

Abstract
Nef is an HIV-1 virulence factor that promotes viral pathogenicity by altering host cell signaling pathways. Nef binds several members of the Src kinase family, and these interactions have been implicated in the pathogenesis of HIV/AIDS. However, the direct effect of Nef interaction on Src family kinase (SFK) regulation and activity has not been systematically addressed. We explored this issue using Saccharomyces cerevisiae, a well defined model system for the study of SFK regulation. Previous studies have shown that ectopic expression of c-Src arrests yeast cell growth in a kinase-dependent manner. We expressed Fgr, Fyn, Hck, Lck, Lyn, and Yes as well as c-Src in yeast and found that each kinase was active and induced growth suppression. Co-expression of the negative regulatory kinase Csk suppressed SFK activity and reversed the growth-inhibitory effect. We then co-expressed each SFK with HIV-1 Nef in the presence of Csk. Nef strongly activated Hck, Lyn, and c-Src but did not detectably affect Fgr, Fyn, Lck, or Yes. Mutagenesis of the Nef PXXP motif essential for SH3 domain binding greatly reduced the effect of Nef on Hck, Lyn, and c-Src, suggesting that Nef activates these Src family members through allosteric displacement of intra-molecular SH3-linker interactions. These data show that Nef selectively activates Hck, Lyn, and c-Src among SFKs, identifying these kinases as proximal effectors of Nef signaling and potential targets for anti-HIV drug discovery.
Nef is an accessory protein encoded by the human (HIV-1 and HIV-2)2 and simian immunodeficiency viruses and is an essential mediator of viral pathogenicity (1–3). Experimental deletion within the simian immunodeficiency virus nef gene reduces viral load, delays the onset of AIDS-like disease, and offers immune protection against challenge with pathogenic simian immunodeficiency virus in rhesus macaques (4, 5). Strong selective pressure has been demonstrated for a functional nef gene, because some animals infected with non-pathogenic, nef-mutant simian immunodeficiency virus show in vivo repair of the mutation and progression to AIDS-like disease (5–7). In addition, some HIV-positive individuals that fail to develop AIDS exhibit nef mutations or deletions (8–12), supporting the hypothesis that nef is essential for efficient disease progression.
Nef has no known catalytic function and is believed to promote viral pathogenicity by altering signaling pathways in infected cells through its interactions with cellular proteins. Nef affects several distinct classes of host cell proteins, including immune receptors, protein kinases, trafficking proteins, and guanine nucleotide exchange factors (13–15). Through interactions with these and other signaling proteins, Nef can affect multiple cellular processes leading to enhancement of viral replication, immune evasion, and enhanced survival in T-cells and macrophages (1, 16–18).
Protein kinases are a major class of Nef effector proteins, and members of the Src family of non-receptor protein-tyrosine kinases have been strongly linked to Nef function. Numerous reports have demonstrated that Nef interacts with the isolated Src homology 3 (SH3) domains from Src family members expressed in HIV target cells, including Fyn, Hck, Lck, Lyn, and c-Src itself (19–25). Among these, the interaction of Nef with Hck has been studied in great detail at the cellular and molecular levels. Hck is strongly expressed in cells of the monocyte/macrophage lineage (26, 27), which are essential HIV-1 target cells and viral reservoirs (28–30). X-ray crystallography demonstrates that Nef interacts with the Hck SH3 domain via a bipartite mechanism dependent upon the three-dimensional fold of Nef (31). These contacts include a highly conserved Nef PXXPXR motif, which forms the polyproline type II helix typical of SH3 ligands. In addition, Ile-96 within the RT-loop of the Hck SH3 domain fits into a pocket within the Nef core that is lined with several highly conserved hydrophobic residues. Both of these interactions are necessary, but not independently sufficient, for high affinity binding of Nef to the Hck SH3 domain (22, 32).
Nef-SH3 domain interaction leads to constitutive Hck activation in vitro (33) and in cell-based systems, including fibro-blasts (32), myeloid cell lines (16, 34), and HIV-infected primary macrophages (35). Nef binding is believed to disrupt the normal role of the Hck SH3 domain in suppression of kinase activity (32). When Hck is down-regulated, its SH3 domain associates with the polyproline type II helix formed by the linker connecting the SH2 and kinase domains (36, 37). This interaction is stabilized by the interaction of the SH2 domain with the C-terminal tail, which is phosphorylated on a conserved tyrosine residue by the negative regulatory kinases Csk and Chk (38–40). Binding of Nef to the SH3 domain causes linker displacement (33), resulting in a conformational shift in the kinase domain permissive for ATP binding, target protein access, and phosphotransfer (41). Nef-induced Hck activation does not require tail dephosphorylation or displacement from the SH2 domain, suggesting that Nef may induce a novel signaling conformation of Hck (42).
Although the functional consequences of the Nef-Hck pathway are still under investigation, mounting evidence suggests a key role for this interaction in AIDS progression. In monocyte-derived macrophages, Komuro et al. (35) established a strong positive correlation of high titer replication of macrophage-tropic HIV-1 with Hck expression. In addition, they showed that HIV replication is blocked following suppression of Hck with antisense oligonucleotides. At the whole animal level, targeted expression of Nef to the T-cell and macrophage compartments in transgenic mice induces an AIDS-like phenotype, characterized by CD4+ T-cell depletion, diarrhea, wasting, and uniform mortality (43). In contrast, mice expressing a mutant form of Nef lacking the PXXPXR motif essential for SH3 binding show no evidence of the AIDS-like phenotype (44). Interestingly, when transgenic mice expressing wild-type Nef were crossed into a hck-null background, appearance of the AIDS-like phenotype was delayed with a significant proportion of the mice living normal lifespans (44). The observation that the Nef-induced AIDS-like syndrome is reduced but not eliminated in the absence of Hck suggests that other SFKs may contribute to Nef signaling in this system.
Less is known about the functional interaction of Nef with other Src family members. Lyn is the only Src family member other than Hck with an Ile residue in the SH3 domain RT-loop. Although the Lyn SH3 domain appears to bind as tightly to Nef as the Hck SH3 domain in vitro (19, 22), the effect of Nef on Lyn kinase activity has not been reported. Several reports suggest that HIV-1 Nef binds to Lck and down-regulates its kinase activity (21, 45, 46). However, other work failed to detect a direct interaction of Nef with Lck or provide evidence for an effect on kinase activity in vivo (47, 48). Conflicting reports exist regarding the interaction of Nef with Fyn and its SH3 domain (19, 22, 48–51), whereas the direct effect of Nef on Fyn kinase activity is unknown. Nef also interacts with both full-length c-Src and its SH3 domain (22), although the direct effect of Nef on c-Src activity is not clear (48, 52). Finally, while Fgr and Yes are present in HIV target cells, neither has been tested for interactions with Nef.
One explanation for the conflicting literature regarding the impact of Nef on SFK activity relates to the use of diverse systems for analysis. In addition, Nef is likely to activate multiple kinases in HIV target cells, obscuring its direct effects on individual Src family members. Identification of those SFKs that are directly activated by Nef is the first step toward validation of these kinases as drug discovery targets. Here we address this important issue using a yeast-based expression system, originally developed for the study of c-Src regulation (53–55). Yeast cells represent a useful model for the study of SFK regulation, because they do not express orthologs of c-Src or other mammalian protein-tyrosine kinases. In addition, ectopic expression of c-Src and other mammalian protein-tyrosine kinases has been shown to induce kinase-dependent growth arrest in yeast, providing a convenient end-point for structure-function analysis (56–60). In the case of c-Src, co-expression of the regulatory kinase Csk reverses the growth-inhibitory effect through phosphorylation of the negative regulatory tail, modeling the natural mechanism of down-regulation in mammalian cells (56, 60, 61).
In this report, we first show that regulation of Hck kinase activity by Nef can be faithfully reconstituted in yeast. When expressed alone, Hck was highly active and produced a strong growth-suppressive phenotype. Hck activity and growth suppression were reversed upon co-expression of the negative regulatory kinase, Csk. Introduction of Nef led to re-activation of Hck despite the presence of Csk, closely modeling previous reports in mammalian cells types (16, 32). We then extended the study to include all other SFKs expressed in HIV target cells: Fgr, Fyn, Lck, Lyn, c-Src, and Yes. Like Hck, all of these kinases suppressed yeast cell growth when active, and this phenotype was reversed upon co-expression with Csk. Introduction of Nef led to clear activation of Lyn and c-Src in addition to Hck. In all three cases, activation involved the PXXPXR motif of Nef, suggesting a common SH3-linker displacement mechanism previously described for Hck. In contrast, Nef did not affect Lck or Fyn activity, despite previous reports of Nef binding to these kinases (20, 24, 46, 47, 50, 51). Nef also failed to affect Yes or Fgr activity. These data provide the first complete analysis of direct HIV-1 Nef-SFK interaction in living cells and identify the complexes of Nef with Hck, Lyn, and c-Src as unique targets for anti-HIV drug discovery.
