

Abstract
Neurodegenerative diseases are commonly associated with the accumulation of intracellular or extracellular protein aggregates. Recent studies suggest that these aggregates are capable of crossing cellular membranes and can directly contribute to the propagation of neurodegenerative disease pathogenesis. We propose that, once initiated, neuropathological changes might spread in a ‘prion-like’ manner and that disease progression is associated with the intercellular transfer of pathogenic proteins. The transfer of naked infectious particles between cells could therefore be a target for new disease-modifying therapies.
The deposition of aggregated proteins and ubiquitin into intracellular inclusion bodies is a common neuropathological denominator for most neurodegenerative disorders, including Parkinson’s, Alzheimer’s and Huntington’s diseases, as well as transmissible prion encephalopathies. Each disorder is characterized by the misfolding of a specific protein or proteins: α-synuclein in Parkinson’s disease, β-amyloid and tau in Alzheimer’s disease, huntingtin in Huntington’s disease and prion protein (PrP) in transmissible prion encephalopathies. This suggests that impaired protein homeostasis is a shared pathogenic feature of these otherwise clinically and etiologically diverse diseases.
Protein misfolding can result from native proteins changing their conformations or newly synthesized polypeptides failing to fold properly (FIG. 1). Incompletely or incorrectly folded proteins expose hydrophobic amino acid side chains on their surfaces that are normally buried in the interior of the native state. Thus, they become prone to self-association into aggregates that can function as nuclei that recruit additional monomers (FIG. 1; BOX 1). Such protein aggregates can become infectious and are called prions1,2 if the intermolecular interactions between the constitutive molecules are so strong that aggregation is effectively irreversible, if they resist the cell clearance machinery (BOX 2) and if they propagate from one cell to another (FIG. 2), in which they recruit non-native polypeptide monomers. Mammalian PrP is a plasma membrane protein1, whereas α-synuclein, huntingtin and tau are normally cytosolic. Thus, the propagation and transmission of α-synuclein, huntingtin and tau aggregates probably differ from PrP. The generic molecular basis of prion particle formation and transmission is illustrated in BOX 1. Protein infectivity depends on several factors: irreversibility of non-native protein assemblies, the efficiency by which precursor polypeptides are recruited into aggregates, the resistance of aggregates to the cellular clearance machinery and the efficiency with which aggregates can transfer to neighbouring cells.
Figure 1. Basic mechanisms of protein aggregation.

The folding of newly synthesized polypeptide chains into their native conformations and the unfolding of proteins from their native states proceeds through distinct intermediates. some of these intermediates are able to self-associate to form non-native oligomeric species of different sizes and structures, in which a given molecule interacts through two interfaces with two neighbouring molecules (an intermediate in which longitudinal interactions are established is shown). As the polypeptides involved in prion, Parkinson’s, Alzheimer’s and Huntington’s diseases populate a wide variety of folding intermediates, they have a higher propensity to form such oligomeric species. The stability of these oligomers increases on establishment of supplementary intermolecular interactions with non-native polypeptides through additional interfaces, as a given molecule in the oligomer becomes multivalent. The rate-limiting step in non-native polypeptide aggregation is therefore the formation of stable oligomers. Such oligomers behave as nuclei and grow from their ends by recruiting non-native monomers. As the binding of a molecule to the oligomer generates an incorporation site for another subunit, the growth of the stable nuclei is unlimited. Brownian52 movement and severing and/or disaggregating factors generate increased numbers of ends and increase the likelihood of new subunits being incorporated.
Box 1. Molecular basis of prion particle formation, growth and transmission.
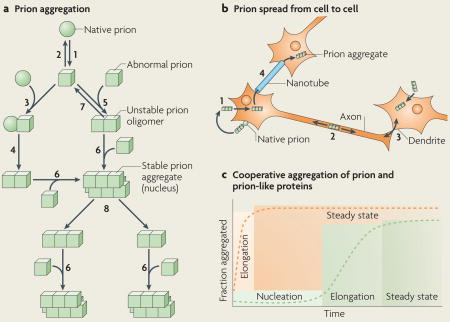
Native (sphere) prion molecules undergo conformational changes that lead to an abnormal (cube) form (see the figure; part a, step 1). This event is unfavourable because the abnormal form is either unstable (part a, step 2) or sensitive to clearance. According to the ‘template assistance’ model, prions in their abnormal form interact with native prions (part a, step 3) and convert them into the abnormal form (part a, step 4). The alternative ‘seeded polymerization’ model proposes that abnormal prions have the ability to interact with molecules in a similar state (part a, step 5). The oligomeric species formed are unstable because the intermolecular interactions do not outweigh the entropic cost of binding. They grow by the incorporation of abnormal prion molecules (part a, step 6) and dissociate (part a, step 7) until a stable nucleus is formed. Such a stable prion aggregate can then grow indefinitely from one or both ends and can also break into smaller fragments (part a, step 8) that act as nuclei (part a, step 6).
Prion aggregates bind to native prion molecules or receptor proteins attached to the cell membrane, and are internalized by endocytosis (part b, step 1). They reach the cytoplasm, by an unknown process, where they grow by the incorporation of cytosolic prions. They can move along the axon in one direction or another (part b, step 2) and can reach neighbour cells through axon–dendrite connections (part b, step 3) and nanotubes (part b, step 4).
