

Abstract
An efficient method for the enantioselective synthesis of (2R, 3S)- and (2S, 3R)-4,4,4-trifluoro-N-Fmoc-O-tert-butyl-threonine (tfT) on multi-gram scales was developed. Absolute configurations of the two stereoisomers were ascertained by X-ray crystallography. Racemization-free coupling conditions for the incorporation of tfT into oligopeptides were then explored. For solution-phase synthesis, tfT racemization was not an issue under conventional coupling conditions. For solid-phase synthesis, the following conditions were identified to achieve racemization-free synthesis: if tfT (3.0 eq.) was not the first amino acid to be linked to the resin (1.0 eq.), the condition is: 2.7 eq. DIC/3.0 eq. HOBt as the coupling reagent at 0 °C for 20 h; if tfT (3.0 eq.) was the first amino acid to be linked to the resin (1.0 eq.), then 1.0 eq. of CuCl2 needs to be added to the coupling reagent.
Keywords: trifluorothreonine, Sharpless AD reaction, solid-phase peptide synthesis, 19F NMR, chiral chromatography, octreotide
Introduction
The introduction of fluorinated amino acids into peptides and proteins often has significant impact on their physicochemical and biological properties, such as increased stability,1 decreased helical propensity,2 preferential association in solution3 and in lipids,4 enhanced membrane binding,5 prolonged in vivo half-life6 and promotion of chemotaxis.7 Furthermore, incorporation of fluorinated amino acids makes it possible to investigate the conformation and metabolism of host peptides or proteins by 19F NMR.8 The multi-faceted impact of fluorinated amino acids on peptides and proteins was discussed in a recent review.9 From a pharmaceutical chemistry standpoint, fluorinated amino acids can be used as pharmacokinetic modulators (via prolonged in vivo half-life, t½) and reporters (via 19F NMR) for peptide-based pharmaceuticals.10
Several methods have been developed for site-specific incorporation of fluorinated amino acids into peptides or proteins, including both solid-1, 2, 3 and solution-7, 11 phase chemical synthesis, protease-catalyzed synthesis12 and in vivo bio-synthesis.13 Among these methods, solid-phase peptide synthesis (SPPS) is still the most commonly used one due to its ease of operation.
Ideally, fluorinated amino acids should be incorporated into peptides or proteins as pure enantiomers in a racemization-free manner. However, only a few reports on the racemization-free incorporation of enantiopure fluorinated amino acids into peptides via SPPS have appeared in the literature, including L-5,5,5,5′,5′,5′-hexafluoroleucine,1d, 1e, 3 4-fluoro-proline14 and 3-(trifluoro-methyl)bicyclopent[1.1.1]-1-ylglycine15. The reason is two fold. First, although the synthesis of fluorinated amino acids is well developed,16 few fluorinated amino acids have been prepared in enantiopure and protected forms suited for SPPS. As a result, many fluorinated amino acids are incorporated into peptides or proteins as racemic mixtures.1a-1c, 5, and 17 Second, even when fluorinated amino acids were prepared in enantiopure and protected forms, racemization can take place during SPPS.18
Our interest in fluorinated amino acids is driven by our effort to modulate and report the pharmacokinetics of octreotide. Octreotide (d-Phe1-c[Cys2-Phe3-d-Trp4-Lys5-Thr6-Cys7]-Thr8) is an octapeptide analog of the natural tetradecapeptide hormone somatostatin. Octreotide is the peptide component of two drugs (Sandostatin® and OctreoScan®) and one drug candidate (OctreoTher®), all for diagnosing and treating neuroendocrine tumors. Compared with somatostatin, octreotide has significantly improved pharmacokinetics (e.g., t½ for somatostatin and octreotide are ca. 2 min and 2 h, respectively).19 Effort to further improve the pharmacokinetics of octretide and other somatostatin analogs is an ongoing research topic that includes chemical conjugation20 and microsphere encapsulation21. Incorporation of fluorinated amino acids presents another angle to improve the pharmacokinetics of octreotide.
From the biology perspective, octreotide provides a unique system to investigate the relationship between fluorination and bioactivity because it contains two threonines, one inside (Thr6) and one outside (Thr8) the receptor-binding site.22 For this reason, we decided to synthesize 4,4,4-trifluorothreonine (tfT) and incorporate it into octreotide. Like threonine, tfT has two chiral centers and hence four stereoisomers: (2S, 3R), (2R, 3S), (2S, 3S) and (2R, 3R). Previous work by Goodman and coworkers has demonstrated clearly that the stereochemistry of Thr6 and Thr8 has critical impact on the bioactivity of octreotide.22 Hence, to achieve desired pharmaceutical benefits, it is absolutely essential that tfT is incorporated into octreotide as pure enantiomers in a racemization-free manner.
From the chemistry standpoint, incorporation of tfT into peptides is a challenging issue because tfT requires side chain protection. Further, since tfT contains two chiral centers (C2 and C3 in FIGURE 1), the probability for racemization during SPPS is greater than fluorinated amino acids with only one chiral center. With two electron-withdrawing groups (-CF3 and -OH) attached to the C3 chiral center, the acidity of both the α- and the β-protons (Hα and Hβ) will be enhanced, thereby making the C2 and C3 chiral centers prone to racemization through enolization during SPPS, especially in the presence of base. 23
FIGURE 1.

Molecular structures of (2R, 3S)-4,4,4-trifluorothreonine (1a) and (2S, 3R)-4,4,4-trifluorothreonine (1b).
In this paper, we report the synthesis of two protected tfT stereoisomers, (2R, 3S)- and (2S, 3R)-4,4,4-trifluoro-N-Fmoc-O-tert-butyl-threonine and the racemization-free incorporation of these two enantiomers into oligopeptides via SPPS.
Enantioselective Synthesis of (2R, 3S)- and (2S, 3R)-4,4,4-Trifluoro-N-Fmoc-O-Tert-Butyl-Threonine
The synthesis of protected Fmoc- and tBu-protected tfT builds on the method developed by Qing and coworkers for the synthesis of free tfT.24 To obtain protected tfT for SPPS, Qing's method was modified to suit the synthesis of orthogonally protected tfT.
The starting material for Qing's method24 is the trifluoromethylation reagent, FSO2CF2CO2Me, which is highly corrosive and rather expensive in the US. We decided to start with the commercially available compounds 4,4,4-trifluoro-3-oxo-butyric acid ethyl ester (3) or ethyl 4,4,4-trifluoro-crotonate (4). Compound 4 can be synthesized from compound 3 (see Supplementary Data for details).
Ester 4 underwent reduction with lithium aluminum hydride in the presence of aluminum chloride25 to give the corresponding alcohol whose hydroxyl group was then protected with a benzoyl group (Bz) to give compound 2 with an 80% yield on a 40-gram scale (SCHEME 1). In Qing's method, the hydroxyl group in 2 was protected by the benzyl (Bn) group.24 The change from Bn to Bz for hydroxyl protection in compound 2 brought certain advantages in downstream synthetic steps. It also altered the stereochemical outcome for the chiral center construction step.
SCHEME 1.

Preparation of starting material 2
With the trifluoromethylated trans-alkene 2 in hand, Sharpless asymmetric dihydroxylation (AD) was employed to construct the two chiral centers simultaneously (SCHEME 2).24 The reaction proceeded smoothly to give the diol 5a or 5b, depending on the catalyst, with excellent yields. The enantiomeric excess (e.e.) values of 5a and 5b (without recrystallization) are both over 99% (see Stereochemical Characterizations). It was found that, with Bz as the hydroxyl protective group in compound 2, the Sharpless AD reaction completed within 12 h with excellent yield even in the absence of methanesulfonamide, a catalyst usually used in Sharpless AD reactions. This is one advantage brought by the Bz protective group.
SCHEME 2.

Construction of chiral centers via Sharpless AD reaction
From recrystallized chiral diol 5a, Fmoc-1a(tBu)-OH was synthesized over 7 steps (SCHEME 3). The same procedures as Qing's were used to proceed from diol 5a to alcohol 7a.24 Treatment of diol 5a with thionyl chloride and triethyl amine afforded a cyclic sulfite intermediate which then underwent oxidation with ruthenium chloride and sodium periodate to give the cyclic sulfate 6a with an 80% yield. Ring opening of the cyclic sulfate 6a with sodium azide followed by hydrolysis provided alcohol 7a with a 98% yield. To get orthogonally protected tfT, the hydroxyl group in alcohol 7a needs to be protected with the tert-butyl group. When sulfuric acid was employed to catalyze this reaction with liquid iso-butylene in a sealed vessel, the tert-butyl ether product 8a was isolated with a 70% yield.26 With the tert-butyl ether 8a in hand, removal of the benzoyl protective group was then carried out. In order to avoid unexpected racemization, a reaction with very mild condition (diisobutylaluminum hydride reduction) was employed and alcohol 9a was isolated with a 93% yield. Such mild cleavage condition is another advantage brought by the Bz protective group. Alcohol 9a was then subjected to palladium-catalyzed hydrogenation of its azido group to give the amine 10a with an 89% yield. Protection of the amino group with 9-fluorenylmethyl chloroformate (FmocCl) yielded alcohol 11a with a 98% yield, which then underwent Jones oxidation to afford the final product Fmoc-1a(tBu)-OH with a 91% yield on a multi-gram scale. The overall yield for the synthesis of Fmoc-1a(tBu)-OH from compound 4 is ca. 30%.
SCHEME 3.

Synthesis of Fmoc-1a(tBu)-OH
By employing the same procedures for the synthesis of Fmoc-1a(tBu)-OH, Fmoc-1b(tBu)-OH was synthesized from recrystallized chiral diol 5b on a multi-gram scale (see Supplementary Data). The overall yield for the synthesis of Fmoc-1b(tBu)-OH from compound 4 is ca. 30%.
Stereochemical Characterizations
Stereochemical characterizations were conducted at three stages. Firstly, after chiral center construction, the enantiopurities of the Sharpless AD reaction products 5a and 5b were verified. Direct determination of the enantiomeric excess (e.e.) values for 5a and 5b by chiral chromatography was unsuccessful.27 Instead, 5a and 5b were transformed into their Mosher's esters 12a (over 99% yield) and 12b (over 99% yield), respectively (SCHEME 4). As 12a and 12b are diastereoisomers, their 19F NMR signals are distinct. The e.e. values of 5a and 5b were inferred from the diastereomeric excess (d.e.) values of 12a and 12b determined by 19F NMR spectroscopy. From the d.e. values of 12a and 12b, the inferred e.e. values of 5a and 5b were over 99% (see Supplementary Data for details).
SCHEME 4.

Synthesis of Mosher's esters
Secondly, the enantiomeric purities of the final products Fmoc-1a(tBu)-OH and Fmoc-1b(tBu)-OH (after silica gel column purification) were determined directly by analytical chiral chromatography.28 High e.e. values were achieved for both enantiomers: 96.8% for Fmoc-1a(tBu)-OH and > 99.5% for Fmoc-1b(tBu)-OH. After recrystallization, the e.e. values of Fmoc-1a(tBu)-OH and Fmoc-1b(tBu)-OH were both higher than 99.5%.