MATERIALS AND METHODS
Yeast Expression Vectors
Coding sequences for human Csk, c-Src, Fyn, Hck, Lck, and Lyn as well as murine Fgr and Yes were amplified by PCR from existing templates to introduce a yeast translation initiation sequence (AATA) immediately 5′ to the ATG start codon. The cDNA clones for HIV-1 Nef (SF2 strain) and the herpesvirus saimiri Tip protein (amino acids 1–187) were similarly amplified and modified. A FLAG epitope tag was added to the N terminus of the Tip coding sequence. All SFK cDNA clones were subcloned downstream of either the Gal1 or Gal10 promoter in the yeast expression vector pESC-Ura (Stratagene). Hck was also subcloned downstream of the Gal10 promoter in the pYC2/CT vector (Invitrogen), which carries the CEN6/ARSH4 sequence for low copy replication. The Csk, Nef, and Tip cDNAs were subcloned downstream of either the Gal 1 or Gal10 promoter in pESC-Trp (Stratagene). cDNA clones for kinase-defective Hck (Hck-K269D), tail-activated Hck (Hck-Y501F), and kinase-defective Csk (Csk-K222D) were created via site-directed mutagenesis (QuikChange XL site-directed mutagenesis kit, Stratagene). The Nef-2PA mutant, in which prolines 72 and 75 are replaced with alanines, has been described elsewhere (32).
Yeast Growth Suppression Assay
Saccharomyces cerevisiae strain YPH 499 (Stratagene) was co-transformed with pESC-Ura (or pYC2/CT) and pESC-Trp plasmids containing the genes of interest via electroporation (Bio-Rad GenePulser II). Yeast cells were selected for 3 days at 30 °C on standard synthetic drop-out plates lacking uracil and tryptophan (SD/-U-T) with glucose as the sole carbon source to repress protein expression. Positive transformants were grown in liquid SD/-U-T medium plus glucose, normalized to A600 = 0.2 in water, and then spotted in 4-fold dilutions onto SD/-U-T agar plates containing galactose as the sole carbon source to induce protein expression. Plates were incubated for 3 days at 30 °C and imaged on a flatbed scanner. Yeast patches appear as dark spots against the translucent agar background. All growth suppression assays were repeated at least three times starting with randomly selected independent transformed clones and produced comparable results; representative examples are shown.
Immunoblotting
Aliquots of the yeast cultures used for the spot assay were grown in SD/-U-T medium plus galactose for 18 h. Cells were pelleted, treated with 0.1 N NaOH for 5 min at room temperature (62), and normalized with SDS-PAGE sample buffer to 0.02 A600 units per microliter. Aliquots of each lysate (0.2 A600 unit) were separated via SDS-PAGE, transferred to polyvinylidene difluoride membranes, and probed for protein phosphotyrosine content with a combination of the anti-phosphotyrosine antibodies PY99 (Santa Cruz Biotechnology, Santa Cruz, CA) and PY20 (BD Transduction Laboratories). Protein expression was verified by immunoblotting with antibodies to Csk (C-20, Santa Cruz Biotechnology), Fgr (C1, Santa Cruz Biotechnology), FLAG (M2, Sigma), Fyn (FYN3, Santa Cruz Biotechnology), Hck (N-30, Santa Cruz Biotechnology), Lck (2102, Santa Cruz Biotechnology), Lyn (44, Santa Cruz Bio-technology), Src (N-16, Santa Cruz Biotechnology), and Yes (3, Santa Cruz Biotechnology). Nef antibodies (monoclonal EH1 and Hyb 6.2) were obtained from the National Institutes of Health AIDS Research and Reference Reagent Program.
Expression and Purification of Recombinant SFKs and Nef
Human Hck, Lyn, and c-Src clones were modified on their C-terminal tails to encode the sequence Tyr-Glu-Glu-Ile-Pro. This modification promotes autophosphorylation of the tail and permits high yield purification of the down-regulated form of each kinase without the need for co-expression of Csk (37). The N-terminal unique domain of each kinase was replaced with a hexahistidine tag, and each construct was used to produce a recombinant baculovirus in Sf9 insect cells using BaculoGold DNA and the manufacturer’s protocol (BD Pharmingen). Recombinant SFKs were purified from 1 liter of infected Sf9 cell culture using a combination of ion-exchange and affinity chromatography as originally described by Schindler et al. for Hck (37). The purity and concentration of each kinase preparation were confirmed by SDS-PAGE and densitometry. The SF2 allele of HIV-1 Nef was similarly expressed and purified with an N-terminal hexahistidine tag.
In Vitro Kinase Assays
Tyrosine kinase assays were performed in 384-well plates using the FRET-based Z′-lyte kinase assay system and the Tyr2 peptide substrate (Invitrogen). Reactions (10 μl) were conducted in kinase buffer (50 mM Hepes, pH 7.5, 10 mM MgCl2, 1 mM EGTA, 0.01% Brij-35). Assay conditions were first optimized to determine the amount of each kinase and the incubation time necessary to phosphorylate 20–30% of the Tyr2 peptide in the absence of Nef. To assess the effect of Nef on SFK activity, Hck (20 ng), Lyn (50 ng), and Src (50 ng) were incubated at room temperature for 5 min with a 5-or 10-fold molar excess of Nef. ATP (50 μM final) and Tyr2 substrate (2 μM final) were then added to the reaction followed by a 1-h incubation (45 min for Hck). Development reagent, containing a protease that digests non-phosphorylated peptide, was then added to the reaction for an additional 60 min at room temperature, at which time the reaction was terminated with the proprietary stop reagent. Fluorescence was assessed at an excitation wavelength of 400 nm; coumarin fluorescence and the fluorescein FRET signal were monitored at 445 and 520 nm, respectively. The coumarin emission excites fluorescein by FRET in the phosphorylated (uncleaved) substrate peptide only. Reactions containing unphosphorylated peptide and kinase in the absence of ATP served as 0% phosphorylation control, whereas a stoichiometrically phosphorylated peptide was used as a 100% phosphorylation control. Raw fluorescence values were corrected for background, and reaction endpoints were calculated as emission ratios of coumarin fluorescence divided by the fluorescein FRET signal. These ratios were then normalized to the ratio obtained with the 100% phosphorylation control. Each condition was assayed in quadruplicate, and results are presented as the mean ± S.D. The entire experiment was repeated twice with comparable results.
RESULTS
Active Hck Suppresses Yeast Growth in a Kinase-dependent Manner
Before reconstituting the interaction of Hck with HIV-1 Nef in yeast, we first determined whether Hck produced a similar growth-suppressive phenotype as described previously for c-Src (56, 58, 60). To accomplish this, we used a plate-based assay to visualize the effects of Hck expression on yeast cell growth. Yeast cultures transformed with galactose-inducible Hck and Csk expression vectors were spotted as a dilution series on galactose-agar plates. As shown in Fig. 1, expression of wild-type Hck induced growth suppression relative to control cultures transformed with empty expression plasmids. Growth suppression was reversed when Hck was co-expressed with wild-type Csk, the kinase responsible for down-regulation of SFKs in mammalian cells (63). Kinase-dead Csk (Csk-K222D) was unable to reverse Hck-induced growth suppression, consistent with negative regulation of Hck by Csk-mediated tail tyrosine phosphorylation as observed previously in fibroblasts (42).
FIGURE 1. Hck induces yeast growth suppression in a kinase-dependent manner.
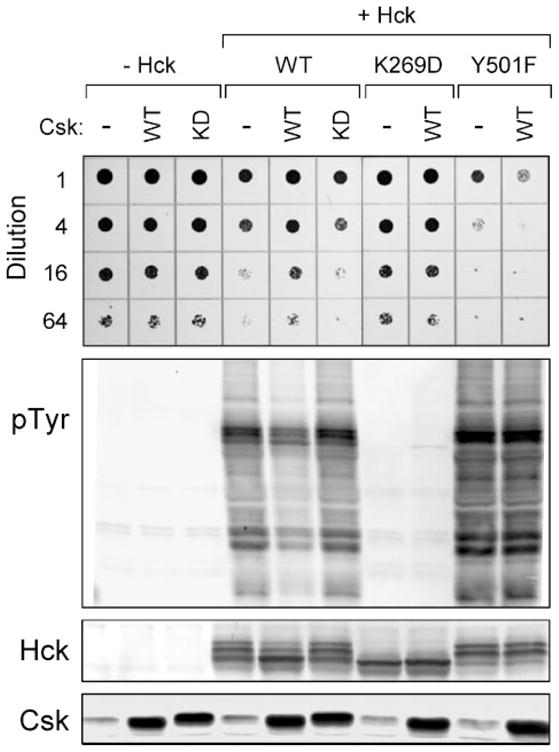
Yeast cultures were transformed with galactose-inducible expression plasmids for wild-type Hck (WT), a kinase-dead mutant (K269D), a mutant lacking the C-terminal Csk phosphorylation site (Y501F), or the empty expression plasmid (−Hck). Cells were co-transformed with galactose-inducible vectors for wild-type (WT) or kinase-dead (KD) Csk as indicated or with the empty vector as a negative control (−). Top: liquid cultures were grown with glucose as the sole carbon source to repress protein expression and normalized to equal densities. Cells were then spotted onto agar selection plates containing galactose as the sole carbon source and incubated for 3 days at 30 °C. Cultures were spotted in 4-fold dilutions to enhance visualization of the growth-suppressive phenotype. Plates were scanned, and yeast patches appear as dark circles. Lower panels: immunoblots from cultures shown at the top. Transformed cells were grown in liquid culture in the presence of galactose at 30 °C for 18 h. Protein extracts were separated via SDS-PAGE and immunoblotted for tyrosine-phosphorylated proteins (pTyr) as well as for Hck and Csk.