Prions and polypeptides involved in Parkinson’s, Alzheimer’s and Huntington’s diseases form aggregates that resist protein denaturation treatments (see the figure, part c). This process can be monitored experimentally. Stable oligomer formation is thermodynamically unfavourable and this is reflected by a nucleation phase. The stable oligomers elongate in an exponential manner until the soluble protein concentration reaches the critical concentration, above which assembly occurs. These events give a sigmoidal shape to the assembly kinetic (see the figure, part c; green curve). The nucleation phase is abolished (see the figure, part c; orange curve) when preformed aggregates that act as seeds are added to the protein solution.
Box 2. Cellular defences against protein aggregation.
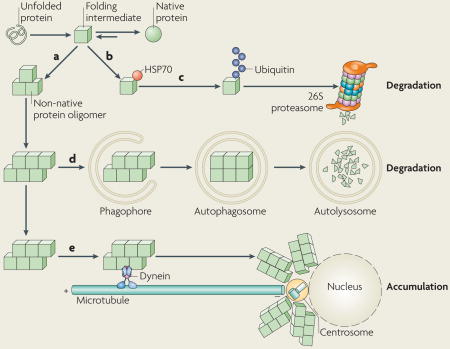
Protein folding proceeds through intermediates, which expose hydrophobic amino acid side chains on their surfaces that are normally buried in the interior of the native state, and are therefore prone to self-associate into non-native oligomers (see the figure, part a). Molecular chaperones of the heat shock protein 70 (HSP70) family interact transiently with these aggregation-prone surfaces (see the figure, part b). They either compete with self-association or favour the formation of native contacts and help maintain intermediates in a monomeric, folding-competent state. Mutations or polymorphisms can destabilize the native state or decrease the efficiency of folding. Insufficient amounts of molecular chaperones also affect folding efficiency. Incompletely folded proteins are tagged with polyubiquitin chains, which direct them to the 26S proteasome for degradation (see the figure, part c). Molecular chaperones contribute to this process by maintaining the proteins in a state that can be unfolded by the 26S proteasome. Proteins that escape degradation by the 26S proteasome can be degraded by lysosomes through macroautophagy (see the figure, part d). Autophagic degradation begins with the capture of the substrate proteins (or aggregates) into phagophores that mature into autophagosomes — a vesicular structure enclosed by two concentric lipid bilayer membranes. Autophagosomes fuse with organelles of the endosomal and lysosomal pathways to form autolysosomes that, endowed with acidic pH and lysosomal hydrolases, are able to degrade proteins without the need to unfold them. Finally, the interaction of protein aggregates with the minus end-directed microtubule motor cytoplasmic dynein results in their accumulation around the centrosome or microtubule organizing centre (see the figure, part e). This process may facilitate the capture of aggregates by macroautophagy or may serve to concentrate potentially toxic aggregation nuclei in a defined region of the cell.
Figure 2. Potential pathways of uptake and release of protein aggregates by cells.

a | Protein aggregates formed in cells can be passively released from cells by membrane rupture or damage, perhaps accompanying cell death. Alternatively, cytoplasmic aggregates can be actively released by exocytosis, possibly following capture by macroautophagy and mis-sorting or incomplete digestion in the endosomal and lysosomal systems. b | Likewise, protein aggregates that bind passively to cell membrane components (phospholipids or protein receptors) can enter the cell either passively by physical rupture of the plasma membrane or actively through endocytosis. Aggregates taken up by endocytosis must cross a biological membrane to reach the cytoplasm, where they can elongate by incorporation of their constituting proteins. c | Protein aggregates, either formed in cells or taken up by cells, can also actively propagate from cell to cell, possibly by cytoskeletal components such as molecular motors and nanotubes (see also BOX 1).
In this Opinion article we highlight how the neuropathologies of Parkinson’s, Alzheimer’s and Huntington’s diseases develop over time and space, and describe recent findings supporting the idea that pathogenic proteins can transfer between cells. We propose that intercellular protein transfer contributes to the progression of neurodegenerative diseases and, therefore, can constitute a target for future disease-modifying therapies.
Patterns of neuropathology spread
In neurodegenerative diseases, such as Parkinson’s, Alzheimer’s and Huntington’s diseases, pathological changes typically develop in the nervous system following specific anatomical patterns that are characteristic for each disorder (FIG. 3). These patterns indicate that the pathology is not only simply propagated between neighbouring cell bodies, but that it also spreads along axonal pathways either away from (anterogradely) or towards (retrogradely) the cell body (FIG. 3a).
Figure 3. Principles for progression of neuropathological changes.