Thirdly, to determine the absolute configurations of both a- and b-series of chiral molecules, amino alcohols 10a and 10b were transformed into their camphorsulfonamides 13a and 13b, respectively (SCHEME 5), for X-ray crystallographic analysis. Treatment of 10a with (1R)-(-)-camphor-10-sulfonyl chloride in the presence of 4-dimethylaminopyridine (DMAP) afforded camphor-sulfonamide 13a with a 48% yield. In the same way, camphorsulfonamide 13b was isolated with a 52% yield after treating 10b with (1S)-(+)-camphor-10-sulfonyl chloride. Therefore a pair of enantiomers, 13a and 13b, were obtained. During the workup, small amounts of 14a and 14b were also isolated with a 15% yield and a 13% yield, respectively.
SCHEME 5.

Synthesis of camphorsulfonamides
Single crystals of compounds 13a and 13b were collected by slow evaporation of their respective dichloromethane/hexanes solutions. With the aid of single crystal X-ray diffraction, the absolute configurations of compounds 13a and 13b were determined to be (2S, 3S) and (2R, 3R) respectively (see Supplementary Data for details). Therefore, Sharpless AD reaction of trans-alkene 2 with (DHQD)2PHAL gives the chiral diol 5a with the (2R, 3S) configuration, while the reaction of trans-alkene 2 with (DHQ)2PHAL gives the chiral diol 5b with the (2S, 3R) configuration. Note that the stereochemistry results of Sharpless AD reaction of compound 2 in our synthesis is opposite to the results of Qing's method24, presumably due to replacing Bn with Bz in compound 2, the substrate of Sharpless AD reaction.
Hydrophobicity Fmoc-tfT vs. Fmoc-Thr
Mindful of the possibility that fluorination might actually decrease the hydrophobicity of a molecule29 and hence reduce membrane permeability30, we set out to determine the hydrophobicity of Fmoc-tfT(tBu)-OH relative to their non-fluorinated counterparts. To this end, we employed the latest reversed-phase liquid chromatography (RPLC) method for amino acid hydrophobicity determination developed by Hodges and coworkers, who determined the relative hydrophobicity of 22 L-amino acids and their D-enantiomers.31 As pointed out by Hodges and coworkers, a criterion for measuring true hydrophobicity of an amino acid is that the D/L enantiomers should give the same retention time, tR. FIGURE 2 shows the co-injection of Fmoc-1a(tBu)-OH (denoted as allo-D-tfT), Fmoc-1b(tBu)-OH (denoted as allo-L-tfT), their non-fluorinated counterparts: (2R, 3R)-N-Fmoc-O-tert-butyl-threonine (denoted as allo-D-Thr), (2S, 3S)-N-Fmoc-O-tert-butyl-threonine (denoted as allo-L-Thr), as well as the other two stereoisomers of Fmoc-protected threonine: (2R, 3S)-N-Fmoc-O-tert-butyl-threonine (denoted as D-Thr) and (2S, 3R)-N-Fmoc-O-tert-butyl-threonine (denoted as L-Thr). Clearly, all the enantiomeric pairs, allo-D-Thr/allo-L-Thr (tR = 7.2 min), D-Thr/L-Thr (tR = 9.2 min) and allo-D-tfT/allo-L-tfT (tR = 11.6 min), co-elute, meeting the criterion established by Hodges and coworkers.31 In RPLC, the larger the retention time, the more hydrophobic a molecule is. The allo-D-tfT/allo-L-tfT pair is more retentive than the allo-D-Thr/allo-L-Thr pair (ΔtR = 11.6 - 7.2 = 4.2 min). This proves that replacing the -CH3 group in Thr by -CF3 in tfT indeed renders the molecule more hydrophobic. To put matters into perspective, the retention time difference between allo-D-tfT/allo-L-tfT and allo-D-Thr/allo-L-Thr (ΔtR = 4.2 min) is larger than that between D-Ala/L-Ala and Gly (ΔtR = 2.8 min), comparable to that between D-Cys/L-Cys and D-Ala/L-Ala (ΔtR = 4.8 min), and smaller than that between D-Val/L-Val and D-Ala/L-Ala (ΔtR = 10.6 min).31 The impact on hydrophobicity brought by replacing Thr with tfT in unprotected peptides will be evaluated in the context of octreotide and the results will be presented in a future report.
FIGURE 2.
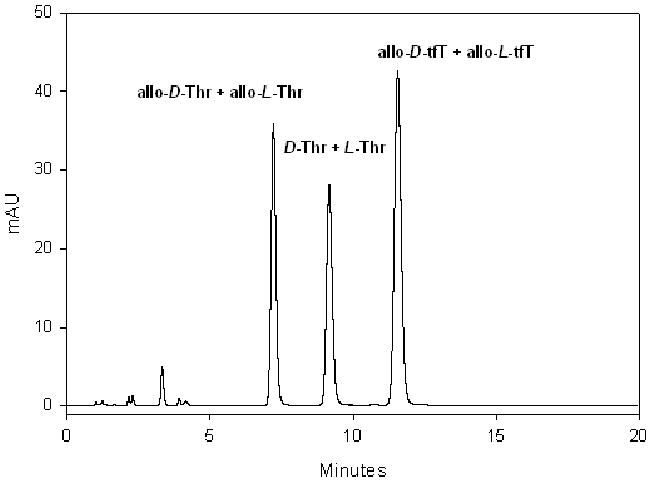
HPLC chromatogram of co-injection of Fmoc-1a(tBu)-OH (allo-D-tfT), Fmoc-1b(tBu)-OH (allo-L-tfT), allo-D-Thr, allo-L-Thr, D-Thr and L-Thr. Chromatography runs followed exactly the same conditions used by Hodges and coworkers31. The HPLC conditions are as follows: column, Kromasil C18 (150 mm × 2.1 mm I.D., 5 μm, 100 Ǻ pore size); mobile phase, A: 0.2% TFA in water, B: 0.2% TFA in CH3CN; condition, linear AB gradient (0.25% CH3CN/min, starting from 55% B); 0.3 mL/min, 280 nm, r.t. Fmoc-1a(tBu)-OH and Fmoc-1b(tBu)-OH were purified by silica-gel column chromatography. The six protected threonines were dissolved in mobile phase B together before injection.
Racemization-Free Incorporation of (2R, 3S)- and (2S, 3R)-4,4,4-Tri-Fluoro-N-Fmoc-O-Tert-Butyl-Threonine into Oligopeptides
To explore racemization-free incorporation of tfT into peptides, a set of 12 oligopeptides were designed (Table 1). The dipeptide Fmoc-tfT(tBu)-Phe-OMe is used to explore coupling conditions for solution-phase synthesis. Then, to explore coupling conditions for solid-phase synthesis, a set of oligopeptides, based on the C-terminal three residues of octreotide (Thr-Cys-Thr), was designed. Detailed design rationales are given in Table 1.
Table 1.
Oligopeptide sequences and design rationales
| Peptide Sequencesi | Design Rationales |
|---|---|
| Fmoc-tfT(tBu)-Phe-OMe | To explore coupling conditions in solution-phase synthesis. |
| H-tfT-NH2ii | To explore coupling conditions in solid-phase synthesis when tfT is the first residue attached to the resin. |
| H-Cys-tfT-NH2ii | |
| H-tfT-Gly-NH2ii | To explore coupling conditions in solid-phase synthesis when tfT is the second residue attached to the resin. |
| H-tfT-Cys-tfT-NH2ii | To explore coupling conditions in solid-phase synthesis when more than one tfT is attached to the resin. |
Because tfT has two isomers (1a and 1b), an oligopeptide containing 1 tfT residue has 2 variants while an oligopeptide containing 2 tfT residues has 4 variants. Hence, there are a total of 12 oligopeptides.
Each oligopeptide synthesized via SPPS contains one positive charge at the N-terminus while the C-terminal negative charge is abolished through amidation. This feature facilitates their purification via cation-exchange chromatography.
Solution-Phase Synthesis of Dipeptides
A pair of dipeptides, Fmoc-1a(tBu)-L-Phe-OCH3 and Fmoc-1b(tBu)-L-Phe-OCH3, were synthesized in solution phase with high yields (92%). Fmoc- and tBu-protected 1a or 1b (1.0 eq.) was coupled with L-Phenylalanine methyl ester (1.6 eq.) smoothly at room temperature in the presence of 2-(1H-benzotriazole-l-yl)-1,1,3,3-tetra-methyluronium tetrafluoroborate (TBTU, 0.9 eq.), N-hydroxy-benzotriazole (HOBt, 1.0 eq.) and N,N-diisopropylethylamine (DIPEA, 2.0 eq.) (SCHEME 6), with dimethylforamide (DMF) as the solvent. FIGURE 3 presents the 19F NMR spectra of Fmoc-1a(tBu)-L-Phe-OCH3 and Fmoc-1b(tBu)-L-Phe-OCH3, in comparison with Fmoc-1a(tBu)-OH and Fmoc-1b(tBu)-OH. From FIGURE 3B and 3D, it can be seen that Fmoc-1a(tBu)-OH and Fmoc-1b(tBu)-OH give identical 19F signal, as expected for a pair of enantiomers. In contrast, 19F signals of Fmoc-1a(tBu)-L-Phe-OCH3 and Fmoc-1b(tBu)-L-Phe-OCH3 are distinct (FIGURE 3A and3C), as expected for a pair of diastereomers. Based on 19F NMR signals, it can be concluded that racemization is negligible under the coupling conditions employed for solution-phase synthesis. This provides the starting point for detailed investigation of solid-phase synthesis conditions.
SCHEME 6.

Solution-phase synthesis of dipeptides Fmoc-tfT(tBu)-L-Phe-OCH3
FIGURE 3.
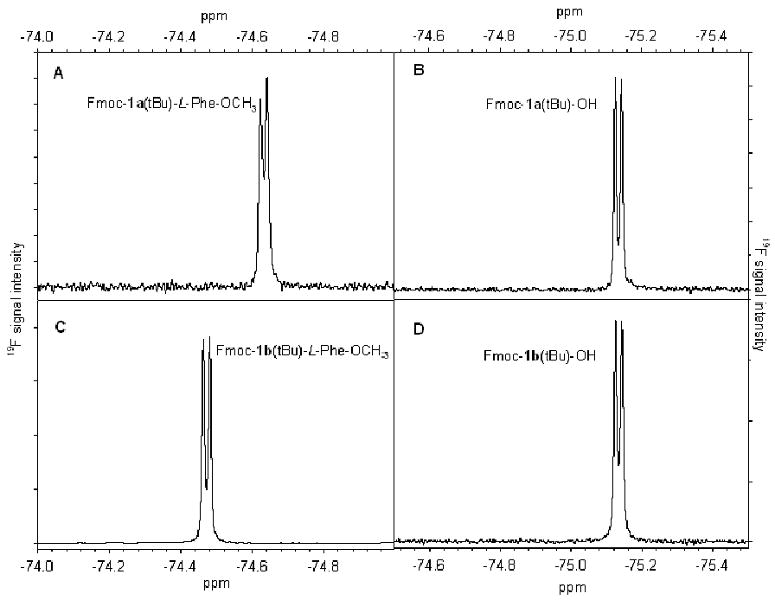
19F NMR spectra (in CDCl3, C6F6 as internal standard) of starting materials (Fmoc-tfT(tBu)-OH, panels B & D) and purified products (Fmoc-tfT(tBu)-L-Phe-OCH3, panels A & C) of solution-phase peptide synthesis.
