

Abstract
Chemoprevention by dietary agents/supplements has emerged as a novel approach to control various malignancies, including colorectal cancer (CRC). This study assessed dietary grape seed extract (GSE) effectiveness in preventing azoxymethane (AOM)-induced aberrant crypt foci (ACF) formation and associated mechanisms in Fischer 344 rats. Six-week old rats were injected with AOM, and fed control diet or the one supplemented with 0.25% or 0.5% (w/w) GSE in pre- and post-AOM or only post-AOM experimental protocols. At 16 weeks of age, rats were sacrificed and colons were evaluated for ACF formation followed by cell proliferation, apoptosis and molecular analyses by immunohistochemistry. GSE-feeding caused strong chemopreventive efficacy against AOM-induced ACF formation in terms of upto 60% (P<0.001) reduction in number of ACF and 66% (P<0.001) reduction in crypt multiplicity. Mechanistic studies showed that GSE-feeding inhibited AOM-induced cell proliferation but enhanced apoptosis in colon including ACF, together with a strong decrease in cyclin D1, COX-2, iNOS and survivin levels. Additional studies showed that GSE-feeding also decreased AOM-caused increase in β-catenin and NF-κB levels in colon tissues. Compared to control animals, GSE alone treatment did not show any considerable change in these biological and molecular events in colon, and was non-toxic. Together, these findings show the chemopreventive efficacy of GSE against the early steps of colon carcinogenesis in rats via likely targeting of β-catenin and NF-κB signaling, and suggest its potential usefulness for the prevention of human CRC.
Keywords: Colorectal cancer, grape seed extract, aberrant crypt foci, chemoprevention
Introduction
Colorectal cancer (CRC) continues to be one of the leading causes of cancer mortality in western countries including the United States (1). It has been estimated that more than 146,970 new cases of CRC will be diagnosed in the United States in 2009 alone and nearly 49,920 patients would die from this disease in the same year (2). Surgical resection remains the only curative treatment for CRC; however, this approach is often unsatisfactory at an advanced stage and thus the search for effective additional strategies is an important and urgent endeavor. One of the recent and complementary approaches to control CRC is chemoprevention. Evidence from epidemiological studies suggests that a high consumption of fruits and vegetables and intake of certain non-nutrients that are present in foods reduce the risk of CRC (3). The search for effective, naturally occurring, food-derived chemopreventive regimen has resulted in identifying several plant-derived compounds with antitumor activity (3, 4). In this regard, plant phenolics are a class of antitumor agents whose beneficial effects have been extensively characterized in several cell culture and animal cancer models (4, 5).
Grape seed extract (GSE) is one such agent which has long been marketed as a dietary supplement in the United States owing to its wide range of health benefits (6). GSE is rich in dimers, trimers and other oligomers of flavan-3-ols named proanthocyanidins, which are strong free radical scavengers and also known to exert wide spectrum of biological, pharmacological and therapeutic activities (6). Moreover, GSE is most notable for its promising chemopreventive and anticancer potential in different in vitro and in vivo cancer models (7–13). Studies from our laboratory and others have shown that GSE reduces the incidence of carcinogen-induced mammary tumors in rats and skin tumors in mice, and inhibits the growth of DU145 and HT29 xenografts in nude mice (8–13). More recently, we have also reported the chemopreventive effect of GSE on prostate tumor growth and progression in the transgenic adenocarcinoma of the mouse prostate model (14).
Aberrant crypt foci (ACF), as first described by Bird (15), are characterized by having altered luminal openings, exhibiting thickened epithelia and are larger than adjacent normal crypts. ACF are putative preneoplastic lesions induced in colon of rodents after subsequent treatment with chemical carcinogen such as azoxymethane (AOM), and are found in most colons of human CRC patients (15). Thus, AOM-induced ACF in rodents are considered as surrogate biomarker to examine the chemopreventive potential of naturally occurring and synthetic agents (16, 17). Aberrant β-catenin expression is a common feature of experimental and human CRC (18), and AOM treatment enhances β-catenin and NF-κB levels in colon (19). The enhanced β-catenin and NF-κB signaling induces the expression of a variety of growth and inflammatory response genes, including cyclin D1, proliferation cell nuclear antigen (PCNA), inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2), and cell survival genes including survivin that favor neoplastic growth in the development of CRC (18, 19). In order to assess the chemopreventive efficacy of GSE on early stages of colon carcinogenesis, here we investigated its effect on the development of AOM-induced ACF formation in F344 rats. Further, we examined the effect of GSE on AOM-induced cell proliferation and cell death, and the expression levels of iNOS, COX-2, cyclin D1, survivin, β-catenin and NF-κB in colon. Our findings show that GSE significantly prevents AOM-induced ACF formation together with a reduction in cell proliferation and inflammatory regulators but increased apoptosis, and suppression of β-catenin and NF-κB signaling.
Materials and Methods
Animals and treatment protocol
Male F344 rats were purchased from the Jackson laboratories (Bar Harbor, ME) at the age of 5 weeks, and maintained in animal facility at the University of Colorado Denver, Aurora. Animal care was strictly in accordance to the institutional as well as NIH guide lines for animal care and use for experimental purposes. GSE-standardized preparation, constituting 89.3% (w/w) procyanidins, 6.6% monomeric flavonols, 2.24% moisture content, 1.06% protein and 0.8% ash (20), was a kind gift from its commercial vendor Kikkoman Corp (Noda City, Japan). Herein, we have used GSE in the powdered diet that was changed at least twice a week. In our experience, GSE stored at 40 exerts same extent of biological effects in cell culture as well as animal studies, even after one year of storage. Animals were maintained at 12 h light/dark cycles with free access to water and food (AIN-76A powder diet from Dyets Inc., Bethlehem, PA). As shown in Figure 1A, after one week of acclimatization, animals were randomly divided into 7 groups of 7 animals each, and fed AIN-76A control powder diet (groups 1 and 2) or diet supplemented with 0.25% or 0.5% (w/w) GSE (groups 3 and 4) using a powder diet feeder till the end of the experiment. One week later, rats in groups 2–6 were given sub-cutaneous injection of AOM (15 mg/kg body weight) once a week for 2 weeks. Four weeks after the last AOM injection, rats in groups 5 and 6 were fed with diet containing 0.25% or 0.5% GSE, respectively. Rats in group 7 were fed with diet containing 0.5% GSE alone throughout the study. At 16 weeks of age, rats were sacrificed and colons were evaluated for ACF followed by molecular marker analyses.
Fig. 1.

Dietary GSE feeding inhibits cell proliferation and induces apoptosis in colon tissues from AOM-treated rats. (A) Experimental design for AOM and GSE treatments as described in Materials and Methods. Groups 3 and 4 represent protocol 1 (pre & post-AOM), and groups 5 and 6 represent protocol 2 (post-AOM) for GSE treatments. Representative photographs for IHC staining of PCNA (B) and TUNEL (C) positive cells in AOM alone and GSE + AOM treated groups, respectively, are shown at 400x magnification. The percentages of PCNA (B) and TUNEL (C) positive cells assessed by quantification of IHC stained rat colonic epithelium are calculated from 5 randomly selected fields from each tissue sample in each group as detailed in Methods, and represent mean ± SE value from 7 rats in each group. Representative colonic tissues from each group were also analyzed by immunoblotting for PCNA expression. Values of band intensity were adjusted with β-actin. AOM, azoxymethane; GSE, grape seed extract.