a | Intracellular protein aggregates can be released from neurons by exocytosis or cell death. The aggregates are taken up by, for example, adjacent neuronal cell bodies and are either retained in the cell soma (local spread of pathology) or transported anterogradely by axons. Alternatively, they are taken up by axon terminals and transported retrogradely to the cell soma. The protein aggregates can spread between brain regions by axonal transport. b–d | Three drawings propose principles for how neuropathological changes in Parkinson’s, Alzheimer’s and Huntington’s diseases spread spatiotemporally during disease progression. The earlier the neuropathology develops in a given brain region, the darker the shading in the diagram. As only one view (mid-sagittal for Parkinson’s and Alzheimer’s diseases; lateral for Huntington’s disease) of the brain is depicted for each disorder, not all relevant anatomical structures and details of the spreading patterns (indicated by arrows) are presented. b | in Parkinson’s disease, α-synuclein aggregates (Lewy neurites and Lewy bodies) are suggested to first appear in the dorsal motor nucleus of the vagal nerve in the brainstem and anterior olfactory structures (darkest green), and then to spread stereotypically to finally occupy large parts of the brain4,5. c | in Alzheimer’s disease, neurofibrillary tangles first appear in the hippocampus (and closely associated structures), the basal nucleus of Meynert and the brainstem15–18 (darkest green). They spread to other brain regions, including the neocortex, in a stereotypical manner, correlating with symptomatic progression. d | in Huntington’s disease, the putamen and caudate nucleus, and related basal ganglia structures deep inside the brain (darkest green), have been suggested to degenerate first25–27. However, recent imaging studies suggest that primary motor and sensory cortices already undergo atrophy in pre-symptomatic gene carriers28. Therefore we propose that cortical involvement precedes basal ganglia pathology.
Parkinson’s disease
The neuropathological hallmarks of the movement disorder Parkinson’s disease are Lewy bodies and Lewy neurites, which are protein aggregates in the cell body and neuronal processes, respectively. The most abundant protein in the aggregates is α-synuclein. This 140 amino acid pre-synaptic protein is natively unfolded (that is, it lacks a well-defined stable tertiary structure when isolated), interacts with multiple proteins as well as lipids and membranes, and has been suggested to play a part in vesicular transport3. Although most Parkinson’s disease cases are idiopathic (of an unknown cause), mutations in the α-synuclein gene underlie rare, inherited forms.
Braak and co-workers hypothesized that brainstem and anterior olfactory structures are afflicted by α-synuclein aggregates very early in Parkinson’s disease4. Indeed, these changes are suggested to occur several years before involvement of the substantia nigra — the midbrain region, the degeneration of which is associated with motor dysfunction in Parkinson’s disease4,5. α-Synuclein aggregates are suggested to progress in a topographically predictable manner as the pathology spreads through anatomical connections throughout the brainstem, limbic and autonomic systems and neocortex4,5 (FIG. 3b). Braak and co-workers propose that this spread primarily follows pathways consisting of long unmyelinated axons and that it starts in the olfactory system and gut6. Olfactory dysfunction is now considered an early sign of Parkinson’s disease and in advanced disease people often exhibit dementia, depression and autonomic nervous system dysfunction, possibly owing to the spreading of α-synuclein aggregates6. Although the concept of Braak’s neuropathological stages has gained much attention7,8, it is not unanimously supported. For example, different brain regions have been suggested to vary in their susceptibility to the unknown underlying disease trigger and this could contribute to the stereotypical pattern9,10. Some investigators have also questioned the reproducibility of the pattern between patients and state that it does not always follow the same temporal order or anatomical distribution9,10.
Alzheimer’s disease
Alzheimer’s disease is characterized by the loss of neurons and synapses in the cerebral cortex and certain subcortical regions. Neurofibrillary tangles and amyloid plaques are the classical neuropathological hallmarks of Alzheimer’s disease11,12. Neurofibrillary tangles are cytoplasmic inclusion bodies that are rich in a hyperphosphorylated form of the microtubule-associated protein tau13. Normally, tau, which is a natively unfolded protein like α-synuclein, is a soluble protein that interacts with tubulin to stabilize microtubules and promote microtubule assembly in the brain. Hyperphosphorylated tau tends to aggregate to form intracellular tangles of paired helical and straight filaments. Amyloid plaques, however, are extracellular and are primarily made up of insoluble aggregates of β-amyloid — a 39–43 residue proteolytic cleavage product of amyloid precursor protein (APP), which is a transmembrane protein of unknown function14. Most cases of Alzheimer’s disease, like Parkinson’s disease, are idiopathic, although mutations in the gene encoding APP or in the enzymes that sequentially cleave it cause inherited forms of Alzheimer’s disease14.
In Alzheimer’s disease, neurofibrillary tangles progressively spread throughout the brain in an anatomically stereotypical manner. Some of the first regions affected by tangles, possibly preceded by olfactory areas15, are the hippocampus (and closely associated structures), the basal nucleus of Meynert and the brainstem, whereas the neocortex is not involved until the disease is more advanced (FIG. 3c)16,17. Even in the hippocampus and related structures, the development of tau pathology has been suggested to follow connections anterogradely18. By contrast, the spreading of amyloid pathology does not progress through such an anatomically strict pattern in Alzheimer’s disease and correlates poorly with the level of cognitive decline11,19.
Huntington’s disease
Mutations in the gene encoding huntingtin underlie the autosomal dominant inheritance of Huntington’s disease, which is characterized by involuntary movements, personality changes and dementia. Huntingtin is made up of 3,144 amino acids, is expressed widely throughout the body and has numerous interacting protein partners. Its normal functions are not fully understood; it has been implicated in anti-apoptosis, neuronal gene transcription, synaptic function and vesicle and axonal transport20. Expansion of a CAG repeat in exon 1 of the huntingtin gene above a critical threshold of 35–40 CAG repeats causes Huntington’s disease. The mutation encodes an expanded polyglutamine tract which makes the protein (or a fragment of the protein) prone to aggregate and to form intraneuronal inclusion bodies21,22. Huntingtin fragments bearing fewer than 35 glutamines do not aggregate and fragments with more than 40 aggregate readily23. The longer the polyglutamine tract, the more rapid the aggregation23 and the earlier the disease onset24, showing that huntingtin aggregation is intimately linked to disease pathogenesis.
