

Abstract
Factors contributing to reduced magnesium-induced neuronal action potential bursting were investigated in primary hippocampal cell culture at high and low culture density. In nominally zero external magnesium medium, pyramidal neurons from high-density cultures produced recurrent spontaneous action potential bursts superimposed on prolonged depolarizations. These bursts were partially attenuated by the NMDA receptor antagonist D-APV. Pharmacological analysis of miniature excitatory postsynaptic currents (EPSCs) revealed 2 components: one sensitive to D-APV and another to the AMPA receptor antagonist DNQX. The components were kinetically distinct. Participation of NMDA receptors in reduced magnesium-induced synaptic events was supported by the localization of the NR1 subunit of the NMDA receptor with the presynaptic vesicular protein synaptophysin. Presynaptically, zero magnesium induced a significant increase in EPSC frequency likely attributable to increased neuronal hyperexcitability induced by reduced membrane surface charge screening. Mean quantal content was significantly increased in zero magnesium. Cells from low-density cultures did not exhibit action potential bursting in zero magnesium but did show increased EPSC frequency. Low-density neurons had less synaptophysin immunofluorescence and fewer active synapses as determined by FM1-43 analysis. These results demonstrate that multiple factors are involved in network bursting. Increased probability of transmitter release presynaptically, enhanced NMDA receptor-mediated excitability postsynaptically, and extent of neuronal interconnectivity contribute to initiation and maintenance of elevated network excitability.
INTRODUCTION
Fast excitatory neurotransmission in the hippocampus is mediated primarily by glutamate acting on 3 types of ionotropic receptors named for their preferred agonists: kainate, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), and N-methyl-D-aspartate (NMDA) (Dingledine et al. 1990). All 3 types of receptor incorporate ion channels which, when activated, become cation permeable. Excitatory glutamatergic synaptic interactions are maintained in in vitro preparations of hippocampal slice (Parfitt and Madison 1993) and pyramidal cell culture. Hippocampal neuronal cultures establish dense neuritic networks with multiple synaptic contacts (Banker and Cowan 1977; Fletcher et al. 1991; Hoch and Dingledine 1986; Muller and Seifert 1982). AMPA and NMDA receptors are increasingly expressed and localized to synaptic sites with time (Bekkers and Stevens 1989; Rao et al. 1998). These anatomic studies have been bolstered by physiological findings of functionally intact excitatory synaptic interactions (Cummings et al. 1996; Wilcox et al. 1994).
Over the course of several years, study of physiological and pathophysiological aspects of glutamatergic neurotransmission has led to the development of techniques by which the excitability level of synaptic interactions can be modulated. One such technique involves the manipulation of divalent ions. The role of calcium in exocytotic neurotransmitter release has been well documented (see Poage and Meriney 2002 for recent review). Reduced extracellular calcium has been used for many years as a standard tool for inhibiting synaptic transmission. Magnesium also has marked effects on synaptic interactions. Dodge and Rahamimoff (1967) first demonstrated that quantal release at the frog neuromuscular junction decreased steeply as Mg2+ concentration increased. In the hippocampus, elevated magnesium has been found to attenuate or abolish the prolonged neuronal depolarizations and synchonized action potential bursts emblematic of epileptiform activity (Schwartzkroin and Prince 1978). Conversely, reducing magnesium below physiological levels induces enhanced excitabilty in both hippocampal slice (Walther et al. 1986) and cultures (Sombati and DeLorenzo 1995).
The mechanism of reduced magnesium enhancement of excitatory neurotransmission has been presumed to be related to the ions’ physiological blockade of NMDA receptor channels. It has long been recognized that physiological magnesium provides a voltage-dependent steric blockade of the cation permeable pore of NMDA receptors and thus renders them largely inactive at normal membrane potentials (Coan and Collingridge 1985; Collingridge et al. 1983; Hablitz and Langmoen 1986; Mayer and Westbrook 1984). Several studies have demonstrated that synchronized spontaneous neuronal activity induced by eliminating extracellular magnesium is reduced or abolished by NMDA receptor antagonists (Albowitz et al. 1997; Coan and Collingridge 1987; Collingridge et al. 1988; Gulyas-Kovács et al. 2002; Gutierrez et al. 1999; Tancredi et al. 1990). These studies suggested that increased excitabilty induced by magnesium reduction was primarily a postsynaptic phenomenon, caused by increased flux of depolarizing ions, mostly calcium, through NMDA receptor channels.
Other studies have suggested additional excitatory mechanisms associated with extracellular magnesium reduction. Magnesium reduction (vs. removal) induced enhanced excitatory responses in the CA1 region of the hippocampus that were not reversed by NMDA receptor antagonists (Hamon et al. 1987). A careful study by Mody and colleagues (1987) suggested that reducing magnesium may produce a reduction in the electric field across the neuronal membrane by reduced surface charge screening causing what is, in essence, a membrane depolarization.
However, modeling studies in the hippocampus have suggested that enhanced excitability, although necessary, is not sufficient for the occurrence of bursting in a network of pyramidal neurons. A model network simulation of hippocampal bursting demonstrated that bursting can occur, even in the presence of neuronal inhibition, only if connectivity between neurons in the circuit is adequately dense. Low connectivity prevents the development of bursting (Traub et al. 1984, 1987).
We have used electrophysiological and immunocytochemical techniques to examine the factors affecting low magnesium-induced bursting in networks of cultured hippocampal pyramidal neurons. Specifically, we sought to identify the contributions of presynaptic and postsynaptic processes to enhanced network excitability and to test model-driven assumptions of adequate connectivity required for synchronized bursting.
METHODS
Treatment of animals followed guidelines set by the University of Virginia Health Sciences Center Animal Research Committee. All efforts were made to minimize animal stress and discomfort.
Hippocampal cultures
Neuronal hippocampal/glial cocultures were prepared according to the method of Goslin et al. (1998). Neurons and glia were cultured on separate surfaces then combined to form a tissue-culture “sandwich.” This approach allowed the preparation of relatively low density hippocampal cultures while still allowing access of hippocampal neurons to glia-derived neurotrophic substances. Each segment of the preparation is summarized.
Preparation of coverslips
FisherBrand coverslips (12-545-86-1D; 25 mm) were placed in 10% nitric acid for ≥18 h. Coverslips were then rinsed in 6 changes of distilled water (20 min each rinse) and sterilized with dry heat (225° for 6 h). After cooling, coverslips were coated with poly-L-lysine (1 mg/ml in borate buffer), placed in a 37° incubator overnight, then rinsed twice with sterile distilled water (30 min each rinse). Coverslips were than layered with minimum essential medium (MEM) plus 10% horse serum (HS: see Goslin et al. for detailed constituents) and placed in a 37° incubator. Fresh MEM/HS was added the day of the coculture procedure (see following text).
Primary glial culture preparation
Glial cell cultures were prepared 10 days before coculturing with hippocampal neurons. Before glial isolation, 3 drops of sterile, melted paraffin were applied at the vertices of an equilateral triangle in several sterile 60-mm tissue-culture dishes. The paraffin provided the spacing necessary to keep the hippocampal neuron-containing cover-slips from directly contacting glial cells during coculturing (see following text). Alternatively, spacing was provided by longitidinally cut Teflon O-rings (3 mm width, 22 mm ID; Small Parts, Miami Lakes, FL). In a laminar flow hood, neonatal Sprague-Dawley rat pups were decapitated after being placed on ice for 2–3 min. Brains were removed into cold HEPES-buffered Hank’s balanced salt solution (HEPES/HBSS). Cerebral hemispheres were isolated and the meninges removed. Excess HEPES/HBSS was removed and the tissue chopped as finely as possible with scissors. The tissue was placed in 12 ml fresh HEPES/HBSS to which 1.5 ml 2.5% trypsin and 1.5 ml DNase (1%) were added. The tissue was incubated at 37° for 15 min with continuous slow speed stirring. The supernatant was passed through sterile nylon mesh (215 m) and diluted with an equal volume of 10% HS in MEM. Cell suspension was centrifuged at 800–1,000 rpm for 5 min. Pellets were resuspended in 10 ml MEM/10% HS. Cells were counted with a hemocytometer. The yield was generally 6 × 106–1 × 107 cells per brain. Cells were diluted to approximately 30,000 cells/ml in 10% HS/MEM; 3 ml was placed in each of several 60-mm tissue-culture dishes. Medium was completely replaced the day after plating; thereafter, medium was replaced every 4 days with 10% HS/MEM.