Determination of ACF
ACF analysis was done according to Bird (15), where the colons were longitudinally opened, rinsed with 0.9% NaCl solution and fixed flat between two pieces of filter paper in 10% buffered formalin for a minimum of 24 h. The colons were then cut into 2 cm segments, starting at the anus, and stained with 0.2% methylene blue in Krebs-Ringer solution for 5 to 10 min, and were then placed mucosal side up on a microscope slide and counted through a light microscope at 400X magnification. Aberrant crypts were distinguished from the surrounding normal crypts by their increased size, the significantly increased distance from lamina to basal surface of cells, and the easily discernible pericryptal zone. The variables used to assess the aberrant crypts were their occurrence and multiplicity. Crypt multiplicity was determined as the number of crypts in each focus. Multicrypts were categorized as those containing up to four or more aberrant crypts/focus.
Immunostaining for PCNA, cyclin D1, survivin, iNOS, COX-2,β-catenin, and NF-κB
Colon tissue samples were fixed in 10% phosphate-buffered formalin for 10 h at 4°C, dehydrated in ascending concentrations of ethanol, cleared with xylene and embedded in PolyFin (Triangle Biomedical Sciences, Durham, NC). Paraffin-embedded tissue blocks were cut with a rotary microtome into 4-μm sections and processed for immunohistochemical staining. Overall, the standard and published procedures have been used in the current study for the IHC methodology related to the expression of PCNA, cyclin D1, beta-catenin, iNOS, and COX-2 (12–14,17). Briefly, after deparaffinization and rehydration, the sections were treated with 0.01 M sodium citrate buffer (pH 6.0) in a microwave for 60 min at full power for antigen-retrieval. The sections were then quenched of endogenous peroxidase activity by immersing in 3% hydrogen peroxide for 5 min at room temperature. The sections were either incubated with antibody against PCNA mouse monoclonal (1:400 dilution, Dako, Carpinteria, CA), cyclin D1 rabbit polyclonal (1:200 dilution; Santa Cruz Biotechnology Inc., CA), survivin (1:200 dilution; Novus Biologicals, Littleton, CO), iNOS rabbit polyclonal (1:200 dilution; Abcam, Inc, Cambridge, MA), COX-2 rabbit polyclonal (1:100 dilution; Cell Signaling Technologies, San Diego, CA) β-catenin (1:100 dilution; SantaCruz Biotechnology Inc., CA) or NF-κB(p65) (1:100 dilution; SantaCruz Biotechnology Inc., CA) in PBS for 2 h at room temperature in a humidity chamber followed by overnight incubation at 4°C. In all the immunohistochemical staining, to rule out the nonspecific staining allowing better interpretation of specific staining at the antigenic site, negative staining controls were used in which sections were incubated with N-Universal Negative Control mouse or rabbit antibody (Dako) under identical conditions. The sections were then incubated with appropriate biotinylated secondary antibody for 1 h at room temperature followed by 30 min incubation with HRP-conjugated streptavidin. The specific immunoreactivity was visualized using 3, 3′-diaminobenzidine (DAB) for 10 min at room temperature. The sections were counterstained with Harris Hematoxylin, dehydrated and mounted. The IHC quantification for nuclear or non-nuclear stains for PCNA-, cyclin D1-, survivin-, iNOS-, COX-2-, nuclear β-catenin-, and NF-κB(p65)-positive cells was done in blinded manner, and respective positivity was determined for the brown color beyond the background level immunoreactivity in each case. For each sample, five random fields involving crypts were analyzed for the quantification of the staining as percent positive cells, by counting brown-stained cells among total number of cells. The percent positivity was determined as the number of brown positive cells x 100/total number of cells. Cytoplasmic β-catenin was quantified by immunoreactivity (represented by intensity of brown staining) and scored as 0 (no staining), +1 (very weak), +2 (weak), +3 (moderate), and +4 (strong) at 5 randomly selected fields at 400x magnification in each sample.
TUNEL staining for in situ apoptotic cells
Apoptotic cells were detected using the DeadEnd Colorimetric TUNEL system (Promega, Madison, WI) following manufacturer’s protocol with some modifications. In brief, the tissue sections after deparaffinization and re-hydration were permeabilized with proteinase K (30 μg/ml) for 1 h at 37°C. Thereafter, the sections were quenched of endogenous peroxidase activity using 3% hydrogen peroxide for 10 min. After thorough washing with 1x PBS, the sections were incubated with equilibration buffer for 10 min, and then TdT reaction mixture was added to the sections, except for the negative control, and incubated at 37°C for 1 h. The reaction was stopped by immersing the sections in 2x saline-sodium citrate buffer for 15 min. The sections were then added with streptavidin-HRP (1:500) for 30 min at room temperature, and after repeated washings, the sections were incubated with substrate DAB until color development (5–10 min). The sections were mounted after dehydration, and observed under 400x magnification for TUNEL-positive cells (brown color). Apoptotic index was calculated as number of apoptotic cells x 100/total number of cells.
Western blot analysis
The representative colonic tissue lysates from each group were analyzed by immunoblotting as previously described (17). Densitometric analyses of bands are adjusted with β-actin as loading control.
Immunohistochemical and statistical analyses
All microscopic analyses were done using Zeiss Axioscop 2 microscope (Carl Zeiss, Jena, Germany). The pictures were taken by Kodak DC290 camera under 400x magnification and processed by Kodak Microscopy Documentation System 290 (Eastman Kodak Company, Rochester, NY). The mean ± SE values were obtained from the evaluation of multiple fields in each group, and the data represent the results from all 7 rats in each group. For statistical significance of the difference, the data were analyzed using the SigmaStat 2.03 software (Jandel Scientific, San Rafael, CA). The statistical significance of difference between control and AOM-treated group, and AOM-treated versus GSE plus AOM- or AOM plus GSE-treated groups was determined by one way analysis of variance (one-way ANOVA) followed by paired t-test for multiple comparisons. P<0.05 was considered statistically significant.
Results
General observations
GSE feeding did not show any gross sign of toxicity or possible adverse side effects as measured by two profiles, namely body weight and food consumption (g/day/rat) throughout the study protocol. There was no considerable change in body weight gain and diet intake among control and GSE-fed groups till the end of the study (data not shown). Furthermore, at necropsy, no pathological alternations were found in any organs by gross observation, including liver, lung and kidney.
