

Abstract
Huntington's disease (HD) is an inherited neurodegenerative disease caused by the expansion of a polyglutamine tract in the protein huntingtin (htt). HD brains are characterized by the presence of ubiquitin‐positive neuronal inclusion bodies, suggesting that disturbances in the distribution of cellular ubiquitin may contribute to disease pathology. The fact that several neurodegenerative diseases are caused by mutations in ubiquitin‐processing enzymes and that the polyubiquitin genes are required for resistance to cellular stress led us to investigate the effect of perturbing the ubiquitin system in HD. We crossed R6/2 transgenic HD mice with heterozygous polyubiquitin Ubc knockout mice (Ubc+/−) and assessed the effect on the R6/2 neurological phenotype. Although the R6/2 phenotype was largely unaffected, surprisingly we observed some subtle improvements in various behavioural activities correlating with heterozygous Ubc knockout. Interestingly, immunoblot analysis revealed that the levels of monoubiquitylated histone H2A (uH2A), a modification associated with gene repression, were significantly increased in the brains of R6/2 mice. Furthermore, the reduction of Ubc expression in R6/2; Ubc+/− mice largely prevented this increase in uH2A levels. However, we were not able to show by the use of a limited number of quantitative RT‐PCR assays that changes in the amount of uH2A in the R6/2‐Ubc mice had an effect on disease‐associated transcriptional abnormalities. These results suggest that the expression of aggregation‐prone mutant htt causes disturbances to the ubiquitin system, which may contribute to disease due to the diverse and important roles of ubiquitin.
Keywords: polyglutamine, Huntington's disease, ubiquitin, aggregation, histone modification, neurodegeneration
Introduction
Huntington's disease (HD) is an autosomal dominant neurodegenerative disorder caused by the expansion of a polyglutamine tract in the N‐terminal of huntingtin (htt), a 350‐kD protein with incompletely defined function [1]. Individuals with 40 or more contiguous glutamine repeats will go on to develop HD within a normal lifespan, whereas the disease shows incomplete penetrance in those with 36–39 repeats. At least eight other neurodegenerative diseases are caused by a polyglutamine expansion in unrelated proteins, including several spinocerebellar ataxias, dentatorubral pallydoluysian atrophy and Kennedy's disease.
The accumulation of aggregated mutant N‐terminal htt fragments in transgenic HD mice [2] and post‐mortem HD neurons [3] is consistent with the ability of mutant polyglutamine proteins to aggregate in vitro[4]. Full‐length htt is believed to undergo proteolytic cleavage leading to a highly toxic and aggregation prone N‐terminal fragment in HD brains [5]. The aggregation of mutant htt fragments has been proposed to mediate toxicity through various mechanisms including aberrant interactions and subsequent coaggregation of essential cellular proteins such as CBP and TBP [6, 7], disturbing protein folding homeostasis [8] and impairment of the ubiquitin‐proteasome system (UPS) [9].
Although investigations into UPS impairment as a prerequisite for polyglutamine disease pathogenesis have revealed conflicting results [9, 10, 11, 12, 13], it is now becoming clear that alterations in the partitioning of ubiquitin into Lys‐conjugated chains is tightly associated with disease progression [14]. Because essential cellular processes such as transcription, DNA repair and endocytosis are all regulated to a considerable extent by ubiquitin, perturbations in ubiquitin levels or its homeostasis could contribute to cellular dysfunction in polyglutamine disease. In addition, there is strong evidence that mutations in ubiquitin‐processing enzymes lead to neurodegenerative disease in humans. For example, mutations in the E3 ubiquitin ligase Parkin lead to autosomal recessive juvenile Parkinson's disease [15], whereas distinct point mutations in the deubiquitylating enzyme UCH‐L1 gene have been associated with both increased and decreased susceptibility to Parkinson's disease [16, 17]. Furthermore, knocking this gene out in mice leads to the neurodegenerative condition gracile axonal dystrophy [18].
Ubiquitin is transcribed from four loci in mammals, and in each case it is translated as a precursor protein. The Uba genes are translated as one ubiquitin monomer fused to either the large ribosomal subunit L40 (Uba52) or the small ribosomal protein S27a (Uba80) [19]. The remaining ubiquitin genes Ubb and Ubc encode stress‐inducible polyubiquitin precursor proteins of around 3–4 or 9–11 monomers, respectively [20, 21], which are cleaved into monomeric ubiquitin by cellular deubiquitylating enzymes. Studies in yeast suggest that the ribosome‐fusion genes provide the bulk of cellular ubiquitin under normal circumstances, whereas the single polyubiquitin gene is required for resistance to various cellular stresses [22, 23].
We set out to investigate the regulation of ubiquitin in a mouse model of HD and test the hypothesis that the mammalian polyubiquitin genes would be protective against the toxic effects of pathogenic polyglutamines. To investigate this, Ubc heterozygous knockout mice (Ubc+/−) were generated [20] and crossed to R6/2 transgenic HD mice. Contrary to expectations, the loss of one Ubc allele actually improved performance of certain R6/2 behavioural activity phenotypes (rearing and centre‐rearing) while having no significant effect on other outcome measures. Levels of free monomeric and total ubiquitin were normal in the R6/2 brain, but the loss of one Ubc allele was sufficient to cause a small but significant decrease in total brain ubiquitin levels. Interestingly, we found that the levels of monoubiquitylated histone H2A (uH2A) were dramatically increased in R6/2 brains, but not in R6/2 brains heterozygous for the Ubc allele. This suggests that the modest decrease in total brain ubiquitin levels was sufficient to significantly reduce increased uH2A levels in R6/2; Ubc+/− mice. However, we were unable to detect alterations in the expression of various transcriptionally dysregulated disease genes between R6/2 and R6/2; Ubc+/− brains. These findings show that although a polyubiquitin stress response is not activated in R6/2 mice, the mutant htt‐induced alterations in ubiquitin partitioning may be important in disease pathogenesis.
Materials and methods
Mouse husbandry and behavioural assessment
Mice were housed and all experimental procedures were performed in accordance with Home Office regulations. Mice had an unlimited supply of water and number 3 rodent breeding chow (Special Diet Services, Witham, UK) and were subject to a 12‐hr light and a 12‐hr dark cycle. Mice were housed 5 per cage with environmental enrichment as described previously [24]. The R6/2 mouse colony was maintained by backcrossing R6/2 males to (C57BL/6JxCBA) F1 females (B6CBAF1/OlaHsd, Harlan Olac, Bichester, UK), and R6/2 mice were identified by PCR genotyping as described previously [24]. Ubc heterozygous knockout mice were generated on mixed (129/sv(J) × c57BL/6J) background and genotyped by PCR as described previously [20]. Rotarod analysis and grip strength assessment were performed as previously described [11], and mice were weighed weekly to the nearest 0.1 g. Exploratory activity of mice was performed as previously described [25].