Hippocampal neuronal culture preparation
Cultures were prepared from E-18 Sprague-Dawley rat fetuses. Fetuses were decapitated and brains removed and placed in HEPES-HBSS. Hippocampi were removed under a dissecting microscope and collected in a small petri dish in HEPES-HBSS. Hippocampi from a single litter were placed in a 15-ml centrifuge tube; HEPES-HBSS was aspirated off and replaced with 5 ml of 0.25% trypsin. The preparation was incubated at 37° for 15 min. Trypsin solution was replaced with 5 ml HEPES-HBSS. Rinsing with HEPES-HBSS was repeated twice more at 5-min intervals. Hippocampi were triturated until no fragments of tissue remained. Cell density was determined with a hemocytometer and was usually approximately 5 × 105 cells/hippocampus. Two densities of hippocampal neurons were prepared. For high-density cultures, 100,000 cells were added to each of the dishes containing the polylysine-coated coverslips prepared previously; for low-density cultures, 10,000 cells were added. After 3– 4 h, coverslips were transferred to dishes containing glial cell monolayers in serum-free MEM with N2 supplement. Coverslips were turned so that hippocampal neurons faced glial cells and placed on paraffin or Teflon spacers; spacing was about 2 mm between the cell layers. N2 supplement (1 ml) was added every 10 days.
Electrophysiology
Patch electrodes were pulled from 1.5 mm (OD) × 1.1 mm (ID) borosilicate micropipettes on a horizontal Flaming-Brown microelectrode puller (model P-97, Sutter Instruments) using a 2-stage pull protocol. Electrode resistances were 5– 8 MΩ. Electrode tips were filled with an internal stock recording solution consisted of (in mM) CsCl 153.3, MgCl2 1.0, HEPES 10.0, and ethylene glycol-bis(β-aminoethyl ether) N,N,N′,N′-tetraacetic acid (EGTA) 5.0, pH 7.3 (with sterile-filtered CsOH), osmolarity 275–280 mOsm. Internal solution was sterile filtered before use. The electrode shank solution contained an ATP regeneration consisting of 3 mM ATP (disodium salt), 19 mM phosphocreatine, and 50 U/ml creatine phosphokinase.
Before electrophysiological experimentation, coverslips containing hippocampal neurons were removed from culture medium and placed in a 30 × 10-mm polystyrene culture dish containing external recording consisting of NaCl (142), CaCl2 (1.0), CsCl (8.1), MgCl2 (2.1), glucose (10.0), and HEPES (10.0), pH 7.4 (NaOH). The osmolarity was adjusted to 320–325 mOsm with sucrose. External recording solution was sterile filtered before use. For nominally zero magnesium experiments, MgCl2 was replaced by an osmotically equivalent concentration of NaCl. Experiments in hippocampal slices have implicated elevated external cesium in the induction of spontaneous recurrent bursting activity in CA3 and dentate gyrus but only in the presence of the calcium channel blocker cadmium (Xiong and Stringer 2001). To determine whether the elevated cesium in our external solution contributed to neuronal bursting in culture and to replicate an external solution used previously by Sombati and De-Lorenzo (1995) in examining the electrophysiological activity of cultured neurons exposed to zero magnesium, KCl (2.5 mM) replaced CsCl in some experiments. NaCl was increased to 145 mM in these experiments. We observed no difference in the frequency of recurrent bursting in these 2 external solutions; results obtained under the 2 conditions were combined. Cultured neurons were viewed on the stage of an inverted Nikon microscope. Microelectrodes were interfaced with the amplifier by an Axon Instruments CV-4 headstage and maneuvered into recording position with a PCS 5000 series micromanipulator (Burleigh Instruments, Fishers, NY).
Whole cell recordings were made at room temperature with an Axopatch 1-D patch clamp amplifier (Axon Instruments, Union City, CA) and low-pass filtered at 2 kHz with an 8-pole Bessel filter before digitization. Data were recorded to a personal computer with Axoscope 7.0 data acquisition software using a Digidata 1200 interface (Axon Instruments). Voltage-clamp recordings of excitatory postsynaptic currents (EPSCs) were digitized at 10–20 kHz; current-clamp recordings of neuronal membrane potential were digitized at 20 kHz. EPSCs were analyzed using MiniAnalysis software (Synaptosoft, Decatur, GA).
Immunocytochemistry
Coverslips used for NMDA receptor subunit NR1, AMPA receptor subunit GluR1, or synaptophysin immunostaining were fixed with 4% paraformaldehyde (PFA)/4% sucrose in phosphate-buffered saline (PBS) for 20–30 min at room temperature. Rao and Craig (1998) reported that NR1 receptor subunit staining required methanol fixation; however, we were able to attain satisfactory staining with PFA. Neurons were permeabilized with 0.25% Triton X-100 in PBS for 5 min. Coverslips were washed twice with PBS, blocked with 10% rabbit serum in PBS for 30 min, and exposed to primary antibodies in 3% rabbit serum in PBS.
The mouse monoclonal antibody 54.1 to NR1 (PharMingen, San Diego, CA) was used at a concentration of 2 μg/ml and rabbit polyclonal antibody to GluR1 (Chemicon International, Temecula, CA) was used at 1 μg/ml. Either mouse anti-synaptophysin (Chemicon; for use in double-labeling experiments with GluR1) or rabbit anti-synaptophysin (Zymed Laboratories, San Francisco, CA; for use with NR1) was used at a concentration of 2.5 μg/ml.
Primary antibodies were visualized with fluorochrome-conjugated secondary antibodies (3–4 μg/ml, Molecular Probes, Eugene, OR). The fluorochromes used were Alexa Fluor 488 (GluR1) and Alexa Fluor 594 (NR1). Synaptophysin primary antibody was labeled with secondary antibodies conjugated to either Alexa Fluor 488 or Alexa Fluor 594. Coverslips were mounted in glycerol. Fluorescent images were captured on a Nikon inverted Eclipse TE 200 microscope equipped with a Photometrics CoolSnap cf digital camera using a 40× oil immersion objective (1.0 numerical aperture). Images were captured using Metamorph imaging software and prepared for printing with AdobePhotoshop 6.0.
For whole cell visualization, neurons were first filled with Biotin (Vector Laboratories; 0.5 mg/ml in internal pipette solution), then incubated with strepatavidin conjugated with Alexa Fluor 350 (Molecular Probes).
FM1-43 visualization
Cells were loaded by exposure to 10 mM potassium external solution (osmolarity maintained by concomitant decrease in sodium concentration) with 8 μM FM1-43 for 10 min. Cells were then washed 3–4 times with normal external solution. For destaining and visualization, cells were exposed to nominally zero-magnesium external.
Data analysis
All data are presented as means + SE unless otherwise noted in the text.
RESULTS
Neuronal properties
Morphologically identified pyramidal-shaped neurons in culture for 13–17 days were studied by the whole cell patch-clamp technique. In initial experiments, pyramidal cells and their processes were filled with biocytin during electrophysiological recording and visualized with fluorescent dye after fixation. Large- and small-diameter dendrites emerged from the apex and base of pyramidal neurons and branched into secondary and tertiary dendrites (Fig. 1). The complex dendritic arbor of pyramidal neurons extended several hundreds of microns from the cell soma. Previous studies have shown that nonpyramidal interneurons are morphologically distinct bipolar or multipolar neurons whose cell soma stains intensely with the γ-aminobutyric acid synthetic enzyme glutamic acid decarboxylase (GAD; Esclapez and Houser 1999). Passive and active membrane properties of these pyramidal neurons were examined in current clamp mode in magnesium-containing external medium to establish that neuronal properties conformed to those for healthy, viable cells. Mean resting membrane potential was −58.3 ± 2.4 mV (n = 23), in good agreement with previous studies on cultured pyramidal neurons (Coulter et al. 1992).
FIG. 1.
