium involve the increase in levels of pro-angiogenic and inflammatory cytokines (i.e., VEGF, LIF and IL-1), their cognate receptors and adhesion molecules (i.e., β3 integrin) (9, 25, 30, 31). In this respect, leptin signaling could be essential for angiogenesis and inflammation required for endometrial cell adhesion, proliferation, survival and migration (1-3, 6, 9, 25, 30-34).
In spite of the increasing evidence supporting leptin's role as an important factor in endometrial biology, there is a lack of information on leptin-mediated signaling mechanisms in benign versus malignant cells. To this end, we have investigated how leptin induced signaling impacts levels of important cytokines/receptors for angiogenesis and inflammation in benign and malignant EEC. Our results suggest leptin triggers specific signaling pathways in a hierarchal fashion and often differentially with respect to the regulation of VEGF, LIF, IL-1 and their cognate receptors in benign compared to malignant EEC. Our results further suggest that malignant-EEC are more sensitive to leptin induced stimulatory effects of pro-inflammatory/pro-angiogenic molecules. The differential responses of malignant EEC to leptin maybe related to a shift in the versatility of endometrial cancer cells.
MATERIALS AND METHODS
Materials
Antibodies for non-phosphorylated (F-2) and phosphorylated STAT3 (p-STAT3, B-7), non-phosphorylated and phosphorylated pAKT1, VEGF-R2 (flk-1 or KDR), all OB-R isoforms (extracellular domain NH2 end) and OB-Rb (long isoform intracellular COOH end), their blocking peptides for competition studies, non-specific species-matched IgGs, HeLa and RAW 264.7 lysates for Western blot positive controls were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Anti phospho-p42/44 MAPK and anti p42/44 MAPK antibodies were purchased from Cell Signaling Technology, Inc (Danvers, MA). Anti β actin antibodies (ab8226) were purchased from Abcam Inc (Cambridge, MA). Specific antibodies IR-Dye-800nm and IR-Dye-800nm were purchased from Rockland (Gilbertsville, PA). Normal serum from mouse, goat, rabbit and horse and biotinylated antibodies were obtained from Biomeda (Foster City, CA) and Vector Laboratories (Burlingame, CA), respectively. ALI 3409 biotinylated antibody was obtained from BioSource International (Camarillo, CA). Human recombinant leptin was obtained from R&D Systems Inc. (Minneapolis, MN). Fetal bovine serum (FBS) was obtained from Gemini Bioproducts (Woodland, CA), and DMEM/F-12 and antibiotic-antimycotic mixtures were purchased from Gibco BRL Products (Gaithersburg, MD). Wortmannin was purchased from Upstate (Charlottesville, VA). Tryphostin AG 490, PD 98,059, Rapamycin and other chemicals were obtained from Sigma (St. Louis, MO). To assess specificity of leptin-induced effects a pan-inhibitor of leptin signaling, leptin receptor antagonist (LPrA1), was synthesized and purified as described elsewhere (26). The peptide was dissolved in sterile filtered vehicle solution [0.04% dimethylsulfoxide (DMSO)-PBS] and kept at 4°C until use.
Endometrial cells
Human endometrial epithelial adenocarcinoma cells (Ishikawa, from ECACC 9832301, Wiltshire, UK grade 1), SK-UT2 (grade 3), An3Ca (grade 3, derived from a lymph node; ATCC), HES (a cell line derived from a benign proliferative endometrium, provided by Dr. Douglas Kniss, Ohio State University, Columbus, OH) and primary EEC cells were used to assess leptin-induced signaling pathways and molecular targets. All primary endometrial samples were collected in accordance with an approved IRB at Massachusetts General Hospital, Boston, MA. The endometrial epithelial cells were isolated from discarded hysterectomy tissue of benign etiologies as previously described (35). Briefly, endometrial tissue was minced and treated with collagenase I (0.1%)–DNAse I, 0.005% for 1 h at 37°C. After gland sedimentation, primary EEC were purified of endometrial stromal cells (ESC) and macrophage contaminants by repeated incubation at 37°C in Falcon flasks. Dispersed epithelial cells were counted with a hemocytometer and cell viability was assessed by optical microscopy using the Trypan Blue exclusion method. The mean cell viability was ≥ 95%. Cell homogeneity (∼92%) was checked in smears using antibodies against cytokeratin (Ck; EEC+), vimentin (Vm; ESC+) and CD45 (leukocyte +) (35).
Treatments
Primary EEC and cell lines were cultured on uncoated flat-bottomed plastic twelve-wells plates for 4 and 7 days, respectively, in DMEM-MCDB105 medium containing 10% FBS; 5 μg/ml insulin, 1% amphotericin B, 100 μg /ml streptomycin and 100 U/ml penicillin until 80% confluence. To eliminate any cytokine effect from the FBS supplement, the cells were washed twice with phosphate-buffered saline (PBS)-2% bovine serum albumin (BSA) and cultured an additional 2 days in DMEM-MCDB105−2% BSA (without serum or insulin; basal medium). Next, the cells were washed out three times with the culture medium and were then incubated for up to 24 h (times indicated in figure legends) in basal medium containing leptin (0, 0.62, 6.25 or 62.5 nM) plus or minus kinase inhibitors (AG 490 for JAK2/STAT3, 30 μM; PD 98,059 for MAPK/ERK1/2, 30 μM; Wortmannin for PI-3K/AKT1, 20 μM (6); and Rapamycin 20 nM for mTOR) (4).
Dependence of phosphorylation and signaling intermediaries on leptin binding to OB-R was assessed in cells cultured in basal medium containing leptin and a pan-inhibitor of leptin signaling (leptin peptide receptor antagonist-1, LPrA1, 300 nM) (26). Negative controls for inhibitor effects included vehicle solutions, either 0.05% ethanol for kinase inhibitors or 0.04% DMSO for LPrA1. After treatment, the cells were washed with ice-cold PBS and homogenized on ice with lysis buffer (20 nM Tris (pH 7.4) containing 137 nM NaCl, 2 mM EDTA, 10% glycerol, 50 nM β-glycerol phosphate, 1% Nonidet P-40, and a mixture of protease and phosphate inhibitors composed of 100 μM antipain, 0.1 mg/ml trypsin inhibitor, protease inhibitor cocktail 1:50 (Sigma), 50 nM NaF, 2 mM phenyl-methylsulfonyl fluoride, and 2 mM sodium orthovanadate). Cellular lysates were centrifuged at 2400 × g at 4°C for 10 min. Lysates were kept at −20°C until ready for use in Western blot analysis. Duplicate wells were run for each treatment and the experiments were repeated at least three times with different cell preparations. Protein concentrations in cell lysates were determined by the Bradford method (Bio-Rad Laboratories, Hercules, CA) (36). BSA was used as standard. Results were expressed as mg/ml.
