

Abstract
Purpose of review
G protein-coupled receptor (GPCR) signaling machinery can serve as a direct target of reactive oxygen species (ROS), including superoxide (O2-), hydrogen peroxide (H2O2) as well as reactive nitrogen species (RNS) including nitric oxide (NO) and S-nitrosothiols (SNOs). NADPH oxidase is one of the major sources of O2- produced following GPCR activation in vasculature. NO is generated by three isoforms of NO synthase (NOS). This review will summarize the recent progress on GPCR signaling modulation by NADPH oxidase-derived ROS and NOS-derived SNOs.
Recent findings
ROS and RNS play an important role in GPCR signaling involved in various physiological functions such as cell growth, migration, gene expression as well as pathophysiologies. NADPH oxidase-derived ROS activate specific redox signaling events involved in cardiovascular diseases. SNOs can modulate GPCR signaling and internalization through S-nitrosylation of the scaffolding protein β-arrestin, the G protein-coupled receptor kinases (GRKs), and dynamin, a GTPase responsible for endocytosis.
Summary
NADPH oxidase-derived ROS and NOS-derived SNOs are now recognized as important second messengers to regulate GPCR signaling, thereby contributing to various biological and pathophysiological functions. Understanding the molecular mechanism of how ROS, NO and SNOs might modulate GPCR signaling is essential for development of novel therapeutic approaches.
Keywords: G protein-coupled receptor, reactive oxygen species, NADPH oxidase, nitric oxide, S-nitrosothiols, redox signaling, S-nitrosylation, vascular smooth muscle cells, endothelial cells
Introduction
G-protein-coupled receptors (GPCRs) are fundamentally involved in mammalian physiology, while GPCR dysfunction/dysregulation is a major contributor to the pathophysiologies of disease. Reactive oxygen species (ROS), including superoxide (O2-) and H2O2 as well as reactive nitrogen species (RNS) including nitric oxide (NO) and its biological metabolites S-nitrosothiols (SNOs) are generated following GPCR activation in vascular cells. The major source of ROS is a NADPH oxidase in the vasculature, and NO is generated by NO synthase (NOS). This review will summarize the recent information on how ROS and SNOs regulate GPCR signaling and trafficking, which is implicated in vascular biology and pathophysiologies.
Role of ROS and NO/SNOs as signaling molecules
GPCRs such as β-adrenergic receptor (β-AR) and angiotensin II type1 receptor (AT1R), transduce signals through the activation of heterotrimeric G proteins [1-4]. Evidence reveals that ROS and/or NO production is increased following GPCR activation depending on agonist and cell types, and that they function as signaling molecules. Physiological level of ROS produced in response to GPCR agonists plays an important role in normal cell signaling to mediate cell growth, migration and gene expression. Excess amount of ROS produced during hypertension, diabetes, ischemia-reperfusion injury and heart failure contributes to cell death and apoptosis. In contrast, NO and SNOs not only activate cGMP-dependent signaling pathways but also directly modify the SH residues of proteins through S-nitrosylation, which has emerged as an important posttranslational modification regulating NO-mediated signaling. Under physiological condition, NO might provide protection to cells from oxidative stress by S-nitrosylation of some critical protein thiols, preventing them from further oxidative modification by ROS. Increased oxidative stress and the formation of peroxynitrite (OONO-) via reaction of NO with O2-, which can irreversibly damage cells by oxidation of free thiols and nitration of tyrosine residues, are implicated in the pathogenesis of cardiovascular diseases.
Role of ROS in GPCR AT1 receptor signaling
Angiotensin II (Ang II) is a pleuripotential hormone in vascular smooth muscle cells (VSMCs) and is a potent mediator of vascular hypertrophy, a hallmark of vascular wall remodeling and hypertension. These effects are mediated, in large part, through the GPCR AT1 receptors (AT1Rs). Many growth-related outputs of the AT1R are dependent upon the production of NAPDH oxides-derived ROS [5]. The proto-type phagocyte NADPH oxidase consists of membrane-bound gp91phox (now also termed Nox2) and p22phox which comprise the flavocytochrome b558, together with the cytosolic p47phox, p67phox p40phox and the small GTPase Rac. Non-phagocytic NADPH oxidase (Nox) consists of several homologs of Nox2 including Nox1, Nox3, Nox4, and Nox5, as well as the Dual oxidases (Duox) [6-9]. In VSMCs, Nox1 and Nox4 are mainly responsible for ROS generation [10]. Targets of NADPH oxidase-derived ROS include protein tyrosine kinases, protein tyrosine phosphatases (PTPs), protein kinase C, MAP kinases, Ca2+ channels, and transcription factors including NF-kB, activator protein-1, and Nrf2 [11, 12].