To determine whether growth suppression induced by Hck correlated with Hck kinase activity, yeast cell lysates were probed with anti-phosphotyrosine antibodies. Expression of Hck alone correlated with strong phosphorylation of many yeast proteins (Fig. 1). Co-expression with Csk led to a marked decrease in protein phosphotyrosine content, consistent with down-regulation of Hck kinase activity. Kinase-dead Csk was unable to inhibit Hck activity, establishing the role of Csk kinase activity in the control of Hck. Expression of Csk alone produced no growth suppressive effect or yeast protein-tyrosine phosphorylation, demonstrating the exquisite specificity of Csk activity for the tail region of Hck. These findings show that co-expression with Csk is sufficient to down-regulate Hck in yeast, consistent with previous observations for c-Src (56, 60, 61).
Immunoblots of yeast lysates with Hck antibodies revealed that Hck consistently migrated as three distinct bands (Fig. 1). Interestingly, this banding pattern changed when Hck was co-expressed with wild-type but not kinase-dead Csk, with three bands shifting to a single high mobility band. This effect of Csk suggests that the variation in Hck mobility reflects different activation states, with the highest mobility form representing the down-regulated conformation.
To demonstrate that growth suppression is dependent upon Hck kinase activity, we transformed yeast with a kinase-dead form of Hck, Hck-K269D. Hck-K269D failed to induce either growth suppression or tyrosine phosphorylation of yeast proteins (Fig. 1). Co-expression with Csk led to tyrosine phosphorylation of Hck-K269D, presumably on the tail tyrosine residue. In addition, Hck-K269D runs as a single high mobility band on the anti-Hck immunoblot, providing further evidence that the high mobility form of Hck corresponds to the inactive conformation.
Csk down-regulates Hck activity in mammalian cells by phosphorylating Tyr-501 on the C-terminal tail (42). To confirm this mechanism of Csk-induced down-regulation of Hck in yeast, we co-expressed a Hck mutant lacking the regulatory tail tyrosine (Hck-Y501F) in the presence or absence of Csk. As shown in Fig. 1, Hck-Y501F markedly suppressed yeast growth and heavily phosphorylated yeast proteins. Csk was unable to alleviate the growth suppression or the protein-tyrosine phosphorylation induced by Hck-Y501F. Interestingly, Hck-Y501F migrated as the two lower mobility bands on anti-Hck immunoblots, and co-expression with Csk had no effect on this pattern. These results show that inhibition of Hck by Csk in yeast requires both Csk kinase activity and the Hck tail tyrosine residue (Tyr-501), thus faithfully modeling the mechanism in mammalian cells.
Nef activates Hck in a PXXP-dependent Manner
Previous studies have established that HIV-1 Nef binds tightly to the SH3 domain of Hck, leading to constitutive kinase activation both in vitro and in mammalian cells (32, 33, 42). To determine if Nef activates Hck in yeast through a similar mechanism, we first co-expressed Nef with Hck in the absence of Csk (Fig. 2). Interestingly, Hck-induced growth arrest and protein-tyrosine phosphorylation were both markedly increased in the presence of Nef. We then repeated the experiment in the presence of Csk and found that Nef completely reversed the inhibitory effect of Csk, leading to growth suppression and protein-tyrosine phosphorylation very similar to that observed when Hck is co-expressed with Nef in the absence of Csk. These observations suggest that Nef may generate a unique highly active conformation of the kinase (see “Discussion”). Control cultures show that Nef, either alone or when co-expressed with Csk, has no effect on cell growth or protein-phosphotyrosine content.
FIGURE 2. HIV-1 Nef activates Hck in a PXXP-dependent manner.
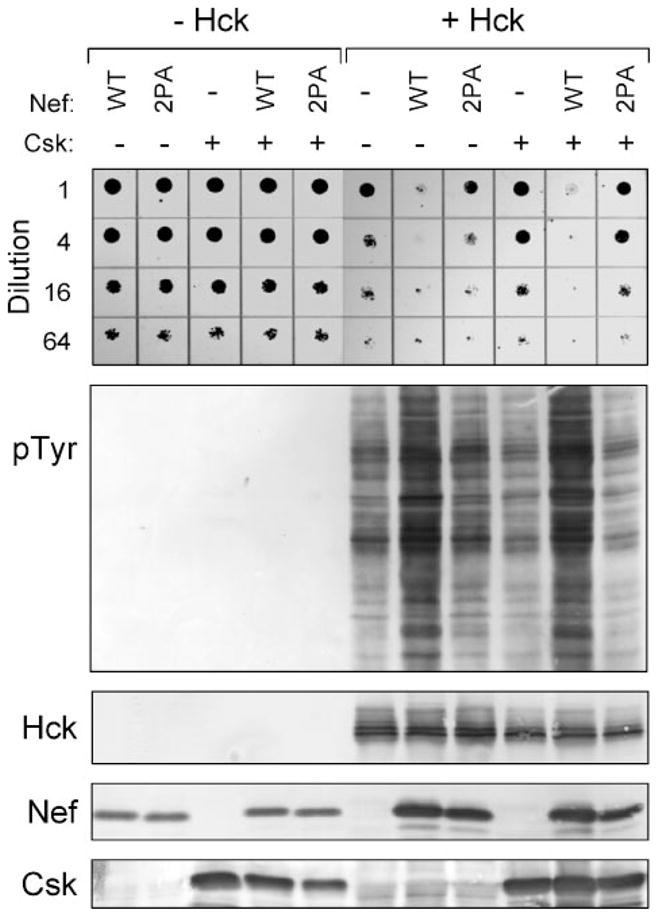
Yeast cultures were transformed with galactose-inducible expression plasmids for wild-type Hck (+Hck) in the absence (−) or presence of wild-type (WT) or PXXP mutant (2PA) forms of HIV-1 Nef. Cells were co-transformed with galac-tose-inducible expression vectors for Csk or the corresponding empty vector as indicated. Control cultures without Hck are shown on the left (−Hck). Top: liquid cultures were grown with glucose as the sole carbon source to repress protein expression and normalized to equal densities. Cells were then spotted onto agar selection plates containing galactose as the sole carbon source and incubated for 3 days at 30 °C. Cultures were spotted in 4-fold dilutions to enhance visualization of the growth-suppressive phenotype. Plates were scanned and yeast patches appear as dark circles. Lower panels: Immunoblots from cultures shown at the top. Transformed cells were grown in liquid culture in the presence of galactose at 30 °C for 18 h. Protein extracts were separated via SDS-PAGE and immunoblotted for tyrosine-phosphorylated proteins (pTyr) as well as for Hck, Nef, and Csk.
To determine if Nef-induced Hck activation is dependent upon the Nef SH3-binding function, we mutated the PXXP motif of Nef to AXXA (Nef-2PA). As shown in Fig. 2, Nef-2PA failed to enhance growth suppression by Hck or increase protein-tyrosine phosphorylation in the presence or absence of Csk. These results support an allosteric mechanism in which Nef-induced activation of Hck requires SH3 binding and displacement of the SH2-kinase linker.
Suppression of Yeast Cell Growth Is a Shared Property of SFK
Before evaluating the effect of Nef expression on other members of the Src kinase family, we first determined whether other SFKs produced the same growth suppressive effect as c-Src and Hck. Fgr, Fyn, Lck, Lyn, and Yes were each expressed in yeast with or without Csk and spot assays performed to measure growth suppression. Hck and c-Src were also included for comparison. As shown in Fig. 3, all seven SFKs suppressed yeast growth and co-expression of Csk reversed the growth-inhibitory phenotype.
FIGURE 3. Csk reverses growth suppression of yeast by SFKs.
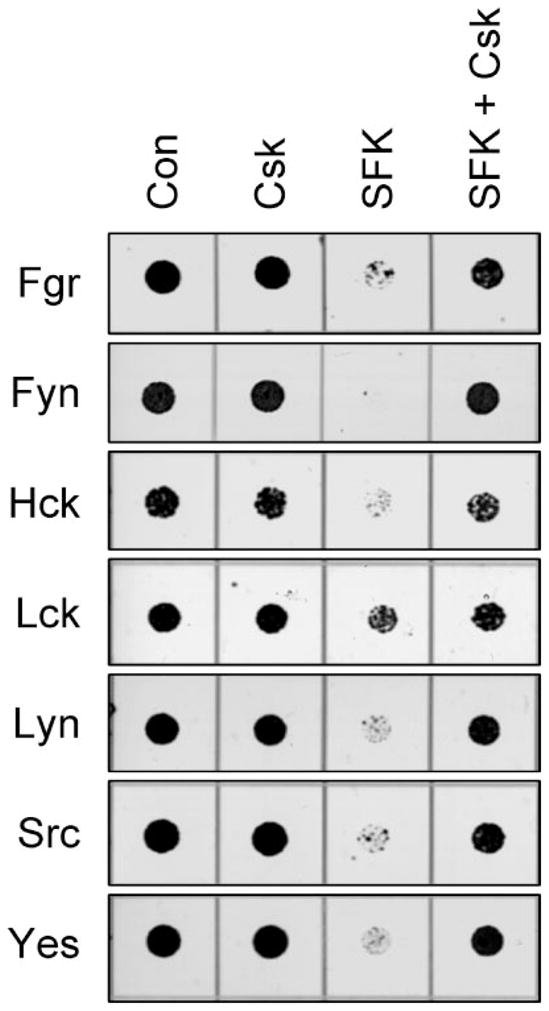
Yeast cultures were transformed with the SFKs indicated on the left either alone (SFK) or in the presence of Csk (SFK + Csk). Cells transformed with empty vectors (Con) or with Csk alone (Csk) were included as controls in each experiment. Liquid cultures were grown with glucose as the sole carbon source to repress protein expression and normalized to equal densities. Cells were then spotted onto agar selection plates containing galactose as the sole carbon source and incubated for 3 days at 30 °C. Plates were scanned, and yeast patches appear as dark circles. Cultures were spotted in 4-fold dilutions to enhance visualization of the growth-suppressive phenotype. The dilutions showing the clearest differences in growth in the presence and absence of Csk are presented.
