

Abstract
Aurora kinases are key regulators of cell mitosis and have been implicated in the process of tumorigenesis. In recent years, the Aurora kinases have attracted much interest as promising targets for cancer treatment. Here we report on the roles of Aurora A and Aurora B kinases in clear cell renal cell carcinoma (ccRCC). Using genomewide expression array analysis of 174 patient samples of ccRCC, we found that expression levels of Aurora A and B were significantly elevated in ccRCC compared to normal kidney samples. High expression levels of Aurora A and Aurora B were significantly associated with advanced tumor stage and poor patient survival. Inhibition of Aurora kinase activity with the drug VX680 (also referred to as MK-0457) inhibited ccRCC cell growth in vitro and led to ccRCC cell accumulation in the G2/M phase and apoptosis. Growth of ccRCC xenograft tumors was also inhibited by VX680 treatment, accompanied by a reduction of tumor microvessel density. Analysis of endothelial cell lines demonstrated that VX680 inhibits endothelial cell growth with effects similar to that seen in ccRCC cells. Our findings suggest that VX680 inhibits the growth of ccRCC tumors by targeting the proliferation of both ccRCC tumor cells and tumor-associated endothelial cells. Aurora kinases and their downstream cell cycle proteins have an important role in ccRCC and may be potent prognostic markers and therapy targets for this disease.
Keywords: Aurora kinase, Aurora, renal cell carcinoma, VX680, MK-0457
Introduction
In 2009 in the United States, an estimated 57,760 people will be diagnosed with, and 12,980 deaths will be attributed to, cancers of the kidney and renal pelvis [1]. The vast majority of these cases will be clear cell renal cell carcinoma (ccRCC) [2]. Although surgery offers a chance to cure localized ccRCC, most patients who experience recurrence after surgery, or who have metastatic disease at the time of diagnosis, will ultimately die of their disease. New agents targeting the tumor endothelium and their supporting stromal elements (including pericytes) have recently been approved by the FDA for ccRCC therapy; however, it appears that all patients eventually develop resistance to these therapies [3, 4]. Thus, there remains a critical need for effective and specific targets for early diagnosis and treatment; new therapies that target not only the ccRCC tumor– associated endothelium but also the tumor cells may be particularly effective.
In recent years, Aurora kinases have attracted much interest as promising targets for cancer treatment [5, 6]. The Aurora kinases are a family of serine-threonine kinases that function as conserved mitotic regulators. Mammals express three members of this family: Aurora A, Aurora B, and Aurora C. Aurora A and Aurora B are the best characterized, and regulate distinct processes in mitosis. During mitosis, Aurora A localizes to the centrosomes and spindle poles, and is thought to regulate centrosome maturation and separation, and assembly of the mitotic spindle. In contrast, Aurora B localizes to the centromeres during the early stages of mitosis, and plays an important role in the attachment of chromosomes to microtubules, the spindle checkpoint, and cytokinesis. Much less is known about the function of Aurora C. Expression of Aurora C is restricted to germ cells, where it is believed to regulate spermatogenesis. Recently, along with cyclins and cyclindependent kinases, Aurora A has been reported to link to the G2/M transition of the cell cycle. Several reports have demonstrated that Aurora kinases interact with and regulate the activities of many important cellular proteins associated with cell cycle and cell division, including p53, cyclin B, and Cdc2 (also known as Cdk1) [5-7].
Aurora A is overexpressed in many human tumors, including primary breast cancer, colorectal cancer and ovarian cancer. Aurora B has also been found to be overexpressed in a number of cancers [5, 6, 8]. Aurora kinase dysregulation and overexpression are frequently found correlated with chromosomal instability and clinical aggressiveness in malignancies. Highcopy amplification of the gene for Aurora-A has been detected in many tumor types, and polymorphisms in the Aurora-A gene have been associated with cancer risk and clinical outcome [5]. A number of small molecule drug inhibitors of Aurora kinases are currently under development or testing for the treatment of cancer. One of these, the pan Aurora kinase inhibitor VX680 (also referred to as MK-0457), has entered clinical trials. However, the functional significance and mechanism(s) of Aurora kinases in ccRCC have not been fully investigated, and whether Aurora kinases inhibitors have activity against ccRCC has not been clarified.
We wanted to assess Aurora kinases as potential biomarkers and therapeutic targets in human ccRCC. Our microarray analysis of primary kidney tumors revealed that Aurora A and B were highly expressed in the majority of ccRCC cases tested, and that expression of both Aurora A and B was correlated with poor patient survival. We observed that Aurora A and B kinases were active in both ccRCC cell lines and endothelial cells, and that the proliferation of these cells was directly inhibited by VX680 via arrest of cells in the G2/M phase and apoptosis. Moreover, the growth of ccRCC xenografts was inhibited by VX680, and this inhibition was accompanied by significantly reduced tumor microvessel density (MVD). Both in vitro and in vivo studies showed that VX680 treatment resulted in upregulation of p53 and decreased expression of cyclin B/Cdc2 concomitant with inhibition of Aurora kinases. Our results indicate that simultaneously targeting ccRCC cells and endothelial cells in tumors through Aurora kinases inhibition may be an effective strategy for the treatment of ccRCC.