Activation of AT1R induces ROS-dependent transactivation of EGF receptor [13], which serves as a scaffold for the assembly of signaling complexes including cSrc and downstream adaptors Shc/GRB2 [14]. Transactivation of EGF receptor is necessary for activation of ERK1/2 and Akt, which are involved in vascular hypertrophy [14]. How ROS mediate Ang II-induced EGFR transactivation? Lipid rafts are cholesterol- and sphingolipid-rich, plasma membrane domains, and ‘caveolae’ constitute a distinct subset of lipid rafts with cell surface flask-shaped invaginations that contain caveolin-1 (Cav1) [15]. Caveolae/lipid rafts concentrate multiple signaling molecules including GPCRs, receptor tyrosine kinases such as EGF receptor, Src family kinases, and G proteins to form signaling platforms, which is necessary for rapid and efficient activation of downstream signaling pathways [16-18]. Thus, these specialized plasma membrane microdomains play an important role in activation of specific ROS-dependent redox signaling events [19-21]. In VSMCs, NADPH oxidase Nox1 is found in Cav1-containing membranes under unstimulated conditions [22], and Ang II stimulation promotes AT1R binding to Cav1 as well as trafficking of AT1R from high density non-caveolar membrane fractions into caveolae/lipid rafts [23]. This in turn promotes Rac1 translocation into caveolae/lipid rafts to increase localized ROS production [24], which is required for transactivation of EGF receptor [13]. Tyrosine phosphorylated EGF receptor and Cav1 through ROS-dependent cSrc activation subsequently appear at focal adhesions where NADPH oxidase Nox4 and paxillin localize, thereby forming redox signaling platforms. Either inhibition of NADPH oxidase or Cav1 knockdown blocks Ang II-induced increase in vascular hypertrophy [23, 25, 26]. These findings indicate that caveolae/lipid rafts are signaling domains in which AT1R couples with NADPH oxidase to increase ROS locally, thereby forming active redox signaling platforms involved in EGFR transactivation and vascular hypertrophy [21](Figure 1).
Figure 1. Caveolae-dependent AT1R-mediated redox signaling in VSMCs.
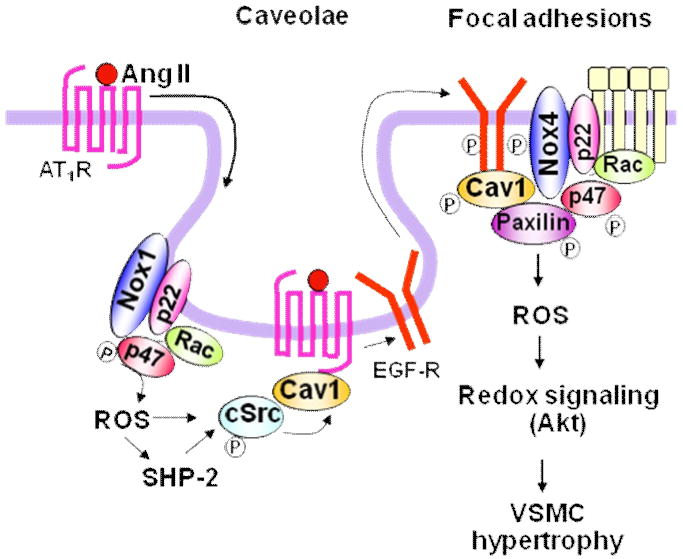
Ang II stimulation promotes AT1 receptor trafficking into caveolin1-enriched membrane fractions where Nox1 is found. This causes localized ROS production and ROS-cSrc dependent transactivation of the EGFR and its egress from caveolae. Tyrosine phosphorylated EGFR and caveolin-1 subsequently appear at focal adhesions where Nox4 and paxillin localize, thereby forming redox signaling platforms. These events are essential for activation of specific redox signaling pathways such as Akt involved in vascular hypertrophy. Note that SHP-2 is target of Nox1-derived ROS, which is involved in Akt activation, thereby contributing to AT1R-mediated hypertension.
Protein tyrosine phosphorylation is controlled by the tightly regulated balance between protein tyrosine kinases and PTPs [27]. The reversible oxidative inhibition of PTPs by ROS is an important mechanism through which ROS increase tyrosine phosphorylation signaling events [27-30]. PTP activity is dependent on the reactive Cys residues (Cys-SH) with a low pKa at their active site that are readily susceptible to reversible oxidation by H2O2 [31]. Tabet et al. [32] recently demonstrated that Ang II inactivates SHP-2 PTPs through Nox1-derived ROS-mediated oxidation and phosphorylation. In spontaneously hypertensive rats (SHRs) Ang II-stimulated SHP-2 oxidation/inactivation is enhanced, while its phosphorylation is blunted. These SHP-2 actions are associated with augmented Akt signaling, indicating the importance of Nox1-SHP2-Akt axis for Ang II-mediated hypertension in SHRs (Figure 1). Given that several PTPs including SHP-2 are localized in caveolae/lipid rafts [33], localization of AT1R, NADPH oxidase and its targets signaling molecules or kinases in caveolae may play an important role in specific activation of ROS-mediated GPCR signaling. Whether this ROS-dependent organized mechanism can apply to other GPCR signal transduction remains unknown.
Role of S-nitrosylation in GPCR signaling
Accumulating evidence indicates that GPCRs signaling is also regulated by S-nitrosylation of Cys residues at active or allosteric sites of proteins via NO and SNOs [34, 35]. The activation of neuronal NMDA receptors produces NO, which S-nitrosylates both the receptor itself as well as associated regulatory and effector proteins that play roles in NMDA receptor trafficking and signaling [34, 36-39]. Cys residues that are S-nitrosylated by NO have been identified in the β2-AR [40] and AT1R [41]. Other NO-mediated S-nitrosylated proteins include, Akt, caspases, cyclooxygenase-2, eNOS, Hsp90, estrogen receptor, hypoxia-inducible factor-1, NF-kB [42], adenylate cyclase [43], and calcium channel [44].