Cell lysates from each of the cultures in Fig. 3 were then immunoblotted for tyrosine-phosphorylated proteins, as well as for expression of each SFK and Csk (Fig. 4). In each case, co-expression with Csk resulted in a decrease in the intensity of the anti-phosphotyrosine signal. These results show that all SFKs are constitutively active following ectopic expression in yeast, and provide direct evidence that Csk alone is sufficient to down-regulate the activity of each member of the Src kinase family.
FIGURE 4. Csk suppresses SFK activity in yeast.
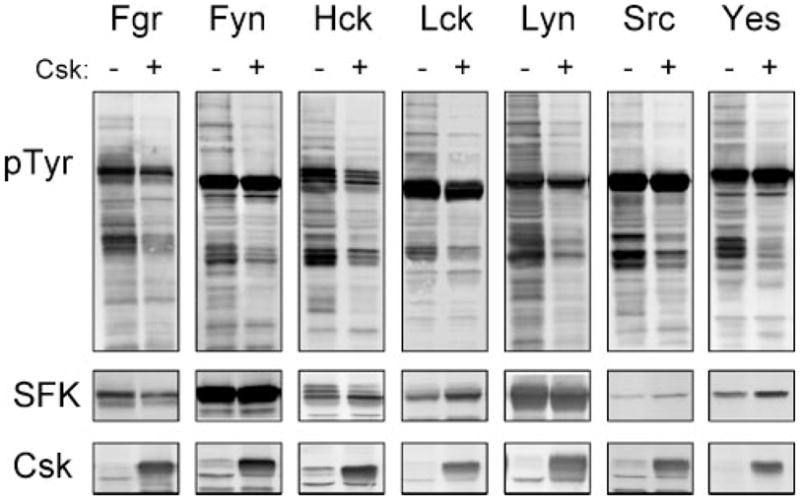
Yeast cultures were transformed with expression vectors for each of the SFKs shown at the top in the presence (+) or absence (−) of Csk. Cells were grown in liquid culture in the presence of galactose at 30 °C for 18 h. Protein extracts were separated via SDS-PAGE, and immunoblotted for tyrosine-phosphorylated proteins (pTyr) as well as for each Src family member (SFK) and Csk.
Nef Selectively Activates a Subset of Src Family Kinases
We next investigated whether co-expression of HIV-1 Nef was sufficient to activate SFKs other than Hck in the yeast model system. The SFKs were also co-expressed with Csk to determine whether interaction with Nef is sufficient to activate the down-regulated form of each kinase. Growth suppression data for each SFK in the presence or absence of Csk and Nef are shown in Fig. 5. In the absence of Csk, Nef enhanced the growth suppression observed with Hck but did not affect growth suppression by the other SFKs. However, Nef readily restored growth suppression by Csk-down-regulated c-Src and Lyn in addition to Hck. Csk-down-regulated Fyn, Fgr, Lck, and Yes were not affected by Nef.
FIGURE 5. HIV-1 Nef selectively induces growth suppression in yeast co-expressing down-regulated forms of Hck, Lyn, and c-Src.
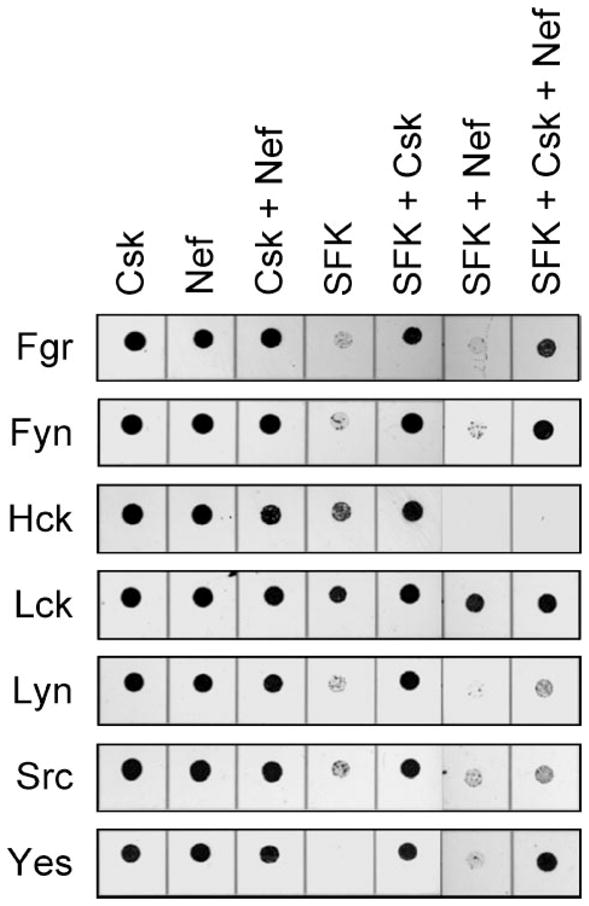
Yeast cultures were transformed with the SFKs indicated on the left either alone (SFK) or in the presence of Csk (SFK + Csk), HIV-1 Nef (SFK + Nef), or both (SFK + Csk + Nef). Cells transformed with Csk alone (Csk), Nef alone (Nef) or both (Csk + Nef) were included as controls in each experiment. Liquid cultures were grown with glucose as the sole carbon source to repress protein expression and normalized to equal densities. Cells were then spotted onto agar selection plates containing galactose as the sole carbon source and incubated for 3 days at 30 °C. Plates were scanned, and yeast patches appear as dark circles. Cultures were spotted in 4-fold dilutions to enhance visualization of the growth-suppressive phenotype. The dilutions showing the greatest differences in growth are presented.
To evaluate the effects of Nef on SFK activity, lysates were prepared from each of the transformed cultures shown in Fig. 5 and immunoblotted with anti-phosphotyrosine antibodies. As shown in Fig. 6, expression of each SFK alone induced strong tyrosine phosphorylation of multiple yeast cell proteins, and this effect was markedly dampened upon co-expression with Csk. Co-expression of Nef with Hck in the absence of Csk led to an even greater degree of protein-tyrosine phosphorylation, consistent with the effect of Nef on Hck-induced growth suppression (Fig. 5). Nef reversed down-regulation of Hck, Lyn, and c-Src kinase activity by Csk, consistent with the growth suppression obtained with Nef and these three Csk-down-regulated SFKs (Fig. 5). In contrast, no detectable changes in the protein-tyrosine phosphorylation patterns or signal intensity were observed upon co-expression of Nef with Fgr, Fyn, Lck, or Yes in the presence or absence of Csk, providing strong evidence that these Src family members are not direct targets for HIV-1 Nef in vivo.
FIGURE 6. HIV-1 Nef selectively activates Hck, Lyn, and c-Src in yeast.
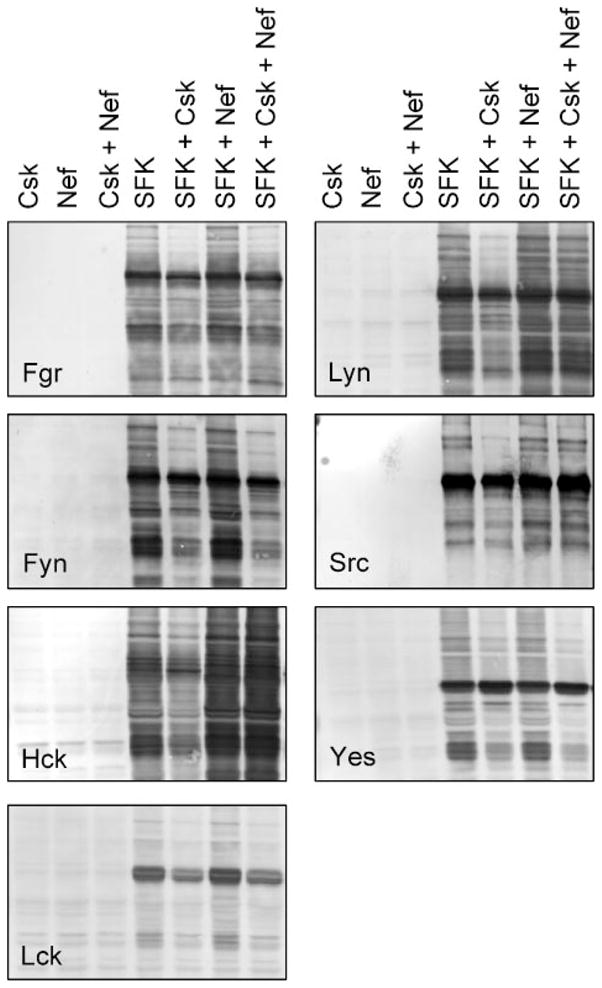
Yeast cultures were transformed with expression vectors for each of the SFKs indicated either alone (SFK) or in the presence of Csk (SFK + Csk), HIV-1 Nef (SFK + Nef), or both (SFK + Csk + Nef). Cells transformed with Csk alone (Csk), Nef alone (Nef), or both (Csk + Nef) were included as negative controls. Cells were grown in liquid culture in the presence of galactose at 30 °C for 18 h. Protein extracts were separated via SDS-PAGE and immu-noblotted for tyrosine-phosphorylated proteins (pTyr). Control immuno-blots confirmed expression of each SFK, Csk, and Nef (data not shown).