Western blot analysis
Mice were sacrificed by cervical dislocation and brains were quickly dissected and frozen in isopropanol chilled with dry ice. Half brains were homogenized in 1 ml of sodium phosphate buffer (20 mM sodium phosphate, 1% sodium dodecyl sulphate [SDS] and complete protease inhibitors [Boehringer Mannheim, Mannheim, Germany]) and sonicated for 30 sec. on ice (amplitude 40, Vibracell sonicator). Protein concentration of the lysates was determined using the BCA assay kit (Perbio, Cramlington, UK) and 20 μg protein were boiled for 1 min. then loaded onto 12.5% SDS gels, separated and blotted onto PVDF membrane. For ubiquitin immunoblotting, membranes were boiled for 30 min. in dH2O. Membranes were blocked in 5% non‐fat fried milk (NFDM) for 1 hr at room temperature and probed with primary antibody in NFDM for 2 hrs at room temperature. Blots were then washed four times in PBS and then incubated with secondary antibody diluted in NFDM. Blots were washed again in PBS and proteins were detected by chemiluminescence (ECL kit, GE Healthcare, Amersham, UK) according to the manufacturer's instructions. Protein levels were quantified on a Bio‐Rad GS‐800 Calibrated Densitometer using Quantity‐One® Software, and four brains of each genotype were used for quantification. Primary antibodies and dilutions were as follows: ubiquitin (Dako, Ely, UK) (rabbit pAb, 1:1000) and α‐tubulin (Sigma‐Aldridge, Dorset, UK) (mAb, 1:2000). Secondary antibody dilutions were: HRP‐linked anti‐rabbit (Dako) (1:3000) and anti‐mouse (Vector Laboratories, Peterborough, UK) (1:5000). Membranes were stripped by incubation in stripping buffer (100 mM 3‐mercaptoethanol, 2% SDS, 62.5 mM Tris‐HCl pH 6.7) for 20 min. at 50°C with occasional agitation.
Indirect competitive enzyme‐linked immunosorbent assay
Brain hemispheres were homogenized in 1 ml hypotonic buffer (10 mM sodium phosphate, 0.1% digitonin and complete protease inhibitors) and incubated on ice for 20 min. Protein concentration was measured using the BCA kit (Perbio, Cramlington, UK) and adjusted to 5 μg/μl. Brain ubiquitin concentrations in untreated and Usp2‐cc‐treated samples were calculated using a previously published protocol described in [26].
Quantitative real‐time RT‐PCR analysis
Mice were sacrificed by cervical dislocation and brains rapidly removed. Striatum, cortex and cerebellum were dissected and flash‐frozen in liquid nitrogen and stored at –80°C until use. RNA was prepared from dissected striatum, cortex and cerebellum from animals at 14 weeks of age using the RNeasy lipid mini kit (Qiagen, Crawley, UK) according to manufacturer's instructions. Quality and quantity of RNA was assessed using the RNA nanochip method on a BioAnalzer (Agilent Technologies, South Queensferry, UK). Reverse transcription of total RNA (5 μg from cerebellum and cortex, 2 μg from striatum) was performed with 14 U/μl MMLV RTase (Invitrogen, Paisley, UK) in 50 mM KCl, 10 mM Tris‐HCl (pH 9.0), 0.1% Triton X‐100, 6.5 mM MgCl2, 10 mM DTT, 1 mM dNTPs, 10 ng/μl random hexamers with 0.35 U/μl RNasin (Promega, Southampton, UK) at 10′@ 23°C, 40′@ 37°C, 5′@ 94°C, 15′@ 4°C then stored at –20°C. The RT reaction was diluted 1:10 in nuclease‐free water (Sigma, Dorset, UK). Taqman real‐time PCR reactions were performed in triplicate with 5 μl of cDNA template in a 25‐μl reaction containing Precision Mastermix (PrimerDesign), 300 nM primers and 200 nM probe using the Opticon 2 real‐time PCR machine (MJ Research, GMI Inc., Ramsey, MN, USA). Crossing threshold data were obtained during the logarithmic phase of amplification where efficiency was close to 100%. Primer and probe sequences used were: Uba52 Fwd 5′‐GTCAGCTTGCCCAGAAGTAC‐3′, Rev 5′‐ACTTCTTCTTGCGGCAGTTG‐3′ (Sybr green); Uba80: Fwd 5′‐GGCAAAATTAGCCGACTTCGT‐3′, Rev 5′‐TCAAAGTGGCTTCCCATGAAAA‐3′ probe 6‐FAM‐TGTCCTTCTGATGAATGTGGTGCTGGA‐TAMRA; Ubb: Fwd 5′‐CACTGAGTGACGAGAGGCTTG‐3′, Rev 5′‐CCAGCGCGTCGAAACA‐3′, probe 6‐FAM‐CCGGTTCGGCGGTCTTTCTGTGA‐TAMRA Ubc: Fwd 5′‐ATGCAGCTACCGCCTGGTT‐3′, Rev 5′‐GCAGCTGTCCTTCACATTCCA‐3′, probe 6‐FAM‐AGGCGACATCAATTTCCTGGCGTG‐TAMRA. All other assays used are described in [27]. Relative quantification analyses were performed using the 2−ΔΔCt method [28], using the geometric mean of three reference genes as a calibrator [29]. All reference genes are available as premixed reactions from Primer Design (Southampton, UK). Striatal expression was normalized to the geometric mean of Atp5b, Eif4a2 and Actb (fl‐actin); cerebellar expression was normalized to the geometric mean of Atp5b, Eif4a2 and Rpl13a.
Statistics
General Linear Model ANOVA and post hoc analysis was performed on Excel and Minitab for behavioural, ELISA and real‐time assays. For Western blot quantification, Student's t‐test was performed.
Results
Ubiquitin gene expression in R6/2‐Ubc mice
In order to investigate the potential role of the polyubiquitin stress response on the progression of an HD mouse model, Ubc heterozygous knockout mice were crossed with R6/2 transgenic HD mice. Although complete Ubc knockout mice die in mid‐gestation, heterozygotes survive into adulthood and are phenotypically normal [20]. R6/2 males of breeding age were mated with Ubc+/− females to generate progeny of four genotypes: WT, R6/2, Ubc+/− and R6/2; Ubc+/− in an equal ratio. Before assessing the affect of Ubc heterozygosity on the mutant polyglutamine‐induced R6/2 neurological phenotype, expression of the ubiquitin genes in striatum and cerebellum was carried out on all four genotypes. It was found that expression of all four ubiquitin genes was normal in these end‐stage (14 week) R6/2 brain regions (Fig. 1), and expression of Uba52, Uba80 and Ubb was normal in mice heterozygous for Ubc. Importantly, the expression of Ubc was reduced in both Ubc+/− and R6/2; Ubc+/− brains at 14 weeks (Fig. 1). However, although this was significant in cerebellum (GLM ANOVA: F (1,33)= 11.95, P= 0.002) and cortex (data not shown), it fell out of statistical significance in striatum (GLM ANOVA: F (1,31)= 2.22, P= 0.147). Because heterozygous knockout of Ubc appears to cause reduction in Ubc mRNA transcript, it was possible to test the effect of reduction of Ubc expression on the neurological phenotype of R6/2.
Figure 1.
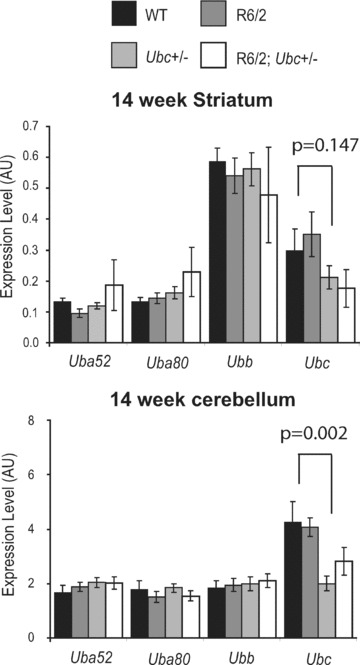
Expression of the ubiquitin genes in R6/2‐Ubc mice. Expression of the four ubiquitin genes was normal in 14 week (end‐stage) R6/2 striatum and cerebellum. Knockout of one Ubc allele can be seen to reduce the levels of Ubc expression in R6/2 and non‐R6/2 striatum and cerebellum. Although significant in cerebellum (P= 0.002) and cortex (data not shown), this reduction of Ubc is not significant in the striatum (P= 0.147). Error bars show standard error of the mean.