Solid-Phase Synthesis of Oligopeptides
Exploration of racemization-free SPPS conditions for tfT
Fmoc-1a(tBu)-OH was used to explore racemization-free coupling conditions for SPPS. At first, the coupling condition (2.7eq. TBTU/3.0 eq. HOBt/6.0 eq. DIPEA, r.t., 2h) adopted from the aforementioned solution-phase synthesis was used to couple Fmoc-1a(tBu)-OH (3.0 eq.) to the MBHA rink-amide resin (1.0 eq.) with DMF as the solvent. While Fmoc-1a(tBu)-NH2 was still attached to the resin (i.e., Fmoc-1a(tBu)-NH-resin), 19F NMR spectroscopy was employed to detect racemization. There were two totally separated peaks in the 19F NMR spectrum of Fmoc-1a(tBu)-resins (FIGURE 4A), indicative of extensive racemization. Fmoc-1a(tBu)-NH-resin was then deprotected and cleaved to give H-1a-NH2. After workup, 19F NMR spectrum of the crude peptide was collected. As shown in FIGURE 4A′, there is a triplet-like peak, again indicative of extensive racemization. Judged by 19F signal heights and shapes in FIGURE 4A & 4A′, it is apparent that 1a has completely racemized under the first coupling condition (TBTU/HOBt/DIPEA at r.t.).
FIGURE 4.
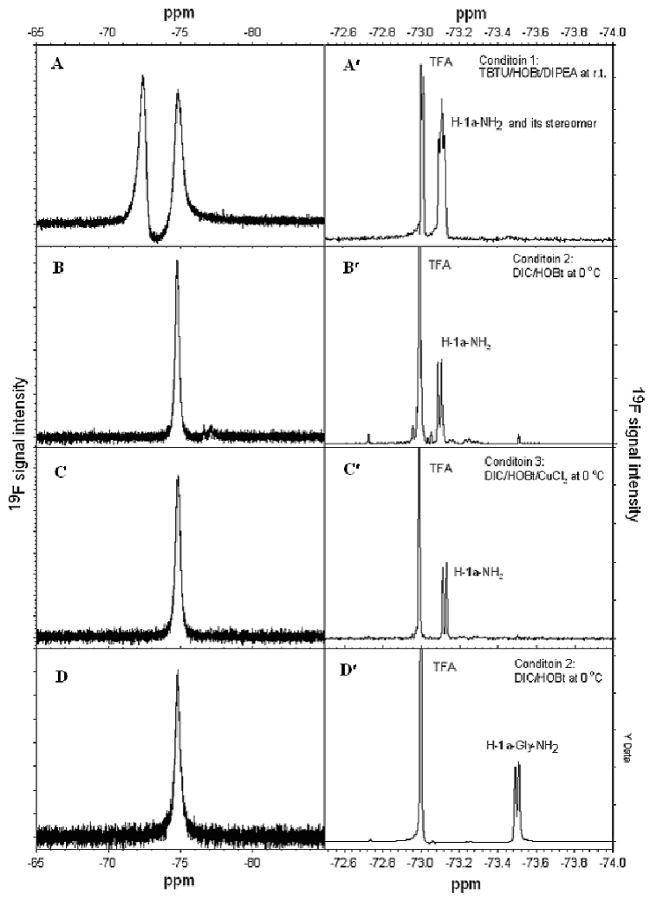
19F NMR spectra of Fmoc-1a(tBu)-resin (A, B and C), Fmoc-1a(tBu)-Gly-resin (D) (CDCl3, C6F6 as internal standard), crude samples of H-1a-NH2 (A′, B′ and C′) and crude sample of H-1a-Gly-NH2 (D′) (8 % D2O PBS solution with 0.1 % TFA). A and A′ coupling condition: 3.0 eq. Fmoc-1a(tBu)-OH/2.7 eq. TBTU/3.0 eq. HOBt/6.0 eq. DIPEA in DMF at r. t. for 2 h; B and B′ coupling condition: 3.0 eq. Fmoc-1a(tBu)-OH/2.7 eq. DIC/3.0 eq. HOBt in DMF/DCM (1:1) at 0 °C for 20 h; C and C′ coupling condition: 3.0 eq. Fmoc-1a(tBu)-OH/2.7 eq. DIC/3.0 eq. HOBt/ 1.0 eq. CuCl2.2H2O in DMF/DCM (1:1) at 0 °C for 20 h; D and D′ coupling conditions: for Gly, 3.0 eq. Fmoc-Gly-OH/3.0 eq. DIC/3.0 eq. HOBt in DMF and DCM (1:1) at r. t. for 2h; for tfT, 3.0 eq. Fmoc-1a(tBu)-OH/2.7 eq. DIC/2.7 eq. HOBt in DMF/DCM (1:1) at 0 °C for 20 h.
Then it is necessary to find a better combination of coupling conditions for tfT. N,N′-diisopropylcarbodiimide (DIC) and HOBt have been found to be effective to suppress racemization in SPPS.23b, 32 Also, base added to the coupling reagent will accelerate not only acylation but also racemization. Barany and coworkers explored base-free coupling conditions and found that racemization can be reduced to negligible extent in the absence of base.23b
Therefore, we tested the coupling reagents DIC (2.7 eq.) and HOBt (3.0 eq.) with no base added. A less polar solvent system, DMF/DCM (1:1), is adopted as it has been reported to suppress racemization.23b, 34 Low coupling temperature (0°C) was also applied to get better result and prolonged reaction time (20h) is necessary to complete the coupling reaction.
Racemization has been effectively suppressed under this second coupling condition, as judged by the 19F NMR spectrum of Fmoc-1a(tBu)-NH-resin (FIGURE 4B) and that of crude free monopeptide H-1a-NH2 (FIGURE 4B′).
To further suppress racemization, copper (II) chloride (CuCl2) was added to the coupling reagent, based on the work by Kuwata33 and Hallberg34. The addition of CuCl2 indeed abolished racemization completely as there are no detectable minor peaks in the 19F NMR spectra of Fmoc-1a(tBu)-resin (FIGURE 4C) and of crude H-1a-NH2 (FIGURE 4C′). It is found that without adding CuCl2, there was no detectable racemization if tfT is the second residue attached to the resin when H-1a-Gly-NH2 was synthesized (FIGURE 4D & 4D′). Hence, it can be concluded that tfT is more prone to racemization when it is the first amino acid to be coupled to the resin.
In summary, the racemization-free SPPS condition we identified for tfT is: 2.7 eq. DIC/3.0 eq. HOBt at 0°C, 20 h for 3.0 eq. tfT and 1.0 eq. resin. When tfT is the first residue to be linked to the resin, 1.0 eq. of CuCl2 needs to be added to the coupling reagent. No base (e.g., DIPEA) was added to the coupling reagent.
Solid-phase synthesis and HPLC purification of oligopeptides
Using the optimized coupling conditions, a family of eight oligopeptides was synthesized, including two monopeptides (H-1a-NH2 and H-1b-NH2), two dipeptides (H-Cys-1a-NH2 and H-Cys-1b-NH2) and four tripeptides (H-1a-Cys-1a-NH2, H-1b-Cys-1b-NH2, H-1a-Cys-1b-NH2 and H-1b-Cys-1a-NH2). Barany's base-free method was applied for Cysteine coupling with slight modification.23b
None of these oligopeptides was retentive on either RPLC or normal-phase (NP) HPLC columns. Fortunately, due to the N-terminal positive charge, all oligopeptides were retentive on strong cation-exchange (SCX) columns. Hence, all oligopeptides were purified by SCX liquid chromatography (SCXLC) with their molecular weights and purities verified by analytical SCXLC and mass spectrometry, respectively.
Characterization of Oligopeptide Enantiopurity
FIGURE 5 shows the 19F NMR spectra of the eight oligopeptides in both the resin-bound and purified free forms. FIGURE 6 shows the SCX and chiral chromatograms of co-injected tripeptides.
FIGURE 5.

19F NMR spectra of Fmoc-tfT(tBu)-resin (A), Fmoc-Cys(Trt)-tfT(tBu)-resin (B), Fmoc-tfT(tBu)-Cys(Trt)-tfT(tBu)-resin (C) (CDCl3, C6F6 as internal standard), purified H-tfT-NH2 (A′), H-Cys-tfT-NH2 (B′) and H-tfT-Cys-tfT-NH2 (C′) (8 % D2O PBS solution with 0.1 % TFA). In panels A′-C′, 19F spectrum of each oligopeptide was acquired separately and then spectra of different mono-, di-, or tri-peptides were plotted in the same frame with peak intensity normalized to roughly the same height. Since the 19F spectra of the two monopeptides, H-1a-NH2 and H-1b-NH2, were collected separately, their 19F signals do not exactly coincide (Δδ19F = 0.04 ppm) due to slight fluctuation of experimental conditions, even though they are enantiomers. However, when their 19F spectra were collected simultaneously from a mixture of the two monopeptides, their 19F signals exactly coincide (FIGURE S5 in Supplementary Data), as one would expect for a pair of enantiomers.
FIGURE 6.
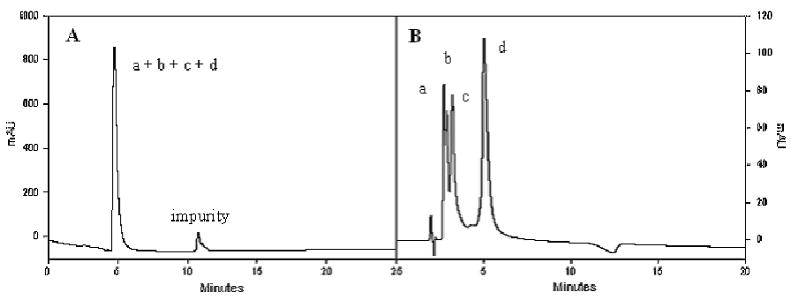
Analytical SCXLC (A) and chiral (B) chromatograms of co-injected tripeptides. a: H-1a-Cys-1b-NH2; b: H-1a-Cys-1a-NH2; c: H-1b-Cys-1a-NH2; d: H-1b-Cys-1b-NH2. The SCXLC chromatogram was acquired after the tripeptides have been stored in dried form at -20 °C for about 10 weeks. Such prolonged storage at -20 °C apparently led to peptide degradation because the impurity peak in panel A was nonexistent in SCXLC chromatograms acquired for each individual tripeptide before such prolonged storage (FIGURE S8 in Supplementary Data). For suggestions on peptide storage, see the EXPERIMENTAL section.
In the resin-bound form, oligopeptides containing a single tfT residue (mono- and di-peptides) give a single broad 19F signal while oligopeptides containing two tfT residues (tripeptides) give two broad 19F signals, with no readily identifiable minor peaks. 19F NMR spectra of SCXLC-purified oligopeptides show similar results, i.e., oligopeptides containing a single tfT residue (mono- and di-peptides) give a single split 19F signal while oligopeptides containing two tfT residues (tripeptides) give two split 19F signals. Note that although SCXLC can separate an oligopeptide from other impurities, it cannot separate an oligopeptide from its stereoisomers, as shown by FIGURE 6A. Hence, the lack of identifiable minor peaks in 19F NMR spectra of either resin-bound or SCXLC-purified free oligopeptides indicates that racemization has been effectively suppressed during the synthesis of these oligopeptides.