Lck Is Activated by Herpesvirus saimiri Tip but Not HIV-1 Nef in Yeast
Data presented above show that Lck exhibited relatively low basal kinase activity in yeast and induced weak growth suppression as a consequence. Neither parameter was influenced by Nef, suggesting that these proteins fail to interact in vivo. As an additional control to support this conclusion, we performed an experiment with Tip, a herpesvirus saimiri protein shown previously to bind and activate Lck (64–68). As shown in Fig. 7, co-expression with Tip led to very strong activation of Lck, inducing marked growth suppression that correlated with enhanced protein-tyrosine phosphorylation. In contrast, co-expression of Lck with Nef did not affect yeast growth or enhance basal kinase activity, consistent with the results presented in Figs. 5 and 6. These data show that co-expression with a known activator enhances Lck kinase activity and induces growth suppression in yeast, thus validating the negative result with Nef.
FIGURE 7. Lck is activated by herpesvirus saimiri Tip but not HIV-1 Nef in yeast.
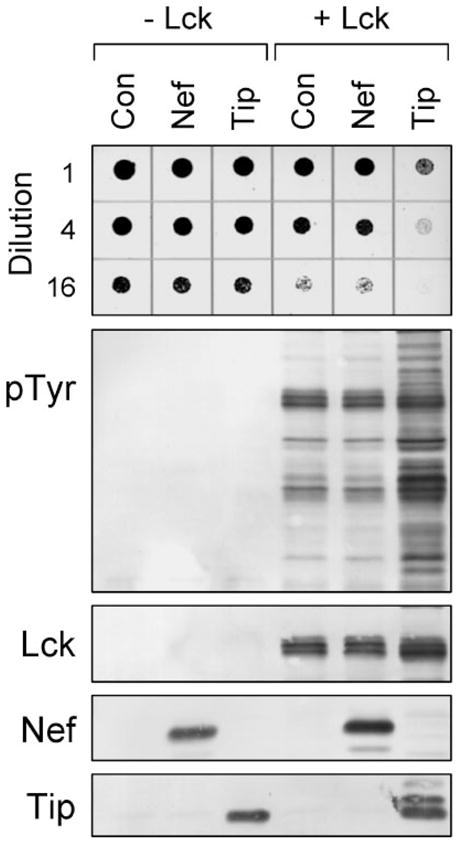
Yeast cultures were transformed with galactose-inducible expression plasmids for Lck (+Lck) or the empty expression plasmid as negative control (−Lck). Cells were co-transformed with galactose-inducible vectors for HIV-1 Nef or FLAG-tagged herpervirus saimiri Tip as indicated or with the empty vector (Con). Top: liquid cultures were grown with glucose as the sole carbon source to repress protein expression and normalized to equal densities. Cells were then spotted onto agar selection plates containing galactose as the sole carbon source and incubated for 3 days at 30 °C. Cultures were spotted in 4-fold dilutions to enhance visualization of the growth-suppressive phenotype. Plates were scanned, and yeast patches appear as dark circles. Lower panels: immunoblots from cultures shown at the top. Transformed cells were grown in liquid culture in the presence of galactose at 30 °C for 18 h. Protein extracts were separated via SDS-PAGE and immunoblotted for tyrosine-phosphorylated proteins (pTyr) as well as for Lck, Nef, and Tip. Tip was visualized using an anti-FLAG antibody and runs as multiple bands in the presence of Lck due to tyrosine phosphorylation.
Nef-mediated Activation of Lyn and c-Src Is PXXP-dependent
We next investigated whether Nef-mediated activation of Lyn and c-Src employs the SH3-binding function of Nef, as is the case with Hck (Fig. 2). For these experiments, we again employed the Nef-2PA mutant, in which proline residues in the conserved PXXPXR motif critical for SH3 engagement are replaced with alanines. Lyn and c-Src were co-expressed with Csk and either wild-type Nef or the Nef-2PA mutant. While wild-type Nef induced strong activation of Lyn and c-Src in terms of growth suppression and protein-tyrosine phosphorylation, both effects were nearly reversed with Nef-2PA (Fig. 8). These results indicate that activation of Lyn and c-Src by HIV-1 Nef requires interaction with the SH3 domains of these kinases, identifying SH3-linker displacement as a common mechanism of SFK activation by Nef.
FIGURE 8. HIV-1 Nef-mediated activation of Lyn and c-Src is PXXP-dependent.
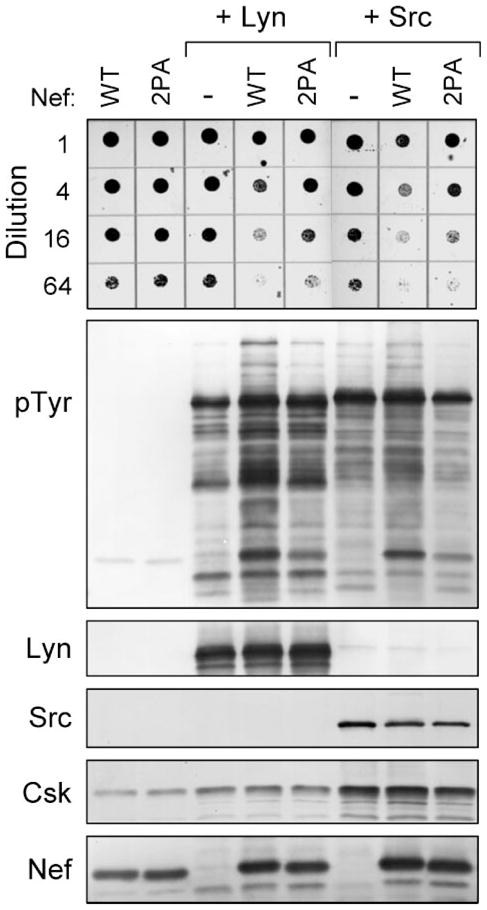
Yeast cultures were transformed with galactose-inducible expression plasmids for Lyn (+Lyn) and c-Src (+Src) in the absence (−) or presence of wild-type (WT) or PXXP mutant (2PA) forms of HIV-1 Nef. Cells expressing the Nef proteins alone were included as a negative control. Top: liquid cultures were grown with glucose as the sole carbon source to repress protein expression and normalized to equal densities. Cells were then spotted onto agar selection plates containing galactose as the sole carbon source and incubated for 3 days at 30 °C. Cultures were spotted in 4-fold dilutions to enhance visualization of the growth-suppressive phenotype. Plates were scanned, and yeast patches appear as dark circles. Lower panels: immunoblots from cultures shown at the top. Transformed cells were grown in liquid culture in the presence of galactose at 30 °C for 18 h. Protein extracts were separated via SDS-PAGE and immunoblotted for tyrosine-phosphorylated proteins (pTyr) as well as for Lyn, Src, Csk, and Nef.
Nef Activates Hck and Lyn but Not c-Src in Vitro
In a final series of studies, we investigated whether the presence of Nef was sufficient for SFK activation, or whether co-expression in a cell-based system is essential. To accomplish this, Hck, Lyn, and c-Src, the three SFKs activated by Nef in yeast, were purified to homogeneity in their inactive forms (see “Materials and Methods”). Each of the kinases was then assayed in vitro with a peptide substrate either alone or in the presence of a 5- or 10-fold molar excess of purified recombinant Nef. As shown in Fig. 9, Hck was strongly activated by Nef under these conditions, supporting the idea that SH3-linker displacement is sufficient for Hck activation as described in previous studies both in vitro and in vivo (33, 42). Similar to Hck, Lyn was also strongly stimulated by Nef in this system, suggesting that SH3-linker displacement is also sufficient for Lyn activation. In contrast to Hck and Lyn, Nef failed to activate c-Src under these conditions. This observation suggests that binding of Nef to the Src SH3 domain is not sufficient for kinase activation in vitro, and that myristoylation and localization to the plasma membrane may also have a role in the activation mechanism observed in vivo (see “Discussion”).
FIGURE 9. Activation of SFKs by HIV-1 Nef in vitro.