Western blots
Thirty μg of protein in Laemmli buffer containing β-mercaptoethanol were loaded on 10% SDS-PAGE precast gels (Bio-Rad). Electrophoresis was performed at 220 V for 5 min followed by 165 V for 40 min (Bio-Rad, electrophoresis apparatus) in Tris-glycine buffer (pH 8.4). Electroblotting onto 0.2 μm nitrocellulose membranes was performed at 22 V overnight at 4 °C in 48 nM Tris-39 nM glycine buffer containing 0.037% SDS and 20% methanol. Membranes were washed with 20 mM Tris, 137 mM NaCl (pH 7.4) buffer containing 0.15% Tween 20 (vol/vol) (wash buffer) and incubated for 1 h at room temperature (RT) in blocking buffer (wash buffer containing 5% wt/vol non-fat milk). The membranes were subsequently incubated for 1 h at RT with 1 μg/ml of specific antibodies in wash buffer containing 2.5% horse, goat or mouse normal serum. After washing (four times 5 min/each), membranes were incubated for 1 h at RT with biotinylated secondary antibody. Antigen detection was carried out by incubation in streptavidin-horseradish peroxidase conjugate (Amersham, Piscataway, NJ) diluted 1:2000 in wash buffer for 30 min at RT. Specific bands in the blots were visualized using ECL-chemiluminescent reagent (Amersham) and Imagetek-B film (American X-ray & Medical Supply, Rancho Cordoba, CA). Simultaneous detection of the above antigens with antibodies linked to IR-Dye-800nm (0.2 μg/ml; IRDye800™) and IR-Dye-700nm (0.2 μg/ml; Cy5.5) for Odyssey InfraRed Imagining System (LI-COR, Inc., Lincoln, NE) was also carried-out. Quantitative analyses of Western blot results for antigen expression were evaluated with the NIH Image 1.62 program or Li-COR Odyssey Software and expressed as percent of controls. Electrophoresis and Western blot analysis were repeated three times using cell lysates from at least three independent experiments. Antibodies to anti-β-actin and non-phosphorylated proteins were used as loading controls.
IL-1, VEGF and LIF determinations
Conditioned media from EEC cultured for 24 h were used to quantify the effects of leptin and inhibition treatment on the levels of IL-1β, VEGF and LIF by ELISA (Quantikine, R&D Systems, Minneapolis, MN). Experiments were repeated at least three times. Antigen concentrations from conditioned media were calculated as pg/ml/mg of total protein or expressed as percentage of control. IL-1β, VEGF and LIF concentrations were within the dynamic range of the standard curve. Standards, controls and samples were assayed in duplicate. Intra- and inter-assay coefficients of variation were between 1−2% and 5−8%, respectively. According to the manufacturer the performance characteristics of ELISAs for IL-1β, VEGF and LIF were as follows: 100% specificity; sensitivity of 0.1 pg/ml, <5.0 pg/ml and <8 pg/ml, no significant cross-reactivity or interference with other cytokines and growth factors for both natural and recombinant human IL-1β, VEGF and LIF, respectively.
Data Analysis
A one-way ANOVA test with Dunnet error protection and a confidence interval of 95% was used from the Analyze-it software (Microsoft Excel, Leeds, UK; http://www.analyse-it.com) for data analysis. All data are presented as mean ± SEM. Values for P <0.05 were considered statistically significant. The model included the main effects of treatments and replicates.
RESULTS
Expression of short isoforms and OB-Rb by endometrial epithelial cells
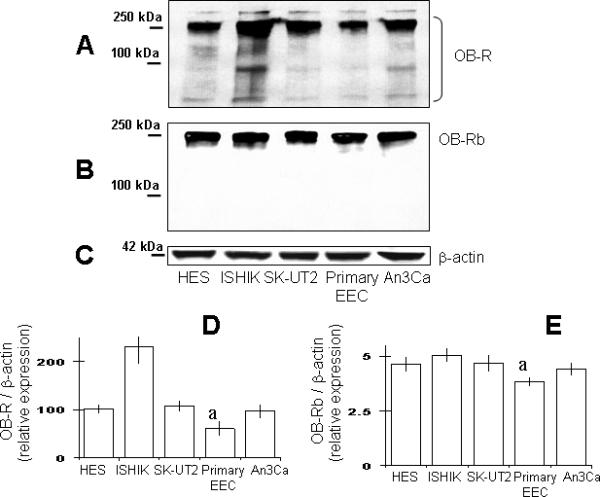
Figure 1 depicts the results from Western blot determination of OB-R isoforms detected with antibodies for extracellular domain (NH2 end) and intracellular (COOH end) domains in extracts from the EEC tested. Analysis of blots showed that all EEC expressed, to some extension, the full-length OB-Rb isoform together with others several shorter OB-R isoforms (see Fig 1A and B). Densitometric analysis suggests that immortalized and cancer EEC express higher detectable quantities of total OB-R (Fig 1D) compared to benign primary EEC. Ishikawa cells showed the highest level of total OB-R. In contrast, primary EEC expressed the lower levels of OB-Rb (Fig 1B and E).
Signaling pathways activated by leptin in endometrial epithelial cells
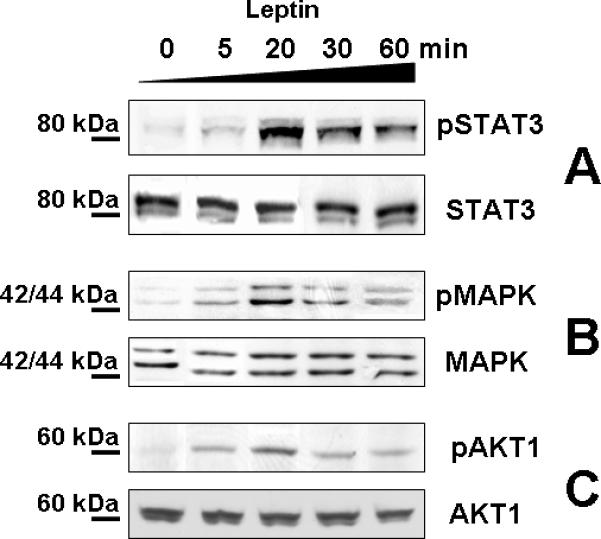
Benign and malignant EEC responded to leptin stimulation by differentially activating the major (canonical) leptin signaling pathways (JAK2/STAT3, MAPK/ERK1/2, PI-3K/AKT). Time-course studies in EEC showed that leptin-induced phosphorylation of STAT3, ERK1/2 and AKT1 was observed by 5 min, peaked by 20 min and decreased after 60 min (Fig. 2). Therefore, further experiments tested leptin effects at 20 min.
The leptin effects were found dose-dependent in all cell tested. Fig 3 shows that addition of increasing concentrations of leptin to the incubation medium of EEC produced a dose-related stimulation of STAT3, ERK1/2 and AKT1 phosphorylation. Maximal increase for different kinases was obtained with 62.5 nmol/l leptin but varies between EEC (130 to 240 %). These leptin induced effects were abrogated with the pan-inhibitor for leptin signaling, LPrA1 (See Fig 3A and B). Kinase inhibition studies using AG490 suggest that leptin-mediated JAK2 phosphorylation was upstream to MAPK and PI-3K phosphorylation (Fig 3C and D). Incubation of EEC with the JAK2 inhibitor alone did not change the basal levels of phosphorylated MAPK (ERK1/2) or AKT1. However, co-incubation of EEC with leptin plus AG490 simultaneously decreased the levels of leptin-induced pSTAT3, pERK1/2 and pAKT1 (See Fig 3C and D). Additional experiments were completed with 62.5 nM leptin.