Classical descriptions of Huntington’s disease emphasize that degeneration simultaneously progresses in two defined anatomical directions in the striatum25. Striatal projection neurons, particularly those expressing encephalin, are among the first affected in Huntington’s disease26. Brain imaging studies reveal that the basal ganglia are shrunken even before symptoms involving involuntary movements emerge27. Recent brain imaging studies, however, show that various cortical regions involved in motor, sensory and visual functions already undergo thinning in asymptomatic huntingtin gene carriers, and cortical areas that subserve more advanced brain functions are afflicted later28. Thus, cortical degeneration in Huntington’s disease follows a topographically predictable pattern. Taken together, degenerative phenomena follow distinct patterns in Huntington’s disease, even if the precise temporal and topographical maps of degeneration are still not well established (FIG. 3d).
Prion-like aggregate transmission
Several recent experimental findings and clinical observations have suggested that protein aggregates associated with Parkinson’s, Alzheimer’s and Huntington’s diseases, might move from affected to unaffected areas of the brain, suggesting that prion-like transmission of these diseases contribute to the anatomical spread of disease pathology.
Grafted cells develop Lewy bodies
A series of autopsies of Parkinson’s disease patients who had received transplants of healthy embryonic neurons over one decade earlier have provided novel insight into mechanisms that might underlie the progression of Parkinson’s disease pathology29. A subset (2–5% over 5 years) of the grafted neurons displayed aggregates containing α-synuclein29–33. These inclusion bodies were positive for all the classical markers of Lewy bodies and exhibited protein fibrils at the ultrastructural level. Studies on Parkinson’s disease patients dying sooner (1–5 years) after transplant surgery did not reveal any protein aggregates in the grafted neurons, showing that Lewy bodies develop slowly or with a long delay in previously healthy embryonic neurons29,30. These data suggest that the Parkinsonian brain promotes conversion of soluble α-synuclein into an insoluble form, but do not reveal the nature of the agent of this conversion. Recently, mouse neural stem cells tagged with green fluorescent protein (GFP) were reported to develop intracellular α-synuclein immunoreactivity and occasional α-synuclein-positive inclusion bodies when injected into the hippocampus of transgenic mice expressing human α-synuclein34. These studies suggest that host-derived α-synuclein can enter transplanted neural cells, analogous to the findings in grafted Parkinson’s disease patients.
α-Synuclein is internalized by cells in vitro. Additional support for the hypothesis that α-synuclein can move between neurons has come from in vitro studies in which GFP-labelled neural stem cells34 or SH-SY5Y cells (a dopaminergic cell line)35 were cultured together with pure α-synuclein. After 24–48 hrs of incubation, the added α-synuclein, marked with a fluorescent tag, could be seen in the cytoplasm of the cells. α-Synuclein could also be seen in GFP-labelled neural stem cells that were cultured together with neuronal cells over-expressing human α-synuclein, suggesting that α-synuclein is transferred between cells in culture34,35. Another study reports that GFP-tagged α-synuclein oligomers added to culture media can be internalized by primary cortical neurons engineered to express α-synuclein tagged with red fluorescent protein36,37. Moreover, the addition of extracellular GFP–α-synuclein induced the formation of inclusion bodies in the cytoplasm of the recipient cells that were labelled with both fluorophores, suggesting that the internalized protein can act as a seed to recruit endogenous α-synuclein into aggregates34,36,37.
Taken together, results from human autopsies, cell cultures and transgenic mice suggest that intercellular α-synuclein transfer can contribute to the spreading of neuropathology in Parkinson’s disease, which could explain why the pathology progresses stereotypically in accordance with the stages described by Braak4. Specifically, non-native forms of α-synuclein can migrate from cell to cell along axonal pathways, leading to progressive propagation of cellular pathology and gradually affecting greater parts of the central nervous system.
Alzheimer’s disease and protein seeding
Two series of experiments support the notion that protein seeding (whereby a misfolded protein acts as a ‘seed’ that initiates aggregate formation by recruiting additional unfolded or oligomeric species of the same protein) contributes to the development of Alzheimer’s disease neuropathology. In experiments on mice overexpressing APP, injections of β-amyloid extracts derived from brains of Alzheimer’s disease patients or aged APP transgenic mice caused the deposition of β-amyloid38. Importantly, these experiments involved the extracellular deposition of β-amyloid and not transmembrane uptake and seeding, making them fundamentally different from the other models we discuss. By contrast, another series of experiments examined the ability of aggregated tau to enter cells in culture39. Fluorescently tagged aggregates of a tau fragment were internalized by C17.2 neural cells and HEK293 cells, either when the pure aggregates were added to the culture media or when the cells were co-cultured with cells expressing an aggregation-prone tau fragment39. These in vitro experiments raise the possibility that tau, like α-synuclein, can move from one brain cell to another. Once inside, the aggregated tau could seed the aggregation of the endogenous protein39, in analogy to prion propagation (BOX 1). This is supported by observations that mutant human tau injected into mouse brains induces the aggregation of the endogenous wild-type mouse protein, and that pathology spreads from the injection site to neighbouring brain regions40.