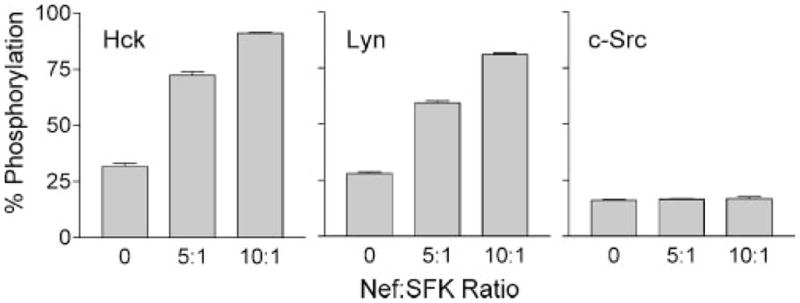
Recombinant Hck, Lyn, and c-Src were purified from Sf9 insect cells in their down-regulated forms and assayed for kinase activity with a peptide substrate in vitro either alone or in the presence of a 5- or 10-fold molar excess of purified recombinant Nef. Details of the FRET-based tyrosine kinase assay used for this experiment can be found under “Materials and Methods.” Each condition was repeated in quadruplicate, and the extent of phosphorylation is expressed as mean percent phosphorylation relative to a control phosphopeptide ± S.D. The overall experiment was repeated twice with comparable results.
DISCUSSION
A growing body of evidence identifies SFKs as important targets for HIV-1 Nef in vivo. Some of the strongest evidence exists for the macrophage Src family member Hck, which is constitutively activated by Nef (32, 33) and has been implicated in disease progression in a mouse model of AIDS (43, 44). At the molecular level, Nef has been shown to bind to isolated SFK SH3 domains and the corresponding full-length kinase proteins in some cases (see the introduction). However, no comparative study of the impact of Nef on full-length SFK activity has been conducted. Here we provide the first complete analysis of the direct effects of Nef on the activities of all SFKs expressed in HIV target cells (T cells and macrophages). Using a yeast-based expression system, we show for the first time that c-Src and Lyn, in addition to Hck, are directly activated by HIV-1 Nef in vivo. Activation occurs in the presence of the physiological SFK regulator, Csk. In contrast, Fgr, Fyn, Lck, and Yes are not activated by Nef, despite previous reports describing the interaction of Nef with several of these kinases or their SH3 domains in vitro (see below). Our observation that Nef selectively activates Hck, Lyn, and c-Src among SFKs may explain why wild-type Nef induces a partial AIDS-like phenotype in Hck-null mice, whereas Nef lacking the PXXPXR motif essential for SFK binding fail to develop AIDS-like disease (44). In the former case, activation of c-Src and Lyn by Nef may functionally compensate for the lack of Hck. Indeed, up-regulation of Lyn kinase activity has been reported in Hck-null macrophages, consistent with a possible compensatory mechanism (69). In contrast, Nef-induced activation of Hck, Lyn, and c-Src requires the PXXPXR motif, supporting the idea that Nef signaling through all three of these Src family members may be essential for development of AIDS-like disease in this model.
We validated our yeast system by demonstrating appropriate regulation of Hck kinase activity by Csk and Nef as reported previously in mammalian cells (16, 32, 42). Yeast cells provide a very useful tool for the study of protein-protein interactions in SFK regulation in vivo, because they lack orthologs of mammalian protein-tyrosine kinases. Here we show that wild-type Hck, when expressed alone, exerts a growth-suppressive effect in yeast consistent with previous reports for c-Src (53, 55, 56). Co-expression with Csk markedly suppressed Hck kinase activity and reversed the growth-inhibitory effect. Suppression of Hck activity required catalytically active Csk as well as an intact Hck tail tyrosine residue, previously established as an essential site for down-regulation of Hck in mammalian cells (42). HIV-1 Nef strongly activated down-regulated Hck, and this effect required the Nef PXXPXR motif responsible for Hck SH3 binding (31, 32). These data support a model in which Nef engages Hck through its SH3 domain and displaces its negative regulatory interaction with the SH2-kinase linker. Demonstration that Nef can overcome Csk-induced down-regulation of Hck in yeast strongly supports a direct activating effect of Nef on Hck in vivo and supports the use of yeast to faithfully model this interaction.
We next extended our study to other members of the Src kinase family expressed in HIV target cells. Fgr, Fyn, Lck, Lyn, and Yes were all active in yeast and induced growth suppression in a manner analogous to c-Src and Hck. Co-expression with Csk reversed growth suppression and reduced endogenous protein-tyrosine phosphorylation, providing direct evidence that Csk alone is sufficient to induce down-regulation of each SFK tested. Introduction of Nef into this system revealed for the first time that Lyn and c-Src are direct targets for Nef-induced activation in vivo. In contrast, Nef had no apparent effect on the activity of Fgr, Fyn, Lck, or Yes. These results are surprising in light of previous reports of Nef interaction with Lck and Fyn, which are discussed in more detail below.
Lck is selectively expressed in T-lymphocytes and plays an essential role in T-cell receptor signal transduction and in thymocyte development (70). Numerous studies have reported that Nef can interact with Lck through its SH2 and SH3 domains (19, 21, 24, 46, 51, 71), and may repress Lck kinase activity and signaling (24, 45, 72). In contrast, our work suggests that, although Nef-Lck interactions can be demonstrated in vitro, these proteins might not interact directly in cells. Unlike Hck, co-expression with Nef had no impact on Lck kinase activity in yeast, in terms of either activation or inhibition. Interestingly, Lck did not suppress yeast growth as strongly as the other SFKs tested, which is most likely a reflection of its relatively low basal kinase activity. To provide a positive control for Lck activation, we co-expressed Lck with the herpesvirus saimiri Tip protein, which has been previously established as an Lck-specific binding protein and activator (64–68). In contrast to Nef, Tip readily activated Lck and induced strong growth arrest and tyrosine phosphorylation of yeast cell proteins.
Like Lck, Fyn is also important in T-lymphocyte antigen responsiveness and development, although its expression pattern is more broad than that of Lck (70). Because of its functional role in T-cells, Fyn has attracted attention as an HIV-1 Nef target protein. The Fyn SH3 domain has been shown to interact with Nef in vitro, although with lower affinity than the Hck and Lyn SH3 domains (23). The structural basis for this difference has been attributed to the lack of an Ile residue in the Fyn SH3 domain RT-loop (31, 49). Indeed, substitution of the Arg residue at this position with Ile converts the Fyn SH3 domain from a low to a high affinity binding partner for Nef (49). The ability of full-length Fyn to interact with Nef is more controversial and may reflect the different experimental approaches used for evaluation of the interaction (22, 51). Here we show that Nef failed to activate Fyn in yeast, both in the growth suppression assay and by anti-phosphotyrosine immunoblotting. Together, these data suggest that, although Nef may interact with the isolated SH3 domain of Fyn in vitro, Nef does not directly affect Fyn kinase activity in vivo.
Like Hck, Lyn is also expressed in macrophages, an important HIV target cell and viral reservoir. At the structural level, only Lyn and Hck have the SH3 domain Ile residue essential for high affinity Nef binding (49). This Ile residue interacts with a hydrophobic pocket within the Nef core (31) and together with the conserved PXXPXR motif is essential for Nef-induced activation of Hck in a rodent fibroblast model (22, 32). Here we show for the first time that Nef activates Lynbya similar PXXPXR-dependent mechanism and can overcome the negative regulatory influence of Csk in doing so. This finding implies that Lyn is regulated by a similar mechanism as Hck, with the SH2-kinase linker engaging the SH3 domain in the down-regulated conformation of the kinase (73). High affinity binding of Nef to the SH3 domain may be sufficient to displace the linker, relieving its inhibitory effect on the kinase domain. Our observation that Nef can drive Lyn activation in vitro also supports this mechanism (Fig. 9). Although Nef readily activates Lyn in vitro and in yeast, we did not observe activation of Lyn by Nef in a previous study using a fibroblast transformation model (48). This difference may relate to localization of Lyn and Nef to different subcellular compartments in fibroblasts, where a portion of the Lyn molecules may localize to the nucleus (74). Alternatively, additional cellular factors may be present in fibroblasts that interfere with downstream signaling by the Nef-Lyn complex.
Although c-Src lacks the SH3 domain Ile residue essential for high affinity Nef binding, we and others have observed Nef interaction with the isolated Src SH3 domain as well as full-length c-Src, although with lower relative affinity than Hck (22, 23). Here we show that co-expression of Nef is sufficient to overcome Csk-induced down-regulation of c-Src in the yeast model system. Both growth suppression and protein-tyrosine phosphorylation by c-Src were dependent upon the Nef PXX-PXR motif, strongly suggesting an SH3-based activation mechanism. Consistent with our findings, He et al. (52) recently showed that Nef augments c-Src kinase activity and induces proliferation in immortalized podocytes in a PXXPXR-dependent manner. Activation of c-Src by Nef led to activation of signal transducers and activators of transcription 3 (STAT3) and extracellular signal-regulated kinase (ERK) signaling downstream, as previously observed in other cell types (16, 17). Nef-Src interaction may contribute to HIV-1-associated nephropathy, the most common cause of chronic renal failure in HIV-seropositive patients (52).
