

SUMMARY
Mitochondrial dysfunction is a hallmark of many neurodegenerative diseases, yet its precise role in disease pathology remains unclear. To examine this link directly, we subtly perturbed electron transport chain function in the Drosophila retina, creating a model of Leigh Syndrome, an early-onset neurodegenerative disorder. Using mutations that affect mitochondrial complex II, we demonstrate that mild disruptions of mitochondrial function have no effect on the initial stages of photoreceptor development, but cause degeneration of their synapses and cell bodies in late pupal and adult animals. In this model, synapse loss is caused by reactive oxygen species (ROS) production, not energy depletion, as ATP levels are normal in mutant photoreceptors, and both pharmacological and targeted genetic manipulations that reduce ROS levels prevent synapse degeneration. Intriguingly, these manipulations of ROS uncouple synaptic effects from degenerative changes in the cell body, arguing that mitochondrial dysfunction activates two genetically separable processes, one that induces morphological changes in the cell body, and another that causes synapse loss. Finally, by blocking mitochondrial trafficking into the axon using a mutation affecting a mitochondrial transport complex, we find that ROS action restricted to the cell body is sufficient to cause synaptic degeneration, demonstrating that ROS need not act locally at the synapse. Thus, alterations in electron transport chain function explain many of the neurodegenerative changes seen in both early and late-onset disorders.
INTRODUCTION
Defining the molecular mechanisms underlying synapse loss presents a critical challenge to understanding neurodegenerative disease pathology. Recent studies have linked mitochondrial dysfunction to a number of adult-onset disorders, including Amyotrophic lateral sclerosis, Parkinson’s disease, Alzheimer’s disease and Huntington’s disease (reviewed in Lin and Beal, 2006). By contrast, mutations that directly affect mitochondrial metabolism cause early-onset neurodegeneration in a number of developmental disorders, including Leigh Syndrome, Leber’s Hereditary Optic Neuropathy and Kearns-Sayre Syndrome (reviewed in Vogel, 2001). In Leigh Syndrome, for example, patients appear normal at birth, but become broadly ataxic, display nystagmus and spasticity, and ultimately die during childhood (Leigh, 1951). These behavioral deficits are associated with lesions in several sub-cortical structures, as well as optic atrophy (Leigh, 1951). However, no animal model that recapitulates the neurodegenerative changes associated with these early-onset diseases has been described, and the molecular mechanisms that would link mitochondrial dysfunction to degeneration are incompletely understood. Here we establish an animal model of Leigh Disease in which mitochondrial function is specifically altered in a particular neuron type, and define the molecular mechanisms necessary and sufficient for synapse loss.
These early onset encephalopathies, including Leigh Syndrome, are often caused by mutations in genes directly involved in the electron transport chain (ETC) (reviewed in Hart et al., 2002). Intriguingly, these patients generally display only mild disruptions in oxidative phosphorylation (as the causal mutations are not null) (reviewed in Hart et al., 2002). Two broad classes of mechanisms could account for the neurodegenerative effects of such mutations. In one view, the high metabolic activity of neurons might cause them to be sensitive to even small reductions in the ATP-generating capacity of the cell. In the alternate view, impairing the ETC might cause production of a toxic metabolite, particularly reactive oxygen species (ROS), generated through electrons leaving the ETC. Of course, both mechanisms may play important roles. Here we determine the relative contributions of each to our animal model, and examine the cellular site of action of dysfunctional mitochondria.
The fly visual system provides a powerful model both for understanding neurodevelopment, as well as defining the mechanisms that underlie a number of human neurodegenerative diseases (reviewed in Marsh and Thompson, 2006; Clandinin and Zipursky, 2002). In particular, the synaptic connections between photoreceptor axon terminals and their post-synaptic targets have been extensively described, and many molecular components of the synapse have been identified (Prokop and Meinertzhagen, 2006; Hiesinger et al., 2005; Zinsmaier et al., 1994). In addition, Drosophila photoreceptors recapitulate the cellular pathologies associated Parkinson’s Disease and polyglutamine repeat diseases like Huntington’s Disease, and have provided a powerful platform for examining the genetic interactions that influence disease progression (Greene, et al., 2003, Jackson, et al., 1998). Finally, the Drosophila retina has also been used to examine the molecular mechanisms that regulate mitochondrial trafficking and activity, and thus provides a wealth of reagents for examining mitochondria (Stowers et al., 2002; Gorska-Andrzejak et al., 2003; Mandal et al., 2005).
Here we demonstrate that mild disruption of mitochondrial function is sufficient to induce degeneration in Drosophila photoreceptors. Using a forward genetic screen, we identified mutations in the flavoprotein subunit (sdhA) of complex II, the succinate dehydrogenase (SDH) complex, as affecting photoreceptor function. Using eye-specific mosaics, we show that sdhA mutant photoreceptors form normal synaptic terminals with the appropriate synaptic partners, which then degenerate in the pupal and adult fly. The morphology of mitochondria in sdhA mutant R cells is abnormal at the ultrastructural level. We show that the synaptic degeneration in R cells is not due to energy depletion, but rather is caused by the excessive production of ROS. Lastly, using trafficking mutants that restrict mitochondria from R cell synaptic terminals, we demonstrate that dysfunctional mitochondria in the cell body can act at a distance to cause synapse loss.
Materials and Methods
Genetics
Fly stocks were maintained on standard media, at 25°C. Chemical mutagenesis was performed using ethylmethanesulfonate under standard conditions, and screened using the FLP/FRT system, placing the recombinase under the control of the Eyeless promoter (Ashburner, 1989; Grigliatti, 1986; Lee et al., 2001). Optomotor assays were conducted as previously described (Lee et al., 2001). To generate flies in which retinas were almost completely homozygous for sdhA, while the rest of the fly was heterozygous, we used the FLP/FRT system, expressing the FLP recombinase under the control of two different eyeless promotors, paired with a recessive cell lethal (Newsome et al., 2000; Chotard et al., 2005). To generate retinas both homozygous for sdhA and expressing CuZnSOD, we used the same system to generate eye-specific mosaics, and incorporated Act5a FRT STOP FRT CuZnSOD flip-out transgene (Sun and Tower, 1999, Sun et al., 2002). To generate retinas homozygous for both sdhA and miro, we generated stocks that contained both mutations on FRT chromosomes (and a corresponding control stock), a stock that contained two appropriate cell lethals, and a FLP source (under the control of the eyeless promoter). The following mutants and transgenes were used: sdhA1110, sdhA1404 (both from this work); sdh5 and sdh7 (P. Lawrence), mirosd32 (T.L. Schwarz); and UAS-Buffy (N. Bonini).
Histology
Fly retinas and brains were dissected and stained as described (Clandinin, et al 2001). We visualized R cells using the monoclonal antibody, mAb24B10, αchaoptin at 1:50, synaptic vesicles using mAb1G12 αCysteine String Protein at 1:10, active zones with mAbnc82 at 1:50 (all from the Developmental Studies Hybridoma Bank at the University of Iowa). We visualized mitochondria with the monoclonal antibody, MS507 αComplex V at 1:500 (Mitosciences). 3rd instar larval eye disc were assessed using mAb24B10 (1:50) and αBar (1:100; Higashijima et al., 1992). 3rd instar optic lobe development was assessed using mAb24B10 (1:50), Rat αelaV (DSHB) (1:100), Goat αHRP-FITC (1:100) (Jackson ImmunoResearch), and mouse αRepo (1:100; DSHB). Secondary antibodies were obtained from Invitrogen. Fluorescence images were collected on a Leica TCS SP2 AOBS confocal microscope, visualized using Imaris (Bitplane), and mounted using Adobe Photoshop. SDH activity was assessed in 3rd instar larval eye discs made homozygous for either sdhA or a control chromosome using a modified EGUF method (Newsome et al., 2000). Eye discs were stained using Nitro-Blue Tetrazolium (Pearse, 1972). Sections from adult fly retinas were prepared as described (Sullivan et al, 2000). Transmission electron microscopic analysis was performed as described, except that heads were mounted in Epon (Pesah et al., 2004). Counts were made from 3 flies of each genotype, based on analysis of 10-15 R cell terminals per fly.