Efforts were also made to characterize the enantiopurity of purified oligopeptides by chiral chromatography. Mono- and di-peptides were not retentive on ChiraDex, Chirobiotic T or Ultron ES-Pepsin chiral columns. Fortunately, tripeptides were retentive on Ultron ES-Pepsin chiral column. Hence, the enantiopurity of each tripeptide was also verified by chiral chromatography, in addition to 19F NMR spectroscopy. The chiral chromatogram of each individual tripeptide is given in Supplementary Data. FIGURE 6B presents the elution profile of co-injected tripeptides, which were eluted in the order of H-1a-Cys-1b-NH2 < H-1a-Cys-1a-NH2 < H-1b-Cys-1a-NH2 < H-1b-Cys-1b-NH2. Since none of the oligoeptides has gone through chrial purification (SCXLC can not separate stereoisomers, as demonstrated by FIGURE 6A), high enantiopurity of the tripeptides provides strong evidence that racemization was suppressed to negligible extent using the optimized coupling conditions.
Conclusion
In summary, an efficient method has been developed for the enantioselective synthesis of (2R, 3S)- and (2S, 3R)-4,4,4-trifluoro-N-Fmoc-O-tert-butyl-threonine on multi-gram scales. The final products Fmoc-1a(tBu)-OH and Fmoc-1b(tBu)-OH were obtained with high e.e. values (96.8% for Fmoc-1a(tBu)-OH and > 99% for Fmoc-1b(tBu)-OH before recrystallization; > 99.5% for both enantiomers after recrystallization). Absolute configurations of these chiral molecules were ascertained by X-ray crystallography. Elevated hydrophobicity of fluorinated amino acids in comparison with their non-fluorinated counterparts was ascertained by RPLC.
Coupling conditions that lead to racemization-free incorporation of tfT into oligopeptides have been identified. For solution-phase synthesis, tfT racemization was not an issue under conventional coupling conditions (TBTU/HOBt/DIPEA at r.t. for 30 min). For solid-phase synthesis, conventional coupling conditions led to extensive racemization. To achieve racemization-free synthesis, no base (e.g., DIPEA) should be added to the coupling reagent. Specifically, if tfT (3.0 eq.) was not the first amino acid residue to be linked to the resin (1.0 eq.), the condition is: 2.7 eq. DIC/3.0 eq. HOBt as the coupling reagent at 0 °C for 20 hr; if tfT (3.0 eq.) was the first amino acid to be linked to the resin (1.0 eq.), then 1.0 eq. of CuCl2 needs to be added to the coupling reagent.
Experimental
General Procedures
allo-D-N-Fmoc-O-tert-butyl-Thr and allo-L-N-Fmoc-O-tert-butyl-Thr were purchased from BACHEM (Bachem California Inc., Torrance, CA). Other Fmoc amino acids and rink amide MBHA resins (0.65 mmol/g) were purchased from Novabiochem (EMD Biosciences Inc., San Diego, CA). Other chemical reagents and solvents were purchased from commercial suppliers and were used without further purification.
NMR spectra were recorded on a 400 MHz Varian NMR spectrometer (1H 400 MHz, 19F 367 MHz, 13C 100.4 MHz). Oligopeptides were dissolved in PBS solution (8 % D2O in H2O, 50 mM sodium phosphate, 100 mM NaCl, 0.1 % trifluoroacetic acid (TFA), pH = 7.0, r.t.). 1H chemical shifts were referenced to tetramethylsilane (TMS in CDCl3, δ = 0 ppm) or water (H2O in PBS, δ = 4.8 ppm). 19F chemical shifts were referenced to hexafluorobenzen (C6F6 in CDCl3, δ = -164.5 ppm) or trifluoroacetic acid (TFA in PBS, δ = -73.0 ppm). 13C chemical shifts were referenced to tetramethylsilane (TMS in CDCl3, δ = 0 ppm) or trifluoroacetic acid (TFA in PBS, δ = -116.6 ppm). The diastereomeric excess (d.e.) values of Fmoc-1a(tBu)-Phe-OMe, Fmoc-1b(tBu)-Phe-OMe, H-Cys-1a-NH2 and H-Cys-1b-NH2 were determined by 19F NMR. d.e. values of tripeptides were determined by chiral HPLC.
For peptide purification and analysis, Agilent 1100 chromatograph system was used.
Benzoic acid (E)-4,4,4-trifluoro-but-2-enyl ester, 2
To a suspension of anhydrous AlCl3 (34.1 g, 0.26 mol) in diethyl ether (80 mL) at 0 °C was added a solution of LiAlH4 (28.5 g, 0.75 mol) in diethyl ether (500 mL). The resulting mixture was then stirred at 0 °C for 15 min. A solution of compound 4 (52.0 g, 0.31 mol) in diethyl ether (40 mL) was added at 0 °C and stirring was continued for another 4 h. Then at 0 °C, water (28 mL), NaOH aqueous solution (5.7 g NaOH in 57 mL water) and another portion of water (86 mL) were added in sequence slowly to the reaction mixture. The resulting solution was filtered and condensed using rotary evaporation under atmospheric pressure. The residue was dissolved in a solution of pyridine (54.0 mL, 0.67 mol) and CH2Cl2 (800 mL). Then benzoyl chloride (65.0 mL, 0.56 mol) was added dropwise at 0 °C. The resulting solution was stirred overnight at room temperature. Then the reaction mixture was washed with 2 N HCl aqueous solution. The organic phase was collected and the aqueous phase was extracted with diethyl ether (3 × 100 mL). The combined organic phase was washed with brine, dried over MgSO4 and concentrated under vacuo. The residue was purified by column chromatography on silica gel (hexanes : ethyl acetate = 30 : 1) to give a colorless oil (57.2 g, 80%). 1H NMR (CDCl3, 400 MHz): δ 7.97-7.30 (m, 5H), 6.44 (m, 1H), 5.87 (m, 1H), 4.83 (m, 2H); 19F NMR (CDCl3, 376.4 MHz): δ −67.24 (d, J = 6.1 Hz); MS (CI): m/z 231 ([M + 1]+, 40).
(2R, 3S)-Benzoic acid 4,4,4-trifluoro-2, 3-dihydroxy-butyl ester, 5a
To a stirred mixture of tert-butyl alcohol (400 mL) and water (400 mL) were added (DHQD)2PHAL (1.3 g, 1.67 mmol), K3Fe(CN)6 (81.5 g, 247 mmol), K2CO3 (34.2 g, 247 mmol), and OsO4 (6.5 mL of 0.1 M aqueous solution, 0.65 mmol) at room temperature. After the solid was dissolved, the solution was cooled to 0 °C. Olefin 2 (19.0 g, 82.5 mmol) was added in one portion, and the heterogeneous slurry was stirred vigorously at room temperature overnight. Then Na2SO3 (81 g, 643 mmol) was added to the resulting yellow solution. The mixture was stirred for 30 min and its color turned into dark brown. The upper organic phase was collected. The lower aqueous solution was extracted with ethyl acetate (5 × 150 mL). The combined organic phase was washed with saturated KHSO4 aqueous solution (100 mL) and saturated K2SO4 aqueous solution (100 mL) to recover some of (DHQD)2PHAL. Then the organic solution was dried over anhydrous MgSO4, filtered and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (hexanes : ethyl acetate = 5 : 1) to give 5a as a white solid (20.2 g, 93% yield). 5a was recrystallized from hexanes and CH2Cl2 to achieve higher purity: [α]20D = -12.7 (c 0.82, CHCl3); mp. 96 °C; 1H NMR (CDCl3, 400 MHz): δ 8.05-7.46 (m, 5H), 4.48 (m, 2H), 4.37 (m, 1H), 4.03 (m, 1H), 3.24 (d, J = 8.8 Hz, 1H), 2.80 (d, J = 5.2 Hz, 1H); 19F NMR (CDCl3, 376.4 MHz): δ -79.83 (d, J = 5.2 Hz); 13C NMR (CDCl3, 100.4 MHz): δ 167.0, 133.9, 130.0, 129.4, 128.8, 124.6 (q, J = 283.2 Hz), 69.5 (q, J = 30.1Hz), 67.0, 65.4; MS (CI): m/z 265 ([M + 1]+, 100); HRMS (CI): Calcd for C11H12F3O4 265.0688, found 265.0674.
(4R, 5S)-Benzoic acid 2,2-dioxo-5-trifluoromethyl-2λ6-1,3,2-dioxathiolan-4-ylmethyl ester, 6a
To a solution of recrystallized diol 5a (10.0 g, 37.9 mmol) and triethylamine (15.3 g, 151.6 mmol, 21.0 mL) in CH2Cl2 (200 mL) was slowly added thionyl chloride (9.0 g, 75.8 mmol, 5.5 mL) at 0 °C over 20 min. The reaction mixture was stirred for another 60 min at 0 °C and then diluted with cold diethyl ether (100 mL). Then cold water (100 mL) was added to the resulting deep brown organic solution. The organic phase was collected and the aqueous phase was extracted with cold diethyl ether. The combined organic phase was washed with cold brine and dried over anhydrous MgSO4. After removing solvent through rotary evaporation below 30 °C, the residue was purified by a short pad of silica gel to give the cyclic sulfite. The cyclic sulfite was then dissolved in water (90 mL), CH3CN (60 mL) and CCl4 (60 mL). Then NaIO4 (9.7 g, 45.5 mmol) and RuCl3 (20 mg) were added to the solution and the resulting mixture was vigorously stirred for 2 h at room temperature. Diethyl ether (100 mL) and saturated NaHCO3 solution (100 mL) were added to the reaction mixture. The organic phase was collected and the aqueous phase was extracted with diethyl ether. The combined organic phase was washed with brine, dried over anhydrous MgSO4 and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (hexanes : ethyl acetate = 10 : 1) to give 6a as a white solid (9.8 g, 80%). [α]20D = +5.0 (c 0.98, CHCl3); mp. 57 °C; 1H NMR (CDCl3, 400 MHz): δ 8.08-7.47 (m, 5H), 5.29 (m, 1H), 5.12 (m, 1H), 4.74 (dd, J = 12.8, 4.0 Hz, 2H); 19F NMR (CDCl3, 376.4 MHz): δ -79.72 (d, J = 6.8 Hz); 13C NMR (CDCl3, 100.4 MHz): δ 165.8, 134.3, 130.2, 129.0, 128.4, 121.4 (q, J = 280.2 Hz), 77.8, 75.9 (q, J = 36.1 Hz), 61.7; MS (CI): m/z 327 ([M + 1]+, 100); HRMS (CI): Calcd for C11H9F3O6S 326.0072, found 326.0074.
(2S, 3S)-Benzoic acid 2-azido-4,4,4-trifluoro-3-hydroxy-butyl ester, 7a
The solution of cyclic sulfate 6a (7.7 g, 23.6 mmol) and sodium azide (3.1 g, 47.2 mmol) in DMF (100 mL) was stirred for 4 h at 80 °C. The solvent was carefully removed by distillation under reduced pressure below 80 °C. Then THF (200 mL), water (1.0 mL), and sulfuric acid (3.0 mL, 96%) were added. The resulting suspension was stirred for 1 h and excess NaHSO3 solid was then added to neutralize the solution. The reaction mixture was stirred for an additional 20 min and filtered through a pad of silica gel. The filtrate was concentrated in vacuo and the residue was purified by column chromatography on silica gel (hexanes : ethyl acetate = 8 : 1) to give 7a as a white solid (6.7 g, 98%). [α]20D = +20.9 (c 1.83, CHCl3); mp. 74 °C; 1H NMR (CDCl3, 400 MHz): δ 7.94-7.32 (m, 5H), 4.63 (m, 1H), 4.52 (m, 1H), 4.08 (m, 1H), 3.97 (m, 1H), 3.88 (m, 1H); 19F NMR (CDCl3, 367.4 MHz): δ -78.85 (d, J = 5.1 Hz); 13C NMR (CDCl3, 100.4 MHz): δ 167.2, 134.0, 130.1, 129.1, 128.9, 124.3 (q, J = 282.5 Hz), 70.1 (q, J = 28.1 Hz), 64.2, 59.7; MS (CI): m/z 290 ([M + 1]+, 100); HRMS (CI): Calcd for C11H11F3N3O3 290.0753, found 290.0742.