Dietary GSE feeding prevents AOM-induced ACF formation in F344 rat colon
As summarized in Table 1, all the rats in groups 2 through 6, which were injected AOM, developed ACF. The mean number of ACF/colon in the animals administered AOM alone (group 2) was 157 ± 9. However, the dietary feeding of GSE at both the doses (0.25% & 0.5%) given as pre- & post-initiation (groups 3 & 4) or post-initiation alone (groups 5 & 6) significantly (P<0.001) reduced the AOM-induced ACF formation in a dose-dependent manner compared to the group 2 animals. Among the two different GSE treatment protocols, the percentage of inhibition was stronger in animals where GSE feeding was started prior to AOM-initiation (50–60% inhibition) than those where it was given as post- AOM-initiation only (34–37% inhibition). More importantly, the number of ACF consisting of 1–4 crypts or more than 4 crypts was also decreased by GSE-feeding as compared to AOM alone-treated group (Table 1). Overall, the number of ACF was decreased by up to 66% (P<0.001) for one crypt, 65% (P<0.001) for two crypts, 52% (P<0.001) for three crypts, and 33% (P<0.05) for four crypts when GSE feeding started prior to AOM initiation (Table 1). The stronger efficacy of GSE in first protocol versus second protocol may also account for duration of GSE treatment that was 10 weeks in pre- & post-initiation protocol versus only 4 weeks in post-initiation protocol.
Table 1.
GSE inhibits AOM-induced aberrant crypt foci incidence and multiplicity in F344 male rat colon
| Groups | Treatments | Incidence of ACF formation (%) | No. of ACF/colon | Crypt multiplicity of ACF | |||
|---|---|---|---|---|---|---|---|
| 1 crypt | 2 crypts | 3 crypts | ≥ 4 crypts | ||||
| 1 | Control | 0/7 (0) | 0 | 0 | 0 | 0 | 0 |
| 2 | AOM | 7/7 (100) | 157 ± 9 | 66 ± 4 | 55 ± 4 | 23 ± 2 | 12 ± 2 |
| 3 | 0.25% GSE + AOM | 7/7 (100) | 79 ± 6* | 31 ± 2* | 30 ± 3* | 11 ± 2* | 8 ± 2† |
| 4 | 0.5% GSE + AOM | 7/7 (100) | 63 ± 3* | 22 ± 1* | 19 ± 3* | 13 ± 1† | 9 ± 2 |
| 5 | AOM + 0.25% GSE | 7/7 (100) | 104 ± 5* | 31 ± 1* | 43 ± 3† | 18 ± 3 | 11 ± 1 |
| 6 | AOM + 0.5% GSE | 7/7 (100) | 99 ± 10* | 27 ± 1* | 44 ± 5† | 19 ± 4 | 8 ± 2† |
| 7 | 0.5% GSE | 0/7 (0) | 0 | 0 | 0 | 0 | 0 |
Data are shown as mean ± SE of 7 samples from individual rats in each group. Groups 3 and 4 represent protocol 1 (pre & post-AOM), and groups 5 and 6 represent protocol 2 (post-AOM) for GSE treatments. ACF, aberrant crypt foci; AOM, azoxymethane; GSE, grape seed extract.
, P<0.001;
, P<0.05 versus group 2.
Dietary GSE feeding inhibits cell proliferation and induces apoptosis
Since increased rate of proliferation and evasion of apoptosis are hallmarks of ACF formation, we next examined the effect of GSE feeding on these two biomarkers by PCNA immunostaining and TUNEL staining, respectively, in colonic samples from all rats in each group (Fig. 1B & C). PCNA is a well-known marker of cellular proliferation (21). Microscopic examination of colonic tissue sections clearly showed a decreased level of PCNA-positive cells in GSE-fed groups compared to AOM alone treated group (Fig. 1B). The quantitative analyses of all the PCNA-stained sections (Fig. 1B) showed a significant (8-fold, P<0.001) increase in the percentage of PCNA-positive cells in the colonic mucosa of AOM-treated rats (40 ± 4) compared to control rats (5 ± 1). The dietary administration of GSE at both the dose levels (0.25 or 0.5%) started either pre-initiation or post-initiation significantly (P<0.001) decreased the AOM-induced PCNA labeling index in the colonic mucosa (Fig. 1B). The decrease in PCNA labeling index by GSE was more profound when GSE treatment started at preinitiation level (52–57% inhibition) than at post-initiation level (28–33% inhibition) (Fig. 1B). These results were further confirmed by immunoblot analysis (Fig. 1B) where densitometric analyses of bands (adjusted with β-actin as loading control) showed 23–60% decrease in PCNA expression in colonic tissues of GSE-treated rats when compared with AOM alone treated positive controls.
With regard to apoptotic response, we observed proapoptotic effect of dietary GSE in the colonic tissue of AOM-injected rats as shown in the representative photographs for TUNEL-positive cells in AOM alone- and GSE plus AOM-treated groups (Fig. 1C). Quantification of the stained sections showed a significant (P<0.001) increase in the number of TUNEL-positive cells by GSE at both dose levels in AOM-injected rats compared to control diet-fed AOM-injected rats (Fig. 1C). A dose-dependent induction, by 2.9 to 4.0-fold (P<0.001) and 2.4 to 3.4-fold (P<0.001), in TUNEL-positive cells was observed in the groups in which GSE feeding started pre-initiation or post-initiation, respectively (Fig. 1C). The percentages of PCNA and TUNEL positive cells were almost comparable between control diet (group 1) and 0.5% GSE diet alone fed rats (group 7).
Dietary GSE feeding suppresses the expression of cyclin D1 and survivin
Microscopic examination of tissue sections showed that compared to controls (group 1), AOM alone treated rats have significantly increased number of cyclin D1-positive cells in the colonic mucosa that is strongly decreased in GSE plus AOM-treated rats (Fig. 2A). The quantification of the staining showed 27 ± 2% (P<0.001) cyclin D1-positive cells in AOM-treated rats as compared to 4 ± 1% in control rats (Fig. 2B). A significant decrease (P<0.001) in cyclin D1-positive cells was observed in the rats that received GSE given either pre- & post-initiation or post-initiation alone compared to AOM-alone rats (Fig. 2B). In quantitative analysis, GSE decreased AOM-induced cyclin D1-positive cells by 38–53% and 26–47% in pre- & post- and post-AOM initiation protocols, respectively (Fig. 2B). Similar to cyclin D1, microscopic examination of colonic tissue sections clearly showed a decreased level of survivin-positive cells in GSE-fed groups compared to AOM alone treated group (Fig. 2C). The quantitative analysis of survivin-stained sections (Fig. 2D) showed a significant (P<0.001) increase in the percentage of survivin-positive cells in the colonic mucosa of AOM-treated rats (40 ± 2) compared to control rats (6 ± 1). The dietary administration of GSE (0.25 or 0.5%) in both protocols significantly (P<0.001) reduced the AOM-induced survivin-positive cells in colonic mucosa. The suppressive effect of GSE was more profound in rats given GSE at pre-& post-initiation (46–57%) than in the rats given GSE at post-initiation alone (36–49%, Fig. 2D). GSE alone (0.5%) did not show any considerable effect on cyclin D1 and survivin levels in colonic mucosa compared to control group (Fig. 2B & D). The IHC results were further confirmed by immunoblot analysis of colonic mucosa which showed that GSE feeding decreases cyclin D1 expression by 23–50% and survivin expression by 12–38% when compared with AOM alone treated positive controls, respectively (Fig. 2B & D).
Fig. 2.