Materials and methods
Tissue collection and gene expression profiling
mRNA expression from the Aurora A and Aurora B genes was examined in 174 clear cell renal tumors and 15 normal kidney samples. Tissue samples were obtained and profiled using Affymetrix HGU133 Plus 2.0 microarrays, as described [9]. Expression values were generated by using Microarray Suite (MAS) v5.0 software (Affymetrix). The probes were filtered according to the study of Dai M et al [10]. The hybridizations were normalized by using the robust multichip averaging (rma) algorithm in Bioconductor package affy (http://www.bioconductor.org/) to obtain summary expression values for each probe set [11-13]. This resulted in more than 17,000 genes, each of which then had one number to represent its relative gene expression intensity in the sample.
Statistical analysis
For statistical analysis of patient survival and gene expression levels, we used survival at five years as the cutoff to separate patient prognosis as good or poor, e.g. patients surviving more than five years following diagnosis were classified in the good prognosis group (C1) and patients surviving less than this were classified as poor prognosis (C2). We used the mean gene expression levels as the cutoff to group patients into either high or low expressers for each gene. The results were similar if the median gene expression levels were used as a cutoff. Cox proportional hazard regression models were fitted to test whether the genes were significant predictors for cancerspecific survival.
For all statistical analysis, values were expressed as mean ± SD. Values were compared using Student’s t test. P < 0.05 was considered significant.
Cell culture
A498, A704, Caki-1, Caki-2, ACHN, 786-O, and 769-P ccRCC cell lines were obtained from the American Type Culture Collection (ATCC). SN12C, UO31, and TK10 cells were kindly provided by Dr. George Vande Woude (Van Andel Research Institute). SKRC39 cells were obtained from Memorial Sloan-Kettering Cancer Center. The cells were maintained in DMEM or RPMI 1640 medium (Invitrogen) supplemented with 10% fetal bovine serum (Invitrogen), 100 IU/mL penicillin, and 100 μg/mL streptomycin (Invitrogen) in a humidified incubator containing 5% CO2 at 37°C. HUAEC, HUVEC, HMVEC and HLMVEC human endothelial cells were obtained from Clonetics and maintained in Clonetics EBM -2 medium supplemented with EGM-2 singlequots (Cambrex).
Cell-cycle analysis
Cells were incubated with either VX680 or dimethyl sulfoxide (DMSO) (control) for 72 h. Cells were collected and analyzed using a cellular DNA flow cytometric analysis kit (Roche). Briefly, cells were collected after treatment and stained with propidium iodide. DNA content was analyzed by flow cytometric anlaysis.
Apoptosis analysis
Cells were incubated with either VX680 or DMSO (control) for 72 h. Apoptotic cells were measured using FITC Annexin V Apoptosis Detection Kit (BD Biosciences). Briefly, cells were collected after treatment and stained with Annexin V-FITC and propidium iodide according to the manufacturer’s protocol, then analyzed by flow cytometry.
Cell synchronization
Cells were synchronized using nocodazole (200 nmol/L) for 16 h. Cells were released from the block in the presence of various concentrations of VX680 or DMSO (control) and incubated for 6 h and 72 h; then proteins were extracted from the collected cells.
Analysis of cell proliferation and viability
Cells were seeded at densities ranging from 1,000 to 3,000 on 96-well plates in DMEM or RPMI 1640 medium supplemented with 10% fetal bovine serum. Cells were treated with DMSO or VX680 for 96 h, and then cell viability was measured by an MTT assay. Briefly, after treatment cells were incubated with fresh media containing MTT solution at 37°C for 2 hours and then cell viability was determined by measurement of absorbance at 540 nm. Percentage of cell viability was calculated as the absorbance of VX680 treated cells divied by DMSO controls.
Tumor implantation and growth in a ccRCC xenograft model
All animal studies were in compliance with VARI Institutional Animal Care and Use Committee policies. Six-week-old male or female BALB/c nu/nu nude mice (Charles River) were used. Ten million Caki-1 cells or five million SN12C cells were subcutaneously implanted in the right flank. Tumor size was measured 2-3 times per week using digital calipers (Mitutoyo, Kawasaki, Japan) that have an accuracy of ±0.02 mm, and tumor volume was calculated as length × width × height × 0.5. Tumor volumes are presented as mean ± SD.
When tumors had grown to an average volume of 100 to 150 mm3, tumorbearing mice were separated into two groups of 9-10 animals. One group received intraperitoneal injections of 50% PEG300 as a vehicle control; one group received intraperitoneal injections of VX680 at 80 mg/kg every day. Mice were euthanized at the end of the treatment period (day 21 in Caki-1 xenograft model and day 30 in SN12C xenograft model). Tumors were removed, cleaned from adjacent tissues, fixed in 4% paraformaldehyde, and paraffinembedded, and then 4-μm-thick sections were prepared. All sections were stained with hematoxylin and eosin and were used for subsequent immunohistochemical analysis. Parts of all sections were stored at – 80°C for Western blotting analysis.
Immunofluorescence of cultured cells
Cell lines were grown on coverslips (Nunc® Lab-Tek® II Chamber Slide™ system) with VX680 diluted in DMSO (0.8 μmol/L) for 72 h. For immunofluorescent staining, the cells were stained with combinations of anti-Aurora A pT288 rabbit antibody (1:50, Cell Signaling) and anti-phosphorylated histone H3 (Ser10, Cell Signaling) mouse monoclonal antibody (1:100, Santa Cruz), followed by addition of a FITC-conjugated (1:100) or TRITC-conjugated (1:300) antibody to rabbit and mouse IgG. DAPI (300 nM) was used to highlight DNA. Fluorescently labeled cells were visualized using a microscope (Zeiss 510 Meta).