ATP assay
Retinas from somatic mosaic adult flies were dissected on ice, and assayed using a luciferin/luciferase-based ATP assay kit (Calbiochem). This dissection tears the retina along the fenestrated membrane at its base, and includes all of the R cell bodies, but excludes all brain tissue. Using this FLP recombinase and cell-lethal combination, less than 1% of retinal tissue is not homozygous. To normalize for differences in the amount of retinal tissue in each sample, the amount of pigment in each specimen was measured at 280nm using a spectrophotometer (Pharmacia), and compared against a standardized curve containing different amounts of retinal tissue. This signal was corrected for non-specific absorbance by subtracting the observed absorbance from that seen in the equivalent number of white mutant retinas. To determine the amount of ATP measured we generated a standard curve using known quantities of ATP. To calculate the cellular concentration of ATP, we directly determined the mass of a large quantity of dissected adult retinas and calculated the volume based on an estimated specific gravity of 1.05. This measured volume was essentially identical to the volume of the retina, calculated based on its physical dimensions and geometry (data not shown).
Antioxidant Treatment
Flies were raised on standard fly food treated with either 200 μg/mL alpha-tocopherol in ethanol, or ethanol alone. Adult flies were transferred to freshly treated food on the day of eclosion.
Results
Isolation and molecular characterization of sdhA mutants
Photoreceptors (R cells) form precise and stereotyped synaptic connections with neurons in the first optic neuropil of the Drosophila visual system, the lamina (Figure S1 in the supplementary material; Meinertzhagen and Hansen, 1993). To identify genes involved in the formation and maintenance of this structure, we conducted a forward genetic screen based on a behavioral assay that depended on the ability of adult flies to respond to motion cues (Clandinin et al., 2001). To identify genes whose loss-of-function phenotypes would otherwise be lethal, we conducted this screen in somatic mosaic animals in which only photoreceptor cells were made homozygous mutant, while the rest of the animal is heterozygous (Stowers et al., 1999; Newsome et al., 2000). Using this approach, we identified two mutations in the succinate dehydrogenase flavoprotein subunit (sdhA, CG17246), mitochondrial complex II, that we designated sdhA1110 and sdhA1404. Two additional alleles, sdhA5 and sdhA7 were identified in separate forward genetic screen for the loss of complex II activity (Lawrence, 1981). All four alleles were recessive lethal, with a lethal phase extending to the first larval stage. All 4 alleles failed to complement one another and a small deficiency uncovering 9 genes for this phenotype. Finally, all trans-heterozygote combinations, both bearing the deficiency, and bearing the reference allele sdhA1110 had lethal phases in late embryogenesis and the first larval stage (Table S1 in the supplementary material). Thus all four alleles are strong reduction-of-function mutations.
The succinate dehydrogenase complex comprises four subunits, catalyzes the oxidation of succinate to fumarate, and transfers electrons into the ETC via ubiquinone (Fig. 1A). This complex is not essential for respiration, as electrons can also enter the ETC via complex I and be passed directly to complex III. Thus, disrupting this complex should allow oxidative phosphorylation to continue. The flavoprotein subunit, sdhA, contains two domains, a FAD binding 2 domain, which covalently binds a flavin adenine dinucleotide (FAD) cofactor, and the succinate dehydrogenase flavoprotein C terminal domain, which forms the enzyme’s catalytic site (Fig. 1B; Yankovskaya et al., 2003). This protein is highly conserved between flies and humans (Fig. 1B). DNA sequence analysis revealed a single missense mutation in each of our sdhA alleles that caused non-conservative changes in SDHA (Fig. 1B). These changes lie in both the FAD binding 2 domain (sdhA1110, sdhA1404, and sdhA7) and the C terminal domain (sdhA5).
Fig. 1. Mutations in sdhA block SDH complex activity.

(A). A schematic illustration of the electron transport chain, complexes I-V. IMS denotes the intermembrane space, IM: inner membrane, M:matrix, Ub: ubiquinone, CytC: cytochrome C. (B) A schematic of the SdhA protein structure and the sequences of identified mutations. (C-E) Eye-specific mosaic 3rd instar larval eye discs visualizing the enzymatic activity of the SDH complex. (C) Wild Type. Chevrons demark the morphogenetic furrow. (D) sdhA mutant. (E) Wild-type disc, stained in the presence of the inhibitor malonate.
To determine how these mutations affect the activity of the succinate dehydrogenase complex, we measured enzymatic assay in situ using a reducible dye that accepts electrons from the ETC (Pearse, 1972). To specifically capture Complex II activity we blocked downstream components of the ETC using cyanide and azide. To directly compare the enzymatic functions of complex II in wild-type and mutant retinas, we generated mitotic clones by expressing the yeast FLP recombinase under the control of the eyeless promoter, and examined eye discs of 3rd instar larvae homozygous for either a wild-type control chromosome (Fig. 1C), or sdhA1110 (Fig. 1D). In wild-type eye discs, SDH activity was relatively low in retinal precursor cells, anterior to the morphogenetic furrow, but increased rapidly in differentiating photoreceptor cells, and outlined developing ommatidial pre-clusters (Fig. 1C). By contrast, the activity of SDH was strongly reduced in retinas homozygous for sdhA1110 compared to eye discs homozygous for a control chromosome. Similar results were obtained with sdhA1404 (data not shown), and had previously been described in other imaginal discs using sdhA5 and sdhA7 (Lawrence, 1981). To confirm that this activity corresponded to the function of complex II, we added the drug malonate, which competes with succinate at the catalytic site of the holoenzyme. Addition of this inhibitor strongly reduced the amount of activity detected, and eliminated all differences in staining intensity across the disc (Fig. 1E). Thus, this assay captures qualitatively the activity of Complex II in eye discs, and confirms the genetic observations, indicating that sdhA1110 and sdhA1404 alleles are reduction of function alleles of sdhA.
sdhA mutant R cells form normal synaptic terminals, which progressively degenerate
Adult eyes in which all R cells are homozygous for sdhA mutations are outwardly indistinguishable from controls, with a smooth, regular array of ommatidia, and normal pigmentation (data not shown). To examine the underlying neural structures, we visualized R cell projections into the first optic ganglion, the lamina (Fig. 2). By late pupal development (72 hours after puparium formation, APF), R cells have formed a regular array of axon fascicles, termed cartridges, each comprising 6 photoreceptor axon terminals surrounding their post-synaptic targets, the lamina neurons (Fig. S1, Fig. 2A). At this stage, the terminals of control R cells, as well as those of R cells homozygous mutant for sdhA expressed high levels of the synaptic vesicle component, Cysteine String Protein 2A, as well as the active zone marker, Bruchpilot (Fig. 2A-D; Zinsmaier et al., 1994; Wagh et al., 2006). In control flies on the day of eclosion, cartridge structure and the localization of synaptic vesicles was regular (Fig. 2E), and the active zone had consolidated into discrete foci (Fig. 2G). This expression pattern was maintained for at least the first 5 days of adult life (Fig. 2I, K). In contrast, on the day of eclosion, R cell terminals mutant for sdhA displayed significant loss of synaptic vesicle staining, and diffuse, irregular labeling of active zones (Fig. 2F, H). This phenotype worsened rapidly: by 5 days after eclosion, R cell terminals lacked almost all labeling of synaptic vesicles, and active zone labeling was highly disrupted (Fig. 2J, L). Finally, this phenotype was maintained when we generated sdhA clones by expressing FLP recombinase under the control of a different retina specific promoter ey3.5, was independent of which cell lethal mutation used to increase clone size, and was seen in all four sdhA alleles (data not shown). Thus R cells mutant for sdhA form normal synaptic terminals during development, as assessed using confocal microscopy, which degenerate later in the pupal and adult fly.