(2S, 3S)-Benzoic acid 2-azido-3-tert-butoxy-4,4,4-trifluoro-butyl ester, 8a
To a solution of compound 7a (23.0 g, 80 mmol) in anhydrous CH2Cl2 (300 mL) was added liquid isobutylene (100 mL) and H2SO4 (1.0 mL, 96%) at -30 °C. The resulting mixture was stirred for 4 days at room temperature in a sealed vessel. After releasing the pressure slowly, saturated Na2CO3 aqueous solution was added and the resulting mixture was stirred for an additional 10 min. The organic phase was collected and the aqueous phase was extracted with CH2Cl2. The combined organic phase was dried over anhydrous Na2SO4 and concentrated under vacuo. The residue was purified by column chromatography on silica gel (hexanes : ethyl acetate = 20 : 1) to give 8a as a yellow oil (19.0 g, 70%) and recovered 7a (6.0 g). [α]20D = -1.8 (c 1.44, CHCl3); 1H NMR (CDCl3, 400 MHz): δ 8.07-7.45 (m, 5H), 4.70 (dd, J = 12.0, 3.2 Hz, 1H), 4.40 (dd, J = 12.0, 8.8 Hz, 1H), 4.10 (m, 2H), 1.20 (s, 9H); 19F NMR (CDCl3, 367.4 MHz): δ -76.49 (d, J = 7.3Hz); 13C NMR (CDCl3, 100.4 MHz): δ 166.3, 133.6, 130.0, 129.6, 128.8, 124.1 (q, J = 289.2 Hz), 78.0, 71.7 (q, J = 30.8 Hz), 64.3, 60.9, 28.5; MS (CI): m/z 346 ([M + 1]+, 100); HRMS (CI): Calcd for C15H19F3N3O3 346.1379, found 346.1393.
(2S, 3S)-2-Azido-3-tert-butoxy-4,4,4-trifluoro-butan-1-ol, 9a
Compound 8a (14.0 g, 41 mmol) was dissolved in anhydrous dichloromethane (200 mL) and the solution was then cooled to -70 °C. Diisobutylaluminum hydride (110 mL 1M solution in hexanes) was added drop wise and the resulting mixture was stirred at -40 °C for 30 min. Then 100 mL ethyl acetate was added. After stirring for another 30 min at room temperature, 1 N HCl aqueous solution was added. The organic phase was collected and the aqueous phase was extracted with diethyl ether. The combined organic phase was washed with brine and dried over anhydrous Na2SO4. After concentrated under vacuo, the residue was purified by column chromatography on silica gel (hexanes : CH2Cl2 = 1 : 1) to give 9a as a colorless oil (9.0 g, 93%). [α]20D = +25.3 (c 1.46, CHCl3); 1H NMR (CDCl3, 400 MHz): δ 4.02 (m, 1H), 3.81-3.68 (m, 3H), 2.54 (b, 1H), 1.21 (s, 9H); 19F NMR (CDCl3, 367.4 MHz): δ -76.08 (d, J = 6.3 Hz); 13C NMR (CDCl3, 100.4 MHz): δ 124.3 (q, J = 283.8 Hz), 78.1, 71.8 (q, J = 29.5 Hz), 63.2, 61.9, 28.3; MS (CI): m/z 242 ([M + 1]+, 100); HRMS (CI): Calcd for C8H15F3N3O2 242.1117, found 242.1114.
(2S, 3S)-2-Amino-3-tert-butoxy-4,4,4-trifluoro-butan-1-ol, 10a
Compound 9a (8.0 g, 33 mmol) was dissolved in methanol (200 mL) and 10% Pd/C powder (1.0 g) was added. This mixture was stirred overnight under a H2 atmosphere at room temperature. After filtration and condensation under vacuo, the residue was purified by column chromatography on silica gel (ethyl acetate : methanol = 4 : 1) to give 10a as a colorless oil (6.4 g, 89%). [α]20D = -4.5 (c 1.23, CHCl3); 1H NMR (CDCl3, 400 MHz): δ 3.90 (m, 1H), 3.58 (m, 2H), 3.02 (m, 1H), 2.19 (b, 3H), 1.19 (s, 9H); 19F NMR (CDCl3, 367.4 MHz): δ -73.92 (d, J = 7.7 Hz); 13C NMR (CDCl3, 100.4 MHz): δ 124.9 (q, J = 283.8 Hz), 77.3, 72.7 (q, J = 25.4 Hz), 63.0, 53.9, 28.4; MS (CI): m/z 216 ([M + 1]+, 100); HRMS (CI): Calcd for C8H17F3NO2 216.1212, found 216.1206.
(2S, 3S)-(2-tert-Butoxy-3,3,3-trifluoro-1-hydroxymethyl-propyl)-carbamic acid 9H- fluoren-9-ylmethyl ester, 11a
Compound 10a (6.0 g, 28 mmol) was dissolved in THF (200 mL) and H2O (200 ml). FmocCl (11.0 g, 42 mmol) and NaHCO3 (7.0 g, 84 mmol) were added at 0 °C and the mixture were stirred for 4 h at room temperature. Then the reaction mixture was extracted with ethyl acetate and the combined organic phase was dried over anhydrous MgSO4. After concentration under vacuo, the residue was purified by column chromatography on silica gel (hexanes : ethyl acetate = 4 : 1) to give 11a as a white solid (12.0 g, 98%). [α]20D = -11.5 (c 1.05, CHCl3); mp. 121 °C; 1H NMR (CDCl3, 400 MHz): δ 7.77-7.29 (m, 8H), 5.71 (d, J = 6.8 Hz, 1H), 4.52-4.41 (m, 2H), 4.32 (m, 1H), 4.20(m, 1H), 4.12 (m, 1H), 3.87 (m, 1H), 3.64 (m, 1H), 2.72 (b, 1H), 1.19 (s, 9H); 19F NMR (CDCl3, 367.4 MHz): δ -75.17 (d, J = 6.6 Hz); 13C NMR (CDCl3, 100.4 MHz): δ 156.1, 144.0, 143.9, 141.6, 128.0, 127.3, 125.2, 125.1, 124.3 (q, J = 284.8 Hz), 120.3, 78.1, 73.0 (q, J = 28.1 Hz), 67.1, 62.2, 50.8, 47.4, 28.5; MS (CI): m/z 438 ([M + 1]+, 78); HRMS (CI): Calcd for C23H27F3NO4 438.1893, found 438.1910.
(2R, 3S)-3-tert-Butoxy-2-(9H-fluoren-9-ylmethoxycarbonylamino)-4,4,4-trifluoro-butyric acid, Fmoc-1a(tBu)-OH
Compound 11a (5.0 g 11.5 mmol) was dissolved in acetone (80 mL) and Jones reagent (11.9 mL, 6.2 N aqueous solution, 71.0 mmol) was added drop wise at 0 °C. The brown solution was stirred at 0 °C for 2 h. Then iso-propanol (60 mL) was added slowly and the mixture was stirred for an additional 10 min. After removing the solvent, the residue was dissolved in water (250 mL) and extracted with ethyl acetate. The combined organic phase was dried over anhydrous MgSO4 and condensed under vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate : methanol = 9 : 1) to give Fmoc-1a(tBu)-OH as a white solid (4.7 g, 91%). Fmoc-1a(tBu)-OH was recrystallized from hexanes and CH2Cl2 to achieve higher purity: [α]20D = +7.4, (c 0.79, CHCl3); mp. 161 °C; 1H NMR (CD3OD, 400 MHz): δ 7.77-7.28 (m, 8H), 4.66 (m, 2H), 4.40 (m, 1H), 4.21 (m, 2H), 3.34 (s, 1H), 1.26 (s, 9H); 19F NMR (CD3OD, 367.4 MHz): δ -73.08 (d, J = 5.6 Hz); 13C (CD3OD, 100.4 MHz): δ 173.7, 157.4, 144.1, 144.0, 141.3, 127.6, 127.0, 125.3, 125.1, 125.0 (q, J = 283.2 Hz), 119.7, 76.9, 70.9 (q, J = 28.4 Hz), 67.1, 57.6, 29.7, 27.0; MS (CI): m/z 452 ([M + 1]+, 25); HRMS (CI): Calcd for C23H25F3NO5 452.1686, found 452.1676.
(2R, 3S)-Benzoic acid 4,4,4-trifluoro-2,3-bis-((R)-3,3,3-trifluoro-2-methoxy-2-phenyl- propionyloxy)-butyl ester, 12a
Compound 5a (27 mg, 0.1 mmol), DCC (N, N- dicyclohexyl-carbodiimde) (63 mg 0.3 mmol) and DMAP (4 mg) were dissolved in CH2Cl2 (3 mL). (R)-(+)-α-methoxy-α-(trifluoromethyl)phenylacetic acid ((+)-MTPA) (72 mg, 0.3 mmol) was added to the solution. The resulting mixture was stirred at room temperature for 16 h (An 19F NMR spectrum was then recorded with 0.5 mL of the reaction mixture and the d.e. value was found to be over 99%, based on the 19F signal of the -CF3 group at the C4 position of 12a (See Supplementary Data, S18-S20). The reaction mixture was concentrated in vacuo. The residue was purified by flash chromatography on silica gel (hexanes : ethyl acetate = 5 : 1) to give 12a as a colorless oil (70 mg, over 99%). [α]20D = +35.7 (c 0.91, CHCl3); 1H NMR (CDCl3, 400 MHz): δ 7.98-7.26 (m, 15H), 5.99 (m, 1H), 5.87 (m, 1H), 4.37 (dd, J = 12.0, 7.6 Hz, 1H), 4.27 (dd, J = 12.0, 6.0 Hz, 1H), 3.46 (s, 3H), 3.44 (s, 3H); 19F NMR (CDCl3, 376.4 MHz): δ −74.51 (s, 3F), -74.67 (s, 3F), -76.25 (d, J = 6.4 Hz, 3F); 13C NMR (CDCl3, 100.4 MHz): δ 165.5, 165.3, 139.3, 136.7, 133.7, 131.0, 130.4, 130.2, 129.8, 128.8, 128.6, 128.5, 127.4, 127.3, 123.0 (q, J = 288.7 Hz), 122.8 (q, J = 287.9 Hz), 120.0 (q, J = 281.7 Hz), 85.1 (q, J = 29.1 Hz), 68.5 (q, J = 32.9 Hz), 68.1, 61.2, 55.5, 55.4, 29.7; MS (CI): m/z: 697 ([M + 1]+, 10); HRMS (CI): Calcd for C31H26F9O8 697.1485, found 697.1467.