Reduction in Ubc does not affect R6/2 rotarod, grip strength or weight loss
To investigate the importance of the Ubc polyubiquitin gene in HD pathogenesis, 8–10 females of the four genotypes (WT, n= 8; Ubc+/−, n= 9; R6/2, n= 9; and R6/2; Ubc+/−, n= 10) were generated. The CAG repeat size of the R6/2 mice was well matched, where CAG repeat means were 206.9 (R6/2) and 205.6 (R6/2; Ubc+/−). These female mice were dispersed carefully between cages to ensure a variety of different genotypes from different mothers were represented in each cage, and all mice were born within 3 days of one another. Mice were then subject to a battery of behavioural tests to assess any alteration of the R6/2 phenotype caused by loss of one Ubc allele.
Firstly, mice were subject to rotarod analysis at 4, 8, 10 and 12 weeks of age. It was found that R6/2 mice performed worse than non‐R6/2 mice at 8 weeks (GLM ANOVA: F (1,35)= 6.73, P= 0.014) and onwards (Fig. 2A). The reduction of one Ubc allele did not have a statistically significant effect on the rotarod performance of R6/2 mice at any time‐point (e.g. at 12 weeks there is no interaction between R6/2 and Ubc genotypes GLM ANOVA: F (1,35)= 0.22, P= 0.22).
Figure 2.
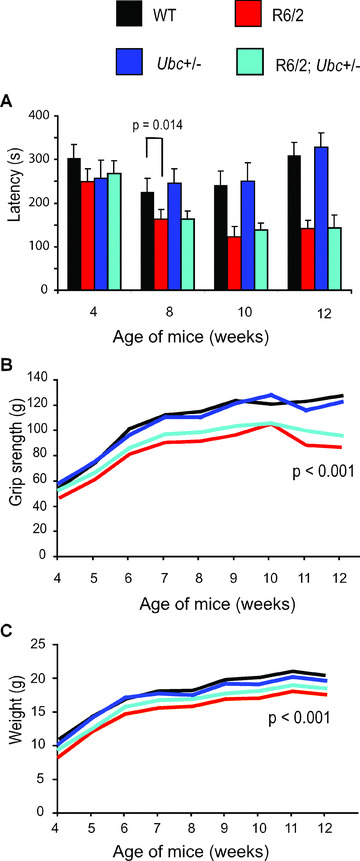
Behavioural characterization of R6/2‐Ubc mice. (A) Rotarod performance of R6/2 mice is significantly worse than that of non‐R6/2 mice from 8 weeks of age onwards. Knocking out one copy of Ubc has no effect on the rotarod performance of R6/2 mice. (B) Grip strength of R6/2 mice is worse than non‐R6/2 mice from 5 weeks onwards. Knocking out one Ubc allele has no significant effect on R6/2 grip strength. (C) R6/2 mice weigh significantly less than non‐R6/2 mice, and genetic reduction of Ubc has no statistically significant effect on this. Error bars represent the standard error of the mean.
R6/2‐Ubc mice were also subject to a weekly analysis of grip strength from 4 until 12 weeks of age. It was found that R6/2 mice performed significantly worse than non‐R6/2 mice from 5 weeks of age onwards (e.g. at 12 weeks, GLM‐ANOVA: F (1,35)= 34.58; P < 0.001) (Fig. 2B). Although the deletion of one copy of Ubc did not significantly affect the R6/2 deficit in grip strength at any time‐point (e.g. at 12 weeks, no interaction between R6/2 and Ubc genotypes GLM ANOVA: F (1,35)= 1.55; P= 0.222), the trend of the graph shows that R6/2; Ubc+/− mice have a small but consistently stronger grip than R6/2 at all time‐points (Fig. 2B).
The weight of R6/2‐Ubc mice was monitored from 4 weeks until 12 weeks. It was found that R6/2 mice weigh significantly less than non‐R6/2 mice from 4 weeks onwards (e.g. at 4 weeks GLM ANOVA: F (1,35)= 4.36; P= 0.045) (Fig. 2C). In agreement with the rotarod and grip strength data, the genetic reduction of Ubc has no statistically significant effect on the R6/2 weight phenotype (e.g. at 12 weeks, no interaction between R6/2 and Ubc genotypes GLM ANOVA: F (1,35)= 1.46; P= 0.236), but again R6/2; Ubc+/− mice appear to consistently weigh slightly more than R6/2 mice at all time‐points (Fig. 2C).
Ubc heterozygosity improves rearing and centre‐rearing activities of R6/2
In addition to rotarod performance, grip strength assessment and weight monitoring, R6/2‐Ubc mice were placed in activity cages and their spontaneous motor activity was monitored over a 1‐hr period (‘exploratory’ activity) every week from 4 weeks until 12 weeks of age. Mice of all genotypes were very active to begin with, and this initial activity gradually declined over the course of the 1‐hr period (Fig. 3). This effect was more pronounced for Activity and Mobility than for Rearing and Centre‐rearing, and is reflected by the large incidence of significant P‐values throughout Table 1 under ‘Time’. From week 7 onwards, R6/2 mice were observed to have less initial activity than non‐R6/2 mice, therefore their activity changes less than non‐R6/2 over the 1‐hr period (Fig. 3). This is reflected by the significant P‐values in Table 1 under ‘Time X R6/2’, but is less obvious for centre‐rearing than the other three parameters however. Ubc genotype has no effect on the time‐dependent decrease in activity (see Table 1 under ‘Time X Ubc’) and loss of one Ubc allele does not affect the change in activity over 1 hr in R6/2 mice (see Table 1 under ‘Time X R6/2; Ubc+/−’).
Figure 3.
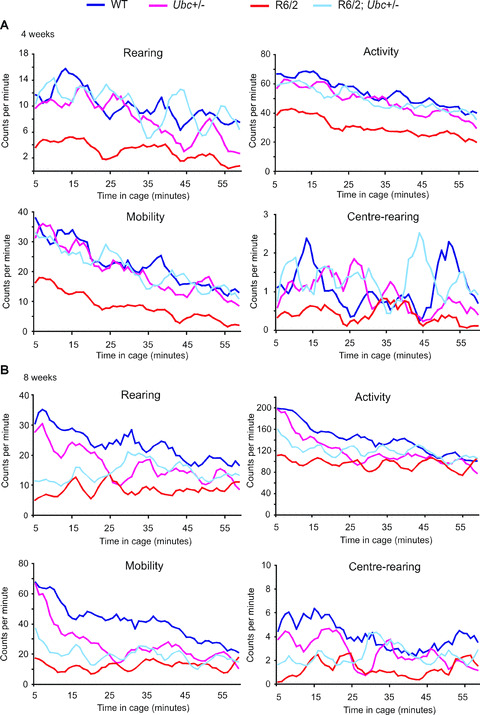
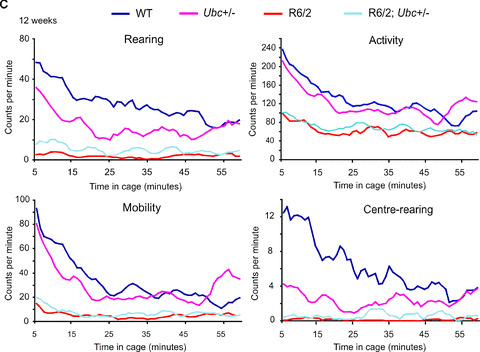
Exploratory activity of R6/2‐Ubc mice. Graphs of exploratory activity, depicting 5‐min. moving averages, for R6/2‐Ubc mice are shown for (A) 4 weeks (B) 8 weeks and (C) 12 weeks of age. All mice tend to be more active when first placed in the cage, and then this initial activity declines over the course of the hour. R6/2 mice display less initial activity than non‐R6/2 from 7 weeks onwards (Table 1). R6/2; Ubc+/− mice were observed to be generally more active than R6/2 mice, and this is statistically significant for rearing and centre‐rearing at all time‐points (Table 2).