Fig. 2. sdhA mutant R cells develop normally, and then degenerate.

(A-L) Cross sections of the lamina neuropil in wild type (A,E,I,C,G,K), and sdh1110 eye-specific mosaic animals (B,F,J,D,H,L), at (A-D) 72h after puparium formation, (E-H) 0 days after eclosion, and (I-L) 5 days after eclosion. Photoreceptor terminals are stained with the R cell specific antibody, anti-chaoptin, mAb24B10 (green), and the synaptic vesicle marker, CSP (magenta; A,B,E,F,I,J), or the active-zone assembly molecule bruchpilot (red; C,D,G,H,K,L). Insets show single cartridges. Cross sections of the retina in wild type (M-O), and sdh1110 eye-specific mosaic animals (Q-S), at 44 hours after puparium formation (M,Q), 0 days after eclosion (N,R), and 5 days after eclosion (O, S). R cells are visualized using mAb24B10 (M,Q), or using phase contrast in plastic sections (N, O, R, S). Inset panels show single ommatidia.
While the external eye was normal in sdhA mutants, photoreceptor cell bodies in the retina degenerated in parallel with their terminals. We first examined the morphology of developing R cells during mid-pupal development (44 hours APF). At this developmental stage, each developing ommatidium contains its full complement of 8 R cells, including 6 outer photoreceptors, R1-R6, which innervate the lamina, and two inner photoreceptors R7 and R8, which innervate a deeper ganglion, the medulla (reviewed in Meinertzhagen and Hansen, 1993). At this stage, retinas homozygous for sdhA mutations were indistinguishable from those of controls (Fig. 2M, Q). We next sectioned adult retinas on the day of eclosion, as well as 5 days later. In control flies, ommatidia form a regular array of facets comprised of R cells and surrounding pigment cells, which can be visualized during the first 5 days of adulthood (Fig. 2N,O). However, in sdhA mutant animals the retina was disrupted at eclosion: while apparently normal rhabdomes were still visible in many R cells, R cell bodies were variable in size, and the array of ommatidia was disordered (Fig. 2R). Five days after eclosion, this phenotype was more severe: many photoreceptor cell bodies were swollen, and many had lost their rhabdomes (Fig. 2S). We conclude that, like their synaptic terminals, mutant photoreceptor structures in the retina form normally and then progressively degenerate. Moreover, since sdhA mutant retinas were of normal morphology and pigmentation, it appears that this disruption affects R cells more severely than the non-neuronal pigment cells (which were also homozygous mutant).
To test whether these phenotypes specifically reflect late-stage degeneration, we systematically examined the early stages of photoreceptor differentiation and visual system development in animals in which photoreceptors were homozygous mutant. In particular, during the 3rd larval stage, we demonstrated that sdhA mutant R cells assembled into ommatidia normally, and expressed fate-appropriate markers. Moreover, the targeting of sdhA mutant R cell axons to appropriate ganglia, the development of their target neurons in the lamina, and the recruitment of their associated glia all occurred normally. Finally, by injecting fluorescent dye into single ommatidia during mid-pupal development, we demonstrated that sdhA mutant R cells almost invariably chose the appropriate post-synaptic partners. Thus, loss of SdhA function in R cells causes little, if any phenotype, until the very late stages of pupal development (Fig. S2 in the supplementary material).
Mitochondrial density and morphology are aberrant in sdhA mutant R cell terminals
We next examined whether deficits in complex II activity were associated with changes in mitochondrial localization within R cell terminals (Fig. 3). To do this, we stained R cell axon terminals with an antibody directed against the alpha subunit of the mitochondrial ATP synthase, Complex V. In control animals, mitochondria were enriched in R cell synaptic terminals during adulthood (Fig. 3A, D). To confirm that our antibody specifically recognized mitochondria we used the mitochondrial trafficking mutant, miro, as a negative control. In particular, in miro mutants, a trafficking defect specific to mitochondria prevents mitochondria from entering pre-synaptic terminals at the neuromuscular junction (Guo et al., 2005). Since miro acts in the same molecular pathway as milton, a gene with well-characterized mitochondrial trafficking defects in R cells (Stowers et al., 2002), we reasoned that miro mutants should have a similar effect in R cells. As expected, mitochondrial complex V staining was absent from miro mutant R cell terminals (Fig. 3B, E). As has previously been reported in milton mutants, the loss of mitochondria from R cell terminals was accompanied by a dramatic increase in mitochondrial staining in surrounding tissues (Gorska-Andrzejak et al, 2003). By comparison, in sdhA mutant R cells, the intensity of mitochondrial staining was reduced at eclosion, and continued to decline such that, by 5 days after eclosion, there was little or no mitochondrial staining within photoreceptor terminals (Fig. 3C, F). Again, as seen in the miro mutant, up-regulation of mitochondria in the surrounding cells was also observed. Finally, while the presence of mitochondria at the synaptic terminal is necessary for the normal formation of synapses at the neuromuscular junction (Guo et al., 2005), our results are consistent with previous studies that demonstrated that synapse-associated mitochondria are not required for either synapse formation or maintenance in R cells (Stowers et al., 2002).
Fig. 3. Mitochondria are lost from sdhA mutant terminals.

Cross section of the lamina neuropil in wild type (A, D), mirosd32 mutant (B, E) sdhA1110 mutant (C, F) eye-specific mosaic animals stained with the R cell specific antibody, mAb24B10 (green), and the mitochondrial complex V subunit alpha (magenta) at 0 (A,B,C) or 5 days (D,E,F) after eclosion. Insets show single cartridges.
As reduced mitochondrial staining could reflect either damage to mitochondria, or loss of mitochondria, or both, we extended our analysis to the ultrastructural level and examined sdhA mutant R cell terminals using electron microscopy. As pre-synaptic terminals in 5 day old sdhA mutants had undergone sufficient degeneration that they could not be reliably identified, we examined the lamina of wild-type and sdhA mutant somatic mosaic flies isolated on the day of eclosion. At this stage, staining of the complex V component is reduced, but not eliminated. In control animals, cartridges comprising R cell axon terminals and their associated lamina neurons formed a highly regular array (data not shown). Within each cartridge, R cell axons were readily identified by their capitate projections, specialized contacts with neighboring glial cells (reviewed in Prokop and Meinertzhagen, 2006). Moreover, as has previously been described, R cell terminals contained numerous mitochondrial profiles with typical membrane structures (Fig. 4A,B; Stowers et al., 2002). By contrast, in sdhA mutant animals, some R cell terminals lacked mitochondrial profiles (Fig. 4G, H), while others contained numerous smaller mitochondria. As a result, the mean ratio of the volume of mitochondrial profiles per R cell terminal volume in sdhA mutants was 0.121±0.018, compared to 0.224±0.015 in control flies (p<0.001; Fig. 4I). We note that the average number of mitochondria per terminal did not change in sdhA mutants (data not shown). Furthermore, while mitochondria in wild-type terminals frequently displayed the characteristic invaginated cristae (Fig. 4C, D), mitchondria in sdhA mutant R cell terminals were frequently abnormal in morphology, displaying irregular invaginations (Fig. 4E, F). Characteristic of early stages of neural degeneration, we also observed multi-lamellar bodies (Fig. 4G). Thus, these ultrastructural studies reveal that sdhA mutations cause defects in mitochondrial volume and structure consistent with our observations of reduced complex V staining in flies at the day of eclosion.