The reaction of compound 9a with (1R)-(-)-camphor-10-sulfonyl chloride to give 13a and 14a
Compound 10a (701 mg, 3.3 mmol) and DMAP (400 mg, 3.3 mmol) were dissolved in anhydrous CH2Cl2 (20 mL). (1R)-(-)-camphor-10-sulfonyl chloride (840 mg, 3.3 mmol) was then added at 0 °C. The resulting solution was stirred overnight at room temperature and washed with 2 N HCl aqueous solution. The organic phase was collected and the aqueous phase was extracted with ether. The combined organic phase were washed with brine and dried over anhydrous MgSO4. After concentration under vacuo, the residue was purified by column chromatography on silica gel (hexanes : ethyl acetate = 5 : 1) to give 13a as a white solid (671 mg, 48%), 14a as a white solid (314 mg, 15%) and some recovered compound 10a (161 mg).
13a
[α]20D = -17.5 (c 1.03, CHCl3); mp. 168 °C; 1H NMR (CDCl3, 400 MHz): δ 5.93 (d, J = 6.8 Hz,1H), 4.40 (m, 1H), 4.01 (m, 1H), 3.80 (m, 2H), 3.60 (AB, J = 15.2 Hz, 1H), 2.99 (AB, J = 15.2 Hz, 1H), 2.52 (dd, J = 8.8, 3.2 Hz, 1H), 2.42 (m, 1H), 2.19 (m, 2H), 2.05 (m, 2H), 1.56 (s, 1H), 1.47 (m, 1H), 1.31 (s, 9H), 1.02 (s, 3H), 0.90 (s, 3H); 19F NMR (CDCl3, 367.4 MHz): δ -75.47 (d, J = 7.7 Hz); 13C (CDCl3, 100.4 MHz): δ 217.4, 124.2 (q, J = 284.0 Hz), 77.9, 74.5 (q, J = 29.1 Hz), 61.6, 59.4, 55.1, 51.0, 48.9, 43.0, 42.7, 28.5, 27.1, 26.6, 19.9, 19.4; MS (CI) m/z: 430 ([M + 1]+, 75); HRMS (CI) Calcd for C18H31F3NO5S 430.1876, found 430.1870.
14a
[α]20D = -23.3 (c 0.95, CHCl3); mp. 144 °C; 1H NMR (CDCl3, 400 MHz): δ 6.08 (d, J = 6.4 Hz, 1H), 4.55 (dd, J = 10.8, 7.2 Hz, 1H), 4.35 (m, 2H), 4.12 (m, 1H), 3.57 (dd, J = 14.8, 3.6 Hz, 2H), 3.02 (dd, J = 18.4, 14.8 Hz, 2H), 2.41 (m, 3H), 2.13 (m, 3H), 2.03 (m, 5H), 1.68 (m, 1H), 1.46 (m, 2H), 1.32 (s, 9H), 1.08 (s, 3H), 1.02 (s, 3H), 0.94 (s, 3H), 0.88 (s, 3H); 19F NMR (CDCl3, 367.4 MHz): δ −75.65 (d, J = 7.7 Hz); 13C (CDCl3, 100.4 MHz): δ 216.7, 215.2, 124.0 (q, J = 284.8 Hz), 77.7, 73.3 (q, J = 29.1 Hz), 68.3, 59.5, 58.0, 54.3, 51.9, 48.9, 48.4, 46.9, 43.0, 42.9, 42.7, 42.6, 29.7, 28.5, 27.0, 26.9, 24.8, 20.0, 19.7, 19.6, 19.5; MS (CI) m/z: 644 ([M + 1]+, 10); HRMS (CI) Calcd for C28H45F3NO8S2 644.2539, found 644.2521.
Solution-phase synthesis of Fmoc-1a(tBu)-Phe-OMe
In a 20 mL dimethylformamide (DMF) solution of Fmoc-1a(tBu)-OH (500 mg, 1.1 mmol) and L-phenylalanine methyl ester hydrochloride (H-Phe-OMe·HCl, 400 mg, 1.8 mmol), TBTU (320 mg, 1.0 mmol), HOBt (149 mg, 1.1 mmol) and DIPEA (0.4 mL, 2.2 mmol) were added. The reaction solution was stirred at room temperature for 30 min. Then 50 mL ethyl acetate was added to the solution. The solution was then washed sequentially with aqueous 0.05 M KHSO4 solution, saturated aqueous NaHCO3 solution, and brine (50 mL each). The organic phase was then dried with Na2SO4 and concentrated under vacuum. The crude residue was purified by flash column chromatography (silica gel, hexaness : EtOAc, 90 : 10 → 70 : 30) to give Fmoc-1a(tBu)-Phe-OMe (620 mg, 1.0 mmol, 92% yield, 99.8% purity by NPLC, 99% d.e. by 19F NMR) as a white solid. 1H-NMR (CDCl3) δ ppm: 7.79-6.72 (m, 13H), 6.73 (d, J = 7.2 Hz, 1H), 5.36 (m, 1H), 4.85 (m, 1H), 4.66 (m, 2H), 4.55 (m, 1H), 4.25 (m, 2H), 3.72 (s, 3H), 3.11 (d, J = 5.6 Hz, 2H), 1.23 (s, 9H); 19F (CDCl3, C6F6) δ ppm: -74.75 (d, J = 6.8 Hz); 13C (CDCl3) δ ppm: 171.23, 166.84, 156.68, 143.60, 143.48, 141.43, 141.28, 135.30, 129.27, 128.62, 127.84, 127.29, 127.17, 125.17, 125.00, 124.58 (q, J = 285 Hz), 120.07, 77.73, 69.71 (q, J = 29 Hz), 67.70, 57.36, 53.06, 52.41, 46.95, 37.82, 27.82. Calculated MW = 612.2447 for C33H35F3N2O6. CI-HRMS: m/z [M + 1]+ 613.25012.
General procedure for solid-phase synthesis of oligopeptides
Solid-phase synthesis of oligopeptides was performed using rink amide MBHA resin, which gives peptide amides after cleavage. Non-fluorinated amino acids (3.0 eq.) were pre-activated with DIC (2.7 eq.) and HOBt (3.0 eq.) in DMF/DCM (1:1) and reacted for 2 h with 1.0 eq. of resin in a solid-phase extraction (SPE) column. Fmoc deprotection was carried out by using 20% (v/v) piperidine in N-methylpyrrolidinone (NMP) (40 min × 3). The extent of reaction was verified by a Kaiser test after each coupling and Fmoc-deprotection. Final deprotection and cleavage were accomplished by treatment with a cleavage cocktail [TFA : H2O : dichloromethane (DCM) : triisopropylsilane (TIS) : 1, 2-ethanedithiol (EDT) = 90 : 2.5 : 2.5 : 2.5 : 2.5] for 2 hr. Afterwards, the solution was drained from SPE column and solvent was removed through rotary evaporation below 40 °C. Water and ethyl acetate were added into the residue. The collected aqueous phase was lyophilized to obtain the crude peptide.
The incorporation of tfT involves the following modification of the above procedure. Fmoc-tfT(tBu)-OH (3.0 eq.) was first mixed with the resin (1.0 eq.) in DMF/DCM (1:1) and cooled to 0 °C (ice-bath) before the coupling reagents (2.7 eq. DIC/3.0 eq. HOBt) were added, and then the reaction mixture was shaken for 20 hours in an icebox. If tfT was the first residue to be linked to the resin, then 1 eq. of CuCl2 was added to the coupling reagent, based on the method developed by Kuwata33 and Hallberg34.
Peptide Purification and Analysis by HPLC
Mono-, di- and tripeptide purification was carried out using SCXLC (Zorbax 300-SCX, 9.4 × 250 mm, 5 μm, Agilent; 100% A in the first 20 min, then 0-100% B over 25 min, A = 0.1% 12.1 N hydrochloride acid in water, B = 0.1% 12.1 N hydrochloride acid in the mixture of water and CH3CN (9:1) with 0.5 M NH4Cl; flow rate: 4.0 mL/min; detection wavelength: 205 nm. r.t.).
To verify peptide purity, Fmoc-1a(tBu)-Phe-OCH3 and Fmoc-1b(tBu)-Phe-OCH3 were analyzed by normal-phase liquid chromatography (NPLC) (TSKgel Amide-80, 4.6 mm I.D. × 250 mm, 5 μm, TOSOH Bioscience; 10-100% B over 45 min, A = hexanes, B = ethyl acetate; 1.5 mL/min, 270 nm, r.t.). Other mono-, di- and tri-peptides were analyzed by SCXLC (ZORBAX 300-SCX, 4.6 mm × 250 mm, 5 μm, Agilent; 100% A in the first 5 min, then 0-100% B over 25 min, A = 0.1% 12.1 N hydrochloride acid in water, B = 0.1% 12.1 N hydrochloride acid in the mixture of water and CH3CN (9:1) with 0.5 M NH4Cl; 1.0 mL/min, 220 nm, r.t.).
Chiral analysis of tripeptides was conducted using a Ultron ES-Pepsin column (4.6 mm × 150 mm, 5 μm, Agilent; 20 mM phosphate in water, pH = 6.0; isocratic elution, 1.0 mL/min, 205 nm, 15 °C).
The oligopeptides should be stored as dried powder in an oxygen-free environment at -80 °C to prevent slow decomposition. Prolonged storage as dried powder at -20 °C led to peptide degradation, as shown in FIGURE 6A for the case of tripeptides.
H-1a-NH2
1H-NMR (PBS) δ ppm: 3.03 (m, 2H), 1.64 (m, 2H), 1.53 (m, 1H); 19F (PBS) δ ppm: -73.09 (d, J = 6.8 Hz); 13C (PBS) δ ppm: 167.65, 128.50 (q, J = 351.2 Hz), 67.72 (q, J = 32 Hz), 52.65. Calculated MW = 172.046 for C4H7F3N2O2. MALDI-HRMS: m/z [M + 1]+ 173.0530; 93.6 % purity.
H-1b-NH2
1H-NMR (PBS) δ ppm: 3.04 (m, 2H), 1.65 (m, 2H), 1.54 (m, 1H); 19F (PBS) δ ppm: -73.07 (d, J = 6.6 Hz); 13C (PBS) δ ppm: 167.65, 128.71 (q, J = 351.4 Hz), 67.65 (q, J = 32 Hz), 52.57. Calculated MW = 172.046 for C4H7F3N2O2. MALDI-HRMS: m/z [M + 1]+ 173.0522; 94.9% purity.
H-1a-Gly-NH2
1H-NMR (PBS) δ ppm: 8.81 (m, 1H), 4.52 (m, 1H), 4.27 (d, J = 6.0 Hz, 1H), 3.86 (dAB, J1 = 17.2 Hz, J2 = 5.2 Hz, 2H); 19F (PBS) δ ppm: -73.44 (d, J = 7.5 Hz); 13C (PBS) δ ppm: 173.60, 166.57, 123.72 (q, J = 283 Hz), 67.98 (q, J = 31 Hz), 52.92, 42.53. Calculated MW = 229.0674 for C6H10F3N3O3. CI-HRMS: m/z [M + 1]+ 230.07421; 99.1% purity.