Dietary GSE feeding suppresses cyclin D1 and survivin expression in colon tissues of AOM-treated rats. Representative photographs for IHC staining of cyclin D1 (A) and survivin (C) positive cells in AOM alone and GSE + AOM treated groups, respectively, are shown at 400x magnification. The percentages of cyclin D1 (B) and survivin (D) positive cells assessed by quantification of IHC stained rat colonic epithelium are calculated from 5 randomly selected fields from each tissue sample in each group as detailed in Methods, and represent mean ± SE value from 7 rats in each group. Representative colonic tissues from each group were also analyzed by immunoblotting for cyclin D1 and survivin expression. Values of band intensity were adjusted with β-actin. AOM, azoxymethane; GSE, grape seed extract.
Dietary GSE feeding decreases the expression of iNOS and COX-2
We also investigated whether the inhibition of AOM-induced ACF formation by dietary GSE is associated with the modulation of iNOS and COX-2 protein levels. The expression levels of iNOS and COX-2 in the colonic tissues were assessed by immunohistochemical staining (Fig. 3A–D), the quantification of which showed an increased number of iNOS- (~9 fold, P<0.001, Fig. 3B) and COX-2-positive cells (~8 fold, P<0.001, Fig. 3D) in AOM alone-treated rats compared to untreated controls (group 1). The representative photographs for the immunohistochemical staining of iNOS (Fig. 3A) and COX-2 (Fig. 3C) positive cells in AOM alone- and GSE plus AOM-treated groups clearly showed a decreased immunoreactivity of these two proteins by GSE treatments. The iNOS expression was significantly (P<0.001) decreased in the animals that received GSE given either pre- & post-initiation or post-initiation alone compared to AOM alone rats. The reduction in AOM-induced iNOS-positive cells was 47–63% and 27–40% in GSE-fed groups during pre- & post-and post-AOM initiation protocols, respectively (Fig. 3B). Similar to iNOS, both the doses of GSE given at pre- & post-initiation or post-initiation phase significantly(P<0.001) decreased the AOM-induced COX-2-positive cells by 45–63% and 19–45%, respectively (Fig. 3D). GSE alone at 0.5% dose level did not show any effect on iNOS and COX-2 levels in colonic mucosa when compared with control diet alone group (Fig. 3B & D). Immunoblot analysis further confirmed that GSE feeding in AOM-treated rats decreases iNOS expression by 30–52% and COX-2 expression by 22–39% when compared with AOM alone-treated positive controls, respectively (Fig. 3B & D). Overall, these results show that GSE selectively decreases the AOM-induced protein levels of these two key inflammatory molecules, iNOS and COX-2, in colonic mucosa.
Fig. 3.

Dietary GSE feeding decreases the protein levels of iNOS and COX-2 in colon tissues from AOM-exposed rats. Representative photographs for IHC staining of iNOS (A) and COX-2 (C) positive cells in AOM alone and GSE + AOM-treated groups, respectively, are shown at 400x magnification. The percentages of iNOS (B) and COX-2 (D) positive cells assessed by quantification of IHC stained rat colonic epithelium are calculated from 5 randomly selected fields from each tissue sample in each group as detailed in Methods, and represent mean ± SE value from 7 rats in each group. Representative colonic tissues from each group were also analyzed by immunoblotting for iNOS and COX-2 expression. Values of band intensity were adjusted with β-actin. AOM, azoxymethane; GSE, grape seed extract; iNOS, inducible nitric oxide synthase; COX-2, cyclooxygenase-2.
Dietary GSE feeding decreases the expression of β-catenin and NF-κB
The β-catenin and NF-κB signaling results in the induction of a variety of oncogenic and growth responsive genes, and overexpression of β-catenin and activation of NF-κB are commonly observed in both experimental and human CRC (18, 19, 22), suggesting that the agents which decrease β-catenin and NF-κB levels or associated activity could be effective against CRC growth and/or in prevention. Based on our above findings that GSE strongly prevents AOM-induced colonic ACF formation and that compared to AOM-alone, GSE-fed and AOM-treated colonic mucosa samples show decreased proliferation and increased apoptosis together with a strong reduction in the levels of cyclin D1, survivin, iNOS and COX-2, all of them regulated by β-catenin and NF-κB, next we analyzed the expression levels of both cytoplasmic and nuclear β-catenin as well as NF-κB(p65) in the rat colonic tissues using immunohistochemical staining. Microscopic examination of colonic tissue sections clearly showed decreased levels of both cytoplasmic and nuclear β-catenin positive cells in GSE-fed and AOM-treated groups compared to AOM alone-treated group (microphotograph not shown). The diffused cytoplasmic staining was quantified at 0–4 scale of increasing positivity/immunoreactivity whereas discrete nuclear staining was quantified as negative or positive nuclei for β-catenin. The quantification showed that GSE (0.25 or 0.5%) given either pre- & post-initiation or post-initiation alone significantly (P<0.001) decreases both cytoplasmic as well as the nuclear expression level of β-catenin in the colonic mucosa of AOM-treated rats compared to AOM alone-treated rats (group 2) rats (Fig. 4A & B). In the first protocol, GSE (0.25 and 0.5%) decreased AOM-induced cytoplasmic β-catenin immunoreactivity by 47 and 53% (P<0.001, Fig. 4A) and nuclear β-catenin-positive cells by 43 and 53% (P<0.001, Fig. 4B). In the animals where GSE (0.25 and 0.5%) was fed only in post-initiation protocol, 32 and 44% reduction (P<0.001) in cytoplasmic β-catenin immunoreactivity (Fig. 4A) and 33 and 46% reduction (P<0.001) in nuclear β-catenin-positive cells (Fig. 4B) was observed as compared to AOM alone-treated animals. Similar to its effect on β-catenin, both the doses of GSE given at pre- & post-initiation or post-initiation phase also significantly (P<0.001) decreased the AOM-induced NF-κB(p65)-positive cells by 46–65% and 26–52%, respectively (Fig. 4C). GSE alone at 0.5% dose level did not show any effect on β-catenin and NF-κB levels in colonic mucosa when compared with control diet alone group (Fig. 4A–C).
Fig. 4.

Dietary GSE feeding decreases the expressions of β-catenin in colon tissues from AOM-treated rats. The positivity of β-catenin in cytoplasm (A), the percentage of nuclear β-catenin positive cells (B), and the percentage of NF-κB(p65) positive cells (C) were assessed by quantification of IHC stained rat colonic epithelium are calculated from 5 randomly selected fields from each tissue sample in each group as detailed in Methods, and represent mean ± SE value from 7 rats in each group. AOM, azoxymethane; GSE, grape seed extract.