Fig. 4. Mitochondrial structure and volume are abnormal in sdhA mutant R cells.
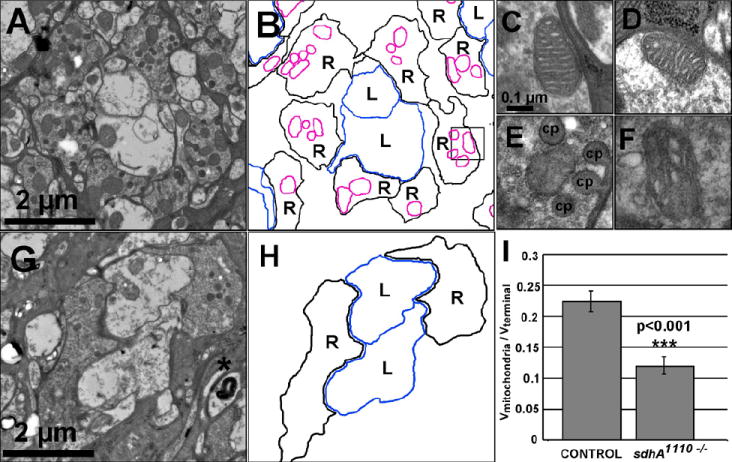
Ultrastructural analysis of wild-type (A) or sdhA1110 mutant (G) R cell terminals in the lamina on the day of eclosion. Asterisk marks a multilamellar body. (B, H) Schematic tracings of R cell terminals (black lines, denoted R), lamina neurons (blue lines, LN), and mitochondrial profiles within R cell terminals (magenta). Profiles of single wild-type (C-D) or sdhA mutant (E-F) mitochondria. Specialized glial invaginations, capitate projections, are denoted (cp). (I) Quantification of mitochondrial volume per terminal volume.
Cells mutant for sdhA are not depleted of ATP
We reasoned that disruption of the ETC in sdhA mutant R cells could impair oxidative phosphorylation sufficiently to cause ATP depletion, which might, in turn, lead to synapse loss and degeneration. To test this possibility we measured ATP levels in control and mutant retinas using a biochemical assay, and observed no difference in the average amount of ATP per retina, both 0 days and 5 days after eclosion (Fig. 5A). To normalize for differences in the mass of retina isolated in each experimental sample we measured the screening pigment absorbance in each specimen at 280 nm using a spectrophotometer, and then compared each measurement to a standard curve generated using known quantities of retina. We found no difference in absorbance between control and sdhA mutant retinas. This standard curve was corrected for non-specific absorbance by retinal tissue by subtracting the observed absorbance from that seen in the equivalent number of white mutant retinas. The amount of pigmentation in wild-type and sdhA mutant retinas was equivalent (Fig. S3 in the supplementary material). By directly measuring the mass of isolated retinas, and using a standard curve generated with known quantities of ATP, we then calculated that the cellular of concentration of ATP we measured using this assay was between 1.5mM and 1.8mM. Given that nearly a third of the mass of the retina is acellular (comprising the lenses of each ommatidium, and the vitreous humor; Franceschini and Kirschfeld, 1971), these values reflect a conservative estimate of the concentration of ATP within R cells. Thus sdhA mutant retinas contain normal levels of ATP in the cell soma. These results are consistent with the fact that R cell differentiation and development in sdhA mutants was normal, as previous observations had demonstrated that mitochondrial mutations that reduce ATP levels by 40% cause dramatic developmental defects in the retina (Mandal et al., 2005).
Fig. 5. R cells are not depleted of ATP.
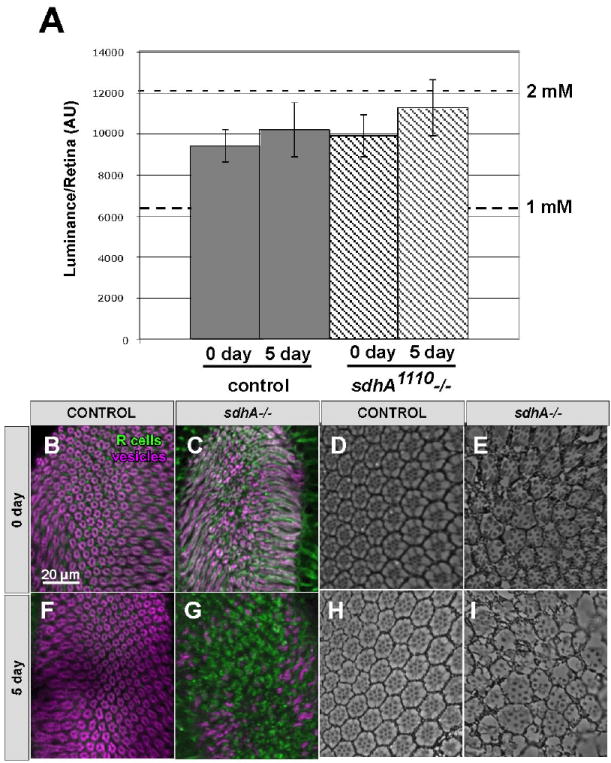
(A) The average luminance and SEM, per retina, as determined using luciferin/luciferase based ATP assays performed on retinas taken from wild type, or sdhA1110 mutant mosaic flies at 0 and 5 days after eclosion. For comparison, the amount of ATP measured in 75% of a retina is displayed. (B-I) Cross sections of the laminas (B, C, F, G) or retinas (D, E, H, I) from animals raised in complete darkness. Laminas are stained with the R cell specific antibody mAb24B10 (green) and CSP (magenta). (B, D, F, H) Wild Type. (C, E, G, I) sdhA1110 mutant. (B-E). 0 days after eclosion. (F- I). 5 days after eclosion.
To further test this hypothesis, we reasoned that if R cells were experiencing small reductions in ATP levels, undetectable by our biochemical assay, reducing the metabolic load of photoreceptors specifically should suppress their degeneration (Fig. 5). The metabolic activity of photoreceptors is directly regulated by light exposure: the difference between complete darkness and normal light exposure corresponds to a more than two-fold difference in photoreceptor ATP consumption (Niven et al., 2007). As dark-rearing has no effect on the development of R cells and their synapses in wild-type flies (Fig. 5 B, D, F, H), we examined whether raising sdhA mutant animals in complete darkness could suppress photoreceptor synaptic terminal degeneration. However, by comparison with flies raised under standard 12:12 light:dark conditions, dark-reared flies showed indistinguishable levels of synaptic degeneration in the brain (Fig. 5 C, G) and damage in the retina (Fig. 5 E, I). Thus, we conclude that the degenerative phenotypes we observe in sdhA mutant animals do not result from ATP depletion.