Immunohistochemistry
Immunohistochemical staining was done on 4-µm formalinfixed, paraffinembedded tissue sections. Endogenous peroxidase activity was blocked with 3% hydrogen peroxide. Antigen retrieval was carried out in citrate buffer (10 mmol/L, pH 6) for 15 min at 100°C in a microwave oven. The slides were incubated with a primary rabbit anti-human Aurora A, rabbit anti-human Aurora B, rabbit anti-phosphorylated human Aurora A (Thr288), rabbit anti-phosphorylated histone H3 (Ser10), rabbit anti-human PCNA, and rabbit anti-mCD34 overnight at 4°C. Sections were then incubated with secondary anti-rabbit IgG (Vector) for 30 min. After washing with 1×TTBS, sections were incubated with Vectastain ABC reagent (Vector). The immune complex was visualized using DAB substrate solution (Vector). For the quantitation of PCNA p-Aurora A, p-histone H3, and CD34, see the description in Huang D et al [9].
Cell lysate and Western blotting analysis
Cell lysates were prepared by washing cells with PBS and then following the methods described [9]. For Western blotting, samples transferred to a nitrocellulose membrane by semiwet electrophoresis (Invitrogen), were incubated with primary antibody (rabbit anti-Aurora A, rabbit anti-Aurora B, rabbit anti-phosphorylated Aurora A (Thr288), mouse anti-phosphorylated histone H3 (Ser10), mouse anti-p53, mouse anti-Cdc2, mouse anti-cyclin B1) overnight at 4°C, detected with horseradish peroxidase–conjugated anti-rabbit or anti-mouse IgG (Santa Cruz), and developed using an ECL Western blotting detection and analysis system (Amersham). Membranes were tested for equal loading by probing for actin.
Results
Overexpression of Aurora A and B were associated with the clinical outcome of ccRCC patients
Microarray gene expression profiling was used to examine expression levels of Aurora A and Aurora B in 174 cases of human ccRCC and 15 normal kidney samples. High expression of Aurora A and B was detected in clinical specimens of ccRCC relative to normal control samples. Advancedstage tumors tended to have higher mRNA levels for Aurora A and B than earlystage tumors (P < 0.01) (Figure 1A, left panel). Moreover, patients with high expression of Aurora kinases (A and B) were more likely to have poor prognosis (P < 0.001) (Figure 1A, left panel). Plotted survival curves showed that patients with high expression of Aurora A and Aurora B had decreased survival times compared to patients with low expression of Aurora kinases (Figure 1A, right panel). Based on these results, we hypothesized that both Aurora A and Aurora B play an important role in the development of ccRCC and that inhibition of Aurora kinase activity would inhibit the growth of ccRCC tumors.
Figure 1.

Expression of Aurora kinases in human ccRCC and growth inhibition by VX680. A, Left panel, Aurora A and Aurora B mRNA expression levels in primary ccRCC classified by T stage and extent of malignancy. C1, patients with good prognosis; C2, patients with poor prognosis. Right panel, survival analyses indicate association between high expression of Aurora A and Aurora B and poor patient survival. The patients were divided into high and low expression groups using as a cutoff value the mean of the mRNA expression level for each gene. Error bars show standard deviation. *P <0.05, **P <0.02, ***P< 0.01 . B, Effect of VX680 on the viability of human ccRCC cell lines. Cells were incubated with different concentrations of VX680 for 96 h, and viability was quantified by an MTT assay. IC50 values for inhibition of proliferation were determined for each cell line and are depicted. Error bars represent standard deviation. C, Growth inhibition curves. A498 and Caki-1 cells were incubated with VX680 for 96 h at the concentrations indicated, and viability was quantified by MTT assay. D, VX680 inhibits Aurora kinase signaling in A498 and Caki-1 cells. Cells were treated with nocodazole for 16 h to induce mitotic arrest (synchronization). Synchronized A498 and Caki-1 cells were released from nocadozole block and treated with indicated concentrations of VX680 for 6 h. “Con” refers to untreated control samples. Separate samples were also treated with DMSO for vehicle control. Synchronized HeLa cells were taken for positive control. Whole-cell lysates were subjected to Western blotting with antibodies against the indicated proteins; Western blotting for actin was used to show equal loading of samples.
Aurora kinase expression in ccRCC cell lines
To test our hypothesis, we first confirmed the expression of Aurora kinases in 11 RCC cell lines (A498, A704, Caki-1, Caki-2, ACHN, 786-O, 769-P, SKRC39, SN12C, TK10, and UO31) by Western blotting. Two of the cell lines tested, Caki-2 and SKRC39, were papillary RCC, while the rest were ccRCC lines. We found that most of the RCC cell lines expressed Aurora A and Aurora B at the protein level (Figure 2). Next, we confirmed the activation of Aurora kinases by examining the phosphorylation status of both Aurora A and histone H3, a direct downstream target of Aurora kinases [14]. Our results showed that the majority of the cell lines expressed pThr288-Aurora A and pSer10-histone H3, indicating that Aurora kinases were activated in those cell lines (Figure 2).
Figure 2.

Expression of Aurora kinases in a panel of human RCC cell lines. Expression of Aurora A and B, phosphorylated Aurora A, and phosphorylated histone H3 in human RCC cell lines as analyzed by Western blotting.