Table 1.
The effect of time on R6/2; Ubc mice activity
| Week | Activity | Rearing | Centre‐rearing | Mobility | |
|---|---|---|---|---|---|
| Time | 4 | <0.001 | 0.004 | 0.604 | <0.001 |
| 5 | <0.001 | 0.004 | 0.107 | <0.001 | |
| 6 | <0.001 | 0.385 | 0.374 | 0.005 | |
| 7 | <0.001 | 0.113 | 0.092 | 0.008 | |
| 8 | <0.001 | 0.017 | 0.192 | <0.001 | |
| 9 | <0.001 | 0.014 | 0.038 | <0.001 | |
| 10 | <0.001 | <0.001 | 0.028 | <0.001 | |
| 11 | <0.001 | 0.001 | 0.071 | <0.001 | |
| 12 | <0.001 | <0.001 | 0.007 | <0.001 | |
| Time X R6/2 | 4 | 0.728 | 0.631 | 0.564 | 0.517 |
| 5 | 0.217 | 0.482 | 0.377 | 0.293 | |
| 6 | 0.118 | 0.501 | 0.635 | 0.271 | |
| 7 | 0.007 | 0.023 | 0.34 | 0.045 | |
| 8 | <0.001 | <0.001 | 0.202 | 0.002 | |
| 9 | 0.001 | 0.026 | 0.286 | 0.002 | |
| 10 | <0.001 | 0.046 | 0.341 | <0.001 | |
| 11 | <0.001 | <0.001 | 0.039 | <0.001 | |
| 12 | <0.001 | <0.001 | 0.003 | <0.001 | |
| Time X Ubc | 4 | 0.869 | 0.417 | 0.408 | 0.735 |
| 5 | 0.642 | 0.1 | 0.342 | 0.356 | |
| 6 | 0.195 | 0.45 | 0.504 | 0.462 | |
| 7 | 0.838 | 0.936 | 0.655 | 0.783 | |
| 8 | 0.332 | 0.586 | 0.616 | 0.274 | |
| 9 | 0.429 | 0.779 | 0.719 | 0.399 | |
| 10 | 0.295 | 0.489 | 0.776 | 0.246 | |
| 11 | 0.478 | 0.193 | 0.132 | 0.466 | |
| 12 | 0.129 | 0.199 | 0.02 | 0.139 | |
| Time X R6/2; Ubc+/− | 4 | 0.715 | 0.711 | 0.463 | 0.668 |
| 5 | 0.437 | 0.315 | 0.332 | 0.351 | |
| 6 | 0.433 | 0.321 | 0.252 | 0.468 | |
| 7 | 0.509 | 0.434 | 0.192 | 0.534 | |
| 8 | 0.737 | 0.468 | 0.538 | 0.668 | |
| 9 | 0.454 | 0.5 | 0.537 | 0.491 | |
| 10 | 0.617 | 0.482 | 0.485 | 0.253 | |
| 11 | 0.69 | 0.815 | 0.221 | 0.577 | |
| 12 | 0.121 | 0.083 | 0.046 | 0.108 |
Table of P‐values showing effect of time on activity of R6/2‐Ubc mice from 4 to 12 weeks of age.
R6/2 mice appear to be less active than non‐R6/2 mice from 4 weeks until 12 weeks (Fig. 3) and this is statistically significant from 8 weeks onwards (see Table 2 under ‘R6/2’). In contrast, Ubc genotype has no effect on activity (see Table 2 under ‘Ubc’). Interestingly, R6/2; Ubc+/− mice appeared to be generally more active than R6/2 mice however (Fig. 3), suggesting that the loss of Ubc allele improves certain polyglutamine‐induced disease phenotypes, albeit modestly. Indeed, this increase in activity was found to be significant for rearing and centre‐rearing from 4 weeks of age until 12 weeks (see Table 2 under ‘R6/2 X Ubc’).
Table 2.
General exploratory activity of R6/2; Ubc mice
| Week | Activity | Rearing | Centre‐rearing | Mobility | |
|---|---|---|---|---|---|
| R6/2 | 4 | 0.135 | 0.249 | 0.693 | 0.1 |
| 5 | 0.169 | 0.131 | 0.026 | 0.165 | |
| 6 | 0.375 | 0.671 | 0.56 | 0.376 | |
| 7 | 0.349 | 0.8 | 0.63 | 0.323 | |
| 8 | 0.112 | 0.001 | 0.034 | 0.011 | |
| 9 | 0.002 | 0.003 | 0.012 | 0.001 | |
| 10 | <0.001 | 0.025 | <0.001 | <0.001 | |
| 11 | <0.001 | <0.001 | <0.001 | <0.001 | |
| 12 | <0.001 | <0.001 | <0.001 | <0.001 | |
| Ubc | 4 | 0.462 | 0.262 | 0.343 | 0.236 |
| 5 | 0.174 | 0.31 | 0.678 | 0.228 | |
| 6 | 0.451 | 0.361 | 0.594 | 0.416 | |
| 7 | 0.343 | 0.074 | 0.066 | 0.388 | |
| 8 | 0.921 | 0.869 | 0.884 | 0.608 | |
| 9 | 0.955 | 0.339 | 0.706 | 0.851 | |
| 10 | 0.782 | 0.715 | 0.148 | 0.935 | |
| 11 | 0.626 | 0.577 | 0.273 | 0.572 | |
| 12 | 0.962 | 0.215 | 0.052 | 0.821 | |
| R6/2 X Ubc | 4 | 0.102 | 0.033 | 0.142 | 0.071 |
| 5 | 0.055 | 0.099 | 0.012 | 0.073 | |
| 6 | 0.192 | 0.022 | 0.021 | 0.179 | |
| 7 | 0.292 | 0.03 | 0.003 | 0.353 | |
| 8 | 0.091 | 0.017 | 0.068 | 0.114 | |
| 9 | 0.797 | 0.033 | 0.016 | 0.677 | |
| 10 | 0.44 | 0.89 | 0.005 | 0.683 | |
| 11 | 0.524 | 0.043 | 0.006 | 0.659 | |
| 12 | 0.463 | 0.029 | 0.016 | 0.502 |
Table of P‐values showing changes in general exploratory activity of R6/2‐Ubc mice from 4 to 12 weeks of age.