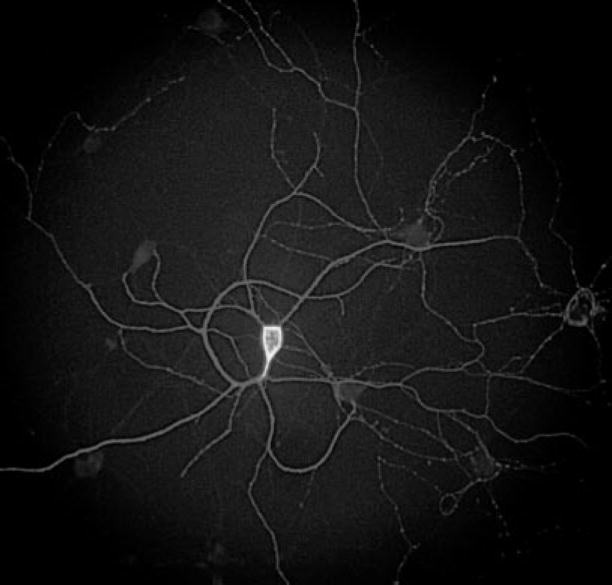
Hippocampal pyramidal neuron cultured for 16 days. Cell was filled with biotin by the recording pipette and then incubated with streptavidin conjugated to a fluorescent probe (see METHODS). Note the outline of a multipolar cell soma at extreme center right of photograph.
Neuronal input resistance was measured in a sampling of cells (n = 7) by injecting 2-s current pulses (−1.0 to 0.3 nA in 0.1-nA increments). Rin was given by the slope of the plot of current versus voltage change (Mangan and Bertram 1998). The Rin was 71.3 ± 4.2 MS, higher than that recorded in pyramidal neurons with sharp microelectrodes in our previous studies (Mangan and Bertram 1998; Mangan and Lothman 1996; Rempe et al. 1995), but in accord with other studies using patch electrodes (Spruston and Johnston 1992; Spruston et al. 1994). Action potential (AP) characteristics were also determined as an indication of active, voltage-dependent membrane function. Membrane potential was varied by current injection. Threshold for AP firing was −59.4 ± 2.2 mV (n = 24). Holding the membrane potential at −58 mV by current injection (usually <100 pA) resulted in occasional spontaneous overshooting APs with amplitude 77.2 ± 3.4 mV. These AP properties were in accordance with previous findings (Rempe et al. 1995). Rarely (4 of 36 neurons tested), depolarization (to about −50 mV) resulted in prolonged action potential bursts. In 3/4 cells, these bursts were not recurrent; in one cell bursting continued with an irregular interburst interval for the duration of the depolarization (about 3 min).
In nominally Mg2+-free external medium (0 [Mg2+]o), most cells exhibited recurrent sustained bursting consisting of multiple (≥) 3 APs with interspike intervals of <200 ms superimposed on a prolonged (500–2,000 ms) depolarization (Fig. 2B). Two pieces of evidence suggested that spike bursting was network-driven rather than an endogenous neuronal property. First, similar activity was absent from cells in low-density cultures (see Fig. 7 below). Second, hyperpolarization to as much as −90 mV by DC current injection often did not terminate bursting. Bursting occurred in 38/43 cells examined and was sustained for the length of the recording (≤20 min). Exposure to 0 [Mg2+]o did not significantly alter resting membrane potential (RMP) (−57.6 ± 3.1 mV; a period of 2–10 s occurred between bursts in all cells; the mean RMP was calculated from this interburst interval), Rin (67.7 ± 3.3 MS; n = 7), or action potential height recorded at a holding potential of −58 mV (79.6 ± 2.1 mV; n = 21). However, the AP firing threshold was shifted significantly to −77.7 ± 4.5 mV after 0 [Mg2+]o exposure. Additionally, treatment resulted in an altered pattern of AP firing. Instead of individually occurring spikes, APs in 0 [Mg2+]o tended to group in discrete repeating epochs (Fig. 2B). Bursting behavior in 0 [Mg2+]o contrasted with action potential firing in normal Mag where 32/36 cells did not burst (Fig. 2A).
FIG. 2.
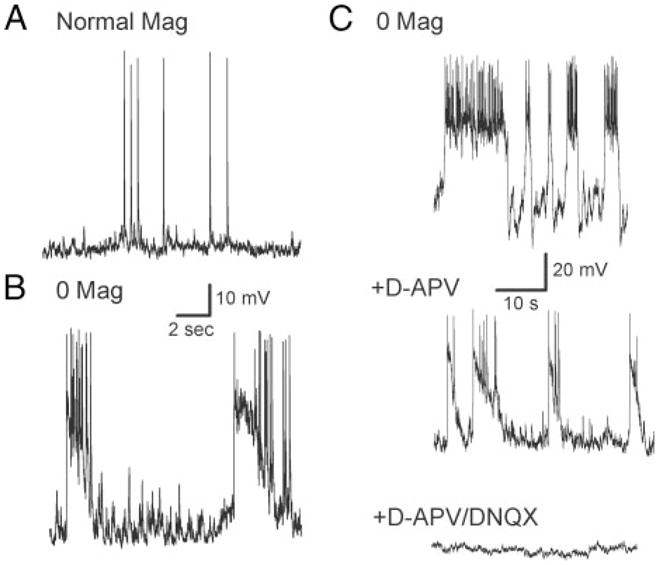
Current clamp recordings from cultured hippocampal pyramidal neurons in normal and nominally zero external magnesium (0 [Mg2+]o). A: recording from a pyramidal cell 16 days in culture. External medium contained 2.1 mM Mg2+. Membrane potential was held at −58 mV by DC current injection. B: recording from a neuron 15 days in culture. Magnesium in the external solution was replaced by equiosmolar Na+. Membrane potential was held at −58 mV. C: pharmacology of pyramidal cell bursting in 0 [Mg2+]o. Top trace: recording from a pyramidal cell 14 days in culture exposed to 0 [Mg2+]o. Middle trace: recording from the same cell 25 min after addition of 40 μM D-APV, an NMDA receptor antagonist. Bursting activity is attenuated but not abolished. Bottom trace: neuronal excitatory activity ceases 15 min after further addition of 20 μM DNQX, an AMPA receptor antagonist. Recordings in A, B, and C are from different cells.
FIG. 7.

Whole cell current and voltage-clamp recordings, in normal and 0 [Mg2+]o, from hippocampal pyramidal neurons cultured at low density. 0 [Mg2+]o treatment did not produce action potential bursting (A); sEPSC frequency did show a slight, but significant increase (B). C: cumulative fraction interevent interval histogram for the cells shown in B. 0 [Mg2+]o condition resulted in significant change in interval distribution (Kolmogorov-Smirnov 2 sample test; d = 0.31, P = 0.04). Normal and 0 [Mg2+]o recordings were from different cells.
Role of NMDA receptors
Removal of magnesium enhances excitatory activity in neuronal networks by abolishing the voltage-dependent blockade of NMDA-type glutamate receptors (Ascher et al. 1988; Mody et al. 1988; Tancredi et al. 1988; Westbrook 1994). We tested whether 0 [Mg2+]o-induced bursts could be blocked by the NMDA receptor antagonist D-2-amino-5-phosphonovaleric acid (D-APV; Fig. 2C). A neuron 17 DIV exhibited recurrent spontaneous action potential bursts in 0 [Mg2+]o external solution. Addition of D-APV (40 μM) reduced the mean number of action potentials per burst and increased the interburst interval but did not return the cell to the intermittent firing of single action potentials observed in normal magnesium. Only on further addition of the AMPA receptor blocker dinitroquinoxaline (DNQX; 20 μM) was bursting behavior eliminated. This observation was repeated in 14 cells from 8 separate culture preparations (each culture prepared from different embryos). In normal external magnesium, synaptically driven action potential firing was abolished by DNQX alone (data not shown); cells were still capable of spiking if depolarized by DC current injection. These studies demonstrated that APV only partially attenuated 0 [Mg2+]o-induced bursting.
One possible explanation for the failure of D-APV to completely block 0 [Mg2+]o-induced bursts was that NMDA receptors were not present at synapses in cultured hippocampal neurons and these did not participate in excitatory neurotransmission. Alternately, 40 μM D-APV may have been insufficient to block NMDA receptor currents in the 0 [Mg2+]o condition. Finally, it is possible that multiple mechanisms enhance synaptic transmission in 0 [Mg2+]o medium. To evaluate these possibilities, voltage-clamp recordings of spontaneous excitatory postsynaptic currents (sEPSCs) were performed at −50 mV in the presence of 5 μM bicuculline methiodide to block GABAA receptor-mediated events. In some experiments, 1 μM tetrodotoxin (TTX) was used to eliminate spontaneous release of multiple neurotransmitter vesicles and produce single postsynaptic quantal events [i.e., miniature EPSCs (mEPSCs).]