Discussion
Cancer chemoprevention is the use of dietary and pharmacological intervention with specific natural or synthetic agents to prevent, suppress, or reverse the process of carcinogenesis before the malignant transformation occurs (23). Preventive approaches such as dietary modulation and chemoprevention could be more practical and effective in the fight against CRC. Epidemiological and experimental evidences have consistently shown that diets rich in fruits and vegetables reduce the risk of gastrointestinal diseases including CRC (24, 25). Currently there has been considerable interest in identifying the compounds in fruits and vegetables since they might provide useful strategies to inhibit CRC with minimal or no toxicity. GSE is one such agent which has been widely consumed as a dietary supplement and possesses anticancer activity against various cancers (10–14). ACF are the putative precancerous lesions induced by AOM and other carcinogens in the colon of rats and mice (17, 26). In particular, AOM-induced ACF have been widely used as a useful biomarker for colon carcinogenesis and to examine anticancer effects of the agents under chemopreventive efficacy evaluation (16, 27). In the present study, our results clearly demonstrated that dietary feeding of GSE given at two different stages during carcinogenesis significantly and dose-dependently inhibits AOM-induced colonic ACF formation in rats. Most importantly, these GSE doses did not show any apparent signs of toxicity to animals in terms of food consumption and body weight gain profiles, suggesting that dietary GSE could efficiently suppress chemically-induced colon carcinogenesis without any toxicity. The suppressive effect of GSE on total colonic ACF formation was also associated with reduction in crypt multiplicity including the multicrypts (more than 4 crypts/focus) which are more likely to persist, increase in size through multiplication and develop into malignant tumors (28). Accordingly, these results suggest that GSE could inhibit the promotion/progression of these foci to tumors of the colon, which are in line with the earlier reports employing other agents (29–31).
Increase in colonocyte proliferation and decrease in apoptosis are hallmarks of ACF. Furthermore, disruption of balance between cell proliferation and apoptosis is the main biological event for enhancement of carcinogenesis during the promotion/progression phase (32). PCNA, a well-known cell cycle marker protein, acts as a DNA sliding clamp for DNA polymerase delta and is an essential component for eukaryotic chromosomal DNA replication and repair (21). Thus, PCNA is regarded as a potential biomarker for cell proliferation and is widely used to evaluate the response of the colonic epithelium to putative chemopreventive agents (33, 34). In this regard, the antiproliferative effect of GSE was evaluated using PCNA labeling index during AOM-induced ACF formation. Our results demonstrated that GSE given at pre- & post-initiation or post-initiation phase significantly reduces the AOM-caused increase in proliferating cells in colonic mucosa. In many cancers, including CRC, the accelerated tumor cell proliferation and tumor progression are mediated through the transcriptional activation of many genes, including cyclin D1 (35), which is an important positive regulator of the G1 to S phase cell cycle transition and is involved in cell growth and proliferation. Increased cyclin D1 expression has been previously reported in CRC patients (36) and in ApcMin/+ mice (37). Dietary administration of GSE also significantly decreased AOM-induced accumulation of cyclin D1 level in colonic mucosa, which might be part of the mechanism by which GSE suppresses AOM-induced cell proliferation in colonic tissue.
In addition to targeting the inhibition of enhanced cell proliferation, the induction of apoptosis by chemopreventive agents is also important to delete the cells carrying mutations. Even in cancer cells, induction of apoptosis is another approach to limit their uncontrolled proliferation. The results of the present study clearly indicated that dietary GSE also induces TUNEL-positive cells or apoptosis in a dose-dependent manner in the colon tissue of AOM-injected rats. Thus, the reduction in cell proliferation and the increase in apoptosis in colonic tissues by GSE seem to correlate with the reduction in aberrant crypt multiplicity. These findings are in concurrence with many in vitro studies showing the growth inhibitory and apoptosis inducing effects of GSE in a variety of human cancer cell lines, including colon cancer cells (11, 12, 38). While exploring the molecular mechanism of apoptosis induction, we observed that GSE down-regulates the AOM-induced colonic level of survivin. Survivin is an apoptosis inhibitor protein that inhibits the activation of caspases, and its overexpression is implicated in the growth and progression of many types of cancers including CRC (39), suggesting that a decrease in survivin should lead to caspases activation and associated apoptotic cell death. Indeed, our recent cell culture study using colon carcinoma cells have shown that GSE induces apoptosis by caspase-3 activation and PARP cleavage (12), providing additional support to the observed in vivo anti-apoptotic effect of GSE and its association with a decrease in AOM-induced survivin levels in colonic mucosa observed in the present study. Taken together our previous findings and the results of present study, it is clear that GSE causes an in vivo caspase-mediated apoptosis selectively in colonic epithelium challenged with a chemical carcinogen as well as colonic carcinoma cells, without affecting the normal colonic epithelium as GSE alone feeding neither inhibits cell proliferation nor induces cell death in colonic tissue as observed in the present study.
It is widely reported that both iNOS and COX-2 are involved in chronic inflammation, which creates a microenvironment conducive for colon carcinogenesis (40), and therefore the suppression of either the synthesis and/or activity of these two enzymes is a target for cancer chemoprevention. Several lines of evidence show that iNOS and COX-2 protein levels are enhanced in colorectal adenomas and adenocarcinomas in humans and chemically-induced colonic tumors in rats (40). Furthermore, it has been reported that overexpression of iNOS and COX-2 leads to DNA damage, mutation, increased proliferation, oxidative stress, resistance to apoptosis, increased tumor vascularity and metastatic potential (41). COX-2, an inducible isoform of COX, regulates the rate-limiting step in prostanoid biosynthesis and is intimately linked to early as well as late stages of colon carcinogenesis (41). Many agents including oleanolic acid, grapefruit pulp or its bioactive compounds naringin and limonin, and red wine and black tea polyphenols are shown to inhibit AOM-induced ACF formation or colon tumorigenesis (42–44). These agents also inhibited COX-2 levels, and suppressed cell proliferation but induced apoptosis in ACF in their antitumor efficacy (42–44). In this regard, selective COX-2 inhibitors, including celecoxib and NS-398 are also shown to suppress AOM-induced colon carcinogenesis (45, 46). COX-2 level is always low in normal colon mucosa which increases with ACF formation and colon tumor development. Thus the present finding with GSE showing decrease in COX-2 levels in ACF could be one of the mechanisms of GSE in inhibiting AOM-induced colon carcinogenesis in rats. iNOS, an inducible form of NOS, is also increased in colon cancer, including AOM-induced tumors, and might also activate COX-2 and further contribute to tumor development or acceleration of the early stage of tumorigenesis (44). A large number of studies suggest that colitis and gastritis or the development of colon cancer involves inflammatory mechanisms in which iNOS-mediated production of nitric oxide plays a critical role. The sustained and elevated levels of iNOS protein expression activate several downstream protein including mitogen-activated protein kinases and IκB kinases which subsequently activate transcription factors such as activator protein-1 and NFκB to enhance the production of pro-inflammatory cytokines (47). Thus, down-regulation of iNOS could have significance for the prevention and treatment of inflammation-associated carcinogenesis. In this regard, inhibitory effect of GSE on iNOS and COX-2 expression could contribute to its anticarcinogenic effect against AOM-induced ACF formation.