Functionally, SNOs treatment prevents tachyphylaxis to β2-AR agonists-induced vasodilation [45] or airway relaxation [45, 46]. Mice with a genetic alteration that impairs breakdown of SNOs are protected from tachyphylaxis [46]. Conversely, β2-AR tachyphylaxis is induced in vivo by the inhibition of NOS; [45], which depletes endogenous SNOs [47, 48]. Kokkola et al. [35] reported that SNOs such as S-nitrosoglutathione (GSNO) and S-nitrosocysteine (CysNO) potentiate M2/M4 muscarinic responses by increasing guanine nucleotide exchange as well as number of high-affinity [35S]GTPγS binding sites for the agonist-activated receptor. In contrast, SNOs inhibit P2Y12 receptor signaling in rat brain and human platelets without affecting human P2Y12 receptor signaling under heterologous expression in CHO cells. This indicates that the cellular signaling partners, rather than the P2Y12 receptor protein itself, are molecular target of SNOs. Furthermore, Nozik-Grayck et al. [49] reported that SNOs inhibit GPCRs 5-hydroxytryptamine- or α1-adrenergic receptor agonist-stimulated pulmonary vasoconstriction via cGMP-independent mechanism involving S-nitrosylation of the receptor or a related protein. Given that the endogenous NOS is located in close proximity with the GPCR signaling complex which is modulated by exogenous SNOs, these findings suggest that GPCR signaling in vivo is likely to be subject to receptor-specific modulation by NO and SNOs.
S-nitrosylation of GRK2, dynamin, and β-arrestin in GPCR β-adrenergic receptor signaling
Transduction through GPCRs is regulated by agonist-induced desensitization, in which receptors are functionally uncoupled from G protein activation, and by internalization, in which receptors undergo endocytosis followed by recycling or degradation [1, 2]. The G protein-coupled receptor kinases (GRKs) phosphorylate agonist-occupied GPCRs and promote their desensitization and internalization [3, 50]. Receptor phosphorylation promotes the recruitment of the scaffolding protein β-arrestins to the activated receptor, where they prevent further activation of heterotrimeric G proteins, thereby desensitizing the receptor while promoting clathrin-mediated receptor endocytosis [1-3]. NO directly regulates desensitization and internalization of GPCR by modulating the function of GPCR trafficking machinery through S-nitrosylation [34, 35, 40, 41, 51, 52]. Activation of the β-AR results in dynamic S-nitrosylation of both dynamin [53] [52] and GRK2 [54], which serves as critical components of GPCR trafficking.
Wang et al. [52] reported that the GTPase dynamin, which regulates endocytic vesicle budding from the plasma membrane and interacts with NOS, is S-nitrosylated at a single Cys607 after stimulation of the β2-AR. S-nitrosylation increases dynamin self-assembly and GTPase activity and facilitates its redistribution to the membrane. In NO-generating cells, expression of mutant dynamin C607A inhibits β2-AR internalization, while NO enhances internalization of β2-AR. Thus, NO regulates endocytic vesicle budding by S-nitrosylation of dynamin. Whalen et al. [54] reported how NO and/or SNOs prevent the loss of β-AR signaling in vivo. In mice, SNOs increase β-AR expression and prevent agonist-stimulated receptor downregulation; and in cells, SNOs decrease GRK2-mediated β-AR phosphorylation and subsequent recruitment of β-arrestin to the receptor, resulting in inhibiting receptor desensitization and internalization. GRK2 S-nitrosylation of Cys340 increases following stimulation of multiple GPCRs with agonists (Figure 2). These findings suggest novel mechanism through which GPCR signaling is regulated by NO and SNOs.
Figure 2. Regulation of agonist-induced β2-AR trafficking by S-nitrosylation of β-arrestin 2, GRK2 and dynamin.