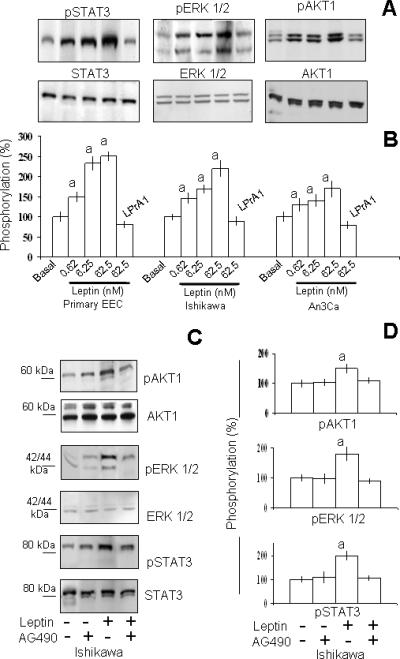
Table 1 shows the relative levels of phosphorylated kinases activated by leptin in EEC. PI-3K/AKT1 and MAPK/ERK1/2 were activated by leptin in all the cells tested. The overall pattern of leptin induced ERK 1/2 or AKT1 phosphorylation did not differ between benign and malignant cells. In primary-EEC leptin induced the greatest increase in pSTAT3. On the other hand, leptin had no effect on pSTAT3 in An3Ca cells (Table 1).
Table 1.
Relative levels of leptin-induced phosphorylated signaling intermediaries in endometrial epithelial cells (EEC)
| Cell type |
pSTAT3 |
pERK 1/2 |
pAKT1 |
|---|---|---|---|
| Primary EEC | 2.4a | 1.9a | 1.3a |
| HES | 1.4a | 2.5a | 1.6a |
| Ishikawa | 1.5a | 2.2a | 1.5a |
| SK-UT2 | 1.4a | 1.3a | 1.4a |
| An3Ca | n.s. | 1.3a | 1.7a |
Note: The data represent quantitative analysis of phosphorylated molecules from benign and cancer EEC after incubation for 20 min with leptin (62.5 nM). Data are derived from densitometry analysis of bands observed on film calculated by the NIH Image program, (http://rsb.info.nih.gov/nih-image) or the Li-COR Odyssey Software. The values were normalized to the non-phosphorylated corresponding protein and expressed on a fold difference from basal expression in vehicle treated cells (control; n ≥ 3). (n.s.) no significant difference with respect to control
P <0.05 significant difference with respect to control.
Leptin induction of VEGF, LIF, IL-1 and their receptors
Leptin signaling differentially impacted on the levels of pro-angiogenic and inflammatory cytokines/receptors in benign compared to malignant EEC (Table 2). Conditioned media from benign cells (primary EEC and HES) had non-detectable amounts of VEGF. Furthermore, treatment with leptin did not affect VEGF levels in conditioned media from cultures of benign EEC (Table 2). In striking contrast, basal levels of VEGF in conditioned media from cancer cells were elevated even under basal conditions when compared to the levels observed in media from benign cells. In cancer cells, leptin induced in a dose-dependent manner a significant increase in VEGF levels (Table 2).
Table 2.
Relative levels of pro-angiogenesis cytokines/receptors induced by leptin in endometrial epithelial cells (EEC)
| Cell type |
VEGF |
VEGFR2 |
LIF |
LIFR |
IL-1 |
IL-1R tl |
|---|---|---|---|---|---|---|
| Primary EEC | n.s. | n.s. | 1.5a | 3.0a | 8.5a | 1.9a |
| HES | n.s. | 1.6a | n.s. | 2.0a | n.s. | 1.4a |
| Ishikawa | 1.7a | 1.8a | 2.2a | 1.5a | n.s. | 2.0a |
| SK-UT2 | 1.5a | 6.2a | n.s. | n.s. | n.s. | 5.0a |
| An3Ca | 1.4a | 1.5a | 4.9a | 7.0a | n.s. | 20a |
Note: The data represent fold differences for quantitative analysis of cytokines determined by ELISA and densitometric calculations from Western blot of cytokine receptors expressed by EEC after incubation for 24 h with leptin (62.5 nM). The Western blot data were normalized to β-actin on a fold difference from control (basal medium; n ≥ 3). (n.s.) no significant difference with respect to control
P <0.05 significant difference with respect to control.
Leptin also induced the expression of VEGFR2 in all cell lines tested except primary-EEC. These leptin-induced effects were dose-dependent (data not shown). Benign-immortalized HES cells responded to leptin stimulation by increasing VEGFR2 expression. This finding suggests that after being immortalized these cells are not entirely normal or that this line has undergone subtle changes during the immortalization process or subsequent continued passages. Notably, leptin induced the most significant increase in VEGFR2 in SK-UT2 cells (more than 6 fold; Table 2).
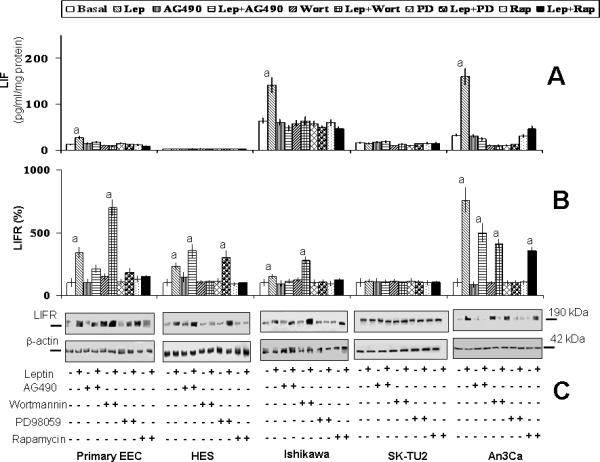
Primary-EEC and two of three cancer EEC tested responded to leptin stimulation as evidenced by elevated levels of LIF (see Table 2). Similarly to the leptin-induced effects on VEGF, cancer cells (Ishikawa and An3Ca) were more responsive to leptin stimulation of LIF than benign cells. In contrast, leptin induced LIFR levels similarly in both benign and malignant cells. An3Ca cells were the most responsive for increasing LIFR expression by leptin actions. However, no significant effects of leptin on LIF or LIFR expression were found in SK-UT2 cells (Table 2).
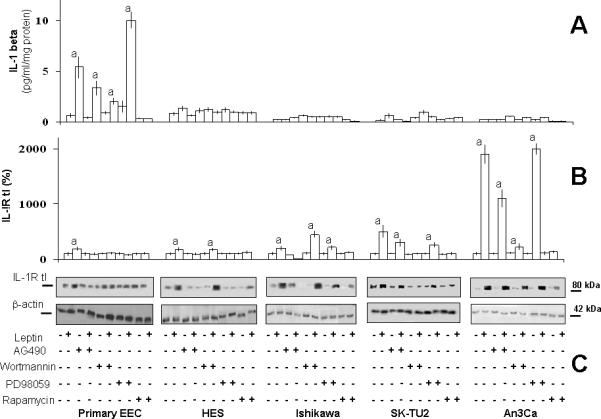
Leptin only significantly increased the levels of IL-1 (>8 fold) in primary EEC (see Table 2). Unlike primary, malignant EEC were refractory to leptin regulation of IL-1 β levels but leptin induced an increase in IL-1R tI in all cell lines. These results suggest that in cancer EEC leptin signaling signature leads to a distinctive increase of VEGF/VEGFR2 and LIF. In contrast, benign primary-EEC mainly responded to leptin by increasing the levels of IL-1β/IL-1R tI and LIFR, and to a slight extent, LIF.