Knocking out one Ubc allele reduces ubiquitin protein levels
The behavioural assessment of R6/2‐Ubc mice demonstrates that knocking out one allele of Ubc actually causes a modest improvement of certain mutant N‐terminal htt‐mediated behavioural activities in R6/2 mice. This was unexpected, as expression of polyubiquitin genes such as Ubc is known to be protective against a wide range of cellular insults in eukaryotes [22].
Therefore, genetically reducing its expression in R6/2 mice was hypothesized to result in a worsening of the disease. In order to investigate possible explanations for this improvement in R6/2 behaviour, total brain hemisphere ubiquitin content was measured across the four genotypes. Quantification of total ubiquitin in tissues is notoriously difficult by immunoblot analysis [30] and is complicated by the fact that ubiquitin is partitioned between its free monomeric state, its covalent attachment to cellular proteins (e.g. monoubiquitylation of histone H2A) and by its polymerization into Lys‐linked polyubiquitin chains. We therefore quantified total ubiquitin in brain samples by converting all ubiquitin isopeptide linkages into monomeric form by treatment of tissues with the ubiquitin‐specific protease Usp2‐cc, followed by quantification by an indirect competitive enzyme‐linked immunosorbent assay (ELISA) [26]. This technique was employed to measure ubiquitin levels in 13‐week‐old brains from the R6/2‐Ubc cross.
Treatment of brain extract with Usp2‐cc followed by Western blot analysis confirms that all visible ubiquitin conjugates were successfully converted into monomeric ubiquitin (Fig. 4A). Six 13‐week‐old brains of all four genotypes were then homogenized and adjusted to the same protein concentration, and total ubiquitin levels were measured in each genotype by treating brain samples with Usp2‐cc prior to performing the ELISA. Untreated samples were also included in parallel to give an indication of what proportion of cellular ubiquitin in mice of different genotypes exists as monomer, as the ELISA is performed using an antibody that preferentially detects monomeric ubiquitin. Commercially available purified ubiquitin protein was attained and two standard curves of known ubiquitin concentration were run alongside the untreated samples on each plate (Fig. 4B). This allowed accurate quantification of total ubiquitin levels in each sample, and it was found that ubiquitin concentrations in the brain were consistent with those for mammalian cells [26] (Fig. 4C). In all genotypes, total tissue ubiquitin levels were only modestly greater than estimates of monomeric ubiquitin in untreated samples (Fig. 4C). The presence of N‐terminal mutant htt did not significantly affect levels of total ubiquitin in the R6/2 brain (no effect of R6/2 genotype on total ubiquitin levels, GLM ANOVA: F(1,39) = 0.8, P= 0.378), whereas knocking out one Ubc allele did cause a statistically significant reduction in ubiquitin, consistent with reduced Ubc expression (Ubc genotype has a significant effect on ubiquitin levels, GLM ANOVA: F (1,68)= 11.16, P= 0.001) (Fig. 4C). Likewise, the levels of total ubiquitin in R6/2; Ubc+/− brains were reduced (Fig. 4C). Therefore, the loss of one Ubc allele causes a reduction in total brain ubiquitin levels in R6/2 and non‐R6/2 mice.
Figure 4.
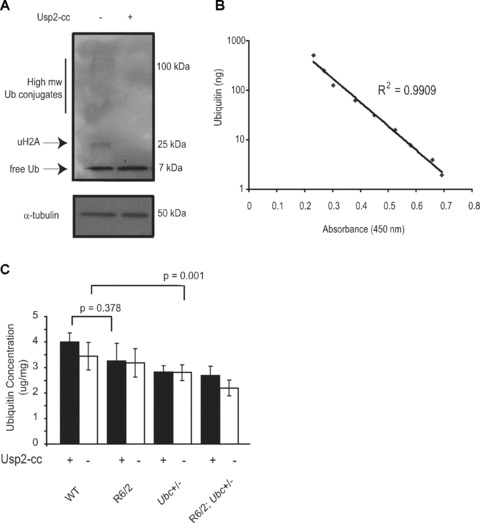
Quantification of ubiquitin levels in R6/2‐Ubc mice by indirect competitive ELISA. (A) Treatment of brain samples with the ubiquitin‐ specific protease Usp2‐cc successfully converts all visible ubiquitin linkages into monomeric ubiquitin. Blots were stripped and reprobed with α‐tubulin to show equal loading of protein. (B) Standard curve using precise ubiquitin concentrations shows the indirect competitive ELISA is linear in the range of 2–500 ng of ubiquitin. (C) Total ubiqutin levels in the R6/2 brain are not significantly different from those in WT, whereas the loss of one Ubc allele does cause a small but significant reduction in ubiquitin levels. Ubiquitin levels are not significantly different between Ubc+/− mice and R6/2; Ubc+/− mice treatment with Usp2‐cc increases measurable ubiquitin as the antibody preferentially detects monomeric ubiquitin. Error bars represent standard errors of the mean.
Monoubiquitylation of histone H2A (uH2A) is altered in R6/2‐Ubc brains
Although ubiquitin quantification showed that levels of total ubiquitin were reduced by loss of one Ubc allele, it was important to examine ubiquitin profiles by Western blot in the brains of R6/2‐Ubc mice. This would highlight any alterations in the partitioning or distribution of ubiquitin caused by the expression of mutant N‐terminal htt. To investigate any changes in the ubiquitin balance in R6/2 mice therefore, ubiquitin profiles of WT and R6/2 brain hemispheres were compared. In keeping with the ELISA results, levels of monomeric ubiquitin in 13‐week‐old R6/2 brains were similar to WT (Fig. 5A). Interestingly, there was a significant increase (P < 0.001) in a prominent 25‐kD band in R6/2 mice (Fig. 5A), which corresponds to monoubiquitylated histone H2A (uH2A). This is the most abundant ubiquitylated protein in the cell and it has been estimated that 5–15% of H2A is ubiquitylated [31, 32]. In light of the finding that uH2A levels are increased in R6/2 brains, it was of interest to determine the abundance of uH2A in R6/2; Ubc+/− brains. Therefore, Western blots were performed to compare ubiquitin profiles between R6/2 mice with one or both copies of the Ubc allele. Although there was no obvious change in high molecular weight ubiquitin conjugates or monomeric ubiquitin between the two genotypes, uH2A levels were found to be significantly less in R6/2; Ubc+/− mice (P= 0.016) (Fig. 5B). Similarly, although increased levels of uH2A was observed in 8‐week‐old R6/2 brains, R6/2; Ubc+/− mice appear to be largely protected against this effect (Fig. 5C). This suggests that the reduction in ubiquitin levels in R6/2; Ubc+/− mice reduces the amount of uH2A which is observed in R6/2 mice. The increase in uH2A levels was not present in 4‐week‐old R6/2 mouse brains (data not shown).
Figure 5.