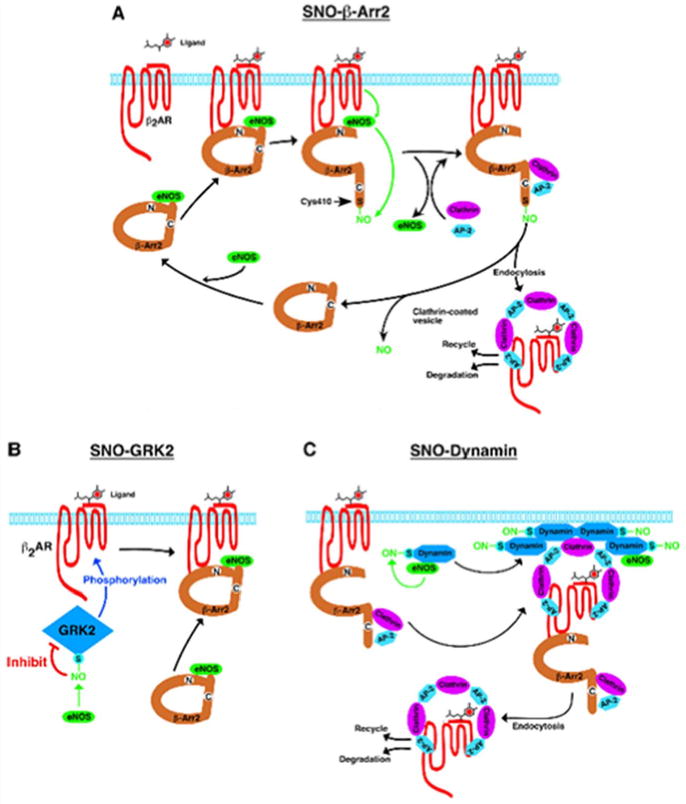
A, β-arrestin2 (β-Arr2) serves as a scaffold to functionally colocalize with eNOS and β-ARs. Ligand (isoproterenol) stimulation results in the activation of eNOS and the S-nitrosylation of β-arrestin2. S-nitrosylation of β-arrestin2 promotes its dissociation from eNOS and its association with clathrin HC/β-adaptin, which facilitates routing of the β2-AR into the clathrin-based endocytotic pathway, and β-arrestin2 is subsequently denitrosylated. B, Ligand-coupled inhibition of GRK2 by S-nitrosylation suppresses agonist-stimulated β-AR phosphorylation, β-arrestin recruitment, and receptor desensitization and downregulation. (Reprinted with permission from Ozawa et al. [55])
Ozawa et al. [55] recently reported that isoproterenol stimulates the S-nitrosylation of β-arrestin 2 by eNOS-derived SNOs. In human umbilical vein endothelial cells, S-nitrosylation of β-arrestin 2 promotes its dissociation from eNOS and its association with clathrin HC/β-adaptin, thereby facilitating internalization of the β2-AR into the clathrin-based endocytotic pathway, which in turn denitrosylating β-arrestin 2. S-nitrosylation seems to play an important role in regulating the β-arrestin-dependent trafficking of GPCRs including the AT1R that are functionally coupled to eNOS [56]. S-nitrosylation is the only known facilitatory modification of β-arrestins, because phosphorylation of β-arrestin 1 or 2 inhibits clathrin-mediated β2-AR internalization [57-59]. Selective S-nitrosylation of β-arrestin 2 versus β-arrestin 1 provides for differential regulation of these widely distributed isoforms. Of note, stimulus-induced dephosphorylation of Ser412 in β-arrestin 1 is prerequisite for clathrin binding and receptor endocytosis [57, 58]. In contrast, β-arrestin 2 does not contain an equivalent C-terminal Ser residue, but has a highly conserved Cys410. The dephosphorylation of Ser412 of β-arrestin 1 promotes clathrin binding, and this effect is recapitulated by S-nitrosylation of Cys410 within β-arrestin 2. Thus, S-nitrosylation of β-arrestin may co-operate with phosphorylation and is important mechanism by which NO influences GPCR function via regulating protein-protein interaction of β-arrestin 2 [52, 54](Figure 2).
β-arrestin-regulated β-AR trafficking is directed primarily through clathrin- rather than caveolar-based pathways [1, 2]. However, eNOS colocalizes with β-AR primarily in caveolae through association with the scaffolding protein Cav1. Since eNOS associates with both dynamin and β-arrestin, and indirectly with GRK2 through Akt [52, 54, 60], S-nitrosylation of these three receptor trafficking elements appear to be mediated by discrete pools of eNOS. Thus, coordinated changes in the S-nitrosylation of multiple, functionally interrelated receptor trafficking machinery subserves dynamic regulation by NO [34]. Whether S-nitrosylation/denitrosylation may influence the class characteristics of GPCRs by altering their affinity for β-arrestin 2, or whether S-nitrosylation differentially regulates signaling dependent upon β-arrestin isotypes [61, 62] are subject of future investigation.
Role of ROS and S-nitrosylation in GPCR signaling in vascular pathophysiologies
In VSMCs, production of ROS and the consequent activation of ROS-mediated GPCR signaling are involved in cell growth, migration, collagen deposition, and altered MMP activity [12]. In endothelial cells, NO/SNO-mediated S-nitrosylation of GPCR machinery plays an important role in normal GPCR trafficking and signaling. NO might provide protection to cells from oxidative stress by S-nitrosylation of some critical protein thiols, preventing them from further oxidative modification by ROS. Oxidative stress stimulates activation of transcription factors NF-kB and AP-1 and pro-inflammatory genes, chemokine production and recruitment of inflammatory cells (monocytes and macrophages), which are involved in vascular inflammation and injury [63]. Further excess amount of ROS impairs endothelium-dependent relaxation, increases GPCR-mediated contractility and alters vascular tone. These effects may be regulated indirectly by reducing NO bioavailability by reaction of O2- and NO to produce ONOO-. Thus, excess ROS production and loss of S-nitrosylation will be potential factor in vascular dysfunction that involves altered GPCR trafficking and signaling linked to hypertension, atherosclerosis, heart failure and diabetes.