Although Nef was able to activate c-Src in yeast and mammalian cells, we found that Nef alone is not sufficient to induce c-Src activation in an in vitro kinase assay under conditions that led to strong Hck and Lyn activation (Fig. 9). The most likely explanation for this difference is the lower affinity of Nef for the c-Src SH3 domain. Arold et al. (23) determined the equilibrium dissociation constants for Nef interaction with various SFK SH3 domains by isothermal titration calorimetry and found that Nef bound the Hck SH3 domain 10- to 20-times more strongly than those of c-Src, Fyn, or Lck. They attributed this difference to the sequence of the Hck RT-loop, which in addition to having the optimal Ile residue is more flexible and able to adopt a conformation favorable for binding to the Nef hydrophobic pocket. The c-Src RT-loop, on the other hand, is more constrained by hydrogen bonds and may be less able to adopt a conformation compatible with Nef binding. Although interaction between the Src SH3 domain and Nef may not be sufficient to activate c-Src in solution, this lower affinity interaction may be enhanced when the two proteins co-localize to the plasma membrane in cells. Another possibility is that the myristoyl group of native Nef may contribute to Src binding via its myristic acid binding pocket (75); note that none of the recombinant proteins tested in Fig. 9 were myristoylated. Future studies will examine the mechanism of Src SH3-Nef interaction in more detail.
The final two SFKs examined in our study were Fgr and Yes, which have not been previously examined with respect to Nef binding or kinase activation. Fgr is strongly expressed in macrophages, and knock-out experiments suggest significant functional overlap with Hck in this cell type (69). Yes expression is more broadly distributed, often mirroring the pattern observed with c-Src (76). Nef did not activate either kinase in yeast, suggesting that these SFKs are not direct targets for Nef in HIV-infected cells. Consistent with this observation, neither the Yes nor the Fgr SH3 domain contains an RT-loop Ile residue, suggesting that the lack of kinase activation may be due to low SH3 affinity as observed for Fyn and Lck.
In summary, our results show that HIV-1 Nef selectively activates Hck, Lyn, and c-Src among the various SFK isoforms expressed in HIV target cells. The mechanism of activation requires binding of Nef via its PXXPXR motif to the SFK SH3 domain, consistent with the SH3-linker displacement mechanism previously described for Hck. The essential role for the Nef PXXPXR motif in the murine AIDS model, together with the finding that Hck knock-out mice are only partially protected from Nef-induced pathogenesis, strongly suggest that redundant activation of Hck, Lyn, and c-Src occurs in HIV-infected cells (43, 44). Future work will address whether small molecule inhibitors targeted to the Nef-SFK complexes affect HIV replication and AIDS progression.
Acknowledgments
We thank Dr. Bart Sefton of the Salk Institute for providing the Tip cDNA. We also acknowledge the National Institutes of Health AIDS Research and Reference Reagent Program for providing antibodies to Nef.
Footnotes
The abbreviations used are: HIV-1, -2, human immunodeficiency virus types 1 and 2; SFK, Src family kinase; FRET, fluorescence resonance energy transfer.
This work was supported by National Institutes of Health Grants AI057083 and CA81398.
References
- 1.Fackler OT, Baur AS. Immunity. 2002;16:493–497. doi: 10.1016/s1074-7613(02)00307-2. [DOI] [PubMed] [Google Scholar]
- 2.Peter F. Immunity. 1998;9:433–437. doi: 10.1016/s1074-7613(00)80626-3. [DOI] [PubMed] [Google Scholar]
- 3.Joseph AM, Kumar M, Mitra D. Curr HIV Res. 2005;3:87–94. doi: 10.2174/1570162052773013. [DOI] [PubMed] [Google Scholar]
- 4.Daniel MD, Kirchhoff F, Czajak SC, Sehgal PK, Desrosiers RC. Science. 1992;258:1938–1941. doi: 10.1126/science.1470917. [DOI] [PubMed] [Google Scholar]
- 5.Kestler HW, III, Ringler DJ, Mori K, Panicali DL, Sehgal PK, Daniel MD, Desrosiers RC. Cell. 1991;65:651–662. doi: 10.1016/0092-8674(91)90097-i. [DOI] [PubMed] [Google Scholar]
- 6.Whatmore AM, Cook N, Hall GA, Sharpe S, Rud EW, Cranage MP. J Virol. 1995;69:5117–5123. doi: 10.1128/jvi.69.8.5117-5123.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sawai ET, Hamza MS, Ye M, Shaw KE, Luciw PA. J Virol. 2000;74:2038–2045. doi: 10.1128/jvi.74.4.2038-2045.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deacon NJ, Tsykin A, Solomon A, Smith K, Ludford-Menting M, Hooker DJ, McPhee DA, Greenway AL, Ellett A, Chatfield C. Science. 1995;270:988–991. doi: 10.1126/science.270.5238.988. [DOI] [PubMed] [Google Scholar]
- 9.Kirchhoff F, Greenough TC, Brettler DB, Sullivan JL, Desrosiers RC. N Engl J Med. 1995;332:228–232. doi: 10.1056/NEJM199501263320405. [DOI] [PubMed] [Google Scholar]
- 10.Learmont JC, Geczy AF, Mills J, Ashton LJ, Raynes-Greenow CH, Garsia RJ, Dyer WB, McIntyre L, Oelrichs RB, Rhodes DI, Deacon NJ, Sullivan JS. N Engl J Med. 1999;340:1715–1722. doi: 10.1056/NEJM199906033402203. [DOI] [PubMed] [Google Scholar]
- 11.Kirchhoff F, Easterbrook PJ, Douglas N, Troop M, Greenough TC, Weber J, Carl S, Sullivan JL, Daniels RS. J Virol. 1999;73:5497–5508. doi: 10.1128/jvi.73.7.5497-5508.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Geffin R, Wolf D, Muller R, Hill MD, Stellwag E, Freitag M, Sass G, Scott GB, Baur AS. AIDS Res Hum Retroviruses. 2000;16:1855–1868. doi: 10.1089/08892220050195810. [DOI] [PubMed] [Google Scholar]
- 13.Arold ST, Bauer AS. Trends Biochem Sci. 2001;26:356–363. doi: 10.1016/s0968-0004(01)01846-1. [DOI] [PubMed] [Google Scholar]
- 14.Geyer M, Fackler OT, Peterlin BM. EMBO Rep. 2001;2:580–585. doi: 10.1093/embo-reports/kve141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Renkema GH, Saksela K. Front Biosci. 2000;5:D268–D283. doi: 10.2741/renkema. [DOI] [PubMed] [Google Scholar]
- 16.Briggs SD, Scholtz B, Jacque JM, Swingler S, Stevenson M, Smithgall TE. J Biol Chem. 2001;276:25605–25611. doi: 10.1074/jbc.M103244200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Choi HJ, Smithgall TE. J Biol Chem. 2004;279:51688–51696. doi: 10.1074/jbc.M410068200. [DOI] [PubMed] [Google Scholar]
- 18.Piguet V, Trono D. Rev Med Virol. 1999;9:111–120. doi: 10.1002/(sici)1099-1654(199904/06)9:2<111::aid-rmv245>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 19.Saksela K, Cheng G, Baltimore D. EMBO J. 1995;14:484–491. doi: 10.1002/j.1460-2075.1995.tb07024.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arold S, Franken P, Strub MP, Hoh F, Benichou S, Benarous R, Dumas C. Structure. 1997;5:1361–1372. doi: 10.1016/s0969-2126(97)00286-4. [DOI] [PubMed] [Google Scholar]
- 21.Greenway AL, Dutartre H, Allen K, McPhee DA, Olive D, Collette Y. J Virol. 1999;73:6152–6158. doi: 10.1128/jvi.73.7.6152-6158.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Choi HJ, Smithgall TE. J Mol Biol. 2004;343:1255–1268. doi: 10.1016/j.jmb.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 23.Arold S, O’Brien R, Franken P, Strub MP, Hoh F, Dumas C, Ladbury JE. Biochemistry. 1998;37:14683–14691. doi: 10.1021/bi980989q. [DOI] [PubMed] [Google Scholar]
- 24.Greenway A, Azad A, Mills J, McPhee D. J Virol. 1996;70:6701–6708. doi: 10.1128/jvi.70.10.6701-6708.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Briese L, Preusser A, Willbold D. J Biomed Sci. 2005;12:451–456. doi: 10.1007/s11373-005-6797-z. [DOI] [PubMed] [Google Scholar]