VX680 directly reduced the viability of human ccRCC cells in vitro
Subsequently, we tested a small-molecule Aurora kinase inhibitor, VX680, which has inhibition constants (Ki) of 0.6, 18, and 46 nM for Aurora A, B, and C, respectively [15]. To determine whether VX680 had a direct antitumor effect on RCC cells in vitro, we treated the 11 RCC cell lines with control media (DMSO) or media containing various concentrations of VX680 for 96 h. The antiproliferation effect was evaluated by examining cell viability using an MTT assay. The proliferation of all 11 RCC lines was significantly attenuated by VX680 in a dose dependent manner. Most of the half-maximal inhibitory concentration (IC50) values were between 0.1 to 10 µmol/. Only one of the 11 lines, A704, had an IC50 greater than 10 µmol/L (Figure 1B). In light of this, it is worth noting that activation of Aurora kinases is barely detectable in A704 cells by Western blotting (Figure 2). A498 and Caki-1 were two of the ccRCC cell lines most sensitive to VX680 (IC50 was 0.367 ± 0.140 µmol/L and 0.482 ± 0.166 µmol/L respectively); for the subsequent studies, the growth curves of these two cells in response to VX680 treatment were tested and plotted (Figure 1C). Based on the results of these growth curves and VX680 IC50 values, we selected VX680 concentrations of 0.05 µmol/L, 0.2 µmol/L, 0.8 µmol/L, or 2.0 µmol/L for further experiments.
VX680 targeted Aurora kinases in ccRCC cells
To confirm that VX680 targets Aurora kinases in ccRCC cells, we examined the phosphorylation status of Aurora A and histone H3 in VX680-treated cells. Consistent with previous reports, we found that basal expression of pThr288 Aurora A and pSer10 histone H3 was relatively weak in asynchronous cell populations, but increased when cells were blocked in G2/M phase with nocodazole treatment (Figure 1D). Six hours of treatment with VX680 was sufficient to inhibit Aurora kinase activity in nocada-zole-synchronized A498 and Caki-1 cells (Figure 1D). Under these treatment conditions, VX680 did not affect overall protein levels of Aurora A or Aurora B. Although basal activity of Aurora kinases is more difficult to detect in asynchronous cell populations, we were also able to show VX680-mediated inhibition of Aurora kinase activity in asynchronous populations of A498 and Caki-1 cells after 72 hours of VX680 treatment (Figure 3A). Interestingly, we noted that extended VX680 treatment of cells for 72 hours resulted in decreased expression of total Aurora A and Aurora B protein, as well as decreased phosphorylation of Aurora kinase substrates (Figure 3B).
Figure 3.

Effects of extended VX680 treatment on the expression of Aurora kinases and cell cycle related proteins in A498 and Caki-1 cell lines. A, 72 hour VX680 treatment of asynchronous cells. Asynchronous A498 or Caki-1 cells were incubated with increasing concentrations of VX680 for 72 hours. “Con” refers to untreated control samples. Separate samples were also treated with DMSO for vehicle control. Synchronized HeLa cells were taken for positive control. Whole-cell lysates were subjected to Western blotting with antibodies against the indicated proteins; blotting for actin was used to show equal loading of samples. B, 72 hour VX680 treatment of nocodazole-blocked ccRCC cells. A498 and Caki-1 cells were treated with nocodazole for 16 h to induce mitotic arrest. Cells were then released from the mitotic block, followed by the addition of DMSO or indicated concentrations of VX680 for 72 h. “Con” refers to untreated control samples; synchronized HeLa cells were taken for positive Western blot control. Sample lysates of asynchronous cells were also run for comparison.
VX680 induced arrest of cells in G2/M phase and apoptotic death
Aurora kinases are essential for proper progression through the cell cycle. We therefore tested the effects of VX680 on cell cycle progression in ccRCC cells. A498 and Caki-1 cells were incubated with VX680 for 72 hours. Analysis by flow cytometry showed that VX680 treatment induced cell cycle arrest at the G2/M phase and polyploidy in A498 and Caki-1 cells (Figure 4A and 4B). Because an important consequence of prolonged G2/M arrest is apoptosis, we also looked at the effects of VX680-treatment on apoptotic cell death. As shown in Figure 4C, VX680-treatment led to increased apoptosis of both A-498 and Caki-1 cells. Our results are consistent with the effects of VX680 in other cell lines and the known functions of Aurora kinases in the cell cycle and apoptosis [6, 15]. We conclude that VX680 inhibits proliferation of ccRCC cells through inhibition of Aurora kinases and resulting cell cycle arrest and apoptotic death.
Figure 4.

VX680 induces arrest of ccRCC cells in G2/M phase and apoptosis. A, B. VX680 induced cell cycle arrest at G2/M and polyploidy. Cells were incubated with VX680 for 72 h, stained with propidium iodide (PI) and analyzed by flow cytometry. Error bars indicate standard deviation. *P < 0.05, **P < 0.01, ***P < 0.001. C, VX680 treatment induced apoptosis of ccRCC cells. Cells were incubated with VX680 for 72 h, stained with Annexin V-FITC, and analyzed by flow cytometry. Error bars indicate standard deviation. *P < 0.05, **P < 0.01.
VX680 injection inhibited the growth of Caki-1 tumor xenografts in nude mice
We next evaluated the effects of VX680 on ccRCC tumor growth in vivo in an established Caki-1 xenograft model. VX680 treatment led to a 75.7% (P < 0.001) decrease in Caki-1 xeno-graft tumor volume (Figure 5A). Treatment with VX680 did not alter animal body weight, peripheral blood counts, or other biological parameters (Figure 6 and data not shown). These results imply that the effect of VX680 on the xenograft model was not due to system toxicity. Three VX680-treated xenograft tumors and four control tumors were selected at random and further analyzed. We found that cell proliferation within VX680-treated tumors was markedly reduced, as assessed by both Western blotting and immunohistochemical staining for PCNA (Figure 5B and 5C). We also evaluated the effect of VX680 on a second ccRCC xenograft model, using SN12C cells. We found that VX680 also inhibited growth of SN12C tumors, with a 33.8% decrease in the size of treated SN12C tumors compared to controls (p < 0.05) (data not shown).