Uptake of huntingtin aggregates
Yang and colleagues first reported that large aggregates composed of fluorescently tagged, synthetic polyglutamine peptides can be taken up by cultured cells41. When these peptide aggregates were appended with a nuclear localization signal, they translocated to the nucleus and became highly toxic, implying that they had become accessible to cytoplasmic nuclear import factors. More recently, it was reported that when similar polyglutamine aggregates or recombinant fragments of mutant huntingtin were added to medium, they were concentrated in juxtanuclear inclusion bodies in cultured HEK293 cells42. To directly assess whether the internalized aggregates had become exposed to the cytoplasm, as opposed to remaining in a vesicular or vacuolar compartment, an intracellular seeding experiment was conducted42. When the recipient cells were engineered to express a fluorescent version of huntingtin encoding 25 glutamines (below the threshold for spontaneous aggregation), the addition of fluorescent huntingtin aggregates containing 44 glutamines to the culture medium altered the distribution of the 25-glutamine reporter from a diffuse to a punctate pattern and caused it to co-localize with the externally added protein. This did not occur when the huntingtin-containing recipient cells internalized aggregates composed of other amyloids, indicating that the changes were probably due to a seeded polymerization process and that some of the internalized material had gained access to the cytoplasm42. Remarkably, when cells that had been exposed to polyglutamine aggregates were maintained in prolonged cell culture, the aggregated phenotype of the 25-glutamine huntingtin reporter persisted for over 80 generations, although the fraction of cells exhibiting this phenotype remained low, possibly owing to unequal mitotic segregation of aggregates in mammalian cells43. This indicates that polyglutamine aggregates, like prions, can replicate, presumably by a seeding nucleation mechanism in which aggregate huntingtin seeds are transmitted to daughter cells during cell division42.
Currently, the relevance of these observations to Huntington’s disease pathogenesis is unclear. Neurons grafted into Huntington’s disease patients’ brains exhibit poor long-term (> 10 years) survival and have been suggested to sustain damage owing to excitotoxicity or inflammation, implicating non-cell-autonomous pathogenetic factors in the disease44. In contrast to the Parkinson’s disease transplant studies, the grafts in Huntington’s disease patients exhibited no morphological evidence that huntingtin with expanded polyglutamine had transferred from host to graft within a decade. These findings do not, however, preclude a role for cell-to-cell transmission of huntingtin aggregates in Huntington’s disease progression. Neurotoxicity in Huntington’s disease might well be due to a combination of excitotoxic and inflammatory damage, and prion-like huntingtin transmission might have a role in the pathogenic cascade, perhaps overcoming the protective effects of the machinery described in BOX 2.
How aggregates move between cells
The spread of protein aggregates in the nervous system is likely to contribute to the advances of clinical symptoms and neuropathological changes in Alzheimer’s, Parkinson’s and Huntington’s diseases. Recent studies have demonstrated cell-to-cell or media-to-cell transfer of aggregates, but have given less insight into the mechanisms by which they are released or taken-up. Below, we describe potential underlying mechanisms.
Requisites for cell-to-cell transmission
In order for a prion-like mechanism to contribute to the progression of a neuro-degenerative disease, four basic requirements must be fulfilled. First and foremost, the protein aggregate must be capable of elongating by the recruitment of soluble polypeptide chains and fragmenting to generate additional elongation sites and amplify aggregation (FIG. 1; BOX 1). This has now been established in vitro, in studies discussed above, for proteins species that aggregate in Parkinson’s disease, tauopathy and Huntington’s disease. Second, cells ‘infected’ by aggregates must continuously synthesize the non-aggregated form. Third, the transmissible aggregate must be released from cells (FIG. 2a). Fourth, aggregates must be able to bind and enter the recipient cell (FIG. 2b).
Mechanisms of aggregate release
Cells can release aggregates either by a vesicle-mediated exocytic process, perhaps resulting from incomplete autophagocytosis45, or by lysosomal exocytosis46 (FIG. 2a). Cultured neuroblastoma cells and neurons secrete α-synuclein monomers and aggregates by a non-classical vesicle-mediated exocytic mechanism; however, the molecular details are still unknown47. Alternatively, aggregates can be passively released from cells either on binding and local rupture of the membrane or after cell lysis, which could occur as a result of the toxicity imposed on cells by the burden of high levels of protein aggregation (FIG. 2a). For example, when cells expressing huntingtin bearing an expanded poly-glutamine tract were selectively killed, the released aggregates caused nucleation of a cytoplasmic polyglutamine reporter protein inside co-cultured cells42. Although both potential release mechanisms (exocytosis and cell rupture due to death) are likely to occur in neurodegenerative disease, their relative importance to pathogenesis has not been studied.