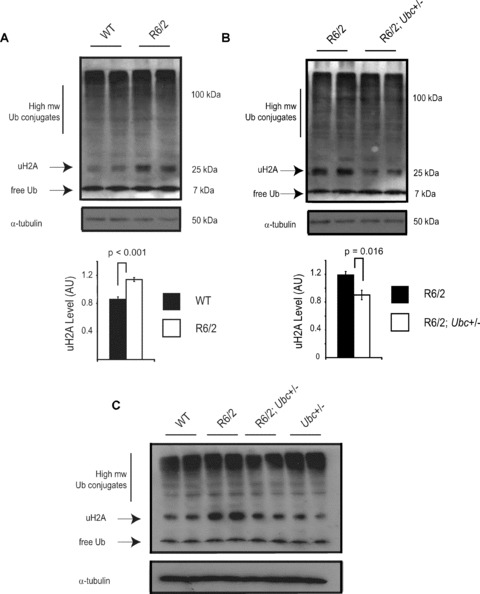
Ubiquitin profiles reveal an increase in monoubiquitylated histone H2A (uH2A) levels in R6/2. (A) Western blot with anti‐ubiquitin antibody confirms there is no difference in free ubiquitin or high molecular weight ubiquitin conjugates in the R6/2 brain. Interestingly, the intensity of the 25‐kD band corresponding to monoubiquitylated histone H2A (uH2A) is significantly increased in R6/2 mice as confirmed by densitometry. Western blotting with a specific H2A antibody showed that non‐ubiquitylated H2A levels were lower in R6/2 mice, in keeping with increased uH2A levels (data not shown). The blot was reprobed with α‐tubulin to confirm equal loading. (B) The increase in uH2A levels in R6/2 brains is reduced in brains of R6/2; Ubc+/− mice as shown by Western blot and densitometry. (C) Increased H2A monoubiquitylation is also observed in 8‐week R6/2 mice but not in R6/2; Ubc+/− mice. Blots were reprobed with α‐tubulin antibody. It should be noted that detection of high molecular weight ubiquitin conjugates in Fig. 5 differs to that depicted in Fig. 4. The lysis buffer used for the Western blot in Fig. 5 is incompatible with Usp2‐cc activity, and immunoprobing with the anti‐ubiquitin antibody does not work as effectively in the buffer used for Usp2‐cc digestion (Fig. 4). Error bars represent standard errors of the mean.
Transcription of disease genes is unchanged in R6/2; Ubc+/− striatum
Histone modifications such as ubiquitylation are believed to affect transcription by altering the association of DNA with proteins that regulate chromatin structure. There was a strong likelihood therefore that increased levels of uH2A in the R6/2 brain was relevant to disease pathology, as transcriptional dysregulation is believed to be a central pathogenic mechanism in HD [33]. The fact that increased uH2A abundance in R6/2 brains is largely prevented in R6/2; Ubc+/− mice raised the possibility that uH2A‐mediated gene expression changes may be related to the observed improvement of certain disease behaviours in these mice.
To investigate this further, quantitative real‐time RT‐PCR analysis was carried out in 13‐week‐old R6/2‐Ubc on several genes expressed in striatum, which are known to be dysregulated in the R6/2 brain [27]. The genes examined were: cannabinoid CB1 receptor (Cnr1), dopamine D2 receptor (Drd2), dopamine and cAMP regulated phosphoprotein (Darpp32) and proenkephalin (Penk1).
These assays were performed on cDNAs from the four genotypes of the R6/2‐Ubc cross (where n= at least 6 per genotype). As expected, expression of all four striatal genes was down‐regulated in R6/2 brains relative to non‐R6/2 (GLM ANOVA: Cnr1 F (1.31)= 88.16, P < 0.000; Drd2 F (1.31)= 15.7, P= 0.000; Darpp32 P (1.31)= 108.75, P < 0.000; Penk1 F (1.31)= 30.2, P < 0.000) (Fig. 6). However, the loss of one Ubc allele did not increase the expression of any of the four genes in R6/2 (no interaction between R6/2 and Ubc genotypes, GLM ANOVA: Cnr1 F (1.31)= 1.22, P= 0.278; Drd2 F (1.31)= 2.45, P= 0.129; Darpp32 F (1.31)= 6.15, P= 0.019; Penk1 F (1.31)= 0.24, P= 0.627). Therefore, the loss of one Ubc allele and prevention of increased uH2A levels in R6/2; Ubc+/− brains did not affect expression of these dysregulated genes.
Figure 6.
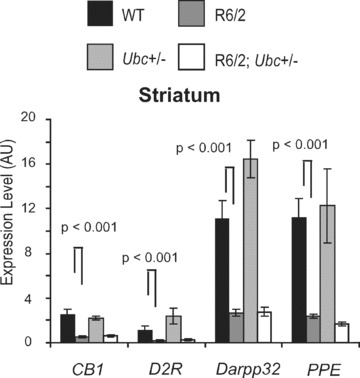
Expression of dysregulated genes in 13‐week R6/2‐Ubc striatum. The striatal expression of dysregulated R6/2 genes was compared between the four genotypes. Although expression of all four genes was reduced in R6/2 mice as expected, the loss of one Ubc allele did not have any effect on R6/2 expression levels. Therefore, the reduction in uH2A levels in R6/2; Ubc+/− mice did not affect expression of these genes. Interestingly, expression of Darpp32 was significantly increased in the Ubc+/− striatum (P= 0.018). In addition, there was an interaction between R6/2 and Ubc genotypes (P= 0.016), which likely reflects the lack of increased Darpp32 expression in R62; Ubc+/− compared to that observed in Ubc+/−. Error bars show the standard error of the mean.
Discussion
In this study, we have set out to investigate the involvement of ubiquitin in the pathogenesis of HD. As well as being central to protein degradation through the UPS, ubiquitin also plays a role in various cellular processes such as transcription, DNA repair and receptor‐mediated endocytosis. Therefore, disturbances in ubiquitin regulation could affect pathogenic processes in HD through a variety of mechanisms. Further highlighting the importance of ubiquitin processing and function to a healthy nervous system is the occurrence of neurological disease in humans and mice caused by mutations in ubiquitin‐processing enzymes [16, 17, 18, 34].
To investigate the role of ubiquitin in HD, we crossed the well‐characterized R6/2 model of HD [35] with Ubc heterozygous knockout mice [20] and evaluated the progeny using various behavioural and biochemical analyses. Ubiquitin gene expression analysis showed that the four ubiquitin genes are expressed normally in R6/2 mice. Knocking out one copy of Ubc caused a subsequent drop in Ubc expression levels in the mouse brain, allowing us to test what effect limiting polyubiquitin gene expression has on the pathogenic polyglutamine phenotype. In yeast, the ribosome‐fusion genes provide the majority of cellular ubiquitin under normal circumstances, and the polyubiquitin gene is required for survival under certain cellular stresses [22].
Therefore, it is likely that under normal mammalian physiological conditions, expression of the Ubb and Ubc genes is required for healthy cellular activity by providing a boost in free ubiquitin levels in response to cellular stress. It was therefore unexpected that knocking out one copy of Ubc in R6/2 mice actually caused an improvement of certain mutant htt‐mediated behaviours, whereby exploratory activity (rearing and centre‐rearing) of R6/2; Ubc+/− mice was increased with respect to R6/2 mice from 4 weeks until 12 weeks.