Figure 5.

VX680 treatment inhibits ccRCC tumor growth and inhibits Aurora kinase signaling in vivo A, Ten million Caki-1 cells were subcutaneous injected into the right flank of each mouse to initiate ectopic tumors. Intraperitoneal injection of 50% PEG300 or VX680 (80 mg/kg) started on day 3 after tumor cell inoculation and continued daily to day 21. The final average tumor volumes (mm3) were measured. Tumor growth curves were plotted for Caki-1 xeno-grafts with intraperitoneal injection of 80 mg/kg VX680 or 50% PEG300 (n = 9-10 mice per group). Error bars represent standard deviation. ***P < 0.001. B, A portion of three or four randomly selected tumors from each group were homogenized for lysate preparation and analyzed by Western blotting for protein expression. C, Immunohisto-chemical staining for p-Aurora A, p-Histone H3, PCNA, and CD34 in Caki-1 xenograft tumors after treatment with VX680 or 50% PEG300. D, Quantification of a proliferation index (PCNA), p-Aurora A staining, and p-Histone H3 staining in the viable rim of Caki-1 xenograft tumors, and quantification of MVD of Caki-1 xenograft tumors treated with VX680 or 50% PEG300. Error bars represented standard deviation. ***P < 0.001.
Figure 6.
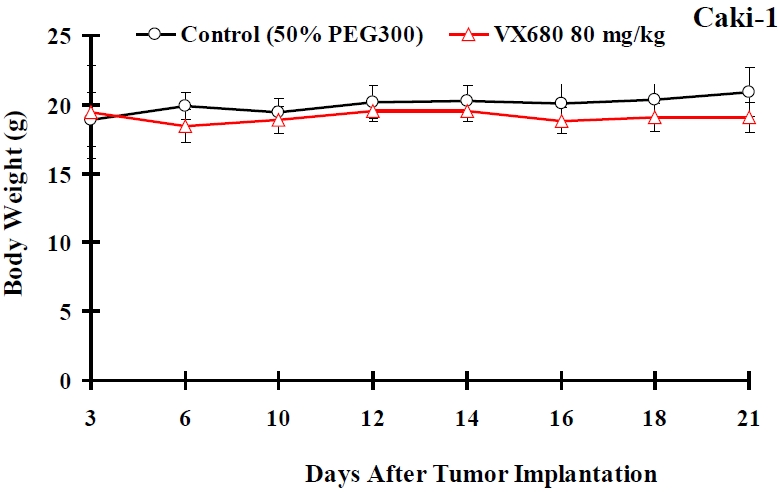
The effects of VX680 on body weight of nude mice carrying Caki-1 xenografts. Xenograft-bearing mice were treated with intraperitoneal injection of 80 mg/kg VX680 or a comparable volume of 50% PEG300 (n = 9-10 mice per group). No effect on weight was observed after VX680 treatment. Error bars represent standard deviations.
VX680 inhibited Aurora kinase activity in vivo
Activity of Aurora kinases was significantly inhibited in VX680-treated tumors. Both Western blotting and immunohistochemical staining showed levels of pThr288 Aurora A and pSer10 histone H3 were decreased in VX680-treated tumors (Figure 5B and 5C). Interestingly, we also saw decreases in overall protein expression of Aurora A and Aurora B after in vivo VX680 treatment (Figure 5B). This is consistent with the decreased Aurora A and Aurora B expression observed in ccRCC cells in vitro after extended VX680 treatment (Figure 3A).
VX680 treatment upregulated p53 expression and downregulated cyclin B1/Cdc2 expression in xenograft tumors
To further characterize mechanisms of VX680 action in ccRCC tumors, we examined VX680-treated xenografts for changes in expression of cell-cycle regulator proteins. Aurora kinases have been shown to regulate the stability and action of p53, a critical regulator of cell cycle arrest and apoptosis [16, 17]. We found that inhibition of Aurora kinases with VX680 led to a marked accumulation of p53 in vivo (Figure 5B). p53 has been previously implicated in cell cycle arrest mediated by Aurora kinase inhibitors [6]. We also looked at the expression of cyclin B1 and Cdc2 (also known as Cdk1), proteins critical for cell cycle progression through G2/M phase. We found that both cyclin B1 and Cdc2 expression is decreased in VX680-treated tumors compared to control tumors (Figure 5B). We observed similar results in vitro after 72 hours VX680 treatment of Caki-1 cells (Figure 3B).
VX680 reduced tumor microvessel density
Tumor MVD is considered an indicator of tumor angiogenesis. The mean MVD in tumors can be evaluated by quantifying the CD34-positive cells using immunohistochemical staining. Tumors harvested from mice treated with VX680 showed a striking reduction in CD34-positive cells (Figure 5C). Quantification of CD34-positive cells in the tumors showed VX680-treated tumors displayed a 66% decrease in MVD compared to control tumors (Figure 5D). Notably, we also saw a similar decrease in microvessel density in VX680-treated SN12C xeno-grafts (data not shown).