β-catenin functions as a transcriptional activator of the Wnt signaling pathway in embryonic development and tumorigenesis (18). In several types of human cancer including CRC, mutation in the β-catenin leads to cytoplasmic/nuclear accumulation of β-catenin resulting in accelerated tumor cell proliferation and tumor progression through the transcriptional activation of target genes including cyclin D1, iNOS and COX-2 (18). Accumulating data from both in vitro and in vivo studies extensively suggested the implication of β-catenin-mediated signaling in tumor initiation and progression in colon (18, 35). Increased β-catenin level has been reported in CRC patients and ApcMin/+ mice (36, 37). In the present study, the increased levels of both cytoplasmic as well as nuclear β-catenin in the colonic tissues of AOM-treated rats were in line with the ACF formation. Dietary GSE feeding, however, significantly decreased the overall AOM-induced accumulation of β-catenin in colon tissue, which might have attenuated the Wnt signaling pathway resulting in reduced transcription of target genes, including cyclin D1, iNOS and COX-2 as observed in colonic tissues from GSE-fed rats. Some recent studies have revealed that overexpression of iNOS and COX-2 can also stimulate β-catenin/T-cell factor–mediated pathway in CRC development (26). Overall, suppression of β-catenin signaling by GSE, in part, could have contributed to the reduction in AOM-induced formation and development of colonic ACF in rats.
NFκB is a well known transcription factor that participates in the regulation of various cellular responses involved in the immediate early processes of the immune and inflammatory responses, apoptosis, and cell proliferation through induction of a large array of target genes (22). NFκB is now considered as an important transcription factor in the tumorigenic process because its constitutive activation exerts strong proliferative and anti-apoptotic functions in cancer cells (22, 48). Furthermore, NFκB acts as a cell survival factor through its regulatory role in the expression of an array of cell proliferation, apoptotic and inflammatory response genes (49). Consistently, constitutive activation of NFκB has been reported in several tumors including colon tumor (49). Thus, inhibition of NFκB signaling pathway provides attractive targets for promising chemopreventive and chemotherapeutic approaches. Interestingly, the results of the present study showed that the increased levels of activated NFκB (p65) in the colonic tissues of AOM-treated rats were significantly decreased by dietary feeding of GSE, which might have contributed to its anticarcinogenic effect against AOM-induced ACF formation through downregulation of PCNA, cyclin D1, iNOS, COX-2 and survivin. Several natural compounds including polyphenols such as resveratrol and epigallocatechin gallate have been reported to suppress NFκB activity and are suggested to be useful for the inhibition of cancer cell growth (50).
Taken together, the results of the present study clearly indicate that dietary feeding of GSE strongly prevents AOM-induced colonic ACF formation and crypt multiplicity in F344 rats, and this effect of GSE was associated with inhibition of cell proliferation and an induction of apoptosis. At molecular level, the suppression of β-catenin and NFκB signaling together with down-regulation of iNOS, COX-2, cyclin D1 and survivin expression by GSE had played a possible contributory role in inhibiting early stages of chemical carcinogen-induced colonic ACF formation and development by GSE (Fig. 5). GSE has been found to inhibit many signaling pathways, including MAPK, AKT and VEGF-receptor pathways in different types of cells (7, 10, 11). A recent report show that antiangiogenic activity of GSE via suppressing VEGF-receptor signaling could also contribute to its anti-tumorigenic activity (7). Thus, if the present study would have been followed for long-term carcinogenesis study, the antiangiogenic mechanisms of GSE could have contributed for its anti-colon cancer efficacy. Accordingly, further investigations on long-term colon cancer chemopreventive potential of GSE are warranted; nevertheless, the results of the present study suggest the clinical usefulness of GSE against CRC growth and development.
Fig. 5.
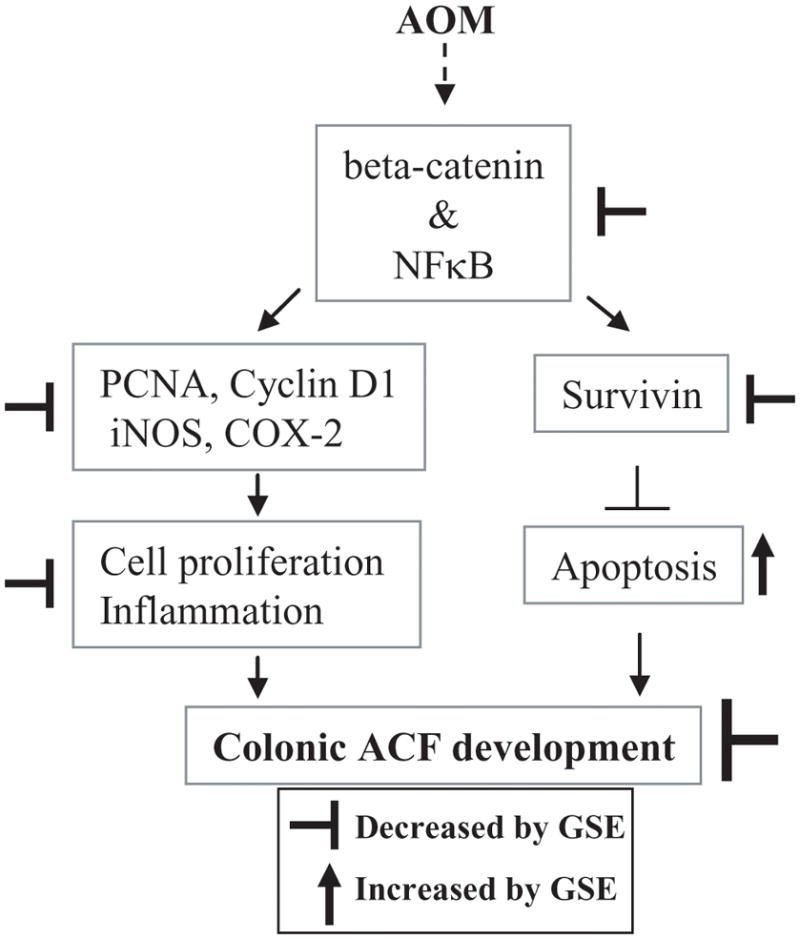
Proposed mechanism of GSE efficacy against AOM-induced colonic ACF formation in F344 rats. GSE suppresses β-catenin and NF-κB signaling and thereby inhibits cell proliferation and induces apoptosis together with down-regulation of cyclin D1, survivin, iNOS and COX-2 during AOM-induced colonic ACF formation in F344 rats. These molecular alterations by GSE could be, in part, underlying mechanisms by which it prevents AOM-induced colonic ACF formation. AOM, azoxymethane; GSE; grape seed extract, ACF, aberrant crypt foci; iNOS, inducible nitric oxide synthase; COX-2, cyclooxygenase-2.