Figure 3 shows an averaged mEPSC consisting of ≥1,000 events from 10 cells under 0 [Mg2+]o conditions. The averaged event had a mean 10–90% rise time of 2.2 ± 0.3 ms and a biexponential decay with τ1 and τ2 of 16.2 ± 1.4 and 80.8 ± 3.2 ms, respectively. After addition of D-APV (Fig. 3, top arrow), neurons produced mEPSCs with mean rise of 1.2 ± 0.3 ms and a monoexponential decay with τ 33.3 ± 2.2 ms (5 cells, 612 events). If, instead of blocking NMDA receptor-mediated events, TTX-treated cells were exposed to the AMPA receptor blocker DNQX (Fig. 3, bottom arrow), the resulting mEPSCs were much slower kinetically, with a rise time of 4.4 ± 0.9 ms and decay τof 64.1 ± 3.7 ms (373 events, 5 cells). These experiments suggested that NMDA receptors were present and active at synapses in the 0 [Mg2+]o condition. They also suggested that all NMDA receptor-mediated currents were blocked by 40 μM S-APV.
FIG. 3.
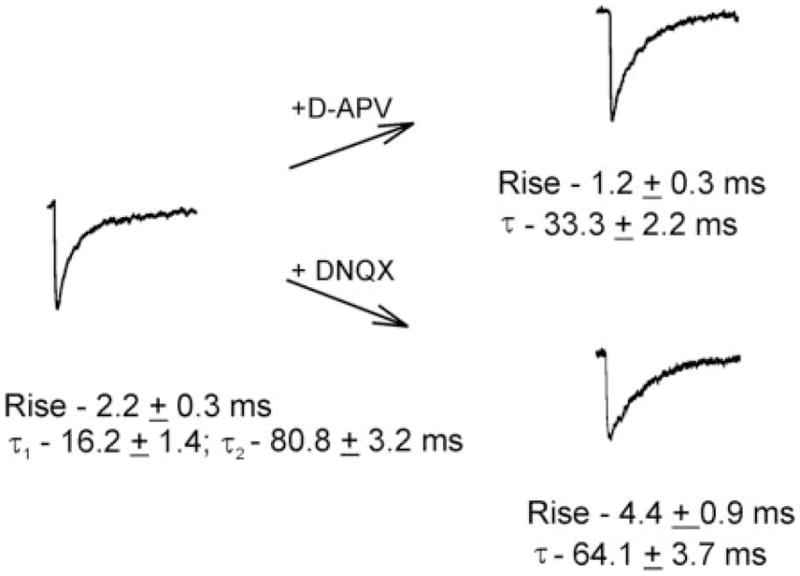
Pharmacology of miniature excitatory postsynaptic currents (mEPSCs) in pyramidal cells exposed to 0 [Mg2+]o. Left-most trace: averaged mEPSC with biexponential decay kinetics. Exposure to D-APV (top arrow) produced an averaged mEPSC with a faster 10–90% rise time (1.2 ± 0.3 ms vs. 2.2 ± 0.3 ms) and a monoexponential decay with τdecay of 33.3 ±2.2 ms. If exposed to DNQX (instead of D-APV; bottom arrow), the resulting mEPSCs have a much slower rise (4.4 ± 0.9 ms) and τdecay (64.1 ± 3.7 ms).
We further confirmed the presence of NMDA receptors at synapses on pyramidal neurons in culture at 13–17 days by immunohistochemical staining. The obligate NMDA receptor subunit NR1 was examined in relation to the presynaptic vesicular protein synaptophysin. In the cell illustrated in Fig. 4, synaptophysin clusters (green fluorescence) were evident in all cellular processes within the field of view (left panels; bottom panel is a higher magnification view of the boxed area in the top panel). NR1 staining was more discrete (red fluorescence) but was widespread in this neuron (middle panels). Overlay of synaptophysin and NR1 fluorescence revealed that most NR1 clusters colocalized with synaptophysin (right panels, orange clusters), suggesting a postsynaptic locus for NMDA receptors at this stage of neuronal development.
FIG. 4.

Hippocampal neuron 14 days in culture labeled with immunofluorescent antibodies to synaptophysin (green) and the NR1 subunit of the NMDA receptor. Labeling of each is extensive proximal and distal to the cell body within the field of view (top row, left and center). Overlay of the 2 images reveals colocalization of synaptophysin and NR1 (top right, orange punta). Box outlined in the top left photo is enlarged in the bottom row of images.
Presynaptic effects of reduced magnesium
In addition to unblocking NMDA receptors, lowered magnesium can induce increased excitability by presynaptic mechanisms. For example, magnesium displaces calcium at the presynaptic membrane surface layer (McLaughlin et al. 1978), thus reducing the amount of calcium available for entry into presynaptic terminals. Removal of magnesium would thus enhance presynaptic release by facilitating influx of calcium into the presynaptic terminal. We therefore examined the effect of magnesium reduction on the frequency of transmitter release. Figure 5, A and B illustrates sEPSCs recorded in normal and reduced magnesium. sEPSC frequency was markedly increased in the neuron recorded in 0 [Mg2+]o; 90% of events occurred with interevent intervals <800 ms (Fig. 5C). In normal magnesium, the comparable interval was 5,200 ms (Fig. 5C). The shift in interevent interval for the cell pictured in Fig. 5 was significant [Kolmogorov–Smirnov 2-sample test; d = 0.58, P < 0.001]. Analysis of 8 cells each in normal and reduced magnesium revealed a similar increase in frequency. In each cell, the 90% interevent interval cutoff value decreased by ≥3-fold.
FIG. 5.

Spontaneous excitatory postsynaptic currents (sEPSCs) in cultured hiipocampal pyramidal cells. sEPSC frequency, recorded in normal external magnesium (A), is markedly increased in zero magnesium medium (B). C: cumulative fraction histogram plot of interevent intervals for the complete 4-min recordings from the cells shown in A and B. Intervals were significantly shorter in zero magnesium (Kolmogorov–Smirnov 2-sample test; d = 0.58, P < 0.001). Recordings in A and B are from 2 different cells.
These data demonstrated an increased frequency of synaptic currents recorded postsynaptically, likely attributable in part to increased neuronal circuit excitability. However, they did not clarify whether magnesium removal also affected presynaptic glutamate release mechanisms. We first examined synaptic activity before and after the application of 1 μM TTX to assess the effect of 0 [Mg2+]o on action potential– dependent transmitter release. Figure 6, A and B shows examples of sEPSC (no TTX) and mEPSC (after TTX application) in cells 16 DIV in normal and 0 [Mg2+]o conditions. For the cellular activity shown, mEPSC frequency was significantly increased in the 0 [Mg2+]o condition (Fig. 6C; Kolmogorov–Smirnov 2-sample test; d = 0.26, P < 0.001). The number of mEPSCs recorded in a 4-min interval was 188 in normal magnesium and 331 in 0 [Mg2+]o. In 6 pairs of cells exposed or not exposed to magnesium, the number of mEPSCs was 57.6 + 12.9% higher in 0 [Mg2+]o conditions versus normal magnesium. This result indicates that action potential–independent neurotransmitter release was enhanced in the 0 [Mg2+]o condition.
FIG. 6.

Effect of tetrodotoxin (TTX) on EPSC amplitude distribution. Cells were recorded for 4 min; 1 μM TTX was then added to the external medium. After 10 min, EPSCs were recorded for another 4-min interval. A: example of sEPSCs in normal (left) and zero (right) external magnesium from a cell 16 days in culture. Addition of TTX caused an overall reduction in EPSC amplitude, producing mEPSCs (B). C: cumulative fraction histogram plot of interevent intervals for the cells shown in A and B. Intervals were significantly shorter in zero 0 [Mg2+]o (Kolmogorov–Smirnov 2-sample test; d = 0.26, P < 0.001). D: amplitude distribution from the complete 4-min recordings from the cells shown in A and B. Most prevalent EPSC amplitude was about 50 pA (recorded at −50 pA) irrespective of magnesium level or the presence of TTX. Note the tail of high-amplitude events in the 0 Mg2+ condition before TTX addition. Ratio of sEPSC and mEPSC amplitudes was used to calculate the mean quantal content of glutamate release (see text).