Aurora kinase expression in endothelial cells
Our in vivo study indicated that VX680 affected the development of blood vessels in tumors. To further elucidate the role of Aurora kinases in endothelial cells, the expression levels of Aurora kinases were examined in four types of human endothelial cells (HUAEC, HUVEC, HLMVEC, and HMVEC). Our results revealed that Aurora A and Aurora B were highly expressed in endothelial cells at the protein level (Figure 7A). Activation of Aurora kinases were also detected in these cell lines (Figure 7A).
Figure 7.

Expression of Aurora kinases in endothelial cells and the effects of VX680 on proliferation and cell cycle arrest in HUAEC cells. A, Western blotting for the expression of Aurora kinases in endothelial cells. B, Growth inhibition curves. Endothelial cells were incubated with VX680 for 96 h at the concentrations indicated, and viability was quantified by MTT assay. C, HUAEC cells were treated with 0.06 or 0.3 µmol/L of VX680 and Caki-1 cells were treated with 0.3 µmol/L VX680 for 72 h. Whole-cell lysates were subjected to Western blotting for detection of Aurora kinase activity and cell cycle proteins; actin was used as a loading control. D, VX680-induced cell cycle arrest at G2/M. HUAEC cells were exposed to VX680 for 72 h and then stained with PI; cell cycle arrest was analyzed by flow cytometry. Error bars represent standard deviation. *P < 0.05, **P < 0.01.
VX680 decreased viability of endothelial cells in vitro through inhibition of Aurora kinases
To verify whether VX680 could inhibit the growth of endothelial cells in vitro, we treated all four endothelial cell lines with control media (DMSO) or media containing various doses of VX680 for 4 days, followed by an MTT assay. As shown in Figure 7B, VX680 significantly inhibited the viability of HUAEC and HLMVEC cells with IC50 values of 0.04 µmol/L and 0.46 µmol/ L respectively. We selected the most sensitive cell line, HUAEC, for an investigation of the mechanism of VX680 in endothelial cells. Western blotting analysis revealed that treatment with VX680 inhibited activation of Aurora kinases in HUAEC cells, and affected the expression of the downstream target proteins, p53, cyclin B1 and Cdc2 (Figure 7C). The effects of VX680 treatment on endothelial HUAEC cells were similar to that on ccRCC Caki-1 cells (Figure 7C).
VX680 induced G2/M arrest in HUAEC cells
In Figure 7D, the percentages of different cell cycle subpopulations of HUAEC—G1, S, and G2/ M—are shown for controls or for VX680 treated cells. After exposure to VX680 at 0.06 µmol/L or 0.3 µmol/L for 72 h, 18.3% and 54.0% of HUAEC cells, respectively, were arrested in G2/ M phase (Figure 7D). Thus, VX680 treatment induces cell cycle arrest in HUAEC cells, similarly as in ccRCC cells. Our results suggest that VX680 targets endothelial cells in a way similar to its targeting of ccRCC cells. Thus, VX680 may inhibit tumor growth through direct targeting of both tumor and surrounding endothelial cells.
Discussion
In this study, we report on the roles of Aurora A and Aurora B in human ccRCC. Analysis of primary kidney tumors using Affymetrix microarrays indicated that the mRNA of Aurora A and B were highly expressed in the majority of ccRCC cases. Highlevel expression of Aurora A and B was correlated with cancer stage and poor prognosis. Inhibition of Aurora kinases by VX680 inhibited ccRCC cell growth in vitro, and led to cell cycle arrest in the G2/M phase and apoptosis. These findings were corroborated by in vivo experiments showing that VX680 treatment inhibits growth of ccRCC xenograft tumors. Inhibition of tumor growth was accompanied by significant decreases in MVD, suggesting that VX680 may also target growth of endothelial cells. We showed that Aurora kinases are active in endothelial cell lines, and that inhibition of Aurora kinases results in endothelial cell cycle arrest, similar to that seen in ccRCC cells. Our findings suggest that Aurora kinases play an important role in the development of ccRCC and that VX680 may inhibit ccRCC growth by targeting of both tumor and endothelial cells.
Aurora kinases are key regulators of cell mitosis, and interact with multiple cell cycle proteins to regulate progression through the G2/M phase. In our studies, we noted that extended inhibition of Aurora kinase activity with VX680 induced changes in expression of the cell cycle proteins cyclin B1, Cdc2 and p53. These observations are consistent with the known biological activities of the Aurora kinases. Aurora A has been demonstrated to control centrosomal activation of the cyclin B1/Cdc2 complex at the start of mitosis [6]. Recently, it was reported that Aurora-A may interact directly with cyclin B1 to promote its stability. Overexpression of Aurora-A was shown to upregulate cyclin B1 expression through enhancement of its stability, while RNAi-mediated knockdown of Aurora-A was shown to reduce cyclin B1 expression [18]. These reported effects were suggested to be dependent upon the kinase activity of Aurora-A, consistent with our finding that inhibition of Aurora kinase activity results in decreased expression of cyclin B1 (Figure 3B, 5B, and 7C). In addition to downregulation of cyclin B1 and Cdc2, we noted that extended VX680 treatment also led to induction of p53 expression in both ccRCC and endothelial cells (Figure 3B, 5B, and 7C). There is a tight functional relationship between Aurora-A and p53, and they have been proposed to act together to regulate cell cycle arrest [6]. Aurora-A has been shown to directly phosphorylate p53, resulting in destabilization and loss of p53 activity [16, 17]. It is therefore unsurprising that inhibition of Aurora-A kinase activity with VX680 should result in increased expression of p53 in our studies. Indeed, Aurora kinase inhibitors have been shown to induce p53 expression in a variety of cell lines [19, 20].