Pharmacological and Genetic Antioxidant treatment suppresses synapse loss
We next tested whether sdhA mutations cause R cell degeneration by increasing ROS production. Mitochondria are a significant cellular source of ROS, which are generated when electrons from the ETC interact with molecular oxygen to form superoxide (Balaban et al, 2005). Notably, impairment of the ETC increases ROS formation (Staniek and Nohl, 2000). We reasoned that if ROS contributes to the degeneration we observe in R cell synaptic terminals, prolonged treatment with the antioxidant alpha-tocopherol should suppress this phenotype. To determine if antioxidants could suppress R cell degeneration, we grew control and sdhA mutant somatic mosaic flies on standard food supplemented with 200 μg/ mL alpha-tocopherol. On the day of eclosion, flies were placed on newly treated food, and then examined 5 days later. Chronic treatment with alpha-tocopherol had no affect on the development of the lamina or retina in control flies (Fig. 6 A, E, I), and treatment with vehicle alone did not suppress the degeneration observed in sdhA mutants (Fig. 6 B, F, J). However, treatment with alpha-tocopherol strongly suppressed the loss of synaptic vesicles (Fig. 6 C, D), as well as loss of mitochondrial staining and disorder of the active zone (Fig. 6 G, H), in sdhA1110 and sdhA1404 mutant R cells at 5 days after eclosion, respectively. Intriguingly, antioxidant treatment failed to suppress the degeneration of photoreceptor cell bodies observed in the retina in these mutants (Fig. 6 K, L). These observations are consistent with the notion that the production of excess ROS is sufficient to cause synapse degeneration, but not degeneration of the cell body, in sdhA mutant R cells. Moreover, they demonstrate that the defects in mitochondrial structure and size seen in sdhA mutant terminals reflect ROS-induced damage, not a direct effect of sdhA mutations on mitochondrial localization per se.
Fig. 6. Chronic antioxidant treatment suppresses synaptic terminal degeneration in sdhA mutant R cells.

Cross sections of the lamina neuropil (A-H) or retinas (A, E, I) from 5 day old wild-type flies grown on food containing 200 μg/mL α tocopherol, (B, F, J) 5 day old sdhA1110 mutant flies treated with vehicle alone. or 5 day old (C, G, K) sdhA1110 and (D, H, L) sdhA1404 mutants treated with 200 μg/mL α tocopherol. . Laminas are stained (A-D) for R cells (green) and CSP (magenta), and (E-F) the presynaptic assembly molecule, bruchpilot (green) and mitochondrial complex V (magenta). Insets show single cartridges (A-H) or ommatidia (I-L).
To exclude the possibility that these protective effects of alpha-tocopherol might reflect a protective function of this molecule that is independent of its effect on ROS levels, we exploited a parallel genetic approach to reducing ROS in photoreceptors. Superoxide dismutase (SOD) is a central component of the cellular defense against ROS, and acts by converting superoxide to less reactive metabolites (McCord, et al., 1969). We therefore tested whether over-expression of SOD in R cells could suppress the degeneration we observed in sdhA mutants (Fig. 7). To over-express SOD specifically in the retina, we generated eye-specific mosaic flies mutant for sdhA, and used the yeast FLP/FRT system to induce eye-specific expression of CuZnSOD under the control of the actin promoter, using Act5a FRT STOP FRT CuZnSOD flip-out transgene (Sun and Tower, 1999, Sun et al., 2002). As in other control flies, over-expression of the SOD transgene in otherwise wild-type R cells did not disrupt R cell morphology in the retina, or the lamina (Fig. 7 A, D, G). By contrast, sdhA mutant R cells displayed progressive degeneration in the adult (Fig. 7 B, E, H). However, when CuZnSOD was over-expressed in sdhA mutant R cells, we observed a dramatic increase in the level of synaptic vesicle staining in many R cell terminals that persisted through 5 days after eclosion (Fig. 7 C, F). Consistent with the effect we observed when sdhA mutants were treated with antioxidants, this suppression was uncoupled from effects on the retina: CuZnSOD expression had no effect on retinal degeneration (Fig. 7 I).
Fig. 7. Overexpression of CuZnSOD suppresses synaptic degeneration in sdhA mutant R cells.

Cross sections of the lamina neuropil (A-F) or retinas G-I) from wild-type flies over-expressing CuZnSOD (A, D, G) sdhA1110 mutants (B, E, H), sdhA1110 mutant over-expressing CuZnSOD (C, F, I) at 0 days (A-C) and 5 days (G-I) after eclosion. Laminas are stained for R cells (green) and CSP (magenta). Insets show single cartridges (A-F).
We conclude that damage caused by increased production of ROS in R cells accounts for much of the synaptic degeneration we observed in sdhA mutants. Moreover, since both antioxidant treatment and CuZnSOD over-expression failed to rescue the degeneration of photoreceptor cell bodies, this degeneration must either be differentially sensitive to ROS damage compared to the synapse, or be caused by an independent mechanism. As mitochondrial complex II is not thought to be a significant source of cellular ROS, we infer that sdhA mutations likely cause increased ROS production indirectly, by affecting the activities of other ETC complexes. Finally, we also tested whether the damage caused by ROS was activating apoptotic pathways by expressing the cell-death suppressor Buffy, specifically in sdhA mutant R1-R6 cells. However, expression of this transgene was unable to alter the severity or extent of the degeneration (Fig. S4 in the supplementary material).
sdhA mutant mitochondria act in the cell body to induce synaptic degeneration
ROS are highly reactive, and hence act very locally with respect to their site of production. We therefore sought to determine whether the synaptic degeneration observed in sdhA mutant R cells reflects damage caused by mitochondria located locally in the synaptic terminal, or could be caused by damage elsewhere in the cell. To distinguish between these alternatives, we excluded mitochondria from the synaptic terminal using the mitochondrial trafficking mutant, miro (Fig. 3B). When the mitochondrial trafficking pathway controlled by Miro is blocked, mitochondria remain restricted to the cell body beginning in the early stages of R cell axonal differentiation and never enter the brain; during adulthood mitochondria in these mutants are at least 20 μm from the terminal (Stowers et al., 2002; Gorska-Andrzejak et al., 2003). We reasoned that if sdhA mutant mitochondria acted locally to damage the synaptic terminal, preventing mitochondria from entering the terminal should suppress degeneration. If, however, damage induced by mitochondria elsewhere in the cell is sufficient to cause degeneration, then preventing entry should have no effect. Using markers for R cell terminal morphology and synapse formation, we first determined that miro mutant R cell terminals were indistinguishable from controls at eclosion, and did not degenerate like sdhA mutant photoreceptors (Fig. 8 A-C, E-G). However, R cells doubly homozygous for mutations in sdhA and miro displayed a degeneration phenotype indistinguishable from that seen in sdhA single mutant cells (Fig. 8 D, H). Thus, removal of abnormal mitochondria from the pre-synaptic terminal is not sufficient to prevent degeneration, demonstrating that ROS-induced damage in the cell body can cause the synapse loss we observe in sdhA mutants. Moreover, these experiments demonstrate the blocking mitochondrial trafficking into the synaptic terminal is not sufficient to cause synapse degeneration, and thus the ROS produced in sdhA mutant photoreceptors cannot simply block mitochondrial transport.
Fig. 8. Mitochondria restricted to the cell body are sufficient to induce synaptic degeneration.