We also examined whether quantal content, and thus the amplitude, of sEPSCs was altered by 0 [Mg2+]o. Although no formal quantal analysis was performed, the mean number of neurotransmitter quanta released per presynaptic action potential was calculated by the direct method in control and 0 [Mg2+]o conditions. The most frequent sEPSC amplitudes were 50–60 pA, regardless of the magnesium condition (Fig. 6D, open bars). The sEPSC amplitude distribution was skewed toward higher amplitude currents in normal and 0 [Mg2+]o; however, higher-amplitude events were more frequent in 0 [Mg2+]o and reached a maximum of about 400 pA for single events versus 150 pA in normal magnesium. These higher-amplitude events presumably represented glutamate from multiple, simultaneously released quanta. After TTX, 50–60 pA mEPSC amplitudes were most prevalent but the tail of higher-amplitude synaptic currents was eliminated (normal magnesium) or greatly reduced in the number of events (0 [Mg2+]o). Quantal content m was calculated from the ratio of mean sEPSC amplitude to mean mEPSC amplitude. In this example, the values were 1.41 in normal magnesium and 2.40 for 0 [Mg2+]o. Direct calculation of m was performed in 6 cells each in normal and reduced magnesium. Mean quantal content in normal magnesium was 1.36 + 0.05 and increased significantly in 0 [Mg2+]o to 2.09 + 0.15 (P < 0.01, t-test).
Effect of cell density on bursting probability
The preceding experiments demonstrated that 0 [Mg2+]o treatment strengthened excitatory synaptic transmission by both presynaptic and postsynaptic means. However, it was unclear whether 0 [Mg2+]o increased the strength of autapses (recurrent synapses from a neuron onto itself) or that of interconnections between pyramidal neurons. In the past, recurrent bursting was demonstrated in microcultures containing as few as 2 excitatory hippocampal neurons (Segal and Furshpan 1990). However, these results may not be applicable to the culture preparation used in our experiments in that cells were exposed to blockers of synaptic activity (kynurenate and elevated Mg2+) for several weeks and exhibited bursting behavior only when cultures were washed free of these blockers. Chronic NMDA receptor blockade has been demonstrated to markedly increase clustering of NMDA receptors and promote their shift to a more synaptic distribution (Rao and Craig 1997). In contrast to this microculture model, the interconnectivity of multiple neurons was found necessary for recurrent bursting induced by penicillin, picrotoxin, or 4-amino pyridine in hippocampal slices (Hablitz 1984; Schwartzkroin and Prince 1978). To test whether interconnections among hippocampal neurons were necessary for recurrent bursting, they were grown at low density (104 neurons/coverslip). Responses to control and 0 [Mg2+]o conditions were compared with those grown at high density (105 neurons/coverslip) after 13–17 days in culture. Figure 7 shows current- and voltage-clamp recordings from pyramidal neurons cultured from the same embryonic hippocampal cells that were divided into low- and high-density groups. In low-density cultures, neurons were able to generate spontaneous action potentials. Exposure to 0 [Mg2+]o did not result in recurrent bursting (0/18 cells) but did increase the frequency of excitatory postsynaptic currents (Fig. 7A). sEPSC frequency increased significantly with 0 [Mg2+]o treatment in 9/13 cells (Fig. 7, B and C; Kolmogorov–Smirnov 2 sample test; d = 0.31, P = 0.04).
Although network excitability was not altered in 0 [Mg2+]o in low-density cultures, alterations in nonaction potential–dependent presynaptic glutamate release probability should still have been enhanced. We examined this possibility by comparing mEPSC frequency in low-density cultures under normal and 0 [Mg2+]o conditions. Figure 8A shows an example of mEPSCs recorded from cells 14 DIV. In normal magnesium, mEPSCs were infrequent; in 0 [Mg2+]o, mEPSC frequency increased significantly. Eighty percent of events occurred with an interevent interval less than about 4,000 ms; in 0 [Mg2+]o, the interval was significantly shorter at 1,550 ms (Kolmogorov–Smirnov test; d = 0.35, P < 0.001).
FIG. 8.

Miniature EPSCs recorded in cells from low-density cultures in normal and 0 [Mg2+]o. A: excerpts from 4-min recordings from 2 different cells. B: analysis of the complete recordings from cells shown in A revealed a significantly shorter interevent interval distribution in 0 [Mg2+]o (Kolmogorov–Smirnov 2-sample test; df = 0.35, P < 0.001).
The finding that neurons from low-density cultures could not be induced to burst suggested that they form fewer active synapses that those cultured more densely. This was tested directly. The presence of synaptic contacts in cells from low-and high-density cultures was compared qualitatively by staining for synaptophysin distribution. In neurons from high-density culture pictured in Fig. 9A, synaptophysin clusters were ubiquitous on all neuronal processes. In several regions, individual puncta could not be distinguished because of their high density. In contrast, synaptophysin distribution was more sparse in the low-density example; individual synaptophysin puncta were easily distinguished on all processes, suggesting that neurons in low-density culture make and receive fewer synaptic contacts.
FIG. 9.

Comparison of synaptophysin (A) and FM1-43 (B) fluorescence in high- vs. low-density hippocampal cultures. Staining for the presynaptic marker synaptophysin and vesicular endocytosis is markedly higher in denser cultures.
To examine active synaptic activity, neurons from high- and low-density cultures were treated with the fluorescent dye FM1-43, which is taken up in endocytosed synaptic vesicles and is detectable on vesicle exocytosis, thus labeling synapses actively undergoing the exocytotic/endocytotic cycle (Cochilla et al. 1999). Figure 9B shows neurons from high- and low-density cultures exposed to FM1-43. The number of fluorescent puncta was markedly higher in the high-density neurons. Quantitative analysis of puncta in 5 cells each from high- and low-density cultures was performed by counting puncta in an area of about 30 × 30 μm. Puncta were counted in 2 areas per cell showing the greatest synaptic activity. In low-density neurons, the mean number of FM1-43 fluorescent puncta was 48.7 ± 5.1; for high-density neurons, activity was significantly higher, 177.2 ± 25.1 (P < 0.001, t-test). These data demonstrate that functional connectivity between cells, as measured by active synaptic contacts, is much reduced in low-density cultures.
DISCUSSION
The major findings of this study are 1) networks of cultured hippocampal neurons were capable of generating recurrent spontaneous action potential bursts very similar to those observed in intact hippocampal slice preparations when exposed to nominally zero magnesium; 2) postsynaptically, bursting behavior was attenuated, but not abolished, by D-APV, suggesting that abolition of magnesium blockade of NMDA receptor channels contributed to bursting onset and maintenance; 3) presynaptically, 0 [Mg2+]o caused an increased probability of glutamate release (analysis suggested a concomitant increase in mean quantal content); 4) reduced magnesium-induced alterations in pre- and postsynaptic neuronal properties were insufficient to induce bursting in lower-density cultures with sparse synaptic connections, indicating that bursting was a network, rather than intrinsic cellular, phenomenon. However, 0 [Mg2+]o did induce an increase in both sEPSC and mEPSC activity in low-density cultures.
Cultured hippocampal neurons as a model for bursting
Recently cell culture has become a tool for the study of synaptic interactions and plasticity (see Salter 2001 for review). Electrophysiological and morphological characterization has extended information on synapse function not amenable to other types of preparations (Gomperts et al. 2000; Stevens and Wesseling 1999). Cell culture has also been a valuable tool for investigation of pathological conditions relating to alteration of normal synaptic function (DeLorenzo et al. 1998; Furshpan 1991).
Our findings regarding reduced magnesium induction of spontaneous bursting activity in cultured hippocampal neurons add to this understanding. They are in agreement with longstanding findings of bursting by magnesium reduction in slice (Anderson et al. 1986; Avoli et al. 1987), slice culture (Gutierrez et al. 1999), and culture preparations (Sombati and DeLorenzo 1995).
Our findings strongly suggest that bursting activity in our preparation results from synaptic interactions rather than intrinsic neuronal properties. However, our data do not rule out the latter. Bursting behavior occasionally occurred in normal external magnesium. It is unlikely that neuronal hyperexcitability in these cases was the result of hypoxia (Rubaj et al. 2003) or pH alteration. Cultures included both CA1 and CA3 pyramidal neurons; the intrinsic pacemaking properties of the latter have been well described and modeled (Traub et al. 1991). It is possible that, given sufficient density and interconnectedness of a particularly CA3 pyramidal cell-rich region of the culture, spontaneous activity could occur. Numerous studies have shown that network-driven recurrent hyperexcitable discharges in intact hippocampus originate in CA3 and propagate to CA1 (Hablitz 1984; Mody et al. 1988).