Mechanisms of aggregate uptake
In cultured cells, internalized α-synuclein aggregates partially co-localized with endosomal and lysosomal markers35. Expression of a dominant-negative form of dynamin — a GTPase required for endocytic membrane fusion — inhibited this uptake. This implies that the endocytic machinery is involved in this process35 (FIG. 2b). Amyloid fibrils, including those associated with systemic amyloid disease48 and those not normally found in eukaryotes49, can be internalized by cultured cells, possibly by breaching the plasma membrane (FIG. 2b). Although these findings raise the possibility that mammalian cells might have a greater ability to internalize ordered fibrillar aggregates than previously thought, further studies are required to understand the molecular mechanisms.
Even if neurons and other mammalian cells take up fibrillar aggregates by endocytosis, in order for them to nucleate aggregation of endogenous cytoplasmic proteins, which is essential for the ‘prion-like’ hypothesis, they must escape the intracellular vesicle and gain access to the cytoplasm. The aggregates investigated in the cell culture studies described above are too polar to diffuse passively across lipid bilayers and too large to pass through transmembrane pores or transporters. Nonetheless, extracellular aggregates containing polyglutamine42, tau39 and α-synuclein34 have now been shown to enter cells and cause seeding of endogenous proteins. Deep-etch electron microscope images of polyglutamine aggregates shortly after internalization into cultured cells revealed ‘naked’ aggregates on the cytoplasmic face of the plasma membrane. There was no evidence of a surrounding membranous structure42, suggesting that aggregates can penetrate the plasma membrane in the absence of vesicular uptake. Studies in artificial systems on α-synuclein oligomers show that they can render lipid bilayer membranes permeable to fluorescent dyes, suggesting that α-synuclein aggregates can intercalate directly into lipid membranes50. This provides a potential means by which aggregates could exit from endosomes or perhaps cross the plasma membrane directly. Finally, it is possible that tunnelling nanotubes — 50–200 nm diameter actin-rich hollow filaments seen between interconnected cells in culture51 — can act as transport conduits for prion-like protein aggregates, as has been suggested for PrP51 (FIG. 2c).
Conclusions and perspectives
Several neurodegenerative diseases have symptomatic onset in mid or late life and often have protracted courses. This is evident in dominantly inherited familial diseases such as Huntington’s disease and rare familial forms of Alzheimer’s disease and Parkinson’s disease, in which the central nervous system copes with the continued synthesis of mutant, aggregation-prone proteins for many decades before eventually exhibiting neuropathology. Cellular defences, such as those described in BOX 2, probably suppress the formation of aggregation nuclei during this latent period. In these diseases, as with the more common idiopathic diseases, the low probability of spontaneous misfolding and aggregation of correctly folded cellular proteins into pathogenic aggregates might explain why disease onset is typically in mid life or later. Following the lag phase, intercellular transfer of proteins might even have a role in the propagation of neuropathology in the genetic neurodegenerative disorders. Thus, once aggregated proteins have appeared in a stochastic manner they may be extruded from an initiating subpopulation of cells and spread to neighbouring neurons.
Whether prion-like transmission mechanisms actively contribute to the pathogenesis of idiopathic neurodegenerative diseases remains unclear. We cannot exclude that disease-related proteins transfer between individuals through the use of contaminated surgical tools or on tissue transplant, but there are no reports to support this. If prion-like transmission has a role, it seems more likely to contribute to the gradual spreading of neuropathological changes in the brains of afflicted individuals. Importantly, the possible existence of extracellular intermediates in the progression of what previously have been considered strictly cell-autonomous intra-cellular disorders, provides a hitherto unappreciated extracellular stage in pathogenesis. This extracellular step in the pathogenesis may represent a more readily accessible target for novel therapeutic intervention.
Acknowledgments
All three investigators are supported by a joint Human Frontier Science Program grant on the topic relevant to this article. In addition, P.B. holds related grants from the MJ Fox Foundation for Parkinson’s Research, Swedish Brain Foundation, Swedish Parkinson Foundation, Söderberg Foundation and the Swedish Research Council. R.R.K. is supported by the Huntington’s disease Society of America Coalition for the Cure, the CHDI Foundation and the National Institute of Neurological Disease and Stroke. R.M. is supported by the Agence Nationale de la Recherche and the Centre National de la Recherche Scientifique. R.M. and P.B. are part of the ERA-net Neuron program MIPROTRAN.
Footnotes
Competing interests statement
The authors declare no competing financial interests.
FURTHER INFORMATION
Patrik Brundin’s homepage: www.med.lu.se/expmed/nesu
ALL LINKS ARE ACTIVE IN THE ONLINE PDF
Contributor Information
Patrik Brundin, Email: patrik.brundin@med.lu.se, Neuronal Survival Unit, Wallenberg Neuroscience Center, Lund University, BMC A10, 221 84 Lund, Sweden.
Ronald Melki, Email: melki@lebs.cnrs-gif.fr, Laboratoire d’Enzymologie et Biochimie Structurales, Centre National de la Recherche Scientifique, 91198 Gif–sur–Yvette, France.
Ron Kopito, Email: kopito@stanford.edu, Department of Biology, Stanford University, Stanford, California 94305–5020, USA.