Conclusion
NADPH oxidase-derived ROS and NOS-derived SNOs are now recognized as important second messengers to regulate GPCR signaling, thereby contributing to various physiological and pathophysiological functions. SNOs and protein S-nitrosylation may serve multiple roles to mitigate oxidation of proteins induced by NADPH oxidase-derived ROS produced following GPCR activation. Dysregulation of GPCR signaling and trafficking due to oxidative stress may contribute to various pathophysiologies in hypertension, atherosclerosis, heart failure and diabetes. Thus, further understand the molecular mechanisms of ROS- and NO-dependent GPCR signaling should provide new therapeutic targets for intervention in cardiovascular diseases.
Acknowledgments
Funding disclosure: NIH R01 HL077524 and AHA Grant-In-Aid 0555308B
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and recommended reading
- 1.Drake MT, Shenoy SK, Lefkowitz RJ. Trafficking of G protein-coupled receptors. Circulation research. 2006;99:570–582. doi: 10.1161/01.RES.0000242563.47507.ce. [DOI] [PubMed] [Google Scholar]
- 2.Reiter E, Lefkowitz RJ. GRKs and beta-arrestins: roles in receptor silencing, trafficking and signaling. Trends in endocrinology and metabolism: TEM. 2006;17:159–165. doi: 10.1016/j.tem.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 3.Pierce KL, Premont RT, Lefkowitz RJ. Seven-transmembrane receptors. Nature reviews. 2002;3:639–650. doi: 10.1038/nrm908. [DOI] [PubMed] [Google Scholar]
- 4.Griendling KK, Ushio-Fukai M, Lassegue B, Alexander RW. Angiotensin II signaling in vascular smooth muscle. New concepts Hypertension. 1997;29:366–373. doi: 10.1161/01.hyp.29.1.366. [DOI] [PubMed] [Google Scholar]
- 5.Griendling KK, Sorescu D, Ushio-Fukai M. NAD(P)H oxidase: role in cardiovascular biology and disease. Circ Res. 2000;86:494–501. doi: 10.1161/01.res.86.5.494. [DOI] [PubMed] [Google Scholar]
- 6.Lambeth JD, Cheng G, Arnold R, Edens WA. Novel homologs of gp91phox. Trends in Biochem Sci. 2000;25:459–461. doi: 10.1016/s0968-0004(00)01658-3. [DOI] [PubMed] [Google Scholar]
- 7.Geiszt M, Kopp JB, Varnai P, Leto TL. Identification of renox, an NAD(P)H oxidase in kidney. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:8010–8014. doi: 10.1073/pnas.130135897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Geiszt M. NADPH oxidases: new kids on the block. Cardiovascular research. 2006;71:289–299. doi: 10.1016/j.cardiores.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 9.Lambeth JD. Nox/Duox family of nicotinamide adenine dinucleotide (phosphate) oxidases. Curr Opin Hematol. 2002;9:11–17. doi: 10.1097/00062752-200201000-00003. [DOI] [PubMed] [Google Scholar]
- 10.Lassegue B, Sorescu D, Szocs K, Yin Q, Akers M, Zhang Y, Grant SL, Lambeth JD, Griendling KK. Novel gp91(phox) Homologues in Vascular Smooth Muscle Cells : nox1 Mediates Angiotensin II-Induced Superoxide Formation and Redox-Sensitive Signaling Pathways. Circulation research. 2001;88:888–894. doi: 10.1161/hh0901.090299. [DOI] [PubMed] [Google Scholar]
- 11.Paravicini TM, Touyz RM. Redox signaling in hypertension. Cardiovascular research. 2006;71:247–258. doi: 10.1016/j.cardiores.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 12.Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol. 2007;292:C82–97. doi: 10.1152/ajpcell.00287.2006. [DOI] [PubMed] [Google Scholar]
- 13.Ushio-Fukai M, Griendling KK, Becker PL, Hilenski L, Halleran S, Alexander RW. Epidermal growth factor receptor transactivation by angiotensin II requires reactive oxygen species in vascular smooth muscle cells. Arteriosclerosis, thrombosis, and vascular biology. 2001;21:489–495. doi: 10.1161/01.atv.21.4.489. [DOI] [PubMed] [Google Scholar]
- 14.Eguchi S, Iwasaki H, Hirata Y, Frank GD, Motley ED, Yamakawa T, Numaguchi K, Inagami T. Epidermal growth factor receptor is indispensable for c-Fos expression and protein synthesis by angiotensin II. Eur J Pharmacol. 1999;376:203–206. doi: 10.1016/s0014-2999(99)00357-x. [DOI] [PubMed] [Google Scholar]
- 15.Harder T, Simons K. Caveolae, DIGs, and the dynamics of sphingolipid-cholesterol microdomains. Curr Opin Cell Biol. 1997;9:534–542. doi: 10.1016/s0955-0674(97)80030-0. [DOI] [PubMed] [Google Scholar]
- 16.Okamoto T, Schlegel A, Scherer PE, Lisanti MP. Caveolins, a family of scaffolding proteins for organizing “preassembled signaling complexes” at the plasma membrane. The Journal of biological chemistry. 1998;273:5419–5422. doi: 10.1074/jbc.273.10.5419. [DOI] [PubMed] [Google Scholar]