(A-H) Cross sections of the lamina neuropil in control (A, E), sdhA1110 mutant (B, F); miro (C, G) and sdh1110; miro double mutant eye-specific mosaic animals (D, H) at 0 days after eclosion (A-D) or 5 days after eclosion (E-H). R cells (green) and CSP (magenta) were labeled.
Discussion
These studies shed light on the molecular mechanisms that underlie neurodegeneration associated with mitochondrial dysfunction, and provide a powerful genetic model of Leigh Syndrome. We demonstrate that disruption of the ETC through mutations in the flavoprotein subunit of succinate dehydrogenase cause synapse loss. This degeneration is not caused by ATP depletion, but is rather induced by the production of ROS by impaired mitochondria. Moreover, ROS-mediated damage to the cell body is sufficient to cause degeneration of the synaptic terminal in the brain. We propose that alterations in ETC function activate at least two independent pathways, one leading to increased ROS production and synapse loss, and the other being critical to degeneration of the cell body.
A fly model of Leigh Syndrome
Our work describes the first animal model of Leigh Syndrome that recapitulates the neurodegenerative changes seen in human patients. Leigh Syndrome is caused by inherited mutations in proteins crucial to electron transport, including mutations in SDH subunits (Bourgeron et al., 1995, Horvath et al., 2006). Children born with such mutations appear normal during the first months of life, but then show signs of psychomotor delay and regression prior to death. These deficits are associated with widespread degeneration in many sub-cortical structures (Leigh, 1951). Notably, optic atrophy is also frequently observed (Birch-Machin et al., 2000; Leigh, 1951). In many respects, our findings in the fly parallel this time course: R cells mutant for sdhA develop completely normally, adopt the correct cell fates, innervate the appropriate synaptic partners, and assemble synapses normally. However, beginning around the time of eclosion, R cells degenerate, progressively losing expression of synaptic markers, and undergoing extensive morphological changes. Thus, our model captures many of the central elements of the human disease and offers the opportunity to examine the molecular mechanisms underlying disease progression.
Genetic Analysis of the Succinate Dehydrogenase Complex
Mutations in other SDH subunits have been described in worms and in flies. The complex is comprised of four subunits: the flavoprotein subunit (sdhA), and an iron-sulfur subunit (sdhB) that together make up the catalytic core of the holoenzyme, as well as two membrane-bound subunits (sdhC and sdhD), which anchor the complex to the inner mitochondrial membrane and transfer electrons to ubiquinone (Ackrell et al., 1990). Mutations in sdhB in Drosophila (Walker et al, 2006), and sdhC (mev-1) in C. elegans (Ishii et al, 1998) shorten life span in a high oxygen environment. In the fly, this sensitivity to hyperoxia manifests as morphological abnormalities in the mitochondria of flight muscles, and behavioral deficits in geotaxis (Walker et al, 2006). In worms, mutations in sdhC increase ROS production under normal oxygen tension (Senoo-Matsuda et al, 2001). These previous findings are consistent with our results in that they link deficits in mitochondrial complex II activity to the production of excess ROS. However, unlike our mutations in sdhA, neither of these mutations are homozygous lethal, suggesting that they cause comparatively weaker effects on the activity of the complex. Moreover, in neither case was gene function examined in the context of degenerative changes in specific neurons.
What are the targets of ROS?
Given the causal role for ROS in neurodegeneration, identifying the cellular site of action of ROS represents a critical challenge. One model is that the primary effect of excessive ROS production is to cause damage to mitochondria, forming a positive feedback loop in which ROS-induced damage further enhances ROS production (reviewed in Lin and Beal, 2006). Indeed, in our system, the damage we observed to synaptic mitochondria in sdhA mutant R cells is ROS dependent, as it can be blocked by antioxidant treatment. However, damage to mitochondria may not be the direct cause of synapse loss because synaptic degeneration could be suppressed by over-expression of the CuZnSOD, a form of SOD that is thought to act primarily in the cytoplasm (O’Brien et al., 2004). That is, while damage to mitochondria is clearly an important aspect of the cellular degeneration we see, excess ROS appears to directly alter the activities of one or more cytosolic components to cause synapse loss. Our observations also demonstrate that degenerative changes in the cell body can be uncoupled from those in the synaptic terminal, as over-expression of CuZnSOD, or addition of an exogenous antioxidant, suppressed the sdhA mutant phenotype in the brain without mitigating the retinal phenotype. This differential sensitivity could reflect quantitative differences in the ability of one structure to withstand damage more than another, or could reflect qualitative differences in the specific cellular targets affected by ROS. One possibility, then, is that mitochondrial dysfunction activates two molecularly distinct pathways, one that is mediated by excess ROS and causes synapse loss, and one that is mediated by as-yet unknown components that leads to degeneration of the cell body. Finally, our demonstration that removal of sdhA mutant mitochondria from the synaptic terminal is not sufficient to prevent synaptic degeneration argues that the critical cellular targets of ROS that affect synapse structure are in the cell body, not the terminal. Regardless of the specific mechanism involved, the powerful genetic model we describe will allow the identification of the cellular targets of ROS in neurons.
A common mechanism of neurodegeneration?
The mechanisms we describe here are likely to underlie important aspects of other neurodegenerative disease pathologies. In particular, alterations in mitochondrial complex II activity have also been linked to Huntington’s Disease (HD) (reviewed in Walker, 2007). A specific decrease in the expression of two SDH subunits, SDHA and SDHB, occurs in striatal neurons of Huntington’s patients, and ectopic expression of mutant Huntingtin causes a similar decrease in neuronal cultures (Benchoua et al., 2006). Moreover, over-expression of these SDH subunits suppresses the cell death induced by mutant Huntingtin protein in both cultured striatal neurons, and in yeast (Benchoua et al, 2006, Solans et al, 2006). In this yeast model, reductions in SDH activity are also associated with increased ROS production (Solans et al., 2006). Finally, chronic administration of 3-nitropropionic acid, an inhibitor of SDH, to rodents and primates, recapitulates many of the characteristic neurological deficits seen in HD patients (Palfi et al., 1996; Guyot et al., 1997; Brouillet et al., 1998). As our studies demonstrate that mutations in sdhA are sufficient to cause neurodegeneration through increased ROS production, we speculate that in striatal neurons, this constitutes one of the central mechanisms underlying neurodegeneration seen in HD. More broadly, our studies are consistent with the notion that the excessive production of ROS that has been detected in other neurodegenerative diseases such as Parkinson’s Disease and ALS might provide a sufficient explanation for at least some of the neurodegenerative changes seen.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
The authors are grateful to T.L. Schwarz, P. Lawrence, J. Tower, I. Salecker, and N. Bonini for providing fly stocks and reagents. We also thank X. Zhao for help with plastic sectioning, and Kang Shen and members of the Clandinin laboratory for helpful suggestions. This work was supported by NIH R01 EY015231 (T.R.C.), and by a gift from the Breetwor Family Fund. TRC was a recipient of a Burroughs-Wellcome Career Development Award, and a Searle Scholar’s Award.