The importance of network interactions in enabling burst activity is supported by several lines of evidence. First, neuronal hyperpolarization of 10–20 mV from the resting potential did not eliminate bursting, although burst duration and frequency were reduced. Second, neurons from low-density cultures did not burst under either normal or 0 [Mg2+]o conditions. Third, pharmacological blockade of synaptic excitation eliminated bursts in 0 [Mg2+]o. Simultaneous dual recordings will be required to definitively address the question of whether neurons are receiving synchronized excitatory barrages.
Reduced magnesium bursting: postsynaptic mechanisms
Hippocampal hyperexcitability induced by magnesium reduction or removal has been noted for several years. The effect is sufficiently pronounced that magnesium deficiency has been used as a model of temporal lobe seizures since the late 1970s (Buck et al. 1978). Numerous reports have noted the dependency of reduced-magnesium bursting on functional NMDA receptor/channels (Albowitz et al. 1997; Collingridge et al. 1988; Gulyas-Kovács et al. 2002; Gutierrez et al. 1999; Hamon et al. 1987; Quilichini et al. 2002; Tancredi et al. 1988). This is presumably attributable to the well-documented postsynaptic effect of magnesium in electrostatically blocking NMDA receptors/channels from the outside of the cell at the resting membrane potential (Dingledine et al. 1990). Our results support these previous studies. First, immunofluorescence studies showed NMDA receptors (the obligate NMDA receptor subunit NR1) were localized with synaptophysin in burst-capable cultures, indicating a synaptic locus. Second, magnesium removal was a requirement for bursting in nearly all experiments, whereas bursting was attenuated by the specific NMDA receptor antagonist D-APV. Thus NMDA receptor activation by reduced magnesium appears crucial to initiation and maintenance of neuronal bursting in our preparation.
However, our data suggest additional excitatory postsynaptic mechanisms may be operative. Excessive neuronal activity (bursts of high-frequency EPSCs) induced by reduced magnesium did not return to normal after blockade of NMDA receptors with D-APV. EPSC bursting still occurred, although inter-burst intervals were increased and burst duration was lessened. Blockade of AMPA-type glutamate receptors was necessary to eliminate activity. These observations suggest that increased AMPA receptor activity as well as activation of NMDA receptors may contribute to neuronal hyperexcitability in 0 [Mg2+]o. One possible explanation for this observation may be a recent hypothesis of the physiological substrate for long-term potentiation, a form of synaptic plasticity that may underlie learning and memory. Liao et al. (2001) demonstrated that activation of NMDA receptors in culture results in rapid recruitment (within minutes) of AMPA receptors to NMDA receptor synaptic sites with a parallel increase in mEPSC frequency. Similar enhancement of mEPSCs occurred after brief, focal application of glycine to activate NMDA receptors (Lu et al. 2001). Augmentation of excitatory transmission was exocytosis dependent and presumably involved cycling of AMPA receptors from remote locales to the site of NMDA activation (Liang and Huganir 2001; Luscher and Frerking 2001; Luscher et al. 1999). Such an influx of AMPA receptors to synaptic sites could result in the D-APV–resistant excitability we have observed.
Reduced magnesium bursting: presynaptic mechanisms
Although enhancement of NMDA receptor-mediated excitabilty has been demonstrated to play a role in reduced magnesium induced bursting, it is unlikely to play a significant role in the presynaptic alterations we have observed in zero magnesium: increased sEPSC frequency and an apparent increase in mean quantal content. These changes suggest that the probability of glutamate release was increased by magnesium reduction.
One possible mechanism was first proposed by Frankenhauser and Hodgkin (1957). They reasoned that reductions in divalent cations could increase neuronal excitabilty by decreasing charge screening at the membrane surface. This would cause a smaller portion of the transmembrane potential to fall across the lipid bilayer (McLaughlin et al. 1971), thus decreasing the electric field sensed by voltage-dependent conductances such as calcium channels in the synaptic terminal. Muller and Finkelstein (1974) later proposed a more magnesium-specific model. They hypothesized that increasing Mg2+ concentration displaces calcium in the surface charge layer adjacent to the negatively charged membrane, thus decreasing the amount of calcium available for influx into synaptic terminals. Reducing or eliminating Mg2+ would have the opposite effect, increasing the effective Ca2+ concentration at the mouth of presynaptic Ca2+ channels. This mechanism may explain the increase in intracellular calcium observed in acutely isolated CA1 pyramidal neurons in the presence of reduced extracellular magnesium, an influx mediated by voltage-dependent calcium channels (Zhang et al. 1996). Thus one presynaptic effect of reducing Mg2+ would be to increase the probability of release as proposed in the Katz model of neurotransmission (Katz 1971). The probability of release reflects whether the release site is occupied and the availability of transmitter (Dobrunz and Stevens 1997; Hanse and Gustaffson, 2002; Staley et al. 1998, 2001) but can be modulated by calcium and magnesium (Bouron 2001). Reduced magnesium may also cause increased glutamate release by facilitating calcium egress from intracellular stores by reduced magnesium inhibition of ryanodine receptors (Masumiya et al. 2001). Whether such a process occurs is problematical given that fluctuations in the extracellular magnesium concentration do not appear to alter intracellular levels (Zhang et al. 1996).
Such mechanisms could explain the marked increased in sEPSC frequency, reflecting increased presynaptic glutamate release, observed in reduced magnesium. It may also explain the calculated increase in mean quantal content. Although we did not attempt a formal quantal analysis for this study, we did use 2 different methods for determining quantal content. These methods did not agree as to the extent of the increase, but in both instances, the increase was significant.
Reduced magnesium bursting: connectivity
Physiological and modeling studies in the hippocampus have suggested that synchronized bursting in some systems of pyramidal neurons may require connectivity among a critical minimum of cells and sufficient excitatory synaptic strength to drive the network (Bains et al. 1999; Traub and Wong 1983; Traub et al. 1995). A model network simulation of hippocampal neuronal bursting demonstrated that bursting can occur, even in the presence of neuronal inhibition, only if connectivity between neurons in the circuit is adequately dense. Low connectivity prevents the development of bursting (Traub et al. 1987). In vitro studies in hippocampus and neocortex suggest that the relative paucity of excitatory synaptic connections likely contributes to neonate rodent seizure resistance (Swann and Hablitz 2000).
We tested this idea by subjecting low-density cultures to the same conditions that produced bursting behavior in high-density cultures. The contrast between the 2 preparations when subjected to 0 [Mg2+]o was evident in 2 regards. No multiple action potential bursts were observed in any low-density neuron tested regardless of the number of days grown in culture and immunofluorescent labeling for synaptophysin indicated many fewer potential synaptic sites. This latter finding was confirmed by labeling of synapses with FM1-43; the number of actively exocytosing synapses was significantly less in low-density cultures. Increases in both sEPSC and mEPSC frequency were evident in 0 [Mg2+]o, indicating that presynaptic mechanisms for increasing glutamate release operate in low-density as well as high-density cultures.
The importance of connectivity in generating aberrant excitatory neuronal activity may be appreciated by an examination of animal models of temporal lobe seizures. The neuronal activity described in this study (multiple action potentials superimposed on prolonged depolarizations) is similar to that observed in these models. On a cellular level, seizure activity is manifested in synchronous excitatory activity among a large population of neurons (Dudek et al. 1999). A prominent anatomic feature in many animal models of chronic temporal lobe seizures is the sprouting of neuronal processes and subsequent formation of aberrant, presumably glutamate-mediated, excitatory synaptic interactions (Lehmann et al. 2001; Sutula et al. 1996; Wuarin and Dudek 1996), although sprouting is not a universal feature of these models (Swann et al. 2001). Recently, Lynch and Sutula (2000) demonstrated aberrant, apparently monosynaptic recurrent excitatory circuitry that likely underlies abnormal neuronal activity in this model.
Acknowledgments
We thank M. Alietta and C. Gregory for expert glial and hippocampal culture preparations.
GRANTS
This research was funded by National Institute of Neurological Disorders and Stroke Grants NS-37192 to P. S. Mangan and NS-02081 and NS-40337 to J. Kapur.