- 17.Simons K, Toomre D. Lipid rafts and signal transduction. Nature reviews. 2000;1:31–39. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- 18.Quest AF, Leyton L, Parraga M. Caveolins, caveolae, and lipid rafts in cellular transport, signaling, and disease. Biochem Cell Biol. 2004;82:129–144. doi: 10.1139/o03-071. [DOI] [PubMed] [Google Scholar]
- 19.Vilhardt F, van Deurs B. The phagocyte NADPH oxidase depends on cholesterol-enriched membrane microdomains for assembly. Embo J. 2004;23:739–748. doi: 10.1038/sj.emboj.7600066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang AY, Yi F, Zhang G, Gulbins E, Li PL. Lipid raft clustering and redox signaling platform formation in coronary arterial endothelial cells. Hypertension. 2006;47:74–80. doi: 10.1161/10.1161/01.HYP.0000196727.53300.62. [DOI] [PubMed] [Google Scholar]
- 21.Callera GE, Montezano AC, Yogi A, Tostes RC, Touyz RM. Vascular signaling through cholesterol-rich domains: implications in hypertension. Curr Opin Nephrol Hypertens. 2007;16:90–104. doi: 10.1097/MNH.0b013e328040bfbd. [DOI] [PubMed] [Google Scholar]
- 22.Hilenski LL, Clempus RE, Quinn MT, Lambeth JD, Griendling KK. Distinct subcellular localizations of Nox1 and Nox4 in vascular smooth muscle cells. Arteriosclerosis, thrombosis, and vascular biology. 2004;24:677–683. doi: 10.1161/01.ATV.0000112024.13727.2c. [DOI] [PubMed] [Google Scholar]
- 23.Zuo L, Ushio-Fukai M, Ikeda S, Hilenski L, Patrushev N, Alexander RW. Caveolin-1 is essential for activation of Rac1 and NAD(P)H oxidase after angiotensin II type 1 receptor stimulation in vascular smooth muscle cells: role in redox signaling and vascular hypertrophy. Arteriosclerosis, thrombosis, and vascular biology. 2005;25:1824–1830. doi: 10.1161/01.ATV.0000175295.09607.18. [DOI] [PubMed] [Google Scholar]
- 24.Zuo L, Ushio-Fukai M, Hilenski LL, Alexander RW. Microtubules regulate angiotensin II type 1 receptor and Rac1 localization in caveolae/lipid rafts: role in redox signaling. Arteriosclerosis, thrombosis, and vascular biology. 2004;24:1223–1228. doi: 10.1161/01.ATV.0000132400.25045.2a. [DOI] [PubMed] [Google Scholar]
- 25.Ushio-Fukai M, Zafari AM, Fukui T, Ishizaka N, Griendling KK. p22phox is a critical component of the superoxide-generating NADH/NADPH oxidase system and regulates angiotensin II-induced hypertrophy in vascular smooth muscle cells. J Biol Chem. 1996;271:23317–23321. doi: 10.1074/jbc.271.38.23317. [DOI] [PubMed] [Google Scholar]
- 26.Zafari AM, Ushio-Fukai M, Akers M, Yin Q, Shah A, Harrison DG, et al. Role of NADH/NADPH oxidase-derived H2O2 in angiotensin II-induced vascular hypertrophy. Hypertension. 1998;32:488–495. doi: 10.1161/01.hyp.32.3.488. [DOI] [PubMed] [Google Scholar]
- 27.Tonks NK. Redox redux: revisiting PTPs and the control of cell signaling. Cell. 2005;121:667–670. doi: 10.1016/j.cell.2005.05.016. [DOI] [PubMed] [Google Scholar]
- 28.Lee SR, Kwon KS, Kim SR, Rhee SG. Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J Biol Chem. 1998;273:15366–15372. doi: 10.1074/jbc.273.25.15366. [DOI] [PubMed] [Google Scholar]
- 29.Finkel T. Signal transduction by reactive oxygen species in non-phagocytic cells. J Leukoc Biol. 1999;65:337–340. doi: 10.1002/jlb.65.3.337. [DOI] [PubMed] [Google Scholar]
- 30.Chiarugi P, Cirri P. Redox regulation of protein tyrosine phosphatases during receptor tyrosine kinase signal transduction. Trends Biochem Sci. 2003;28:509–514. doi: 10.1016/S0968-0004(03)00174-9. [DOI] [PubMed] [Google Scholar]
- 31.Wu Y, Kwon KS, Rhee SG. Probing cellular protein targets of H2O2 with fluorescein-conjugated iodoacetamide and antibodies to fluorescein. FEBS Lett. 1998;440:111–115. doi: 10.1016/s0014-5793(98)01415-x. [DOI] [PubMed] [Google Scholar]
- 32.Tabet F, Schiffrin EL, Callera GE, He Y, Yao G, Ostman A, et al. Redox-sensitive signaling by angiotensin II involves oxidative inactivation and blunted phosphorylation of protein tyrosine phosphatase SHP-2 in vascular smooth muscle cells from SHR. Circulation research. 2008;103:149–158. doi: 10.1161/CIRCRESAHA.108.178608. [DOI] [PubMed] [Google Scholar]; “ This recent paper shows that Ang II inactivates SHP-2 PTPs through Nox1-derived ROS-mediated oxidation and phosphorylation. In spontaneously hypertensive rats (SHRs) Ang II-stimulated SHP-2 oxidation/inactivation is enhanced, while its phosphorylation is blunted. These SHP-2 actions are associated with augmented Akt signaling, indicating the importance of Nox1-SHP2-Akt axis for Ang II-mediated hypertension in SHRs.