H-1b-Gly-NH2
1H-NMR (PBS) δ ppm: 8.75 (m, 1H), 4.53 (m, 1H), 4.27 (d, J = 6.0 Hz, 1H), 3.79 (dAB, J1 = 17.6 Hz, J2 = 5.6 Hz, 1H); 19F (PBS) δ ppm: -73.48 (d, J = 6.8 Hz); 13C (PBS) δ ppm: 173.58, 166.47, 123.69 (q, J = 282 Hz), 67.94 (q, J = 32 Hz), 52.90, 42.56. Calculated MW = 229.0674 for C6H10F3N3O3. FAB-HRMS: m/z [M + 1]+ 230.07565; 95.8% purity.
H-Cys-1a-NH2
1H-NMR (PBS) δ ppm: 8.99 (m, 1H), 7.77 (m, 1H), 7.20 (m, 1H), 4.39 (m, 1H), 4.16 (m, 1H), 2.95 (m, 2H); 19F (PBS) δ ppm: -73.11 (d, J = 7.9 Hz); 13C (PBS) δ ppm: 171.86, 168.18, 124.35 (q, J = 283 Hz), 68.38 (q, J = 31 Hz), 54.69, 53.10, 25.29. Calculated MW = 275.0551 for C7H12F3N3O3S. FAB-HRMS: m/z [M + 1]+ 276.06524; 99.6% purity; > 99% d.e. (determined by 19F NMR).
H-Cys-1b-NH2
1H-NMR (PBS) δ ppm: 8.98 (m, 1H), 7.76 (m, 1H), 7.21 (m, 1H), 4.35 (m, 1H), 4.16 (m, 1H), 2.97 (m, 2H); 19F (PBS) δ ppm: -73.46 (d, J = 6.8 Hz); 13C (PBS) δ ppm: 171.81, 168.13, 124.29 (q, J = 283 Hz), 68.37 (q, J = 31 Hz), 54.52, 53.10, 25.30. Calculated MW = 275.0551 for C7H12F3N3O3S. FAB-HRMS: m/z [M + 1]+ 276.06353; 99.9% purity; > 99% d.e. (determined by 19F NMR).
H-1a-Cys-1a-NH2
1H-NMR (PBS) δ ppm: 8.95 (d, J = 6.4 Hz, 1H), 8.75 (d, J = 8.4 Hz, 1H), 7.72 (s, 1H), 7.19 (s, 1H), 4.53 (m, 1H), 4.39 (m, 1H), 4.32 (m, 1H), 2.81 (m, 2H); 19F (PBS) δ ppm: -73.16 (m); 13C (PBS) δ ppm: 172.12, 171.32, 165.99, 124.36 (q, J = 283 Hz), 123.75 (q, J = 283 Hz), 68.38 (q, J = 31 Hz), 67.91 (q, J = 32 Hz), 56.11, 53.15, 52.84, 25.44. Calculated MW = 430.0746 for C11H16F6N4O5S. CI-HRMS: m/z [M + 1]+ 431.08157; 99.0% purity; 97.4% d.e. (determined by chiral HPLC).
H-1b-Cys-1b-NH2
1H-NMR (PBS) δ ppm: 8.87 (d, J = 8.0 Hz, 1H), 8.63 (d, J = 8.8 Hz, 1H), 7.69 (s, 1H), 7.16 (s, 1H), 4.52 (m, 1H), 4.32 (m, 1H), 4.28 (m, 1H), 2.77 (m, 2H); 19F (PBS) δ ppm: -73.33 (t, J = 6.4 Hz), -73.50 (t, J = 6.8 Hz); 13C (PBS) δ ppm: 171.98, 170.74, 165.62, 124.31 (q, J = 282 Hz), 123.69 (q, J = 283 Hz), 68.42 (q, J = 31 Hz), 67.95 (q, J = 32 Hz), 55.82, 52.93, 52.86, 25.69. Calculated MW = 430.0746 for C11H16F6N4O5S. MALDI-HRMS: m/z [M + 1]+ 431.0789; 99.6% purity; 98.3% d.e. (determined by chiral HPLC).
H-1b-Cys-1a-NH2
1H-NMR (PBS) δ ppm: 8.61 (d, J = 8.8 Hz, 1H), 7.69 (s, 1H), 7.16 (s, 1H), 4.97 (m, 1H), 4.47 (m, 1H), 4.34 (m, 1H), 4.18 (m, 1H), 2.81 (m, 2H); 19F (PBS) δ ppm: -73.08 (t, J = 7.9 Hz), -73.19 (t, J = 6.8 Hz); 13C (PBS) δ ppm: 171.97, 170.86, 167.34, 166.32, 124.36 (q, J = 282 Hz), 123.78 (q, J = 283 Hz), 68.43 (q, J = 31 Hz), 68.13 (q, J = 31 Hz), 55.85, 52.98, 25.45. Calculated MW = 430.0746 for C11H16F6N4O5S. CI-HRMS: m/z [M + 1]+ 431.08285; 99.9% purity; 99.9% d.e. (determined by chiral HPLC).
H-1a-Cys-1b-NH2
1H-NMR (PBS) δ ppm: 8.95 (d, J = 6.8 Hz, 1H), 8.71 (d, J = 8.4 Hz, 1H), 7.73 (s, 1H), 7.19 (s, 1H), 5.17 (b, 1H), 4.59 (m, 1H), 4.52 (m, 1H), 4.36 (m, 1H), 4.32 (m, 1H), 2.82 (m, 2H); 19F (PBS) δ ppm: -73.14 (t, J = 6.8 Hz), -73.35 (t, J = 6.8 Hz); 13C (PBS) δ ppm: 172.05, 171.27, 165.96, 124.34 (q, J = 283 Hz), 123.75 (q, J = 283 Hz), 68.34 (q, J = 31 Hz), 67.85 (q, J = 32 Hz), 56.15, 53.01, 52.89, 25.46. Calculated MW = 430.0746 for C11H16F6N4O5S. MALDI-HRMS: m/z [M + 1]+ 431.0792; 99.7% purity; 94.0% d.e. (determined by chiral HPLC).
Supplementary Material
Table 2.
19F NMR chemical shifts of tfT in oligopeptides from SPPS in PBS
| Peptide Sequence | 19F chemical shifti |
|---|---|
| H-1a-NH2 | -73.09 |
| H-1b-NH2 | -73.10 |
| H-1a-Gly-NH2 | -73.44 |
| H-1b-Gly-NH2 | -73.48 |
| H-Cys-1a-NH2 | -73.11 |
| H-Cys-1b-NH2 | -73.35 |
| H-1a-Cys-1a-NH2 | -73.06, -73.08 |
| H-1b-Cys-1b-NH2 | -73.36, -73.51 |
| H-1b-Cys-1a-NH2 | -73.08, -73.19 |
| H-1a-Cys-1b-NH2 | -73.14, -73.35 |
In tripeptides that contain two tfT residues, no assignment was made to the two 19F signals.
Acknowledgments
This work was supported by grants from the NIH (EB002880 and EB004416) and the Sidney Kimmel Foundation for Cancer Research. YBY is a Kimmel scholar. X-ray crystallographic data were collected and analyzed by Dr. Atta M. Arif of the Chemistry Department at the University of Utah.
List of symbols and abbreviations
- [α]20D
optical rotary power
- AD
asymmetric dihydroxylation
- Bn
benzyl
- Bz
benzoyl
- d.e.
diastereomeric excess
- DCM
dichloromethane
- DIC
N,N′-diisopropylcarbodiimide
- DIPEA
N,N-diisopropylethylamine
- DMAP
4-dimethylaminopyridine
- DMF
dimethylformamide
- DMSO-d6
dimethyl sulfoxide-d6
- e.e.
enantiomeric excess
- EDT
1, 2-ethanedithiol
- FmocCl
9-fluorenylmethyl chloroformate
- HOBt
N-hydroxybenzotriazole
- HPLC
high-performance liquid chromatography
- HRMS
high-resolution mass spectrometry
- I.D.
inner diameter
- MBHA
methylbenzhydrylamine
- mp
melting point
- MTPA
α-methoxy-α-trifluoromethylphenylacetic acid
- NMP
N-methylpyrrolidinone
- NMR
nuclear magnetic resonance
- NP
normal-phase
- NPLC
normal-phase liquid chromatography
- PBS
physiological buffer system (50 mM sodium phosphate, 100 mM NaCl, pH 7.0 in 92%H2O/8 %D2O, v/v)
- RP
reversed-phase
- RPLC
reversed-phase liquid chromatography
- SCX
strong-cation exchange
- SCXLC
strong-cation exchange liquid chromatography
- SPE
solid-phase extraction
- SPPS
solid-phase peptide synthesis
- TBTU
2-(1H-benzotriazole-l-yl)-1,1,3,3-tetra-methyluronium tetrafluoroborate
- tBu
tert-butyl
- TFA
trifluoroacetic acid
- tfT
4,4,4-trifluorothreonine
- THF
tetrahydrofuran
- TIS
triisopropylsilane
- TMS
tetramethylsilane
Footnotes
Supplementary Data Available: Details of experimental procedures for the syntheses of 4, 5b-14b, Fmoc-1b(tBu)-OH; Optical rotary powers of enantiomeric pairs; copies of 1H, 19F, 13C NMR spectra and RPLC and chiral chromatograms for Fmoc-1a(tBu)-OH and Fmoc-1b(tBu)-OH; molecular structures of 13a and 13b; copies of 19F NMR spectra for 12a, 12b and mixture of H-1a-NH2 and H-1b-NH2; SCXLC chromatograms of H-1a-NH2, H-1b-NH2, H-1a-Gly-NH2, H-1b-Gly-NH2, H-Cys-1a-NH2, H-Cys-1b-NH2; SCXLC and chiral HPLC chromatograms of H-1a-Cys-1a-NH2, H-1b-Cys-1b-NH2, H-1a-Cys-1b-NH2 and H-1b-Cys-1a-NH2.