- 26.Quintrell N, Lebo R, Varmus H, Bishop JM, Pettenati MJ, Le Beau MM, Diaz MO, Rowley JD. Mol Cell Biol. 1987;7:2267–2275. doi: 10.1128/mcb.7.6.2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ziegler SF, Marth JD, Lewis DB, Perlmutter RM. Mol Cell Biol. 1987;7:2276–2285. doi: 10.1128/mcb.7.6.2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martin JC, Bandres JC. J Acquired Immune Defic Syndr. 1999;22:413–429. doi: 10.1097/00126334-199912150-00001. [DOI] [PubMed] [Google Scholar]
- 29.Orenstein JM. Immunobiology. 2001;204:598–602. doi: 10.1078/0171-2985-00098. [DOI] [PubMed] [Google Scholar]
- 30.Crowe S, Zhu T, Muller WA. J Leukoc Biol. 2003;74:635–641. doi: 10.1189/jlb.0503204. [DOI] [PubMed] [Google Scholar]
- 31.Lee CH, Saksela K, Mirza UA, Chait BT, Kuriyan J. Cell. 1996;85:931–942. doi: 10.1016/s0092-8674(00)81276-3. [DOI] [PubMed] [Google Scholar]
- 32.Briggs SD, Sharkey M, Stevenson M, Smithgall TE. J Biol Chem. 1997;272:17899–17902. doi: 10.1074/jbc.272.29.17899. [DOI] [PubMed] [Google Scholar]
- 33.Moarefi I, LaFevre-Bernt M, Sicheri F, Huse M, Lee CH, Kuriyan J, Miller WT. Nature. 1997;385:650–653. doi: 10.1038/385650a0. [DOI] [PubMed] [Google Scholar]
- 34.Ye H, Choi HJ, Poe J, Smithgall TE. Biochemistry. 2004;43:15775–15784. doi: 10.1021/bi048712f. [DOI] [PubMed] [Google Scholar]
- 35.Komuro I, Yokota Y, Yasuda S, Iwamoto A, Kagawa KS. J Exp Med. 2003;198:443–453. doi: 10.1084/jem.20022018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sicheri F, Moarefi I, Kuriyan J. Nature. 1997;385:602–609. doi: 10.1038/385602a0. [DOI] [PubMed] [Google Scholar]
- 37.Schindler T, Sicheri F, Pico A, Gazit A, Levitzki A, Kuriyan J. Mol Cell. 1999;3:639–648. doi: 10.1016/s1097-2765(00)80357-3. [DOI] [PubMed] [Google Scholar]
- 38.Chong YP, Ia KK, Mulhern TD, Cheng HC. Biochim Biophys Acta. 2005;1754:210–220. doi: 10.1016/j.bbapap.2005.07.027. [DOI] [PubMed] [Google Scholar]
- 39.Okada M, Nada S, Yamanashi Y, Yamamoto T, Nakagawa H. J Biol Chem. 1991;266:24249–24252. [PubMed] [Google Scholar]
- 40.Klages S, Adam D, Class K, Fargnoli J, Bolen JB, Penhallow RC. Proc Natl Acad Sci U S A. 1994;91:2597–2601. doi: 10.1073/pnas.91.7.2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Adams JA. Biochemistry. 2003;42:601–607. doi: 10.1021/bi020617o. [DOI] [PubMed] [Google Scholar]
- 42.Lerner EC, Smithgall TE. Nat Struct Biol. 2002;9:365–369. doi: 10.1038/nsb782. [DOI] [PubMed] [Google Scholar]
- 43.Hanna Z, Kay DG, Rebai N, Guimond A, Jothy S, Jolicoeur P. Cell. 1998;95:163–175. doi: 10.1016/s0092-8674(00)81748-1. [DOI] [PubMed] [Google Scholar]
- 44.Hanna Z, Weng X, Kay DG, Poudrier J, Lowell C, Jolicoeur P. J Virol. 2001;75:9378–9392. doi: 10.1128/JVI.75.19.9378-9392.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Greenway A, Azad A, McPhee D. J Virol. 1995;69:1842–1850. doi: 10.1128/jvi.69.3.1842-1850.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Collette Y, Dutartre H, Benziane A, Ramos M, Benarous R, Harris M, Olive D. J Biol Chem. 1996;271:6333–6341. doi: 10.1074/jbc.271.11.6333. [DOI] [PubMed] [Google Scholar]
- 47.Baur AS, Sass G, Laffert B, Willbold D, ChengMayer C, Peterlin BM. Immunity. 1997;6:283–291. doi: 10.1016/s1074-7613(00)80331-3. [DOI] [PubMed] [Google Scholar]
- 48.Briggs SD, Lerner EC, Smithgall TE. Biochemistry. 2000;39:489–495. doi: 10.1021/bi992504j. [DOI] [PubMed] [Google Scholar]
- 49.Lee CH, Leung B, Lemmon MA, Zheng J, Cowburn D, Kuriyan J, Saksela K. EMBO J. 1995;14:5006–5015. doi: 10.1002/j.1460-2075.1995.tb00183.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Collette Y, Arold S, Picard C, Janvier K, Benichou S, Benarous R, Olive D, Dumas C. J Biol Chem. 2000;275:4171–4176. doi: 10.1074/jbc.275.6.4171. [DOI] [PubMed] [Google Scholar]
- 51.Cheng H, Hoxie JP, Parks WP. Virology. 1999;264:5–15. doi: 10.1006/viro.1999.9937. [DOI] [PubMed] [Google Scholar]
- 52.He JC, Husain M, Sunamoto M, D’Agati VD, Klotman ME, Iyengar R, Klotman PE. J Clin Invest. 2004;114:643–651. doi: 10.1172/JCI21004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brugge JS, Jarosik G, Andersen J, Queral-Lustig A, Fedor-Chaiken M, Broach JR. Mol Cell Biol. 1987;7:2180–2187. doi: 10.1128/mcb.7.6.2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cooper JA, Runge K. Oncogene Res. 1987;1:297–310. [PubMed] [Google Scholar]
- 55.Kornbluth S, Jove R, Hanafusa H. Proc Natl Acad Sci U S A. 1987;84:4455–4459. doi: 10.1073/pnas.84.13.4455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Murphy SM, Bergman M, Morgan DO. Mol Cell Biol. 1993;13:5290–5300. doi: 10.1128/mcb.13.9.5290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Walkenhorst J, Goga A, Witte ON, Superti-Furga G. Oncogene. 1996;12:1513–1520. [PubMed] [Google Scholar]
- 58.Florio M, Wilson LK, Trager JB, Thorner J, Martin GS. Mol Biol Cell. 1994;5:283–296. doi: 10.1091/mbc.5.3.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Takashima Y, Delfino FJ, Engen JR, Superti-Furga G, Smithgall TE. Biochemistry. 2003;42:3567–3574. doi: 10.1021/bi0272499. [DOI] [PubMed] [Google Scholar]
- 60.Superti-Furga G, Fumagalli S, Koegl M, Courtneidge SA, Draetta G. EMBO J. 1993;12:2625–2634. doi: 10.1002/j.1460-2075.1993.tb05923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nada S, Okada M, MacAuley A, Cooper JA, Nakagawa H. Nature. 1991;351:69–72. doi: 10.1038/351069a0. [DOI] [PubMed] [Google Scholar]
- 62.Kushnirov VV. Yeast. 2000;16:857–860. doi: 10.1002/1097-0061(20000630)16:9<857::AID-YEA561>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 63.Chong YP, Mulhern TD, Cheng HC. Growth Factors. 2005;23:233–244. doi: 10.1080/08977190500178877. [DOI] [PubMed] [Google Scholar]
- 64.Kjellen P, Amdjadi K, Lund TC, Medveczky PG, Sefton BM. Virology. 2002;297:281–288. doi: 10.1006/viro.2002.1419. [DOI] [PubMed] [Google Scholar]
- 65.Hartley DA, Hurley TR, Hardwick JS, Lund TC, Medveczky PG, Sefton BM. J Biol Chem. 1999;274:20056–20059. doi: 10.1074/jbc.274.29.20056. [DOI] [PubMed] [Google Scholar]
- 66.Biesinger B, Tsygankov AY, Fickenscher H, Emmrich F, Fleckenstein B, Bolen JB, Broker BM. J Biol Chem. 1995;270:4729–4734. doi: 10.1074/jbc.270.9.4729. [DOI] [PubMed] [Google Scholar]
- 67.Lund T, Medveczky MM, Medveczky PG. J Virol. 1997;71:378–382. doi: 10.1128/jvi.71.1.378-382.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wiese N, Tsygankov AY, Klauenberg U, Bolen JB, Fleischer B, Broker BM. J Biol Chem. 1996;271:847–852. doi: 10.1074/jbc.271.2.847. [DOI] [PubMed] [Google Scholar]
- 69.Lowell CA, Soriano P, Varmus HE. Genes Dev. 1994;8:387–398. doi: 10.1101/gad.8.4.387. [DOI] [PubMed] [Google Scholar]
- 70.Palacios EH, Weiss A. Oncogene. 2004;23:7990–8000. doi: 10.1038/sj.onc.1208074. [DOI] [PubMed] [Google Scholar]
- 71.Dutartre H, Harris M, Olive D, Collette Y. Virology. 1998;247:200–211. doi: 10.1006/viro.1998.9244. [DOI] [PubMed] [Google Scholar]
- 72.Collette Y, Dutartre H, Benziane A, Olive D. Res Virol. 1997;148:52–58. doi: 10.1016/s0923-2516(97)81914-0. [DOI] [PubMed] [Google Scholar]
- 73.Boggon TJ, Eck MJ. Oncogene. 2004;23:7918–7927. doi: 10.1038/sj.onc.1208081. [DOI] [PubMed] [Google Scholar]
- 74.Kharbanda S, Saleem A, Yuan ZM, Kraeft S, Weichselbaum R, Chen LB, Kufe D. Cancer Res. 1996;56:3617–3621. [PubMed] [Google Scholar]
- 75.Cowan-Jacob SW, Fendrich G, Manley PW, Jahnke W, Fabbro D, Liebetanz J, Meyer T. Structure (Camb) 2005;13:861–871. doi: 10.1016/j.str.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 76.Gessler M, Barnekow A. Biosci Rep. 1984;4:757–770. doi: 10.1007/BF01128817. [DOI] [PubMed] [Google Scholar]