Signaling pathways involved in leptin-induction of VEGF/VEGFR2 in cancer EEC
In order to determine the specific signaling pathways responsible for leptin's increase of VEGF and VEGFR2 in cancer EEC several kinase inhibitors were tested. Inhibition studies using PD98059 show that leptin-mediated activation of MAPK/ERK1/2 was required for increase of VEGF levels in cancer cells (Fig 4A). Inhibition studies from cells incubated with leptin and Wortmannin show that with the exception of Ishikawa cells the activation of PI-3K/AKT1 was not involved in the leptin-mediated increase of VEGF levels. In sharp contrast, Rapamycin-inhibition of mTOR effectively abrogated the leptin-induced increase in VEGF in all cancer cells. These results suggest that in cancer EEC leptin-mediated regulation of VEGF levels does not proceed through the classical signaling pathway PI-3K/AKT1/mTOR. Rather, leptin regulation of VEGF in cancer EEC probably proceeds through a signaling mechanism involving MAPK/ERK1/2/mTOR.
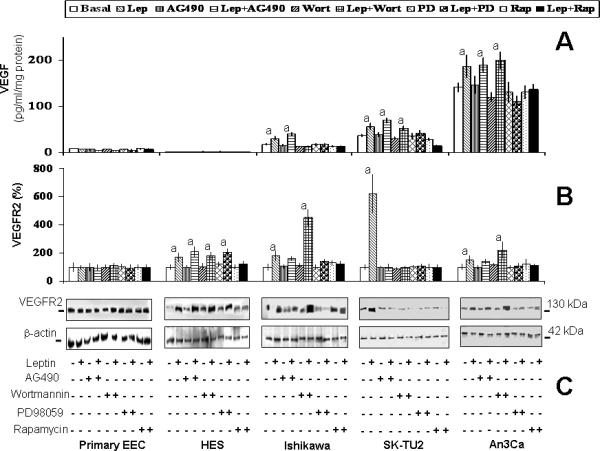
Similar to the findings from leptin regulation of VEGF, leptin did not increase the levels of VEGFR2 in benign primary-EEC. Results from inhibition studies show that JAK2/STAT3, MAPK/ERK1/2 and mTOR were involved in the leptin-mediated increase in VEGFR2 expression in cancer cells. In contrast, none of the signaling pathways appeared to be involved in the regulation of VEGFR2 expression in benign HES cells (see Fig 4B and C) suggesting that leptin regulation of VEGFR2 in immortalized-benign HES cells involves other less common leptin signaling pathways.
Signaling pathways involved in leptin-induction of LIF/LIFR
Leptin induced increase in LIF in primary EEC, Ishikawa and An3Ca was abrogated by all kinase inhibitors tested (Fig 5A). LIFR expression was increased by leptin in all EEC with the exception of SK-UT2 cells (Fig 5B and C). In contrast to the leptin regulation of LIF, activation of JAK2/STAT3 and MAPK/ERK1/2 was mainly involved in leptin-mediated regulation of LIFR expression in primary-EEC, Ishikawa and An3Ca. The activation of mTOR was involved in leptin regulation of LIF and LIFR levels (see Fig 5B and C).
Signaling pathways involved in leptin-induction of IL-1/IL-1R tI
Kinase inhibition studies show that leptin increase of IL-1β levels in primary-EEC was mediated by JAK2/STAT3, PI-3K/AKT1 and mTOR signaling pathways (Fig 6A). However, leptin-mediated increase in IL-1R tI expression in benign primary EEC involved the activation of all the canonical leptin signaling pathways (Fig 6B and C). In contrast, in benign immortalized HES and cancer EEC leptin-mediated increase in IL-1R tI involved the activation of JAK2/STAT3, MAPK/ERK1/2 and mTOR but PI-3K/AKT1 phosphorylation did not show a clear pattern. Notably, the highest levels of IL-1 R tI induced by leptin were seen in SK-UT2 cancer cells.
DISCUSSION
Regulation of energy balance by leptin signaling in the hypothalamus involves the primary activation of JAK2/STAT3 pathway through OB-Rb (full-length isoform). However, other signaling pathways are involved in leptin's pleiotropic effects (angiogenesis, inflammation, proliferation) in peripheral tissues (37). Leptin pro-angiogenesis and pro-inflammatory effects are thought to be critical for endometrial thickness, implantation (25, 26) and endometrial cancer growth (7, 25, 33). However, limited data is available on the signaling mechanisms leading to the leptin-induced effects in EEC. To assess the involvement of leptin-induced signaling pathways in the regulation of levels of target molecules related to angiogenesis and inflammation we investigated time-course and leptin-dose responses in EEC and, used key kinase inhibitors and a pan-inhibitor of leptin/OB-R signaling (LPrA1) (26). Leptin at all doses assayed (0.6−62.5 nM) increases phosphorylation levels of main kinases and levels of cytokines and receptors. Although, 62.5 nmol/l represents a supra physiological dose the data presented were obtained with this concentration to better evaluate the leptin's effects.
Leptin activation of JAK2/STAT3 signaling is associated with boxes 1 and 2, only found in OB-Rb (38-40). Both short OB-Ra and full-length OB-Rb isoforms can activate JAK2 but OB-Rb has the unique capability to activate STAT3 (41). The OB-Ra isoform lacks box 2 and therefore is considered not fully functional, at least with respect to signaling pathway activation. However, OB-Ra is able to activate some intracellular signaling pathways (42). OB-Ra and OB-Rb are able to induce PI-3K and MAPK signaling pathways through box 1 (40, 43).
Benign (13) and cancer EEC express the OB-Rb and several short isoforms (7, 16). In the present investigation primary-EEC showed lower expression of OB-Rb (Fig 1B). Cancer EEC had higher expression of total OB-R (Fig 1A) than benign primary-EEC as was previously reported (16). Leptin-induced phosphorylation of JAK2 in benign and cancer EEC was upstream to the phosphorylation of PI-3K or MAPK signaling pathways (Fig 3) as previous reported (33). The degree of leptin-mediated activation of key kinases in benign or cancer EEC did not always correlate with the levels of induced molecules. The reasons for these facts are unknown but seem unrelated to the levels of expression of OB-Rb or other isoforms. Remarkably, in cancer EEC leptin-mediated activation of mTOR (mainly linked to MAPK) seems to play a central role in the regulation of these factors (Fig 7).