References
- 1.(a) Bilgiçer B, Fichera A, Kumar K. J Am Chem Soc. 2001;123:4393–4399. doi: 10.1021/ja002961j. [DOI] [PubMed] [Google Scholar]; (b) Tang Y, Ghirlanda G, Vaidehi N, Kua J, Mainz DT, Goddard WA, III, DeGrado WF, Tirell DA. Biochemistry. 2001;40:2790–2796. doi: 10.1021/bi0022588. [DOI] [PubMed] [Google Scholar]; (c) Horng JC, Raleigh DP. J Am Chem Soc. 2003;125:9286–9287. doi: 10.1021/ja0353199. [DOI] [PubMed] [Google Scholar]; (d) Lee KH, lee HY, Slutsky MM, Anderson JT, Marsh ENG. Biochemistry. 2004;43:16277–16284. doi: 10.1021/bi049086p. [DOI] [PubMed] [Google Scholar]; (e) Lee HY, Lee KH, Al-hashimi HM, Marsh ENG. J Am Chem Soc. 2006;128:337–343. doi: 10.1021/ja0563410. [DOI] [PubMed] [Google Scholar]
- 2.Chiu HP, Suzuki Y, Gullickson D, Ahmad R, Kokona B, Fairman R, Cheng RP. J Am Chem Soc. 2006;128:15556–15557. doi: 10.1021/ja0640445. [DOI] [PubMed] [Google Scholar]
- 3.(a) Bilgiçer B, Xing X, Kumar K. J Am Chem Soc. 2001;123:11815–11816. doi: 10.1021/ja016767o. [DOI] [PubMed] [Google Scholar]; (b) Bilgiçer B, Kumar K. Tetrahedron. 2002;58:4105–4112. [Google Scholar]
- 4.(a) Bilgiçer B, Kumar K. Proc Natl Acd Sci USA. 2004;101:15324–15329. doi: 10.1073/pnas.0403314101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Naarmann N, Bilgiçer B, Meng H, Kumar K, Steinem C. Angew Chem Int Ed. 2006;45:2588–2591. doi: 10.1002/anie.200503567. [DOI] [PubMed] [Google Scholar]
- 5.Niemz A, Tirrell DA. J Am Chem Soc. 2001;123:7407–7413. doi: 10.1021/ja004351p. [DOI] [PubMed] [Google Scholar]
- 6.Hsieh KH, Needleman P, Marshall GR. J Med Chem. 1987;30:1097–1100. doi: 10.1021/jm00389a021. [DOI] [PubMed] [Google Scholar]
- 7.Koksch B, Dahl C, Radic G, Vocks A, Arnold K, Arnhold J, Sieler J, Burger K. J Pept Sci. 2004;10:67–81. doi: 10.1002/psc.515. [DOI] [PubMed] [Google Scholar]
- 8.(a) Entress RMH, Dancer RJ, O'Brien DP, Try AC, Cooper MA, Williams DH. Chem Biol. 1998;5:329. doi: 10.1016/s1074-5521(98)90171-5. [DOI] [PubMed] [Google Scholar]; (b) Ulrich AS. In: Encyclopedia of Spektroskopy and Spectrometry. Lindon J, Tranter G, Holmes J, editors. Academic Press; New York: 2000. p. 813. [Google Scholar]
- 9.Jäckel C, Koksch B. Eur J Org Chem. 2005:4483–4503. [Google Scholar]
- 10.Jiang ZX, Yu YB. J Org Chem. 2007;72:1464–1467. doi: 10.1021/jo0616308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koksch B, Sewald N, Hofmann HJ, Burger K, Jakubke HD. J Peptide Sci. 1997;3:157–167. doi: 10.1002/(SICI)1099-1387(199705)3:3%3C157::AID-PSC94%3E3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 12.(a) Bordusa F, Dahl C, Jakubke HD, Burger K, Koksch B. Tetrahedron: Asymmetry. 1999;10:307–313. [Google Scholar]; (b) Thust S, Koksch B. J Org Chem. 2003;68:2290–2296. doi: 10.1021/jo020613p. [DOI] [PubMed] [Google Scholar]
- 13.(a) Yoshikawa E, Fournier MJ, Mason TL, Tirrell DA. Macromolecules. 1994;27:5471–5475. [Google Scholar]; (b) Tang Y, Tirrell DA. J Am Chem Soc. 2001;123:11089–11090. doi: 10.1021/ja016652k. [DOI] [PubMed] [Google Scholar]; (c) Tang Y, Ghirlanda G, Petka WA, Nakajima T, DeGrado WF, Tirrell DA. Angew Chem Int Ed. 2001;40:1494–1496. [PubMed] [Google Scholar]; (d) Furter R. Protein Sci. 1998;7:419–426. doi: 10.1002/pro.5560070223. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Wang P, Tang Y, Tirrell DA. J Am Chem Soc. 2003;125:6900–6906. doi: 10.1021/ja0298287. [DOI] [PubMed] [Google Scholar]; (f) Duewel HS, Daub E, Robinson V, Honek JF. Biochemistry. 2001;40:13167–13176. doi: 10.1021/bi011381b. [DOI] [PubMed] [Google Scholar]; (g) Drieden C, Hoeltzli SD, Bann JG. Methods Enzymol. 2004;380:400–415. doi: 10.1016/S0076-6879(04)80018-1. [DOI] [PubMed] [Google Scholar]
- 14.(a) Holmgren SK, Taylor KM, Bretscher LE, Raines RT. Nature. 1998;392:666. doi: 10.1038/33573. [DOI] [PubMed] [Google Scholar]; (b) Hodges JA, Raines RT. J Am Chem Soc. 2003;125:9262–9263. doi: 10.1021/ja035881z. [DOI] [PubMed] [Google Scholar]; (c) Doi M, Nishi Y, Uchiyama S, Nishiuchi Y, Nakazawa T, Ohkubo T, Kobayashi Y. J Am Chem Soc. 2003;125:9922–9923. doi: 10.1021/ja035997v. [DOI] [PubMed] [Google Scholar]; (d) Barth D, Milbradt AG, Renner C, Moroder L. ChemBioChem. 2004;5:79–86. doi: 10.1002/cbic.200300702. [DOI] [PubMed] [Google Scholar]
- 15.Mikhailiuk PK, Afonin S, Chernega AN, Rusanov EB, Platonov MO, Dubinina GG, Berditsch M, Ulrich AS, Komarov IV. Angew Chem Int Ed. 2006;45:5659–5661. doi: 10.1002/anie.200600346. [DOI] [PubMed] [Google Scholar]
- 16.(a) Sutherland A, Willis CL. Nat Prod Rep. 2000;17:621–631. doi: 10.1039/a707503k. [DOI] [PubMed] [Google Scholar]; (b) Sergeeva NN, Golubev AS, Hennig L, Burger K. Synthesis. 2002:2579–2584. [Google Scholar]; (c) Qiu XL, Meng WD, Qing FL. Tetrahedron. 2004;60:6711–6745. [Google Scholar]
- 17.Bilgicer B, Fichera A, Kumar K. J Am Chem Soc. 2001;123:4393–4399. doi: 10.1021/ja002961j. [DOI] [PubMed] [Google Scholar]
- 18.Afonin S, Glaser RW, Berditchevskaia M, Wadhwani P, Gührs KH, Möllmann U, Perner A, Ulrich AS. ChemBioChem. 2003;41:1151–1163. doi: 10.1002/cbic.200300568. [DOI] [PubMed] [Google Scholar]
- 19.Marbach P, Briner U, Schweitzer A, Terasaki T. Metabolism. 1992;41(suppl 2):7–10. doi: 10.1016/0026-0495(92)90024-5. [DOI] [PubMed] [Google Scholar]
- 20.(a) Dasgupta P, Singh AT, Mukherjee R. Pharm Res. 1999;16:1047–1053. doi: 10.1023/a:1018935800052. [DOI] [PubMed] [Google Scholar]; (b) Morpurgo M, Monfardini C, Hofland LJ, Sergi M, Orsolini P, Dumont JM, Veronese FM. Bioconj Chem. 2002;13:1238–1243. doi: 10.1021/bc0100511. [DOI] [PubMed] [Google Scholar]; (c) Na DH, lee KC, DeLuca PP. Pharm Res. 2005;22:743–749. doi: 10.1007/s11095-005-2590-y. [DOI] [PubMed] [Google Scholar]; (d) Wulbrand U, Feldman M, Pfestroff A, Fheman HC, Du J, Hiltunen J, marquez M, Arnold R, Westlin JE, Nilsson S, Holmberg AR. Cancer. 2002;94:1293–1297. doi: 10.1002/cncr.10299. [DOI] [PubMed] [Google Scholar]
- 21.(a) 26 Lancranjan I, Bruns C, Grass P, Jaquet P, Jervell J, Kendall-Taylor P, Lamberts SWJ, Marbach P, Ørskov H, Pagani G, Sheppard M, Simionescu L. Metabolism. 1995;44(suppl 1):18. doi: 10.1016/s0026-0495(96)90087-6. [DOI] [PubMed] [Google Scholar]; (b) Murty SB, Goodman J, Thanoo BC, DeLuca PP. PPAS PharmaSciTech. 2003;4:392–405. [Google Scholar]
- 22.(a) Rueter JK, Mattern RH, Zhang L, Taylor J, Morgan B, Hoyer D, Goodman M. Biopolymers. 2000;53:497–505. doi: 10.1002/(SICI)1097-0282(200005)53:6<497::AID-BIP6>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]; (b) Mattern RH, Zhang L, Rueter JK, Goodman M. Biopolymers. 2000;53:506–522. doi: 10.1002/(SICI)1097-0282(200005)53:6<506::AID-BIP7>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 23.(a) Benoiton NL. Chemistry of Peptide Synthesis. CRC Press: Taylor & Francis Group; Boca Raton: 2006. Section 1.9 and 4.22. [Google Scholar]; b Han Y, Albericio F, Barany G. J Org Chem. 1997;62:4307–4312. doi: 10.1021/jo9622744. [DOI] [PubMed] [Google Scholar]
- 24.Jiang ZX, Qin YY, Qing FL. J Org Chem. 2003;68:7544–7547. doi: 10.1021/jo0344384. [DOI] [PubMed] [Google Scholar]
- 25.Loh TP, Li XR. Eur J Org Chem. 1999:1893–1899. [Google Scholar]
- 26.Zhang XG, Ni WJ, van der Donk WA. J Org Chem. 2005;70:6685–6692. doi: 10.1021/jo051182o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Three different chiral columns, ChiraDex, Chirobiotic T and Ultron ES-Pepsin, were tried. None could resolve 5a and 5b.
- 28.The HPLC conditions are as follows: column, ChiraDex (250 mm × 4.6 mm I.D., 5 μm); mobile phase, 5 mM Na-phosphate in MeOH-H2O (60/40, v/v, pH = 5.6); flow rate, 0.60 mL/min; temperature, 10 °C. Samples were purified by silica-gel column chromatography and dissolved in the mobile phase before injection.
- 29.(a) Böhm HJ, Banner D, Bendels S, Kansy M, Kuhn B, Müller K, Obst-Sander U, Stahl M. ChemBioChem. 2004;5:637–643. doi: 10.1002/cbic.200301023. [DOI] [PubMed] [Google Scholar]; (b) Schweizer E, Hoffmann-Röder A, Schärer K, Olsen JA, Fäh C, Seiler P, Obst-Sander U, Wagner B, Kansy M, Diederich F. ChemMedChem. 2006;1:611–621. doi: 10.1002/cmdc.200600015. [DOI] [PubMed] [Google Scholar]
- 30.Abbruscato TJ, Williams SA, Misicka A, Lipkowski AW, Hryby VJ, Davis TP. J Pharmacol Exp Therapeut. 1996;276:1049–1057. [PubMed] [Google Scholar]
- 31.Kovacs JM, Mant CT, Hodges RS. Biopolymers (Pept Sci) 2006;84:283–297. doi: 10.1002/bip.20417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.(a) Fields GB, Noble RL. Int J Peptide Protein Res. 1990;35:161–214. doi: 10.1111/j.1399-3011.1990.tb00939.x. [DOI] [PubMed] [Google Scholar]; (b) Barlos K, Gatos D. Biopolymers (Pept Sci) 1999;51:266–278. doi: 10.1002/(SICI)1097-0282(1999)51:4<266::AID-BIP3>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 33.(a) Miyazawa T, Otomatsu T, Fukui Y, Yamada T, Kuwata S. J Chem Soc, Chem Commun. 1988:419–420. [Google Scholar]; (b) Miyazawa T, Otomatsu T, Fukui Y, Yamada T, Kuwata S. Int J Peptide Protein Res. 1992;39:308–314. doi: 10.1111/j.1399-3011.1992.tb01590.x. [DOI] [PubMed] [Google Scholar]
- 34.Johansson A, Åkerblom E, Ersmark K, Lindeberg G, Hallberg A. J Comb Chem. 2000;2:496–507. doi: 10.1021/cc000022h. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.