Interestingly, in addition to expected effects on the stability of cell cycle proteins, we found that extended VX680 treatment also led to down-regulation of Aurora A and Aurora B proteins themselves. To our knowledge, this effect has not been previously reported. Because this effect was only seen upon extended 72-hour VX680 treatment, it may have been missed by other groups studying VX680 treatment at shorter time points. The mechanisms behind this downregulation of Aurora A and Aurora B protein expression are currently unknown. Like many cell cycle regulatory proteins, the expression levels of Aurora kinases rise and fall during cell cycle progression in a ubiquitin and protea-some-dependent manner [8]. We speculate that sustained VX680 treatment and subsequent alterations to the cell cycle may result in decreased stability of Aurora kinase proteins. It is possible that this decreased expression of Aurora kinases represents an additional mechanism (in addition to the direct inhibition of Aurora kinase activity) by which VX680 and related compounds may inhibit Aurora kinase function.
Aurora kinases are overexpressed in a number of different cancers, including breast cancer, colorectal cancer, ovarian cancer and gliomas [5, 8]. The established involvement of Aurora kinases in cellular mitosis, along with strong circumstantial evidence suggesting a role for Aurora kinases in tumorigenesis, has led to the development of small-molecule inhibitors of these kinases for the treatment of cancer. VX680 is one of a class of pan-Aurora kinase inhibitors now in clinical testing. VX680 has been shown to suppress tumor growth in a variety of xenograft models, including xenograft models of ovarian cancer, colorectal cancer and leukemia [15, 21]. However, the effects of VX680 or related Aurora kinase inhibitors have not previously been shown for ccRCC. In our study, we demonstrate for the first time that pharmacological inhibition of Aurora kinases significantly inhibits growth of ccRCC xenograft tumors in vivo[22]. Moreover, our study suggests that VX680 may inhibit tumor growth by targeting of both tumor cells and surrounding endothelial cells. Hardwicke et al. recently reported that a novel Aurora kinase inhibitor GSK1070916, suppresses growth of endothelial HUVEC cells in vitro. Our work extends Harwicke’s in vitro results to multiple endothelial cell lines and a distinct Aurora kinase inhibitor. Moreover, ours is the first demonstration that Aurora kinase inhibitors may have anti-angiogenic effects, as well as direct effects on tumor cells. Our results suggest that Aurora kinase inhibitors may have clinical efficacy in the treatment of ccRCC. In light of this, it is worth noting a recent report that the histone deacetylase inhibitor LBH589 induces degradation of Aurora A and Aurora B proteins in ccRCC cells, and also suppresses growth of ccRCC xenograft tumors [23]. Inhibition of Aurora kinases (through either inhibition of kinase activity, suppression of protein expression, or a combination of both approaches) may represent a novel approach toward the treatment of kidney cancer.
Acknowledgments
We thank the Cooperative Human Tissue Network (CHTN) of the National Cancer Institute for providing samples for analysis; Bree Berghuis, Eric Hudson, and J.C. Goolsby, from the Laboratory of Analytical, Cellular, and Molecular Microscopy, Van Andel Research Institute, for technical support in immunohistochemistry staining; Rich West, from the Laboratory of Cell Structure and Signal Integration, Van Andel Research Institute, for technical support in fluorescence-activated cell sorting analysis; and Dawna Dylewski and Lisa DeCamp, from Vivarium Operations, Van Andel Research Institute, for their help with the animal experiments. We also thank David Nadziejka and Vanessa Fogg from the Van Andel Research Institute for technical editing of the manuscript, and Sabrina Noyes, from Van Andel Research Institute, for assistance in preparation and submission of the manuscript.
This study was funded by the National Foundation for Cancer Research. Min-Han Tan’s research is supported by the Singapore Millennium Foundation and the National Kidney Foundation. Hyung Kim is partially funded by the National Institute of Health (R01CA133072-01). The corresponding author would also like to thank the Hauenstein Foundation and the Van Andel Foundation for their continued support.