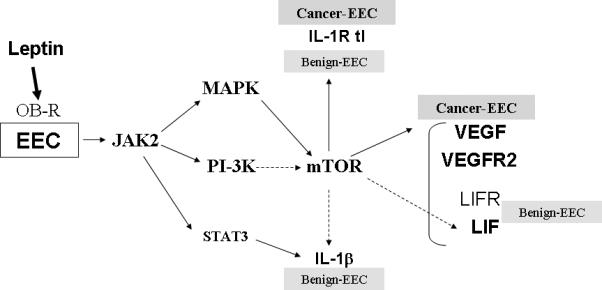
Angiogenesis is involved in hormonal induced endometrial thickening during the menstrual cycle and plays a pivotal role in inflammation and development/progression of cancer (44). Several factors can induce angiogenesis that eventually entails an increase in VEGFR2 levels (45). Leptin, a known mitogenic, inflammatory and angiogenic factor for many tissues (46-48) increases the levels of VEGF/VEGFR2 in cancer cells (6, 49). Consequently, the blockade of leptin signaling decreases the expression of these pro-angiogenic factors and mammary tumor growth (6). Leptin induction of IL-1β/IL-1R tI signaling (50) could lead to the up-regulation of VEGF (51, 52). Leptin mainly increases the levels of VEGF and VEGFR2 in malignant compared to benign EEC (Table 2) and VEGFR2 in immortalized HES. Interestingly, leptin-induction of VEGF levels in cancer-EEC was related to the activation of MAPK/ERK1/2 and mTOR but not to PI-3K/AKT1 signaling pathway (Fig 4A and Fig 7). In particular, An3Ca cells have inactive mutant PTEN (Phosphatase and Tensin homolog, a tumor suppressor and inhibitor of AKT). However, the inhibition of PI-3K (in contrast to the inhibition of mTOR) did not affect leptin-induction of VEGF levels in An3Ca cells. Rapamycin negatively affects phosphorylation of p70S6 kinase and 4E-BP1 protein, cell cycle progression and induces G1 arrest (53). Cytokines/ growth factors may activate mTOR indirectly through PI-3K/AKT1 (54). However, this is not always fully dependent on PI-3K (55, 56) but MAPK/ERK1/2 activity (57). Similarly, in cancer EEC leptin-activation of MAPK/ERK1/2 and mTOR regulate VEGF levels.
Leptin-mediated increase in LIF levels was higher in cancer ECC (3−5 fold) compared to benign primary-EEC (1.5 fold; Table 2). The leptin-induced effects on LIF involve all leptin canonical signaling pathways and mTOR (Fig 5A and Fig 7). However, leptin induction of LIFR was mainly dependent on MAPK/ERK1/2 and mTOR activation (Fig 5B and C; Fig 7). The signaling potential of LIFR may differ depending upon the tissue and cellular response initiated (58). In this respect, leptin-mediated increase of LIFR signaling could oppose the effects of TNF-α (59) and lead to survival and EEC transformation required for cancer growth. LIF, an essential factor for embryo implantation (60), is a marker of endometrial cell transformation (61) involved in inflammation/angiogenesis that induces a variety of disparate biological responses in different cell types. However, the angiogenic effects of LIF reported have been contradicting (62-64).
Leptin only induced IL-1β levels in primary EEC. The increase was mainly dependent on JAK2/STAT3, PI-3K/AKT1 and mTOR signaling (Fig 6A and Fig 7). In contrast, leptin induced IL-1R tI in all EEC through a mechanism that generally includes JAK2/STAT3, MAPK/ERK1/2 and mTOR but no consistent pattern for PI-3K involvement was found (Fig 6B and C; Fig 7). Differential regulation of IL-1 and its receptor by leptin may be instrumental to the development of physiological or pathological processes. Chronic inflammation can induce rapid cell division, increasing the possibility for replication error, ineffective DNA repair, and subsequent mutations that could pave the way to endometrial cancer development (65). IL-1/IL-1R tI signaling could induce leptin and OB-R expression in EEC (30) and LIF/LIFR (25), VEGF (51) and IL-8 expression (66) in cancer EEC. Although, similar levels of IL-1 have been reported in benign and malignant endometrial tissues (67), IL-1 signaling in these tissues could mainly depend on the expression/function of IL-1R tI (68).
The observation that in some cases the inhibition of a particular kinase provoked a further increase in leptin-induced levels of cytokines/receptors (Fig 4B, Fig 5B and Fig 6A) suggests that an active and complex crosstalk between leptin-induced signaling pathways occurs in EEC (6, 69). Leptin activation of JAK2/STAT involves the regulatory effects of CIS family of proteins (SOCS or SSI family) (70). It is conceivable that the inhibition of a particular signaling pathway could have a positive feedback loop for increasing other leptin-induced cytokine signaling pathways by inhibiting the production or levels of negative regulators.
In Summary we have dissected what we believe to be the main signaling mechanisms involved in leptin-mediated increase in pro-angiogenic/pro-inflammatory factors in benign and cancer EEC. Present findings suggest that the regulation of JAK2 and MAPK signaling pathway crosstalk in the activation of mTOR. This could play a central role in leptin pro-angiogenic actions in endometrial cancer. Overall our results suggest that there is a complex level of regulation of leptin signaling pathways required to accomplish leptin's normal biological functions. Among these mechanisms, the relative abundance of OB-R isoforms and crosstalk between key kinases may have a central influence on the hierarchy and effects of leptin-induced signaling in endometrial cells. Overall, deregulation of leptin signaling may contribute to the development of pathological events in EEC. A comprehensive understanding of the complex relationships and regulatory mechanisms of leptin signaling pathways in endometrial cells is needed to help unravel the specific roles of this pleiotropic molecule in physiologic and pathologic events in the endometrium.
FUNDING & DISCLOSURE
This work was supported in part by Grants from the Post-doctoral Program of the Boston Biomedical Research Institute (to C.C.), the Advanced Medical Research Foundation (to B.R.R.), NIH RO1 098333 (to B.R.R.), the Susan G. Komen Foundation for the Cure (to R.R.G.; BC 504370), the Cancer Research and Prevention Foundation (to R.R.G.), the Consortium for Industrial Collaboration in Contraceptive Research (CICCR), a program of Contraceptive Research and Development Program (CONRAD), Eastern Virginia Medical School (to R.R.G.; CIG-02-87; CIG-06-113 and CIG-07-114). All authors wish to declare that they have no conflicts of interest that would prejudice their impartiality and have not received support for the study from any commercial source.