References
- Ackrell BAC, Johnson MK, Gunsalus RP, Cecchini G. Structure and function of succinate dehydrogenase and fumarate reductase. In: Muller F, editor. Chemistry and Biochemistry of Flavoproteins. Vol. 3. CRC Press, Inc.; Boca Raton, FL: 1990. pp. 229–297. [Google Scholar]
- Ashburner M. Drosophila: a Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–95. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Benchoua A, Trioulier Y, Zala D, Gaillard MC, Lefort N, Dufour N, Saudou F, Elalouf JM, Hirsch E, Hantraye P, Deglon N, Brouillet E. Involvement of mitochondrial complex II defects in neuronal death produced by N-terminus fragment of mutated huntingtin. Mol Biol Cell. 2006;17:1652–63. doi: 10.1091/mbc.E05-07-0607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birch-Machin MA, Taylor RW, Cochran B, Ackrell BA, Turnbull DM. Late-onset optic atrophy, ataxia, and myopathy associated with a mutation of a complex II gene. Ann Neurol. 2000;48:330–5. [PubMed] [Google Scholar]
- Bourgeron T, Rustin P, Chretien D, Birch-Machin M, Bourgeois M, Viegas-Pequignot E, Munnich A, Rotig A. Mutation of a nuclear succinate dehydrogenase gene results in mitochondrial respiratory chain deficiency. Nat Genet. 1995;11:144–9. doi: 10.1038/ng1095-144. [DOI] [PubMed] [Google Scholar]
- Brouillet E, Guyot MC, Mittoux V, Altairac S, Conde F, Palfi S, Hantraye P. Partial inhibition of brain succinate dehydrogenase by 3-nitropropionic acid is sufficient to initiate striatal degeneration in rat. J Neurochem. 1998;70:794–805. doi: 10.1046/j.1471-4159.1998.70020794.x. [DOI] [PubMed] [Google Scholar]
- Chotard C, Leung W, Salecker I. glial cells missing and gcm2 cell autonomously regulate both glial and neuronal development in the visual system of Drosophila. Neuron. 2005;48:237–51. doi: 10.1016/j.neuron.2005.09.019. [DOI] [PubMed] [Google Scholar]
- Clandinin TR, Lee CH, Herman T, Lee RC, Yang AY, Ovasapyan S, Zipursky SL. Drosophila LAR regulates R1-R6 and R7 target specificity in the visual system. Neuron. 2001;32:237–248. doi: 10.1016/s0896-6273(01)00474-3. [DOI] [PubMed] [Google Scholar]
- Clandinin TR, Zipursky SL. Making connections in the fly visual system. Neuron. 2002;35:827–41. doi: 10.1016/s0896-6273(02)00876-0. [DOI] [PubMed] [Google Scholar]
- Crapo JD, Oury T, Rabouille C, Slot JW, Chang LY. Copper, Zinc Superoxide Dismutase is Primarily a Cytosolic Protein in Human Cells. Proc Natl Acad Sci U S A. 1992;89:10405–10409. doi: 10.1073/pnas.89.21.10405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson MW, Guo M. Pink1, Parkin, DJ-1 and mitochondrial dysfunction in Parkinson’s disease. Curr Opin Neurobiol. 2007;3:331–337. doi: 10.1016/j.conb.2007.04.010. [DOI] [PubMed] [Google Scholar]
- Franceschini N, Kirschfeld K. Pseudopupil phenomena in the compound eye of Drosophila. Kybernetik. 1971;9:159–82. doi: 10.1007/BF02215177. [DOI] [PubMed] [Google Scholar]
- Gorska-Andrzejak J, Stowers RS, Borycz J, Kostyleva R, Schwarz TL, Meinertzhagen IA. Mitochondria are redistributed in Drosophila photoreceptors lacking milton, a kinesin-associated protein. J Comp Neurol. 2003;463:372–88. doi: 10.1002/cne.10750. [DOI] [PubMed] [Google Scholar]
- Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci USA. 2003;100:4078–4083. doi: 10.1073/pnas.0737556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigliatti T. In: Mutagenesis in Drosophila: A Practical Approach. Roberts DB, editor. Oxford: IRL Press; 1986. pp. 39–48. [Google Scholar]
- Guo X, Macleod GT, Wellington A, Hu F, Panchumarthi S, Schoenfield M, Marin L, Charlton MP, Atwood HL, Zinsmaier KE. The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron. 2005;47:379–93. doi: 10.1016/j.neuron.2005.06.027. [DOI] [PubMed] [Google Scholar]
- Guyot M-C, Hantraye P, Dolan R, Palfi S, Maziére M, Brouillet E. Quantifiable Bradykinesia, gait abnormalities, and Huntington’s disease-like striatal lesions in rats chronically treated with 3-nitropropionic acid. Neuroscience. 1997;79:45–56. doi: 10.1016/s0306-4522(96)00602-1. [DOI] [PubMed] [Google Scholar]
- Hart PE, De Vivo DC, Shapira AH. Clinical Features of the Mitochondrial Encephalomyopathies. In: Shapira AHV, DiMauro S, editors. Mitochondrial Disorders in Neurology 2. USA: Butterworth-Heinemann; 2002. pp. 35–67. [Google Scholar]
- Hiesinger PR, Fayyazuddin A, Mehta SQ, Rosenmund T, Schulze KL, Zhai RG, Verstreken P, Cao Y, Zhou Y, Kunz J, Bellen HJ. The v-ATPase V0 subunit a1 is required for a late step in synaptic vesicle exocytosis in Drosophila. Cell. 2005;121:607–20. doi: 10.1016/j.cell.2005.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashijima S, Michiue T, Emori Y, Saigo K. Subtype determination of Drosophila embryonic external sensory organs by redundant homeo box genes BarH1 and BarH2. Genes Dev. 1992;6:1005–18. doi: 10.1101/gad.6.6.1005. [DOI] [PubMed] [Google Scholar]
- Horvath R, Abicht A, Holinski-Feder E, Laner A, Gempel K, Prokisch H, Lochmuller H, Klopstock T, Jaksch M. Leigh syndrome caused by mutations in the flavoprotein (Fp) subunit of succinate dehydrogenase (SDHA) J Neurol Neurosurg Psychiatry. 2006;77:74–6. doi: 10.1136/jnnp.2005.067041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii N, Fujii M, Hartman PS, Tsuda M, Yasuda K, Senoo-Matsuda N, Yanase S, Ayusawa D, Suzuki K. A mutation in succinate dehydrogenase cytochrome b causes oxidative stress and ageing in nematodes. Nature. 1998;394:694–697. doi: 10.1038/29331. [DOI] [PubMed] [Google Scholar]
- Jackson GR, Salecker I, Dong X, Yao X, Arnheim N, Faber PW, MacDonald ME, Zipursky SL. Polyglutamine-expanded human huntingtin transgenes induce degeneration of Drosophila photoreceptor neurons. Neuron. 1998;21:633–42. doi: 10.1016/s0896-6273(00)80573-5. [DOI] [PubMed] [Google Scholar]
- Lawrence PA. A general cell marker for clonal analysis of Drosophila development. J Embryol Exp Morphol. 1981;64:321–32. [PubMed] [Google Scholar]
- Lee C-H, Herman T, Clandinin TR, Lee R, Zipursky SL. N-cadherin regulates target specificity in the Drosophila visual system. Neuron. 2001;30:437–450. doi: 10.1016/s0896-6273(01)00291-4. [DOI] [PubMed] [Google Scholar]
- Leigh D. Subacute necrotizing ecephalomyelopathy in an infant. J Neurol Neurosurg Psychiatry. 1951;14:216–221. doi: 10.1136/jnnp.14.3.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–95. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, Wang N, Caspersen C, Chen X, Pollak S, Chaney M, Trinchese F, Liu S, Gunn-Moore F, Lue LF, Walker DG, Kuppusamy P, Zewier ZL, Arancio O, Stern D, Yan SS, Wu H. ABAD directly links Aβ to mitochondrial toxicity in Alzheimer’s disease. Science. 2004;304:448–452. doi: 10.1126/science.1091230. [DOI] [PubMed] [Google Scholar]