Acknowledgments
Grant support: This work was supported by the NIH RO1 grant AT003623 from the National Center for Complementary and Alternative Medicine, and the Office of Dietary Supplement, National Institutes of Health, Bethesda, MD
Abbreviations
- AOM
azoxymethane
- GSE
grape seed extract
- ACF
aberrant crypt foci
- CRC
colorectal cancer
References
- 1.Huerta S. Recent advances in the molecular diagnosis and prognosis of colorectal cancer. Expert Rev Mol Diagn. 2008;8:277–288. doi: 10.1586/14737159.8.3.277. [DOI] [PubMed] [Google Scholar]
- 2.American Cancer Society. Colorectal cancer facts and figures. American Cancer Society; Atlanta, GA: 2009. [Google Scholar]
- 3.Half E, Arber N. Colon cancer: preventive agents and the present status of chemoprevention. Expert Opin Pharmacother. 2009;10:211–219. doi: 10.1517/14656560802560153. [DOI] [PubMed] [Google Scholar]
- 4.Veeriah S, Miene C, Habermann N, et al. Apple polyphenols modulate expression of selected genes related to toxicological defense and stress response in human colon adenoma cells. Int J Cancer. 2008;122:2647–2655. doi: 10.1002/ijc.23440. [DOI] [PubMed] [Google Scholar]
- 5.Rudolf E, Andelova H, Cervinka M. Polyphenolic compounds in chemoprevention of colon cancer - targets and signaling pathways. Anticancer Agents Med Chem. 2007;7:559–575. doi: 10.2174/187152007781668670. [DOI] [PubMed] [Google Scholar]
- 6.Bagchi D, Bagchi M, Stohs S, et al. Cellular protection with proanthocyanidins derived from grape seeds. Ann N Y Acad Sci. 2002;957:260–270. doi: 10.1111/j.1749-6632.2002.tb02922.x. [DOI] [PubMed] [Google Scholar]
- 7.Wen W, Lu J, Zhang K, Chen S. Grape seed extract inhibits angiogenesis via suppression of the vascular endothelial growth factor receptor signaling pathway. Cancer Prev Res (Phila Pa) 2008;1:554–561. doi: 10.1158/1940-6207.CAPR-08-0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singletary KW, Meline B. Effect of grape seed proanthocyanidins on colon aberrant crypts and breast tumors in a rat dual-organ tumor model. Nutr Cancer. 2001;39:252–258. doi: 10.1207/S15327914nc392_15. [DOI] [PubMed] [Google Scholar]
- 9.Zhao J, Wang J, Chen Y, Agarwal R. Anti-tumor-promoting activity of a polyphenolic fraction isolated from grape seeds in the mouse skin two-stage initiation-promotion protocol and identification of procyanidin B5-3′-gallate as the most effective antioxidant constituent. Carcinogenesis. 1999;20:1737–1745. doi: 10.1093/carcin/20.9.1737. [DOI] [PubMed] [Google Scholar]
- 10.Agarwal C, Sharma Y, Agarwal R. Anticarcinogenic effect of a polyphenolic fraction isolated from grape seeds in human prostate carcinoma DU145 cells: modulation of mitogenic signaling and cell-cycle regulators and induction of G1 arrest and apoptosis. Mol Carcinog. 2000;28:129–138. doi: 10.1002/1098-2744(200007)28:3<129::aid-mc1>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 11.Singh RP, Tyagi AK, Dhanalakshmi S, Agarwal R, Agarwal C. Grape seed extract inhibits advanced human prostate tumor growth and angiogenesis and upregulates insulin-like growth factor binding protein-3. Int J Cancer. 2004;108:733–740. doi: 10.1002/ijc.11620. [DOI] [PubMed] [Google Scholar]
- 12.Kaur M, Singh RP, Gu M, Agarwal R, Agarwal C. Grape seed extract inhibits in vitro and in vivo growth of human colorectal carcinoma cells. Clin Cancer Res. 2006;12:6194–6202. doi: 10.1158/1078-0432.CCR-06-1465. [DOI] [PubMed] [Google Scholar]
- 13.Kaur M, Agarwal C, Agarwal R. Anti-cancer and cancer chemopreventive potential of grape seed extract and other grape-based products. J Nutr. 2009;139:1806S–1812S. doi: 10.3945/jn.109.106864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raina K, Singh RP, Agarwal R, Agarwal C. Oral grape seed extract inhibits prostate tumor growth and progression in TRAMP mice. Cancer Res. 2007;67:5976–5982. doi: 10.1158/0008-5472.CAN-07-0295. [DOI] [PubMed] [Google Scholar]
- 15.Bird RP. Observation and quantification of aberrant crypts in the murine colon treated with a colon carcinogen: preliminary findings. Cancer Lett. 1987;37:147–151. doi: 10.1016/0304-3835(87)90157-1. [DOI] [PubMed] [Google Scholar]
- 16.Guruswamy S, Swamy MV, Choi CI, Steele VE, Rao CV. S-adenosyl L-methionine inhibits azoxymethane-induced colonic aberrant crypt foci in F344 rats and suppresses human colon cancer Caco-2 cell growth in 3D culture. Int J Cancer. 2008;122:25–30. doi: 10.1002/ijc.23031. [DOI] [PubMed] [Google Scholar]
- 17.Velmurugan B, Singh RP, Tyagi A, Agarwal R. Inhibition of azoxymethane-induced colonic aberrant crypt foci formation by silibinin in male Fisher 344 rats. Cancer Prev Res (Phila Pa) 2008;1:376–384. doi: 10.1158/1940-6207.CAPR-08-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Paul S, Dey A. Wnt signaling and cancer development: therapeutic implication. Neoplasma. 2008;55:165–176. [PubMed] [Google Scholar]
- 19.Kim HS, Kundu JK, Lee JS, Oh TY, Na HK, Surh YJ. Chemopreventive effects of the standardized extract (DA-9601) of Artemisia asiatica on azoxymethane-initiated and dextran sulfate sodium-promoted mouse colon carcinogenesis. Nutr Cancer. 2008;60:90–97. doi: 10.1080/01635580802404170. [DOI] [PubMed] [Google Scholar]
- 20.Yamakoshi J, Saito M, Kataoka S, Kikuchi M. Safety evaluation of proanthocyanidin-rich extract from grape seeds. Food Chem Toxicol. 2002;40:599–607. doi: 10.1016/s0278-6915(02)00006-6. [DOI] [PubMed] [Google Scholar]
- 21.Naryzhny SN. Proliferating cell nuclear antigen: a proteomics view. Cell Mol Life Sci. 2008;65:3789–3808. doi: 10.1007/s00018-008-8305-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baldwin AS., Jr The NF-κB and IκB proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 23.Bonovas S, Tsantes A, Drosos T, Sitaras NM. Cancer chemoprevention: a summary of the current evidence. Anticancer Res. 2008;28:1857–1866. [PubMed] [Google Scholar]
- 24.Marshall JR. Prevention of colorectal cancer: diet, chemoprevention, and lifestyle. Gastroenterol Clin North Am. 2008;37:73–82. doi: 10.1016/j.gtc.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mason JB. Nutritional chemoprevention of colon cancer. Semin Gastrointest Dis. 2002;13:143–153. [PubMed] [Google Scholar]