REFERENCES
- 1.Matarese G, Alviggi C, Sanna V, Howard JK, Lord GM, Carravetta C, Fontana S, Lechler RI, Bloom SR, De Placido G. Increased leptin levels in serum and peritoneal fluid of patients with pelvic endometriosis. J Clin End Metab. 2000;85:2483–7. doi: 10.1210/jcem.85.7.6703. [DOI] [PubMed] [Google Scholar]
- 2.Wu MH, Chuang PC, Chen HM, Lin CC, Tsai SJ. Increased leptin expression in endometriosis cells is associated with endometrial stromal cell proliferation and leptin gene up-regulation. Mol Hum Reprod. 2002;8:456–64. doi: 10.1093/molehr/8.5.456. [DOI] [PubMed] [Google Scholar]
- 3.Petridou E, Belechri M, Dessypris N, Koukoulomatis P, Diakomanolis E, Spanos E, Trichopoulos D. Leptin and Body Mass Index in Relation to Endometrial Cancer Risk. Ann Nutr Metab. 2002;46:147–51. doi: 10.1159/000063081. [DOI] [PubMed] [Google Scholar]
- 4.La Cava A, Matarese G. The weight of leptin in immunity. Nat Rev Immunol. 2004;4:371–9. doi: 10.1038/nri1350. [DOI] [PubMed] [Google Scholar]
- 5.Giudice LC, Kao LC. Endometriosis. Lancet (Seminar) 2004;364:1789–99. doi: 10.1016/S0140-6736(04)17403-5. [DOI] [PubMed] [Google Scholar]
- 6.Gonzalez RR, Chefils S, Escobar M, Yoo JH, Carino C, Styer AK, Sullivan BT, Sakamoto H, Olawaiye A, Serikawa T, Lynch MP, Rueda BR. Leptin signaling promotes the growth of mammary tumors and increases the expression of vascular endothelial growth factor (VEGF) and its receptor type two (VEGFR2). J Biol Chem. 2006;281:26320–8. doi: 10.1074/jbc.M601991200. [DOI] [PubMed] [Google Scholar]
- 7.Koda M, Sulkowska M, Wincewicz A, Kanczuga-Koda L, Musiatowicz B, Szymanska M, Sulkowski S. Expression of leptin, leptin receptor, and hypoxia-inducible factor 1 alpha in human endometrial cancer. Signal Transduction Pathways, PTC. 2007;1095:90–8. doi: 10.1196/annals.1397.013. [DOI] [PubMed] [Google Scholar]
- 8.Styer A, Gonzalez RR, Leavis P, Petrozza J, Rueda BR. In Vivo Disruption of Leptin Receptor Function Impairs the Establishment and Viability of Endometriosis-Like Lesions. Fertil Steril. 2005;84:S389–90. [Google Scholar]
- 9.Styer AA, Sullivan BT, Puder M, Arsenault D, Petrozza JC, Serikawa T, Chang S, Hasan T, Gonzalez RR, Rueda BR. Ablation of leptin signaling disrupts the establishment, development of and maintenance of endometriosis-like lesions in a murine model. Endocrinology. 2007;149:506–14. doi: 10.1210/en.2007-1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maffei M, Halaas J, Ravussin E, Pratley RE, Lee GH, Zhang Y, Fei H, Kim S, Lallone R, Ranganathan S, Kern PA, Friedman JM. Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nature Medicine. 1995;1:1155–61. doi: 10.1038/nm1195-1155. [DOI] [PubMed] [Google Scholar]
- 11.Tartaglia LA. The leptin receptor. J Biol Chem. 1997;272:6093–6. doi: 10.1074/jbc.272.10.6093. [DOI] [PubMed] [Google Scholar]
- 12.Baumann H, Morella KK, White DW, Dembski M, Bailon PS, Kim H, La CF, Tartaglia LA. The full-length leptin receptor has signaling capabilities of interleukin 6-type cytokine receptors. Proc Natl Acad Sci U S A. 1996;93:8374–8. doi: 10.1073/pnas.93.16.8374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gonzalez RR, Caballero-Campo P, Jasper M, Mercader A, Devoto L, Simon C. Leptin and leptin receptor are expressed in the human endometrium and endometrial leptin secretion is regulated by the human blastocyst. J Clin Endocrinol Metab. 2000;85:4883–8. doi: 10.1210/jcem.85.12.7060. [DOI] [PubMed] [Google Scholar]
- 14.Gonzalez RR, Simon C, Caballero-Campo P, Norman R, Chardonnens D, Devoto L, Bischof P. Leptin and reproduction. Hum Reprod Update. 2000;6:290–300. doi: 10.1093/humupd/6.3.290. [DOI] [PubMed] [Google Scholar]
- 15.Kitawaki J, Koshiba H, Ishihara H, Kusuki I, Tsukamoto K, Honjo H. Expression of Leptin Receptor in Human Endometrium and Fluctuation during the Menstrual Cycle. J Clin Endocrinol Metab. 2000;85:1946–50. doi: 10.1210/jcem.85.5.6567. [DOI] [PubMed] [Google Scholar]
- 16.Yuan SS, Tsai KB, Chung YF, Chan TF, Yeh YT, Tsai LY, Su JH. Aberrant expression and possible involvement of the leptin receptor in endometrial cancer. Gynecol Oncol. 2004;92:769–75. doi: 10.1016/j.ygyno.2003.11.043. [DOI] [PubMed] [Google Scholar]
- 17.Hekerman P, Zeidler J, Bamberg-Lemper S, Knobelspies H, Lavens D, Tavernier J, Joost HG, Becker W. Pleiotropy of leptin receptor signalling is defined by distinct roles of the intracellular tyrosines. FEBS Journal. 2005;272:109–19. doi: 10.1111/j.1742-4658.2004.04391.x. [DOI] [PubMed] [Google Scholar]
- 18.Ghilardi N, Ziegler S, Wiestner A, Stoffel R, Heim MH, Skoda RC. Defective STAT signaling by the leptin receptor in diabetic mice. Proc Natl Acad Sci U S A. 1996;93:6231–5. doi: 10.1073/pnas.93.13.6231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Y, Kuropatwinski KK, White DW, Hawley TS, Hawley RG, Tartaglia LA, Baumann H. The full-length leptin receptor has signaling capabilities of interleukin 6-type cytokine receptors. J Biol Chem. 1997;272:16216–23. doi: 10.1074/jbc.272.26.16216. [DOI] [PubMed] [Google Scholar]
- 20.Takahashi Y, Okimura Y, Mizuno I, Iida K, Takahashi T, Kaji H, Abe H, Chihara K. Leptin Induces Mitogen-activated Protein Kinase- dependent Proliferation of C3H10T1/2 Cells. J Biol Chem. 1997;272:12897–900. doi: 10.1074/jbc.272.20.12897. [DOI] [PubMed] [Google Scholar]
- 21.Sweeney G. Leptin signalling. Cell Signal. 2002;14:655–63. doi: 10.1016/s0898-6568(02)00006-2. [DOI] [PubMed] [Google Scholar]
- 22.Onuma M, Bub JD, Rummel TL, Iwamoto Y. Prostate cancer cell-adipocyte interaction: leptin mediates androgen-independent prostate cancer cell proliferation through c-Jun NH2-terminal kinase. J Biol Chem. 2003;278:42660–7. doi: 10.1074/jbc.M304984200. [DOI] [PubMed] [Google Scholar]
- 23.Minokoshi Y, Kim YB, Peroni OD, Fryer LG, Müller C, Carling D, Kahn BB. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature. 2002;415:339–43. doi: 10.1038/415339a. [DOI] [PubMed] [Google Scholar]