Glossary
Abbreviations
- ccRCC
(clear cell renal cell carcinoma)
- RCC
(renal cell carcinoma)
- MVD
(microvessel density)
- robust multichip averaging
(rma)
- dimethyl sulfoxide
(DMSO)
- propidium iodide
(PI)
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer Statistics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Cohen HT, McGovern FJ. Renal-cell carcinoma. N Engl J Med. 2005;353:2477–2490. doi: 10.1056/NEJMra043172. [DOI] [PubMed] [Google Scholar]
- 3.Rini BI, Flaherty K. Clinical effect and future considerations for molecularly-targeted therapy in renal cell carcinoma. Urol Oncol. 2008;26:543–549. doi: 10.1016/j.urolonc.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 4.Reeves DJ, Liu CY. Treatment of metastatic renal cell carcinoma. Cancer Chemother Pharmacol. 2009;64:11–25. doi: 10.1007/s00280-009-0983-z. [DOI] [PubMed] [Google Scholar]
- 5.Gautschi O, Heighway J, Mack PC, Purnell PR, Lara PN, Jr, Gandara DR. Aurora kinases as anticancer drug targets. Clin Cancer Res. 2008;14:1639–1648. doi: 10.1158/1078-0432.CCR-07-2179. [DOI] [PubMed] [Google Scholar]
- 6.Vader G, Lens SM. The Aurora kinase family in cell division and cancer. Biochim Biophys Acta. 2008;1786:60–72. doi: 10.1016/j.bbcan.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 7.Carmena M, Earnshaw WC. The cellular geography of aurora kinases. Nat Rev Mol Cell Biol. 2003;4:842–854. doi: 10.1038/nrm1245. [DOI] [PubMed] [Google Scholar]
- 8.Katayama H, Brinkley WR, Sen S. The Aurora kinases: role in cell transformation and tumori-genesis. Cancer Metastasis Rev. 2003;22:451–464. doi: 10.1023/a:1023789416385. [DOI] [PubMed] [Google Scholar]
- 9.Huang D, Ding Y, Luo WM, Bender S, Qian CN, Kort E, Zhang ZF, VandenBeldt K, Duesbery NS, Resau JH, Teh BT. Inhibition of MAPK kinase signaling pathways suppressed renal cell carcinoma growth and angiogenesis in vivo. Cancer Res. 2008;68:81–88. doi: 10.1158/0008-5472.CAN-07-5311. [DOI] [PubMed] [Google Scholar]
- 10.Dai M, Wang P, Boyd AD, Kostov G, Athey B, Jones EG, Bunney WE, Myers RM, Speed TP, Akil H, Watson SJ, Meng F. Evolving gene/ transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 2005;33:e175. doi: 10.1093/nar/gni179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 13.Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- 14.Crosio C, Fimia GM, Loury R, Kimura M, Okano Y, Zhou H, Sen S, Allis CD, Sassone-Corsi P. Mitotic phosphorylation of histone H3: spatiotemporal regulation by mammalian Aurora kinases. Mol Cell Biol. 2002;22:874–885. doi: 10.1128/MCB.22.3.874-885.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harrington EA, Bebbington D, Moore J, Rasmussen RK, Ajose-Adeogun AO, Nakayama T, Graham JA, Demur C, Hercend T, Diu-Hercend A, Su M, Golec JM, Miller KM. VX-680, a potent and selective small-molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo. Nat Med. 2004;10:262–267. doi: 10.1038/nm1003. [DOI] [PubMed] [Google Scholar]
- 16.Katayama H, Sasai K, Kawai H, Yuan ZM, Bondaruk J, Suzuki F, Fujii S, Arlinghaus RB, Czerniak BA, Sen S. Phosphorylation by aurora kinase A induces Mdm2-mediated destabilization and inhibition of p53. Nat Genet. 2004;36:55–62. doi: 10.1038/ng1279. [DOI] [PubMed] [Google Scholar]
- 17.Liu Q, Kaneko S, Yang L, Feldman RI, Nicosia SV, Chen J, Cheng JQ. Aurora-A abrogation of p53 DNA binding and transactivation activity by phosphorylation of serine 215. J Biol Chem. 2004;279:52175–52182. doi: 10.1074/jbc.M406802200. [DOI] [PubMed] [Google Scholar]
- 18.Qin L, Tong T, Song Y, Xue L, Fan F, Zhan Q. Aurora-A interacts with Cyclin B1 and enhances its stability. Cancer Lett. 2009;275:77–85. doi: 10.1016/j.canlet.2008.10.011. [DOI] [PubMed] [Google Scholar]
- 19.Dreier MR, Grabovich AZ, Katusin JD, Taylor WR. Short and long-term tumor cell responses to Aurora kinase inhibitors. Exp Cell Res. 2009;315:1085–1099. doi: 10.1016/j.yexcr.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 20.Gizatullin F, Yao Y, Kung V, Harding MW, Loda M, Shapiro GI. The Aurora kinase inhibitor VX-680 induces endoreduplication and apoptosis preferentially in cells with compromised p53-dependent postmitotic checkpoint function. Cancer Res. 2006;66:7668–7677. doi: 10.1158/0008-5472.CAN-05-3353. [DOI] [PubMed] [Google Scholar]
- 21.Lin YG, Immaneni A, Merritt WM, Mangala LS, Kim SW, Shahzad MM, Tsang YT, Armaiz-Pena GN, Lu C, Kamat AA, Han LY, Spannuth WA, Nick AM, Landen CN, Jr, Wong KK, Gray MJ, Coleman RL, Bodurka DC, Brinkley WR, Sood AK. Targeting aurora kinase with MK-0457 inhibits ovarian cancer growth. Clin Cancer Res. 2008;14:5437–5446. doi: 10.1158/1078-0432.CCR-07-4922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hardwicke MA, Oleykowski CA, Plant R, Wang J, Liao Q, Moss K, Newlander K, Adams JL, Dhanak D, Yang J, Lai Z, Sutton D, Patrick D. GSK1070916, a potent Aurora B/C kinase inhibitor with broad antitumor activity in tissue culture cells and human tumor xenograft models. Mol Cancer Ther. 2009;8:1808–1817. doi: 10.1158/1535-7163.MCT-09-0041. [DOI] [PubMed] [Google Scholar]
- 23.Cha TL, Chuang MJ, Wu ST, Sun GH, Chang SY, Yu DS, Huang SM, Huan SK, Cheng TC, Chen TT, Fan PL, Hsiao PW. Dual degradation of aurora A and B kinases by the histone deacetylase inhibitor LBH589 induces G2-M arrest and apoptosis of renal cancer cells. Clin Cancer Res. 2009;15:840–850. doi: 10.1158/1078-0432.CCR-08-1918. [DOI] [PubMed] [Google Scholar]