- Mandal S, Guptan P, Owusu-Ansah E, Banerjee U. Mitochondrial regulation of cell cycle progression during development as revealed by the tenured mutation in Drosophila. Dev Cell. 2005;9:843–54. doi: 10.1016/j.devcel.2005.11.006. [DOI] [PubMed] [Google Scholar]
- Marsh JL, Thompson LM. Drosophila in the study of neurodegenerative disease. Neuron. 2006;52:169–78. doi: 10.1016/j.neuron.2006.09.025. [DOI] [PubMed] [Google Scholar]
- McCord JM, Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein) J Biol Chem. 1969;244:6049–55. [PubMed] [Google Scholar]
- Meinertzhagen IA, Hanson TE. In: The Development of Drosophila melanogaster. Bate M, Martinez Arias A, editors. Cold Spring Harbor, NY: Cold Spring Harbour Laboratory Press; 1993. [Google Scholar]
- Newsome TP, Asling B, Dickson BJ. Analysis of Drosophila photoreceptor axon guidance in eye-specific mosaics. Development. 2000;127:851–860. doi: 10.1242/dev.127.4.851. [DOI] [PubMed] [Google Scholar]
- Niven JE, Anderson JC, Laughlin SB. Fly photoreceptors demonstrate Energy-Information trade-offs in neural coding. PLOS Biology. 2007;5:828–840. doi: 10.1371/journal.pbio.0050116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien KM, Dirmeier R, Engle M, Poyton RO. Mitochondrial protein oxidation in yeast mutants lacking manganese-(MnSOD) or copper- and zinc-containing superoxide dismutase (CuZnSOD): evidence that MnSOD and CuZnSOD have both unique and overlapping functions in protecting mitochondrial proteins from oxidative damage. J Biol Chem. 2004;279:51817–51827. doi: 10.1074/jbc.M405958200. [DOI] [PubMed] [Google Scholar]
- Okado-Matsumoto A, Fridovich I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu,Zn-SOD in mitochondria. J Biol Chem. 2001;276:38388–93. doi: 10.1074/jbc.M105395200. [DOI] [PubMed] [Google Scholar]
- Palfi S, Ferrante RJ, Brouillet E, Beal MF, Dolan R, Guyot MC, Peschanski M, Hantraye P. Chronic 3-nitropropionic acid treatment in baboons replicates the cognitive and motor deficits of Huntington’s disease. J Neurosci. 1996;16:3019–25. doi: 10.1523/JNEUROSCI.16-09-03019.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearse AGE. Histochemistry vol II. 3. Churchill Livingston; London: 1972. [Google Scholar]
- Pesah Y, Pham T, Burgess H, Middlebrooks B, Verstreken P, Zhou Y, Harding M, Bellen H, Mardon G. Drosophila parkin mutants have decreased mass and cell size and increased sensitivity to oxygen radical stress. Development. 2004;131:2183–94. doi: 10.1242/dev.01095. [DOI] [PubMed] [Google Scholar]
- Prokop A, Meinertzhagen IA. Development and structure of synaptic contacts in Drosophila. Semin Cell Dev Biol. 2006;1:20–30. doi: 10.1016/j.semcdb.2005.11.010. [DOI] [PubMed] [Google Scholar]
- Senoo-Matsuda N, Yasuda K, Tsuda M, Ohkubo T, Yoshimura S, Nakazawa H, Hartman PS, Ishii N. A defect in the cytochrome b large subunit in complex II causes both superoxide anion overproduction and abnormal energy metabolism in Caenorhabditis elegans. J Biol Chem. 2001;276:41553–41558. doi: 10.1074/jbc.M104718200. [DOI] [PubMed] [Google Scholar]
- Solans A, Zambrano A, Rodriguez M, Barrientos A. Cytotoxicity of a mutant huntingtin fragment in yeast involves early alterations in mitochondrial OXPHOS complexes II and III. Hum Mol Genet. 2006;15:3063–81. doi: 10.1093/hmg/ddl248. [DOI] [PubMed] [Google Scholar]
- Staniek K, Nohl H. Biochim Biophys Acta. 2000;1460:268–275. doi: 10.1016/s0005-2728(00)00152-3. [DOI] [PubMed] [Google Scholar]
- Stowers RS, Megeath LJ, Gorska-Andrzejak J, Meinertzhagen IA, Schwarz TL. Axonal transport of mitochondria to synapses depends on milton, a novel Drosophila protein. Neuron. 2002;36:1063–77. doi: 10.1016/s0896-6273(02)01094-2. [DOI] [PubMed] [Google Scholar]
- Stowers RS, Schwarz TL. A genetic method for generating Drosophila eyes composed exclusively of mitotic clones of a single genotype. Genetics. 1999;152:1631–1639. doi: 10.1093/genetics/152.4.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan W, Ashburner M, Hawley RS. Drosophila Protocols. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2000. [Google Scholar]
- Sun J, Folk D, Bradley TJ, Tower J. Induced overexpression of mitochondrial Mn-superoxide dismutase extends the life span of adult Drosophila melanogaster. Genetics. 2002;161:661–72. doi: 10.1093/genetics/161.2.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Tower J. FLP recombinase-mediated induction of Cu/Zn-superoxide dismutase transgene expression can extend the life span of adult Drosophila melanogaster flies. Mol Cell Biol. 1999;19:216–28. doi: 10.1128/mcb.19.1.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel H. Mitochondrial myopathies and the role of the pathologist in the molecular era. J Neuropathol Exp Neurol. 2001;60:217–27. doi: 10.1093/jnen/60.3.217. [DOI] [PubMed] [Google Scholar]
- Wagh DA, Rasse TM, Asan E, Hofbauer A, Schwenkert I, Durrbeck H, Buchner S, Dabauvalle MC, Schmidt M, Qin G, Wichmann C, Kittel R, Sigrist SJ, Buchner E. Bruchpilot, a protein with homology to ELKS/CAST, is required for structural integrity and function of synaptic active zones in Drosophila. Neuron. 2006;49:833–44. doi: 10.1016/j.neuron.2006.02.008. [DOI] [PubMed] [Google Scholar]
- Walker FO. Huntington’s disease. Lancet. 2007;369:218–28. doi: 10.1016/S0140-6736(07)60111-1. [DOI] [PubMed] [Google Scholar]
- Walker DW, Hájek P, Muffat J, Knoepfle D, Cornelison S, Attardi G, Benzer S. Hypersensitivity to oxygen and shortened lifespan in a Drosophila mitochondrial complex II mutant. Proc Natl Acad Sci U S A. 2006;103:16382–7. doi: 10.1073/pnas.0607918103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yankovskaya V, Horsefield R, Törnroth S, Luna-Chavez C, Miyoshi H, Léger C, Byrne B, Cecchini G, Iwata S. Architecture of Succinate Dehydrogenase and Reactive Oxygen Species Generation. Science. 2003;299:700–704. doi: 10.1126/science.1079605. [DOI] [PubMed] [Google Scholar]
- Zinsmaier KE, Eberle KK, Buchner E, Walter N, Benzer S. Paralysis and early death in cysteine string protein mutants of Drosophila. Science. 1994;263:977–80. doi: 10.1126/science.8310297. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.