- 24.Alfer J, Muller-Schottle F, Classen-Linke I, von Rango U, Happel L, Beier-Hellwig K, Rath W, Beier HM. The endometrium as a novel target for leptin: differences in fertility and subfertility. Mol Hum Reprod. 2000;6:595–601. doi: 10.1093/molehr/6.7.595. [DOI] [PubMed] [Google Scholar]
- 25.Gonzalez RR, Rueda BR, Ramos MP, Littell RD, Glasser S, Leavis PC. Leptin-induced increase in leukemia inhibitory factor and its receptor by human endometrium is partially mediated by interleukin 1 receptor signaling. Endocrinology. 2004;145:3850–7. doi: 10.1210/en.2004-0383. [DOI] [PubMed] [Google Scholar]
- 26.Ramos MP, Rueda BR, Leavis PC, Gonzalez RR. Leptin serves as an upstream activator of an obligatory signaling cascade in the embryo-implantation process. Endocrinology. 2005;146:694–701. doi: 10.1210/en.2004-1186. [DOI] [PubMed] [Google Scholar]
- 27.Tanaka T, Utsunomiya T, Bai T, Nakajima S, Umesaki N. Leptin inhibits decidualization and enhances cell viability of normal human endometrial stromal cells. Int J Mol Med. 2003;12:95–8. [PubMed] [Google Scholar]
- 28.Somasundar P, McFadden DW, Hileman SM, Vona-Davis L. Leptin is a growth factor in cancer. J Surg Res. 2004;116:337–9. doi: 10.1016/j.jss.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 29.Garofalo C, Surmacz E. Leptin and cancer. J Cell Physiol. 2006;207:212–22. doi: 10.1002/jcp.20472. [DOI] [PubMed] [Google Scholar]
- 30.Gonzalez RR, Leavis P. Leptin upregulates β3-integrin expression and interleukin-1β upregulates leptin and leptin receptor expression in human endometrial epithelial cell cultures. Endocrine. 2001;16:21–8. doi: 10.1385/ENDO:16:1:21. [DOI] [PubMed] [Google Scholar]
- 31.Gonzalez RR, Leary K, Petrozza JC, Leavis PC. Leptin regulation of the interleukin-1 system in human endometrial cells. Mol Hum Reprod. 2003;9:151–8. doi: 10.1093/molehr/gag022. [DOI] [PubMed] [Google Scholar]
- 32.Fukuda J, Kae N, Bing S, Sujie S, Yasushi K, Isao M. Effects of leptin on the production of cytokines by cultured human endometrial stromal and epithelial cells. Fertil Steril. 2003;80:783–7. doi: 10.1016/s0015-0282(03)00776-3. [DOI] [PubMed] [Google Scholar]
- 33.Sharma D, Saxena NK, Vertino PM, Anania FA. Leptin promotes the proliferative response and invasiveness in human endometrial cancer cells by activating multiple signal-transduction pathways. Endocr Relat Cancer. 2006;13:629–40. doi: 10.1677/erc.1.01169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bogusiewicz M, Semczuk A, Gogacz M, Skomra D, Jakowicki JA, Rechberger T. Lack of correlation between leptin receptor expression and PI3-K/Akt signaling pathway proteins immunostaining in endometrioid-type endometrial carcinomas. Cancer Lett. 2006;238:61–68. doi: 10.1016/j.canlet.2005.06.028. [DOI] [PubMed] [Google Scholar]
- 35.Gonzalez RR, Palomino A, Boric A, Vega M, Devoto L. A quantitative evaluation of alpha1, alpha4, alphaV and beta3 endometrial integrins of fertile and unexplained infertile women during the menstrual cycle. A flow cytometric appraisal. Hum Reprod. 1999;14:2485–92. doi: 10.1093/humrep/14.10.2485. [DOI] [PubMed] [Google Scholar]
- 36.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–54. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 37.Hubschle T, Thom E, Watson A, Roth J, Klaus S, Meyerhof W. Leptin-induced nuclear translocation of stat3 immunoreactivity in hypothalamic nuclei involved in body weight regulation. J Neurosci. 2001;21:2413–24. doi: 10.1523/JNEUROSCI.21-07-02413.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ihle JN. Cytokine receptor signaling. Nature. 1995;377:591–4. doi: 10.1038/377591a0. [DOI] [PubMed] [Google Scholar]
- 39.Darnel JE. STATs and gene regulation. Science. 1997;277:1630–5. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- 40.Bjorbaek C, Buchholz RM, Davis SM, Bates SH, Pierroz DD, Gu H, Neel BG, Myers MG, Jr, Flier JS. Divergent Roles of SHP-2 in ERK Activation by Leptin Receptors. J Biol Chem. 2001;276:4747–55. doi: 10.1074/jbc.M007439200. [DOI] [PubMed] [Google Scholar]
- 41.Kloek C, Haq AK, Dunn SL, Lavery HJ, Bank AS, Myers MG., Jr Regulation of Jak Kinases by Intracellular Leptin Receptor Sequences. J Biol Chem. 2002;277:41547–55. doi: 10.1074/jbc.M205148200. [DOI] [PubMed] [Google Scholar]
- 42.Murakami T, Yamashita T, Iida M, Kuwajima M, Shima K. A short form of leptin receptor performs signal transduction. Biochem Biophys Res Commun. 1997;231:26–9. doi: 10.1006/bbrc.1996.6030. [DOI] [PubMed] [Google Scholar]
- 43.Kellerer M, Koch M, Metzinger E, Mushack J, Capp E, Haring HU. Diabetologia. 1997;40:1358–62. doi: 10.1007/s001250050832. [DOI] [PubMed] [Google Scholar]
- 44.Ferrara N, Davis-Smyth T. The Biology of Vascular Endothelial Growth Factor. Endocr Rev. 1997;18:4–25. doi: 10.1210/edrv.18.1.0287. [DOI] [PubMed] [Google Scholar]
- 45.Giraudo E, Primo L, Audero E, Gerber HP, Koolwijk P, Soker S, Klagsbrun M, Ferrara N, Bussolino F. Tumor necrosis factor-α regulates expression of vascular endothelial growth factor receptor-2 and of its co-receptor neuropilin-1 in human vascular endothelial cells. J Biol Chem. 1998;273:22128–35. doi: 10.1074/jbc.273.34.22128. [DOI] [PubMed] [Google Scholar]
- 46.Bouloumie A, Hannes C, Drexler A, Lafontan M, Busse R. Leptin, the product of Ob gene, promotes angiogenesis. Circ Res. 1998;83:1059–66. doi: 10.1161/01.res.83.10.1059. [DOI] [PubMed] [Google Scholar]
- 47.Park HY, Kwon HM, Lim HJ, Hong BK, Lee JY, Park BE. Potential role of leptin in angiogenesis: leptin induces endothelial cell proliferation and expression of matrix metalloproteinases in vivo and in vitro. Exp Mol Med. 2001;33:95–102. doi: 10.1038/emm.2001.17. [DOI] [PubMed] [Google Scholar]