References
- Albowitz B, Konig P, Kuhnt U. Spatiotemporal distribution of intracellular calcium transients during epileptiform activity in guinea pig hippocampal slices. J Neurophysiol. 1997;77:491–501. doi: 10.1152/jn.1997.77.1.491. [DOI] [PubMed] [Google Scholar]
- Anderson WW, Lewis DV, Swartzwelder HS, Wilson WA. Magnesium-free medium activates seizure-like events in the rat hippocampal slice. Brain Res. 1986;398:215–219. doi: 10.1016/0006-8993(86)91274-6. [DOI] [PubMed] [Google Scholar]
- Ascher P, Bregestovski P, Nowak L. N-Methyl-D-aspartate-activated channels of mouse central neurones in magnesium-free solutions. J Physiol. 1988;399:207–226. doi: 10.1113/jphysiol.1988.sp017076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avoli M, Louvel J, Pumain R, Olivier A. Seizure-like discharges induced by lowering [Mg2+]o in the human epileptogenic neocortex maintained in vitro. Brain Res. 1987;417:199–203. doi: 10.1016/0006-8993(87)90201-0. [DOI] [PubMed] [Google Scholar]
- Bains JS, Longacher JM, Staley KJ. Reciprocal interactions between CA3 network activity and strength of recurrent collateral synapses. Nat Neurosci. 1999;2:720–726. doi: 10.1038/11184. [DOI] [PubMed] [Google Scholar]
- Banker GA, Cowan WM. Rat hippocampal neurons in dispersed cell culture. Brain Res. 1977;126:397–442. doi: 10.1016/0006-8993(77)90594-7. [DOI] [PubMed] [Google Scholar]
- Banker G, Goslin K, editors. Culturing Nerve Cells. Cambridge, MA: MIT Press; 1998. [Google Scholar]
- Bekkers JM, Stevens CF. NMDA and non-NMDA receptors are colocalized at individual excitatory synapses in cultured rat hippocampus. Nature. 1989;341:230–233. doi: 10.1038/341230a0. [DOI] [PubMed] [Google Scholar]
- Bouron A. Modulation of spontaneous quantal release of neurotransmitters in the hippocampus. Prog Neurobiol. 2001;63:613–635. doi: 10.1016/s0301-0082(00)00053-8. [DOI] [PubMed] [Google Scholar]
- Buck DR, Mahoney AW, Hendricks DG. Preliminary report on the magnesium deficient rat as a model of epilepsy. Lab Anim Sci. 1978;28:680–685. [PubMed] [Google Scholar]
- Coan EJ, Collingridge GL. Magnesium ions block an N-methyl-D-aspartate receptor-mediated component of synaptic transmission in rat hippocampus. Neurosci Lett. 1985;53:21–26. doi: 10.1016/0304-3940(85)90091-6. [DOI] [PubMed] [Google Scholar]
- Coan EJ, Collingridge GL. Characterization of an N-methyl-D-aspartate receptor component of synaptic transmission in rat hippocampal slices. Neuroscience. 1987;22:1–8. doi: 10.1016/0306-4522(87)90192-8. [DOI] [PubMed] [Google Scholar]
- Cochilla AJ, Angleson JK, Betz WJ. Monitoring secretory membrane with FM1-43 fluorescence. Annu Rev Neurosci. 1999;22:1–10. doi: 10.1146/annurev.neuro.22.1.1. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Kehl SJ, McLennan H. Excitatory amino acids in synaptic transmission in the Schaffer collateral-commissural pathway of the rat hippocampus. J Physiol. 1983;334:33–46. doi: 10.1113/jphysiol.1983.sp014478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulter DA, Sombati S, DeLorenzo RJ. Electrophysiology of glutamate neurotoxicity in vitro: induction of a calcium-dependent extended neuronal depolarization. J Neurophysiol. 1992;68:362–373. doi: 10.1152/jn.1992.68.2.362. [DOI] [PubMed] [Google Scholar]
- Cummings DD, Wilcox KS, Dichter MA. Calcium-dependent paired-pulse facilitation of miniature EPSC frequency accompanies depression of EPSCs at hippocampal synapses in culture. J Neurosci. 1996;16:5312–5323. doi: 10.1523/JNEUROSCI.16-17-05312.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLorenzo RJ, Pal S, Sombati S. Prolonged activation of the N-methyl-D-aspartate receptor-Ca2+ transduction pathway causes spontaneous recurrent epileptiform discharges in hippocampal neurons in culture. Proc Natl Acad Sci USA. 1998;95:14482–14487. doi: 10.1073/pnas.95.24.14482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingledine R, McBain CJ, McNamara JO. Excitatory amino acid receptors in epilepsy. Trends Pharmacol Sci. 1990;11:334–388. doi: 10.1016/0165-6147(90)90238-4. [DOI] [PubMed] [Google Scholar]
- Dobrunz LE, Stevens CF. Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron. 1997;18:995–1008. doi: 10.1016/s0896-6273(00)80338-4. [DOI] [PubMed] [Google Scholar]
- Dodge FA, Jr, Rahamimoff R. Co-operative action a calcium ions in transmitter release at the neuromuscular junction. J Physiol. 1967;193:419–432. doi: 10.1113/jphysiol.1967.sp008367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek FE, Patrylo PR, Wuarin JP. Mechanisms of neuronal synchronization during epileptiform activity. Adv Neurol. 1999;79:699–708. [PubMed] [Google Scholar]
- Esclapez M, Houser CR. Up-regulation of GAD65 and GAD67 in remaining hippocampal GABA neurons in a model of temporal lobe epilepsy. J Comp Neurol. 1999;412:488–505. [PubMed] [Google Scholar]
- Fletcher TL, Cameron P, De Camilli P, Banker G. The distribution of synapsin I and synaptophysin in hippocampal neurons developing in culture. J Neurosci. 1991;11:1617–1626. doi: 10.1523/JNEUROSCI.11-06-01617.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankenhaeuser B, Hodgkin AL. Tha action of calcium on the electrical properties of squid axon. J Physiol. 1957;137:218–244. doi: 10.1113/jphysiol.1957.sp005808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furshpan EJ. Seizure-like activity in cell culture. Epilepsy Res. 1991;10:24–32. doi: 10.1016/0920-1211(91)90091-s. [DOI] [PubMed] [Google Scholar]
- Gomperts SN, Carroll R, Malenka RC, Nicoll RA. Distinct roles for ionotropic and metabotropic glutamate receptors in the maturation of excitatory synapses. J Neurosci. 2000;20:2229–2237. doi: 10.1523/JNEUROSCI.20-06-02229.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulyas-Kovács A, Doczi J, Tarnawa I, Detari L, Banczerowski-Pelyhe I, Vilagi I. Comparison of spontaneous and evoked epileptiform activity in three in vitro epilepsy models. Brain Res. 2002;945:174–180. doi: 10.1016/s0006-8993(02)02751-8. [DOI] [PubMed] [Google Scholar]
- Gutierrez R, Armand V, Schuchmann S, Heinemann U. Epileptiform activity induced by low Mg2+ in cultured rat hippocampal slices. Brain Res. 1999;815:294–303. doi: 10.1016/s0006-8993(98)01102-0. [DOI] [PubMed] [Google Scholar]
- Hablitz JJ. Picrotoxin-induced epileptiform activity in hippocampus: role of endogenous versus synaptic factors. J Neurophysiol. 1984;51:1011–1027. doi: 10.1152/jn.1984.51.5.1011. [DOI] [PubMed] [Google Scholar]
- Hablitz JJ, Langmoen IA. N-Methyl-D-aspartate receptor antagonists reduce synaptic excitation in the hippocampus. J Neurosci. 1986;6:102–106. doi: 10.1523/JNEUROSCI.06-01-00102.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamon B, Stanton PK, Heinemann U. An N-methyl-D-aspartate receptor-independent excitatory action of partial reduction of extracellular [Mg2+] in CA1-region of rat hippocampal slices. Neurosci Lett. 1987;75:240–245. doi: 10.1016/0304-3940(87)90304-1. [DOI] [PubMed] [Google Scholar]
- Hanse E, Gustafsson B. Release dependence to a paired stimulus at a synaptic release site with a small variable pool of immediately releasable vesicles. J Neurosci. 2002;22:4381–4387. doi: 10.1523/JNEUROSCI.22-11-04381.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoch DB, Dingledine R. GABAergic neurons in rat hippocampal culture. Brain Res. 1986;390:53–64. doi: 10.1016/0165-3806(86)90151-3. [DOI] [PubMed] [Google Scholar]