- 33.Caselli A, Mazzinghi B, Camici G, Manao G, Ramponi G. Some protein tyrosine phosphatases target in part to lipid rafts and interact with caveolin-1. Biochemical and biophysical research communications. 2002;296:692–697. doi: 10.1016/s0006-291x(02)00928-2. [DOI] [PubMed] [Google Scholar]
- 34.Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nature reviews. 2005;6:150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 35.Kokkola T, Savinainen JR, Monkkonen KS, Retamal MD, Laitinen JT. S-nitrosothiols modulate G protein-coupled receptor signaling in a reversible and highly receptor-specific manner. BMC cell biology. 2005;6:21. doi: 10.1186/1471-2121-6-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chung KK, Thomas B, Li X, Pletnikova O, Troncoso JC, Marsh L, et al. S-nitrosylation of parkin regulates ubiquitination and compromises parkin's protective function. Science (New York, NY. 2004;304:1328–1331. doi: 10.1126/science.1093891. [DOI] [PubMed] [Google Scholar]
- 37.Huang Y, Man HY, Sekine-Aizawa Y, Han Y, Juluri K, Luo H, Cheah J, Lowenstein C, Huganir RL, Snyder SH. S-nitrosylation of N-ethylmaleimide sensitive factor mediates surface expression of AMPA receptors. Neuron. 2005;46:533–540. doi: 10.1016/j.neuron.2005.03.028. [DOI] [PubMed] [Google Scholar]
- 38.Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH. Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nature cell biology. 2001;3:193–197. doi: 10.1038/35055104. [DOI] [PubMed] [Google Scholar]
- 39.Lipton SA, Choi YB, Pan ZH, Lei SZ, Chen HS, Sucher NJ, Loscalzo J, Singel DJ, Stamler JS. A redox-based mechanism for the neuroprotective and neurodestructive effects of nitric oxide and related nitroso-compounds. Nature. 1993;364:626–632. doi: 10.1038/364626a0. [DOI] [PubMed] [Google Scholar]
- 40.Adam L, Bouvier M, Jones TL. Nitric oxide modulates beta(2)-adrenergic receptor palmitoylation and signaling. The Journal of biological chemistry. 1999;274:26337–26343. doi: 10.1074/jbc.274.37.26337. [DOI] [PubMed] [Google Scholar]
- 41.Leclerc PC, Lanctot PM, Auger-Messier M, Escher E, Leduc R, Guillemette G. S-nitrosylation of cysteine 289 of the AT1 receptor decreases its binding affinity for angiotensin II. British journal of pharmacology. 2006;148:306–313. doi: 10.1038/sj.bjp.0706725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sun J, Steenbergen C, Murphy E. S-nitrosylation: NO-related redox signaling to protect against oxidative stress. Antioxidants & redox signaling. 2006;8:1693–1705. doi: 10.1089/ars.2006.8.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ostrom RS, Bundey RA, Insel PA. Nitric oxide inhibition of adenylyl cyclase type 6 activity is dependent upon lipid rafts and caveolin signaling complexes. The Journal of biological chemistry. 2004;279:19846–19853. doi: 10.1074/jbc.M313440200. [DOI] [PubMed] [Google Scholar]
- 44.Sun J, Picht E, Ginsburg KS, Bers DM, Steenbergen C, Murphy E. Hypercontractile female hearts exhibit increased S-nitrosylation of the L-type Ca2+ channel alpha1 subunit and reduced ischemia/reperfusion injury. Circulation research. 2006;98:403–411. doi: 10.1161/01.RES.0000202707.79018.0a. [DOI] [PubMed] [Google Scholar]
- 45.Whalen EJ, Johnson AK, Lewis SJ. Beta-adrenoceptor dysfunction after inhibition of NO synthesis. Hypertension. 2000;36:376–382. doi: 10.1161/01.hyp.36.3.376. [DOI] [PubMed] [Google Scholar]
- 46.Que LG, Liu L, Yan Y, Whitehead GS, Gavett SH, Schwartz DA, Stamler JS. Protection from experimental asthma by an endogenous bronchodilator. Science (New York, NY. 2005;308:1618–1621. doi: 10.1126/science.1108228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gow AJ, Chen Q, Hess DT, Day BJ, Ischiropoulos H, Stamler JS. Basal and stimulated protein S-nitrosylation in multiple cell types and tissues. The Journal of biological chemistry. 2002;277:9637–9640. doi: 10.1074/jbc.C100746200. [DOI] [PubMed] [Google Scholar]
- 48.Liu L, Yan Y, Zeng M, Zhang J, Hanes MA, Ahearn G, McMahon TJ, Dickfeld T, Marshall HE, Que LG, Stamler JS. Essential roles of S-nitrosothiols in vascular homeostasis and endotoxic shock. Cell. 2004;116:617–628. doi: 10.1016/s0092-8674(04)00131-x. [DOI] [PubMed] [Google Scholar]
- 49.Nozik-Grayck E, McMahon TJ, Huang YC, Dieterle CS, Stamler JS, Piantadosi CA. Pulmonary vasoconstriction by serotonin is inhibited by S-nitrosoglutathione. Am J Physiol Lung Cell Mol Physiol. 2002;282:L1057–1065. doi: 10.1152/ajplung.00081.2001. [DOI] [PubMed] [Google Scholar]