- 48.Okumura M, Yamamoto M, Sakuma H, Kojima T, Maruyama T, Jamali M, Cooper DR, Yasuda K. Leptin and high glucose stimulate cell proliferation in MCF-7 human breast cancer cells: reciprocal involvement of PKC-alpha and PPAR expression. Biochem Biophy Acta. 2002;1592:107–16. doi: 10.1016/s0167-4889(02)00276-8. [DOI] [PubMed] [Google Scholar]
- 49.Islami D, Bischof P, Chardonnens D. Modulation of placental vascular endothelial growth factor by leptin and hCG. Mol Hum Reprod. 2003;9:395–8. doi: 10.1093/molehr/gag053. [DOI] [PubMed] [Google Scholar]
- 50.Gonzalez RR, Leavis PC. A peptide derived from the human leptin molecule is a potent inhibitor of the leptin receptor function in rabbit endometrial cells. Endocrine. 2003;21:185–95. doi: 10.1385/ENDO:21:2:185. [DOI] [PubMed] [Google Scholar]
- 51.Salven P, Hattori K, Heissig B, Rafi S. Interleukin-1 promotes angiogenesis in vivo via VEGFR-2 pathway by inducing inflammatory cell VEGF synthesis and secretion. FASEB J. 2002;16:1471–3. doi: 10.1096/fj.02-0134fje. [DOI] [PubMed] [Google Scholar]
- 52.Coxon A, Bolon B, Estrada J, Kaufman S, Scully S, Rattan A, Duryea D, Hu YL, Rex K, Pacheco E, Van G, Zack D, Feige U. Inhibition of Interleukin-1 but Not Tumor Necrosis Factor Suppresses Neovascularization in Rat Models of Corneal Angiogenesis and Adjuvant Arthritis. Arthritis Rheum. 2002;46:2604–12. doi: 10.1002/art.10546. [DOI] [PubMed] [Google Scholar]
- 53.Gera JF, Mellinghoff IK, Shi Y, Rettig MB, Tran C, Hsu JH, Sawyers CL, Lichtenstein LK. AKT activity determines sensitivity to mammalian target of rapamycin (mTOR) inhibitors by regulating cyclin D1 and c-myc expression. J Biol Chem. 2004;279:2737–46. doi: 10.1074/jbc.M309999200. [DOI] [PubMed] [Google Scholar]
- 54.Fruhbeck G. Intracellular signalling pathways activated by leptin. Biochem J. 2006;393:7–20. doi: 10.1042/BJ20051578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wlodarski P, Kasprzycka M, Liu X, Marzec M, Robertson ES, Slupianek A, Wasik MA. A possible linkage between AMP-activated protein kinase (AMPK) and mammalian target of rapamycin (mTOR) signalling pathway. Cancer Research. 2005;65:7800–8. doi: 10.1158/0008-5472.CAN-04-4180. [DOI] [PubMed] [Google Scholar]
- 56.Leseux L, Hamdi SM, al Saati T, Capilla F, Recher C, Laurent G, Bezombes C. Syk-dependent mTOR activation in follicular lymphoma cells. Blood. 2006;108:4156–62. doi: 10.1182/blood-2006-05-026203. [DOI] [PubMed] [Google Scholar]
- 57.Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell. 2005;121:179–93. doi: 10.1016/j.cell.2005.02.031. [DOI] [PubMed] [Google Scholar]
- 58.Starr R, Novak U, Willson TA, Inglese M, Murphy V, Alexander WS, Metcalf D, Nicola NA, Hilton DJ, Ernstet E. Distinct roles for leukemia inhibitory factor receptor alpha-chain and gp130 in cell type-specific signal transduction. J Biol Chem. 1997;272:19982–6. doi: 10.1074/jbc.272.32.19982. [DOI] [PubMed] [Google Scholar]
- 59.Yu C, Kastin AJ, Tu H, Pan W. Opposing effects of proteasomes and lysosomes on LIFR: Modulation by TNF. J Mol Neurosci. 2007;32:80–9. doi: 10.1007/s12031-007-0017-4. [DOI] [PubMed] [Google Scholar]
- 60.Kimber SJ. Leukaemia inhibitory factor in implantation and uterine biology. Reproduction. 2005;130:131–45. doi: 10.1530/rep.1.00304. [DOI] [PubMed] [Google Scholar]
- 61.Mylonas I, Makovitzky J, Shabani N, Richter DU, Kuhn C, Jeschke U, Briese V, Friese K. Leukaemia inhibitory factor (LIF) is immunohistochemically expressed in normal, hyperplastic and malignant endometrial tissue. Eur J Obstet Gynecol Reprod Biol. 2005;118:101–8. doi: 10.1016/j.ejogrb.2004.06.022. [DOI] [PubMed] [Google Scholar]
- 62.Paradis H, Gendron RL. LIF transduces contradictory signals on capillary outgrowth through induction of stat3 and (P41/43) MAP kinase. J Cell Sci. 2000;113:4331–9. doi: 10.1242/jcs.113.23.4331. [DOI] [PubMed] [Google Scholar]
- 63.Ferrara N, Winer J, Henzel WJ. Pituitary follicular cells secrete an inhibitor of aortic endothelial cell growth: Identification as leukemia inhibitory factor. Proc Natl Acad Sci USA. 1992;89:698–702. doi: 10.1073/pnas.89.2.698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vasse M, Pourtau J, Trochon V, Muraine M, Vannier JP, Lu H, Soria J, Soria C. Oncostatin M induces angiogenesis in vitro and in vivo. Arteriosclerosis, Thrombosis, and Vascular Biology. 1999;19:1835–42. doi: 10.1161/01.atv.19.8.1835. [DOI] [PubMed] [Google Scholar]
- 65.Modugno F, Ness RB, Chen C, Weiss NS. Inflammation and endometrial cancer: a hypothesis. Cancer Epidemiology Biomarkers & Prevention. 2005;14:2840–7. doi: 10.1158/1055-9965.EPI-05-0493. [DOI] [PubMed] [Google Scholar]
- 66.Berry KK, Varney ML, Dave BJ, Bucana CD, Fidler IJ, Singh RK. Expression of interleukin-8 in human metastatic endometrial carcinoma cells and its regulation by inflammatory cytokines. Int J Gynecol Cancer. 2001;11:54–60. doi: 10.1046/j.1525-1438.2001.011001054.x. [DOI] [PubMed] [Google Scholar]
- 67.Van Le L, Haskill S, Jaffe GJ, Fowler WG., Jr Expression of interleukin-1 and interleukin-1 receptor antagonists in endometrial cancer. Gynecol Oncol. 1991;42:161–4. doi: 10.1016/0090-8258(91)90338-6. [DOI] [PubMed] [Google Scholar]
- 68.Sims JE, Gayle MA, Slack JL, Alderson MR, Bird TA, Giri JG, Colotta F, Re F, Mantovani A, Shanebeck K, Grabstein KH, Dower SK. Interleukin 1 signaling occurs exclusively via the type I receptor. Proc Natl Acad Sci USA. 1993;90:6155–9. doi: 10.1073/pnas.90.13.6155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ktori C, Shepherd PR, O'Rourke L. TNF-alpha and leptin activate the alpha-isoform of class II phosphoinositide 3-kinase. Biochem Biophys Res Commun. 2003;306:139–43. doi: 10.1016/s0006-291x(03)00933-1. [DOI] [PubMed] [Google Scholar]
- 70.Yasukawa H, Sasaki A, Yoshimura A. Negative regulation of cytokine signaling pathways. Ann Rev Immunol. 2000;18:143–6. doi: 10.1146/annurev.immunol.18.1.143. [DOI] [PubMed] [Google Scholar]