- 26.Takahashi M, Wakabayashi K. Gene mutations and altered gene expression in azoxymethane-induced colon carcinogenesis in rodents. Cancer Sci. 2004;95:475–480. doi: 10.1111/j.1349-7006.2004.tb03235.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Janakiram NB, Steele VE, Rao CV. Estrogen receptor-beta as a potential target for colon cancer prevention: chemoprevention of azoxymethane-induced colon carcinogenesis by raloxifene in F344 rats. Cancer Prev Res (Phila Pa) 2009;2:52–59. doi: 10.1158/1940-6207.CAPR-08-0140. [DOI] [PubMed] [Google Scholar]
- 28.Alrawi SJ, Schiff M, Carroll RE, et al. Aberrant crypt foci. Anticancer Res. 2006;26:107–119. [PubMed] [Google Scholar]
- 29.Feregrino-Perez AA, Berumen LC, Garcia-Alcocer G, et al. Composition and chemopreventive effect of polysaccharides from common beans (Phaseolus vulgaris L.) on azoxymethane-induced colon cancer. J Agric Food Chem. 2008;56:8737–8744. doi: 10.1021/jf8007162. [DOI] [PubMed] [Google Scholar]
- 30.Yasui Y, Miyamoto S, Kim M, Kohno H, Sugie S, Tanaka T. Aqueous and ethanolic extract fractions from the Brazilian propolis suppress azoxymethane-induced aberrant crypt foci in rats. Oncol Rep. 2008;20:493–499. [PubMed] [Google Scholar]
- 31.Kohno H, Tanaka T, Kawabata K, et al. Silymarin, a naturally occurring polyphenolic antioxidant flavonoid, inhibits azoxymethane-induced colon carcinogenesis in male F344 rats. Int J Cancer. 2002;101:461–468. doi: 10.1002/ijc.10625. [DOI] [PubMed] [Google Scholar]
- 32.Trosko JE, Ruch RJ. Cell-cell communication in carcinogenesis. Front Biosci. 1998;3:208–236. doi: 10.2741/a275. [DOI] [PubMed] [Google Scholar]
- 33.Arakaki J, Suzui M, Morioka T, et al. Antioxidative and modifying effects of a tropical plant Azadirachta indica (Neem) on azoxymethane-induced preneoplastic lesions in the rat colon. Asian Pac J Cancer Prev. 2006;7:467–471. [PubMed] [Google Scholar]
- 34.Morioka T, Suzui M, Nabandith V, et al. Modifying effects of Terminalia catappa on azoxymethane-induced colon carcinogenesis in male F344 rats. Eur J Cancer Prev. 2005;14:101–105. doi: 10.1097/00008469-200504000-00005. [DOI] [PubMed] [Google Scholar]
- 35.Wang HL, Wang J, Xiao SY, et al. Elevated protein expression of cyclin D1 and Fra-1 but decreased expression of c-Myc in human colorectal adenocarcinomas over expressing beta-catenin. Int J Cancer. 2002;101:301–310. doi: 10.1002/ijc.10630. [DOI] [PubMed] [Google Scholar]
- 36.Bondi J, Bukholm G, Nesland JM, Bukholm IR. Expression of non-membranous beta-catenin and gamma-catenin, c-Myc and cyclin D1 in relation to patient outcome in human colon adenocarcinomas. APMIS. 2004;112:49–56. doi: 10.1111/j.1600-0463.2004.apm1120109.x. [DOI] [PubMed] [Google Scholar]
- 37.Issa AY, Volate SR, Muga SJ, Nitcheva D, Smith T, Wargovich MJ. Green tea selectively targets initial stages of intestinal carcinogenesis in the AOM-ApcMin mouse model. Carcinogenesis. 2007;28:1978–1984. doi: 10.1093/carcin/bgm161. [DOI] [PubMed] [Google Scholar]
- 38.Sharma G, Tyagi AK, Singh RP, Chan DC, Agarwal R. Synergistic anti-cancer effects of grape seed extract and conventional cytotoxic agent doxorubicin against human breast carcinoma cells. Breast Cancer Res Treat. 2004;85:1–12. doi: 10.1023/B:BREA.0000020991.55659.59. [DOI] [PubMed] [Google Scholar]
- 39.Andersen MH, Svane IM, Becker JC, Straten PT. The universal character of the tumor-associated antigen survivin. Clin Cancer Res. 2007;13:5991–5994. doi: 10.1158/1078-0432.CCR-07-0686. [DOI] [PubMed] [Google Scholar]
- 40.Watanabe K, Kawamori T, Nakatsugi S, Wakabayashi K. COX-2 and iNOS, good targets for chemoprevention of colon cancer. Biofactors. 2000;12:129–133. doi: 10.1002/biof.5520120120. [DOI] [PubMed] [Google Scholar]
- 41.van der Woude CJ, Kleibeuker JH, Jansen PL, Moshage H. Chronic inflammation, apoptosis and (pre-)malignant lesions in the gastro-intestinal tract. Apoptosis. 2004;9:123–130. doi: 10.1023/B:APPT.0000018794.26438.22. [DOI] [PubMed] [Google Scholar]
- 42.Janakiram NB, Indranie C, Malisetty SV, Jagan P, Steele VE, Rao CV. Chemoprevention of colon carcinogenesis by oleanolic acid and its analog in male F344 rats and modulation of COX-2 and apoptosis in human colon HT-29 cancer cells. Pharm Res. 2008;25:2151–2157. doi: 10.1007/s11095-008-9582-7. [DOI] [PubMed] [Google Scholar]
- 43.Vanamala J, Leonardi T, Patil BS, et al. Suppression of colon carcinogenesis by bioactive compounds in grapefruit. Carcinogenesis. 2006;27:1257–1265. doi: 10.1093/carcin/bgi318. [DOI] [PubMed] [Google Scholar]
- 44.Luceri C, Caderni G, Sanna A, Dolara P. Red wine and black tea polyphenols modulate the expression of cycloxygenase-2, inducible nitric oxide synthase and glutathione-related enzymes in azoxymethane-induced f344 rat colon tumors. J Nutr. 2002;132:1376–1379. doi: 10.1093/jn/132.6.1376. [DOI] [PubMed] [Google Scholar]
- 45.Reddy BS, Hirose Y, Lubet R, et al. Chemoprevention of colon cancer by specific cyclooxygenase-2 inhibitor, celecoxib, administered during different stages of carcinogenesis. Cancer Res. 2000;60:293–297. [PubMed] [Google Scholar]
- 46.Yoshimi N, Shimizu M, Matsunaga K, et al. Chemopreventive effect of N-(2-cyclohexyloxy-4-nitrophenyl)methane sulfonamide (NS-398), a selective cyclooxygenase-2 inhibitor, in rat colon carcinogenesis induced by azoxymethane. Jpn J Cancer Res. 1999;90:406–412. doi: 10.1111/j.1349-7006.1999.tb00762.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Murakami A. Chemoprevention with phytochemicals targeting inducible nitric oxide synthase. Forum Nutr. 2009;61:193–203. doi: 10.1159/000212751. [DOI] [PubMed] [Google Scholar]
- 48.Olivier S, Robe P, Bours V. Can NF-κB be a target for novel and efficient anti-cancer agents? Biochem Pharmacol. 2006;72:1054–1068. doi: 10.1016/j.bcp.2006.07.023. [DOI] [PubMed] [Google Scholar]
- 49.Lind DS. NF-κB is upregulated in colorectal cancer. Surgey. 2001;130:363–369. doi: 10.1067/msy.2001.116672. [DOI] [PubMed] [Google Scholar]
- 50.Surh YJ, Chun KS, Cha HH, et al. Molecular mechanisms underlying chemopreventive activities of anti-inflammatory phytochemicals: downregulation of COX-2 and iNOS through suppression of NF-κB activation. Mutat Res. 2001;480:243–268. doi: 10.1016/s0027-5107(01)00183-x. [DOI] [PubMed] [Google Scholar]