- 50.Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by beta-arrestins. Science (New York, NY. 2005;308:512–517. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- 51.Nozik-Grayck E, Whalen EJ, Stamler JS, McMahon TJ, Chitano P, Piantadosi CA. S-nitrosoglutathione inhibits alpha1-adrenergic receptor-mediated vasoconstriction and ligand binding in pulmonary artery. Am J Physiol Lung Cell Mol Physiol. 2006;290:L136–143. doi: 10.1152/ajplung.00230.2005. [DOI] [PubMed] [Google Scholar]
- 52.Wang G, Moniri NH, Ozawa K, Stamler JS, Daaka Y. Nitric oxide regulates endocytosis by S-nitrosylation of dynamin. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:1295–1300. doi: 10.1073/pnas.0508354103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kang-Decker N, Cao S, Chatterjee S, Yao J, Egan LJ, Semela D, Mukhopadhyay D, Shah V. Nitric oxide promotes endothelial cell survival signaling through S-nitrosylation and activation of dynamin-2. Journal of cell science. 2007;120:492–501. doi: 10.1242/jcs.03361. [DOI] [PubMed] [Google Scholar]
- 54.Whalen EJ, Foster MW, Matsumoto A, Ozawa K, Violin JD, Que LG, Nelson CD, Benhar M, Keys JR, Rockman HA, Koch WJ, Daaka Y, Lefkowitz RJ, Stamler JS. Regulation of beta-adrenergic receptor signaling by S-nitrosylation of G-protein-coupled receptor kinase 2. Cell. 2007;129:511–522. doi: 10.1016/j.cell.2007.02.046. [DOI] [PubMed] [Google Scholar]
- 55.Ozawa K, Whalen EJ, Nelson CD, Mu Y, Hess DT, Lefkowitz RJ, Stamler JS. S-nitrosylation of beta-arrestin regulates beta-adrenergic receptor trafficking. Molecular cell. 2008;31:395–405. doi: 10.1016/j.molcel.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]; “ In this recent publication, the authors reported that isoproterenol stimulates the S-nitrosylation of ?-arrestin 2 by eNOS-derived SNOs. In human umbilical vein endothelial cells, S-nitrosylation of ?-arrestin 2 promotes its dissociation from eNOS and its association with clathrin HC/?-adaptin, thereby facilitating internalization of the ?2-AR into the clathrin-based endocytotic pathway, which in turn denitrosylating ?-arrestin 2.
- 56.Dudzinski DM, Igarashi J, Greif D, Michel T. The regulation and pharmacology of endothelial nitric oxide synthase. Annual review of pharmacology and toxicology. 2006;46:235–276. doi: 10.1146/annurev.pharmtox.44.101802.121844. [DOI] [PubMed] [Google Scholar]
- 57.Lin FT, Krueger KM, Kendall HE, Daaka Y, Fredericks ZL, Pitcher JA, Lefkowitz RJ. Clathrin-mediated endocytosis of the beta-adrenergic receptor is regulated by phosphorylation/dephosphorylation of beta-arrestin1. The Journal of biological chemistry. 1997;272:31051–31057. doi: 10.1074/jbc.272.49.31051. [DOI] [PubMed] [Google Scholar]
- 58.Lin FT, Miller WE, Luttrell LM, Lefkowitz RJ. Feedback regulation of beta-arrestin1 function by extracellular signal-regulated kinases. The Journal of biological chemistry. 1999;274:15971–15974. doi: 10.1074/jbc.274.23.15971. [DOI] [PubMed] [Google Scholar]
- 59.Lin FT, Chen W, Shenoy S, Cong M, Exum ST, Lefkowitz RJ. Phosphorylation of beta-arrestin2 regulates its function in internalization of beta(2)-adrenergic receptors. Biochemistry. 2002;41:10692–10699. doi: 10.1021/bi025705n. [DOI] [PubMed] [Google Scholar]
- 60.Liu S, Premont RT, Kontos CD, Zhu S, Rockey DC. A crucial role for GRK2 in regulation of endothelial cell nitric oxide synthase function in portal hypertension. Nature medicine. 2005;11:952–958. doi: 10.1038/nm1289. [DOI] [PubMed] [Google Scholar]
- 61.Lefkowitz RJ, Rajagopal K, Whalen EJ. New roles for beta-arrestins in cell signaling: not just for seven-transmembrane receptors. Molecular cell. 2006;24:643–652. doi: 10.1016/j.molcel.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 62.Rajagopal K, Whalen EJ, Violin JD, Stiber JA, Rosenberg PB, Premont RJ, Coffman TM, Rockman HA, Lefkowitz RJ. Beta-arrestin2-mediated inotropic effects of the angiotensin II type 1A receptor in isolated cardiac myocytes. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:16284–16289. doi: 10.1073/pnas.0607583103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Madamanchi NR, Vendrov A, Runge MS. Oxidative stress and vascular disease. Arteriosclerosis, thrombosis, and vascular biology. 2005;25:29–38. doi: 10.1161/01.ATV.0000150649.39934.13. [DOI] [PubMed] [Google Scholar]