Our finding that histone H2A, the first identified ubiquitin substrate [36], shows a dramatic increase in its monoubiquitylated form in R6/2 brains suggests that the balance of ubiquitin partitioning is disturbed by mutant N‐terminal htt. Histone H2A is the most abundant ubiquitylated protein detectable in the cell, where some 5–15% of H2A is ubiquitylated [31, 32]. Monoubiquitylation of H2A (uH2A) is generally accepted to be involved in gene repression, and indeed has been shown to be required for polycomb gene silencing and X chromosome inactivation [37, 38, 39]. The finding that uH2A levels are robustly increased in the R6/2 brain therefore suggests that there may be a relationship between the ubiquitin system and transcriptional alterations in HD. This link could stem from disturbances in the overall ubiquitin balance as a result of the effort exerted by the protein quality control arm of the ubiquitin system (i.e. the UPS) in dealing with the proteotoxic effects of mutant htt. The link between proteotoxic burden and disturbances in the ubiquitin equilibrium has recently been shown to occur in cells upon heat shock or proteasome inhibition, where uH2A levels were depleted in favour of the formation of high molecular weight ubiquitin conjugates [40]. In addition, the accumulation of polyubiquitin chains in R6/2 mice and human HD patient brains suggests that disturbances to the ubiquitin system may be a global consequence of mutant htt expression [14].
Remarkably, the increase in uH2A observed in R6/2 brains was largely prevented in R6/2; Ubc+/− mice. Interestingly, Kim et al.[41] also recently found increased uH2A levels in R6/2 brains and attributed this to a higher association of Bmi‐1 (a component of the hPRC1L E3 ubiquitin ligase for uH2A) with histones due to its reduced binding to mutant htt. However, mutant N‐terminal htt protein levels, aggregation or transgene expression were not significantly different between R6/2 and R6/2; Ubc+/− mice (data not shown), suggesting decreased Bmi‐1 histone association is not likely to explain the observed decrease in uH2A levels in R6/2; Ubc+/− mice. Although alterations in the levels or activity of specific uH2A deubiquitylases could affect uH2A levels in R6/2‐Ubc mice, the simplest explanation is that the modest reduction in ubiquitin levels caused by Ubc heterozygosity limits the availability of free ubiquitin to the hPRC1L uH2A E3 ligase complex.
It is unclear to what extent increased uH2A in HD mice contributes to toxicity, or indeed how it may contribute to gene repression. Kim et al. recently showed that monoubiquitylation of H2A appears to function upstream of di‐ and tri‐methylation of histone H3 at lys 9, both markers of heterochromatin [41]. In addition, H2A monoubiquitylation has been shown to prevent di‐ and tri‐methylation of histone H3 on lys 4, which is required for transcription initiation (reviewed in [42]). Further cross‐talk between uH2A and histone modifications has been demonstrated whereby histone acetylation was shown to facilitate H2A deubiquitylation [42]. This is particularly intriguing in light of the fact that histones associated with promoters of genes down‐regulated in R6/2 mice are hypoacetylated, and that histone deacetylase inhibitors (HDACs) have been shown to both decrease uH2A levels [43] and in a separate study improve polyglutamine‐induced behavioural abnormalities [44]. It was therefore surprising that we were unable to show that the prevention of increased levels of uH2A in R6/2; Ubc+/− brains were linked to changes in the expression of dysregulated striatal genes. This may be because altered levels of uH2A are not associated with the majority of the specific gene promoters investigated, or that the decrease in levels of uH2A in R6/2; Ubc+/− mice was insufficient to exert a transcriptional effect. In support of the latter case, Kim et al. have shown that increased uH2A levels are associated with the Drd2 promoter in R6/2 striatum [41], whereas the global reduction in uH2A levels in R6/2; Ubc+/− mice did not affect Drd2 expression (Fig. 6). Therefore, the alterations in uH2A levels between R6/2 and R6/2; Ubc+/− are either insufficient to affect gene expression levels in vivo, or the effects are beyond the range of detection in our study. Alternatively, the lack of transcriptional changes in R6/2; Ubc+/− mice may suggest that the modest improvements in behaviour and changes in uH2A levels are in large part due to underlying alterations in protein ubiquitylation and folding. Indeed, it would be interesting to investigate which other cellular proteins show mutant htt‐induced differential mono‐ or polyubiquitylation and how these may affect HD phenotypes. This may add insights into, for example, the growing idea that UPS activity in HD neurons may vary depending on subcellular localization [13].
Given that the expression of N‐terminal mutant htt disturbs the balance of protein folding homeostasis within organisms [8], it would be expected that more misfolded proteins are tagged for proteasomal destruction in R6/2 mice. This extra burden on the UPS, while not necessarily compromising the overall efficiency of the system, may in fact disturb the ubiquitin balance by tagging a higher proportion of proteins with K48‐linked polyubiquitin chains. Indeed, the abundance of K48‐linked polyubiquitin chains is increased in the brains of R6/2 and HD patients [14]. It therefore seems likely that the expression of an aggregation‐prone protein such as mutant htt causes the redistribution of ubiquitin within the cell by disturbing the balance of the cell's protein refolding and degradation machinery. This would doubtless have myriad downstream consequences for the cell, given the diverse nature of the roles and substrates of ubiquitin. Our findings that histone H2A shows an increase in its monoubiquitylated form is possibly just one downstream effect of many as a consequence of a mutant htt‐induced disturbance to the ubiquitin balance, and suggests protein misfolding may induce downstream cellular abnormalities through disturbing the ubiquitin system.
Acknowledgements
This work was supported by the Wellcome Trust (66270, and Prize Studentship to JSB, 73061) and by the Huntington's disease Society of America Coalition for the Cure to GPB and RRK.
References
- 1. Landles C, Bates GP. Huntingtin and the molecular pathogenesis of Huntington's disease. Fourth in molecular medicine review series. EMBO Rep . 2004; 5: 958–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Davies SW, Turmaine M, Cozens BA, et al . Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell . 1997; 90: 537–48. [DOI] [PubMed] [Google Scholar]
- 3. DiFiglia M, Sapp E, Chase KO, et al . Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science . 1997; 277: 1990–3. [DOI] [PubMed] [Google Scholar]
- 4. Scherzinger E, Lurz R, Turmaine M, et al . Huntingtin‐encoded polyglutamine expansions form amyloid‐like protein aggregates in vitro and in vivo . Cell . 1997; 90: 549–58. [DOI] [PubMed] [Google Scholar]
- 5. Graham RK, Deng Y, Slow EJ, et al . Cleavage at the caspase‐6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell . 2006; 125: 1179–91. [DOI] [PubMed] [Google Scholar]
- 6. Kazantsev A, Preisinger E, Dranovsky A, et al . Insoluble detergent‐resistant aggregates form between pathological and nonpathological lengths of polyglutamine in mammalian cells. Proc Natl Acad Sci USA . 1999; 96: 11404–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schaffar G, Breuer P, Boteva R, et al . Cellular toxicity of polyglutamine expansion proteins: mechanism of transcription factor deactivation. Mol Cell . 2004; 15: 95–105. [DOI] [PubMed] [Google Scholar]