References
- 1.Prusiner SB. Prions. Proc Natl Acad Sci USA. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–144. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- 3.Uversky VN. Neuropathology, biochemistry, and biophysics of α-synuclein aggregation. J Neurochem. 2007;103:17–37. doi: 10.1111/j.1471-4159.2007.04764.x. [DOI] [PubMed] [Google Scholar]
- 4.Braak H, Ghebremedhin E, Rub U, Bratzke H, Del Tredici K. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 2004;318:121–134. doi: 10.1007/s00441-004-0956-9. [DOI] [PubMed] [Google Scholar]
- 5.Braak H, et al. Stanley Fahn Lecture 2005: The staging procedure for the inclusion body pathology associated with sporadic Parkinson’s disease reconsidered. Mov Disord. 2006;21:2042–2051. doi: 10.1002/mds.21065. [DOI] [PubMed] [Google Scholar]
- 6.Hawkes CH, Del Tredici K, Braak H. Parkinson’s disease: a dual-hit hypothesis. Neuropathol Appl Neurobiol. 2007;33:599–614. doi: 10.1111/j.1365-2990.2007.00874.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jang H, et al. Highly pathogenic H5N1 influenza virus can enter the central nervous system and induce neuroinflammation and neurodegeneration. Proc Natl Acad Sci USA. 2009;106:14063–14068. doi: 10.1073/pnas.0900096106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jang H, Boltz DA, Webster RG, Smeyne RJ. Viral parkinsonism. Biochim Biophys Acta. 2009;1792:714–721. doi: 10.1016/j.bbadis.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burke RE, Dauer WT, Vonsattel JP. A critical evaluation of the Braak staging scheme for Parkinson’s disease. Ann Neurol. 2008;64:485–491. doi: 10.1002/ana.21541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jellinger KA. Formation and development of Lewy pathology: a critical update. J Neurol. 2009;256:270–279. doi: 10.1007/s00415-009-5243-y. [DOI] [PubMed] [Google Scholar]
- 11.Nelson PT, Braak H, Markesbery WR. Neuropathology and cognitive impairment in Alzheimer disease: a complex but coherent relationship. J Neuropathol Exp Neurol. 2009;68:1–14. doi: 10.1097/NEN.0b013e3181919a48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duyckaerts C, Delatour B, Potier MC. Classification and basic pathology of Alzheimer disease. Acta Neuropathol. 2009;118:5–36. doi: 10.1007/s00401-009-0532-1. [DOI] [PubMed] [Google Scholar]
- 13.Goedert M, Klug A, Crowther RA. Tau protein, the paired helical filament and Alzheimer’s disease. J Alzheimers Dis. 2006;9:195–207. doi: 10.3233/jad-2006-9s323. [DOI] [PubMed] [Google Scholar]
- 14.Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 15.Pearson RC, Esiri MM, Hiorns RW, Wilcock GK, Powell TP. Anatomical correlates of the distribution of the pathological changes in the neocortex in Alzheimer disease. Proc Natl Acad Sci USA. 1985;82:4531–4534. doi: 10.1073/pnas.82.13.4531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 17.Delacourte A, et al. Tau aggregation in the hippocampal formation: an ageing or a pathological process? Exp Gerontol. 2002;37:1291–1296. doi: 10.1016/s0531-5565(02)00141-9. [DOI] [PubMed] [Google Scholar]
- 18.Lace G, et al. Hippocampal tau pathology is related to neuroanatomical connections: an ageing population-based study. Brain. 2009;132:1324–1334. doi: 10.1093/brain/awp059. [DOI] [PubMed] [Google Scholar]
- 19.Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology. 1992;42:631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- 20.Cattaneo E, Zuccato C, Tartari M. Normal huntingtin function: an alternative approach to Huntington’s disease. Nature Rev Neurosci. 2005;6:919–930. doi: 10.1038/nrn1806. [DOI] [PubMed] [Google Scholar]
- 21.Davies SW, et al. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–548. doi: 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- 22.DiFiglia M, et al. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- 23.Scherzinger E, et al. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell. 1997;90:549–558. doi: 10.1016/s0092-8674(00)80514-0. [DOI] [PubMed] [Google Scholar]
- 24.Duyao M, et al. Trinucleotide repeat length instability and age of onset in Huntington’s disease. Nature Genet. 1993;4:387–392. doi: 10.1038/ng0893-387. [DOI] [PubMed] [Google Scholar]
- 25.Vonsattel JP, DiFiglia M. Huntington disease. J Neuropathol Exp Neurol. 1998;57:369–384. doi: 10.1097/00005072-199805000-00001. [DOI] [PubMed] [Google Scholar]
- 26.Deng YP, et al. Differential loss of striatal projection systems in Huntington’s disease: a quantitative immunohistochemical study. J Chem Neuroanat. 2004;27:143–164. doi: 10.1016/j.jchemneu.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 27.Kipps CM, et al. Progression of structural neuropathology in preclinical Huntington’s disease: a tensor based morphometry study. J Neurol Neurosurg Psychiatr. 2005;76:650–655. doi: 10.1136/jnnp.2004.047993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rosas HD, et al. Cerebral cortex and the clinical expression of Huntington’s disease: complexity and heterogeneity. Brain. 2008;131:1057–1068. doi: 10.1093/brain/awn025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brundin P, Li JY, Holton JL, Lindvall O, Revesz T. Research in motion: the enigma of Parkinson’s disease pathology spread. Nature Rev Neursci. 2008;9:741–745. doi: 10.1038/nrn2477. [DOI] [PubMed] [Google Scholar]
- 30.Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nature Med. 2008;14:504–506. doi: 10.1038/nm1747. [DOI] [PubMed] [Google Scholar]
- 31.Kordower JH, Chu Y, Hauser RA, Olanow CW, Freeman TB. Transplanted dopaminergic neurons develop PD pathologic changes: a second case report. Mov Disord. 2008;23:2303–2306. doi: 10.1002/mds.22369. [DOI] [PubMed] [Google Scholar]
- 32.Li JY, et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nature Med. 2008;14:501–503. doi: 10.1038/nm1746. [DOI] [PubMed] [Google Scholar]
- 33.Li JY, et al. Characterization of Lewy body pathology in 12- and 16-year old intrastriatal mesencephalic grafts surviving in a patient with Parkinson’s disease. Mov Disord. 2010 Mar 2; doi: 10.1002/mds.23012. [DOI] [PubMed] [Google Scholar]
- 34.Desplats P, et al. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of α-synuclein. Proc Natl Acad Sci USA. 2009;106:13010–13015. doi: 10.1073/pnas.0903691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee HJ, et al. Assembly-dependent endocytosis and clearance of extracellular α-synuclein. Int J Biochem Cell Biol. 2008;40:1835–1849. doi: 10.1016/j.biocel.2008.01.017. [DOI] [PubMed] [Google Scholar]
- 36.Danzer KM, Krebs SK, Wolff M, Birk G, Hengerer B. Seeding induced by α-synuclein oligomers provides evidence for spreading of α-synuclein pathology. J Neurochem. 2009;111:192–203. doi: 10.1111/j.1471-4159.2009.06324.x. [DOI] [PubMed] [Google Scholar]
- 37.Danzer KM, et al. Different species of α-synuclein oligomers induce calcium influx and seeding. J Neurosci. 2007;27:9220–9232. doi: 10.1523/JNEUROSCI.2617-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meyer-Luehmann M, et al. Exogenous induction of cerebral β-amyloidogenesis is governed by agent and host. Science. 2006;313:1781–1784. doi: 10.1126/science.1131864. [DOI] [PubMed] [Google Scholar]
- 39.Frost B, Jacks R, Diamond M. Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem. 2009;284:12845–12852. doi: 10.1074/jbc.M808759200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Clavaguera F, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nature Cell Biol. 2009;11:907–913. doi: 10.1038/ncb1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang W, Dunlap JR, Andrews RB, Wetzel R. Aggregated polyglutamine peptides delivered to nuclei are toxic to mammalian cells. Hum Mol Genet. 2002;11:2905–2917. doi: 10.1093/hmg/11.23.2905. [DOI] [PubMed] [Google Scholar]
- 42.Ren PH, et al. Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nature Cell Biol. 2009;11:219–225. doi: 10.1038/ncb1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rujano MA, et al. Polarised asymmetric inheritance of accumulated protein damage in higher eukaryotes. PLoS Biol. 2006;4:e417. doi: 10.1371/journal.pbio.0040417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cicchetti F, et al. Neural transplants in patients with Huntington’s disease undergo disease-like neuronal degeneration. Proc Natl Acad Sci USA. 2009;106:12483–12488. doi: 10.1073/pnas.0904239106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vogiatzi T, Xilouri M, Vekrellis K, Stefanis L. Wild type α-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J Biol Chem. 2008;283:23542–23556. doi: 10.1074/jbc.M801992200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jaiswal JK, Fix M, Takano T, Nedergaard M, Simon SM. Resolving vesicle fusion from lysis to monitor calcium-triggered lysosomal exocytosis in astrocytes. Proc Natl Acad Sci USA. 2007;104:14151–14156. doi: 10.1073/pnas.0704935104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee HJ, Patel S, Lee SJ. Intravesicular localization and exocytosis of α-synuclein and its aggregates. J Neurosci. 2005;25:6016–6024. doi: 10.1523/JNEUROSCI.0692-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morten IJ, Gosal WS, Radford SE, Hewitt EW. Investigation into the role of macrophages in the formation and degradation of β2-microglobulin amyloid fibrils. J Biol Chem. 2007;282:29691–29700. doi: 10.1074/jbc.M705004200. [DOI] [PubMed] [Google Scholar]
- 49.Bucciantini M, et al. Prefibrillar amyloid protein aggregates share common features of cytotoxicity. J Biol Chem. 2004;279:31374–31382. doi: 10.1074/jbc.M400348200. [DOI] [PubMed] [Google Scholar]
- 50.van Rooijen BD, Claessens MM, Subramaniam V. Lipid bilayer disruption by oligomeric α-synuclein depends on bilayer charge and accessibility of the hydrophobic core. Biochim Biophys Acta. 2009;1788:1271–1278. doi: 10.1016/j.bbamem.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 51.Gousset K, et al. Prions hijack tunnelling nanotubes for intercellular spread. Nature Cell Biol. 2009;11:328–336. doi: 10.1038/ncb1841. [DOI] [PubMed] [Google Scholar]
- 52.Brown R. A brief account of microscopical observations made in the month of June, July and August, 1827, on the particles contained in the pollen of plants; and on the general existence of active molecules in organic and inorganic bodies. Phil Mag. 1828;4:161–173. [Google Scholar]