- 8. Gidalevitz T, Ben‐Zvi A, Ho KH, et al . Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science . 2006; 311: 1471–4. [DOI] [PubMed] [Google Scholar]
- 9. Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin‐proteasome system by protein aggregation. Science . 2001; 292: 1552–5. [DOI] [PubMed] [Google Scholar]
- 10. Bowman AB, Yoo SY, Dantuma NP, et al . Neuronal dysfunction in a polyglutamine disease model occurs in the absence of ubiquitin‐proteasome system impairment and inversely correlates with the degree of nuclear inclusion formation. Hum Mol Genet . 2005; 14: 679–91. [DOI] [PubMed] [Google Scholar]
- 11. Bett JS, Goellner GM, Woodman B, et al . Proteasome impairment does not contribute to pathogenesis in R6/2 Huntington's disease mice: exclusion of proteasome activator REGgamma as a therapeutic target. Hum Mol Genet . 2006; 15: 33–44. [DOI] [PubMed] [Google Scholar]
- 12. Diaz‐Hernandez M, Hernandez F, Martin‐Aparicio E, et al . Neuronal induction of the immunoproteasome in Huntington's disease. J Neurosci . 2003; 23: 11653–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang J, Wang CE, Orr A, et al . Impaired ubiquitin‐proteasome system activity in the synapses of Huntington's disease mice. J Cell Biol . 2008; 180: 1177–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bennett EJ, Shaler TA, Woodman B, et al . Global changes to the ubiquitin system in Huntington's disease. Nature . 2007; 448: 704–8. [DOI] [PubMed] [Google Scholar]
- 15. Kitada T, Asakawa S, Hattori N, et al . Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature . 1998; 392: 605–8. [DOI] [PubMed] [Google Scholar]
- 16. Leroy E, Boyer R, Auburger G, et al . The ubiquitin pathway in Parkinson's disease. Nature . 1998; 395: 451–2. [DOI] [PubMed] [Google Scholar]
- 17. Maraganore DM, Farrer MJ, Hardy JA, et al . Case‐control study of the ubiquitin carboxy‐terminal hydrolase L1 gene in Parkinson's disease. Neurology . 1999; 53: 1858–60. [DOI] [PubMed] [Google Scholar]
- 18. Saigoh K, Wang YL, Suh JG, et al . Intragenic deletion in the gene encoding ubiquitin carboxy‐terminal hydrolase in gad mice. Nat Genet . 1999; 23: 47–51. [DOI] [PubMed] [Google Scholar]
- 19. Kirschner LS, Stratakis CA. Structure of the human ubiquitin fusion gene Uba80 (RPS27a) and one of its pseudogenes. Biochem Biophys Res Commun . 2000; 270: 1106–10. [DOI] [PubMed] [Google Scholar]
- 20. Ryu KY, Maehr R, Gilchrist CA, et al . The mouse polyubiquitin gene UbC is essential for fetal liver development, cell‐cycle progression and stress tolerance. EMBO J . 2007; 26: 2693–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ryu KY, Sinnar SA, Reinholdt LG, et al . The mouse polyubiquitin gene Ubb is essential for meiotic progression. Mol Cell Biol . 2008; 28: 1136–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Finley D, Ozkaynak E, Varshavsky A. The yeast polyubiquitin gene is essential for resistance to high temperatures, starvation, and other stresses. Cell . 1987; 48: 1035–46. [DOI] [PubMed] [Google Scholar]
- 23. Finley D, Bartel B, Varshavsky A. The tails of ubiquitin precursors are ribosomal proteins whose fusion to ubiquitin facilitates ribosome biogenesis. Nature . 1989; 338: 394–401. [DOI] [PubMed] [Google Scholar]
- 24. Hockly E, Cordery PM, Woodman B, et al . Environmental enrichment slows disease progression in R6/2 Huntington's disease mice. Ann Neurol . 2002; 51: 235–42. [DOI] [PubMed] [Google Scholar]
- 25. Hockly E, Tse J, Barker AL, et al . Evaluation of the benzothiazole aggregation inhibitors riluzole and PGL‐135 as therapeutics for Huntington's disease. Neurobiol Dis . 2006; 21: 228–36. [DOI] [PubMed] [Google Scholar]
- 26. Ryu KY, Baker RT, Kopito RR. Ubiquitin‐specific protease 2 as a tool for quantification of total ubiquitin levels in biological specimens. Anal Biochem . 2006; 353: 153–5. [DOI] [PubMed] [Google Scholar]
- 27. Woodman B, Butler R, Landles C, et al . The Hdh(Q150/Q150) knock‐in mouse model of HD and the R6/2 exon 1 model develop comparable and widespread molecular phenotypes. Brain Res Bull . 2007; 72: 83–97. [DOI] [PubMed] [Google Scholar]
- 28. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) Method. Methods . 2001; 25: 402–8. [DOI] [PubMed] [Google Scholar]
- 29. Vandesompele J, De PK, Pattyn F, et al . Accurate normalization of real‐time quantitative RT‐PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002; 3: RESEARCH0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mimnaugh EG, Bonvini P, Neckers L. The measurement of ubiquitin and ubiquitinated proteins. Electrophoresis . 1999; 20: 418–28. [DOI] [PubMed] [Google Scholar]
- 31. West MH, Bonner WM. Histone 2B can be modified by the attachment of ubiquitin. Nucleic Acids Res . 1980; 8: 4671–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Carlson N, Rogers S, Rechsteiner M. Microinjection of ubiquitin: changes in protein degradation in HeLa cells subjected to heat‐shock. J Cell Biol . 1987; 104: 547–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sadri‐Vakili G, Cha JH. Mechanisms of disease: histone modifications in Huntington's disease. Nat Clin Pract Neurol . 2006; 2: 330–8. [DOI] [PubMed] [Google Scholar]
- 34. Wilson SM, Bhattacharyya B, Rachel RA, et al . Synaptic defects in ataxia mice result from a mutation in Usp14, encoding a ubiquitin‐specific protease. Nat Genet . 2002; 32: 420–5. [DOI] [PubMed] [Google Scholar]
- 35. Mangiarini L, Sathasivam K, Seller M, et al . Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell . 1996; 87: 493–506. [DOI] [PubMed] [Google Scholar]
- 36. Goldknopf IL, Busch H. Isopeptide linkage between nonhistone and histone 2A polypeptides of chromosomal conjugate‐protein A24. Proc Natl Acad Sci USA . 1977; 74: 864–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. de NM, Mermoud JE, Wakao R, et al . Polycomb group proteins Ring1A/B link ubiquitylation of histone H2A to heritable gene silencing and X inactivation. Dev Cell . 2004; 7: 663–76. [DOI] [PubMed] [Google Scholar]
- 38. Wang H, Wang L, Erdjument‐Bromage H, et al . Role of histone H2A ubiquitination in Polycomb silencing. Nature . 2004; 431: 873–8. [DOI] [PubMed] [Google Scholar]
- 39. Baarends WM, Wassenaar E, van der LR, et al . Silencing of unpaired chromatin and histone H2A ubiquitination in mammalian meiosis. Mol Cell Biol . 2005; 25: 1041–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dantuma NP, Groothuis TA, Salomons FA, et al . A dynamic ubiquitin equilibrium couples proteasomal activity to chromatin remodeling. J Cell Biol . 2006; 173: 19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kim MO, Chawla P, Overland RP, et al . Altered histone monoubiquitylation mediated by mutant huntingtin induces transcriptional dysregulation. J Neurosci . 2008; 28: 3947–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vissers JH, Nicassio F, van Lohuizen M, et al . The many faces of ubiquitinated histone H2A: insights from the DUBs. Cell Div. 2008; 3: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sadri‐Vakili G, Bouzou B, Benn CL, et al . Histones associated with downregulated genes are hypo‐acetylated in Huntington's disease models. Hum Mol Genet . 2007; 16: 1293–306. [DOI] [PubMed] [Google Scholar]
- 44. Hockly E, Richon VM, Woodman B, et al . Suberoylanilide hydroxamid acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington's disease. Proc Natl Acad Sci USA . 2003; 100: 2041–6. [DOI] [PMC free article] [PubMed] [Google Scholar]