- Katz B. Quantal mechanism of neural transmitter release. Science. 1971;173:123–126. doi: 10.1126/science.173.3992.123. [DOI] [PubMed] [Google Scholar]
- Lehmann TN, Gabriel S, Eilers A, Njunting M, Kovacs R, Schulze K, Lanksch WR, Heinemann U. Fluorescent tracer in pilocarpine-treated rats shows widespread aberrant hippocampal neuronal connectivity. Eur J Neurosci. 2001;14:83–95. doi: 10.1046/j.0953-816x.2001.01632.x. [DOI] [PubMed] [Google Scholar]
- Liang F, Huganir RL. Coupling of agonist-induced AMPA receptor internalization with receptor recycling. J Neurochem. 2001;77:1626–1631. doi: 10.1046/j.1471-4159.2001.00377.x. [DOI] [PubMed] [Google Scholar]
- Liao D, Scannevin RH, Huganir R. Activation of silent synapses by rapid activity-dependent synaptic recruitment of AMPA receptors. J Neurosci. 2001;21:6008–6017. doi: 10.1523/JNEUROSCI.21-16-06008.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Man H, Ju W, Trimble WS, MacDonald JF, Wang YT. Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron. 2001;29:243–254. doi: 10.1016/s0896-6273(01)00194-5. [DOI] [PubMed] [Google Scholar]
- Luscher C, Frerking M. Restless AMPA receptors: implications for synaptic transmission and plasticity. Trends Neurosci. 2001;24:665–670. doi: 10.1016/s0166-2236(00)01959-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscher C, Xia H, Beattie EC, Carroll RC, von Zastrow M, Malenka RC, Nicoll RA. Role of AMPA receptor cycling in synaptic transmission and plasticity. Neuron. 1999;24:649–658. doi: 10.1016/s0896-6273(00)81119-8. [DOI] [PubMed] [Google Scholar]
- Lynch M, Sutula T. Recurrent excitatory connectivity in the dentate gyrus of kindled and kainic acid-treated rats. J Neurophysiol. 2000;83:693–704. doi: 10.1152/jn.2000.83.2.693. [DOI] [PubMed] [Google Scholar]
- Mangan PS, Bertram EH., 3rd Ontogeny of altered synaptic function in a rat model of chronic temporal lobe epilepsy. Brain Res. 1998;799:183–196. doi: 10.1016/s0006-8993(98)00411-9. [DOI] [PubMed] [Google Scholar]
- Mangan PS, Lothman EW. Profound disturbances of pre- and postsynaptic GABAB-receptor-mediated processes in region CA1 in a chronic model of temporal lobe epilepsy. J Neurophysiol. 1996;76:1282–1296. doi: 10.1152/jn.1996.76.2.1282. [DOI] [PubMed] [Google Scholar]
- Masumiya H, Li P, Zhang L, Chen SR. Ryanodine sensitizes the Ca(2+) release channel (ryanodine receptor) to Ca(2+) activation. J Biol Chem. 2001;276:39727–39735. doi: 10.1074/jbc.M106557200. [DOI] [PubMed] [Google Scholar]
- Mayer ML, Westbrook GL. Mixed-agonist action of excitatory amino acids on mouse spinal cord neurones under voltage clamp. J Physiol. 1984;354:29–53. doi: 10.1113/jphysiol.1984.sp015360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin A, Grathwohl C, McLaughlin S. The adsorption of divalent cations to phosphatidylcholine bilayer membranes. Biochim Biophys Acta. 1978;513:338–357. doi: 10.1016/0005-2736(78)90203-1. [DOI] [PubMed] [Google Scholar]
- McLaughlin SG, Szabo G, Eisenman G. Divalent ions and the surface potential of charged phospholipid membranes. J Gen Physiol. 1971;58:667–687. doi: 10.1085/jgp.58.6.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mody I, Lambert JD, Heinemann U. Low extracellular magnesium induces epileptiform activity and spreading depression in rat hippocampal slices. J Neurophysiol. 1987;57:869–888. doi: 10.1152/jn.1987.57.3.869. [DOI] [PubMed] [Google Scholar]
- Muller HW, Seifert W. A neurotrophic factor (NTF) released from primary glial cultures supports survival and fiber outgrowth of cultured hippocampal neurons. J Neurosci Res. 1982;8:195–204. doi: 10.1002/jnr.490080209. [DOI] [PubMed] [Google Scholar]
- Muller RU, Finkelstein A. The electrostatic basis of Mg++ inhibition of transmitter release. Proc Natl Acad Sci USA. 1974;71:923–926. doi: 10.1073/pnas.71.3.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parfitt KD, Madison DV. Phorbol esters enhance synaptic transmission by a presynaptic, calcium-dependent mechanism in rat hippocampus. J Physiol. 1993;471:245–268. doi: 10.1113/jphysiol.1993.sp019900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poage RE, Meriney SD. Presynaptic calcium influx, neurotransmitter release, and neuromuscular disease. Physiol Behav. 2002;77:507–512. doi: 10.1016/s0031-9384(02)00937-x. [DOI] [PubMed] [Google Scholar]
- Quilichini PP, Diabira D, Chiron C, Ben-Ari Y, Gozlan H. Persistent epileptiform activity induced by low Mg2+ in intact immature brain structures. Eur J Neurosci. 2002;16:850–860. doi: 10.1046/j.1460-9568.2002.02143.x. [DOI] [PubMed] [Google Scholar]
- Rao A, Craig AM. Activity regulates the synaptic localization of the NMDA receptor in hippocampal neurons. Neuron. 1997;19:801–812. doi: 10.1016/s0896-6273(00)80962-9. [DOI] [PubMed] [Google Scholar]
- Rao A, Kim E, Sheng M, Craig AM. Heterogeneity in the molecular composition of excitatory postsynaptic sites during development of hippocampal neurons in culture. J Neurosci. 1998;18:1217–1229. doi: 10.1523/JNEUROSCI.18-04-01217.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rempe DA, Mangan PS, Lothman EW. Regional heterogeneity of pathophysiological alterations in CA1 and dentate gyrus in a chronic model of temporal lobe epilepsy. J Neurophysiol. 1995;74:816–828. doi: 10.1152/jn.1995.74.2.816. [DOI] [PubMed] [Google Scholar]
- Rubaj A, Zgodzinski W, Sieklucka-Dziuba M. The epileptogenic effect of seizures induced by hypoxia. The role of NMDA and AMPA/KA antagonists. Pharmacol Biochem Behav. 2003;74:303–311. doi: 10.1016/s0091-3057(02)00998-x. [DOI] [PubMed] [Google Scholar]
- Salter MW. LTP gets culture. Trends Neurosci. 2001;24:560–561. doi: 10.1016/s0166-2236(00)01882-8. [DOI] [PubMed] [Google Scholar]
- Schwartzkroin PA, Prince DA. Cellular and field potential properties of epileptogenic hippocampal slices. Brain Res. 1978;147:117–130. doi: 10.1016/0006-8993(78)90776-x. [DOI] [PubMed] [Google Scholar]
- Segal MM, Furshpan EJ. Epileptiform activity in microcultures containing small numbers of hippocampal neurons. J Neurophysiol. 1990;64:1390–1399. doi: 10.1152/jn.1990.64.5.1390. [DOI] [PubMed] [Google Scholar]
- Sombati S, Delorenzo RJ. Recurrent spontaneous seizure activity in hippocampal neuronal networks in culture. J Neurophysiol. 1995;73:1706–1711. doi: 10.1152/jn.1995.73.4.1706. [DOI] [PubMed] [Google Scholar]
- Spruston N, Jaffe DB, Johnston D. Dendritic attenuation of synaptic potentials and currents: the role of passive membrane properties. Trends Neurosci. 1994;17:161–166. doi: 10.1016/0166-2236(94)90094-9. [DOI] [PubMed] [Google Scholar]
- Spruston N, Johnston D. Perforated patch-clamp analysis of the passive membrane properties of three classes of hippocampal neurons. J Neurophysiol. 1992;67:508–529. doi: 10.1152/jn.1992.67.3.508. [DOI] [PubMed] [Google Scholar]
- Staley KJ, Bains JS, Yee A, Hellier J, Longacher JM. Statistical model relating CA3 burst probability to recovery from burst-induced depression at recurrent collateral synapses. J Neurophysiol. 2001;86:2736–2747. doi: 10.1152/jn.2001.86.6.2736. [DOI] [PubMed] [Google Scholar]
- Staley KJ, Longacher M, Bains JS, Yee A. Presynaptic modulation of CA3 network activity. Nat Neurosci. 1998;1:201–209. doi: 10.1038/651. [DOI] [PubMed] [Google Scholar]
- Stevens CF, Wesseling JF. Identification of a novel process limiting the rate of synaptic vesicle cycling at hippocampal synapses. Neuron. 1999;24:1017–1028. doi: 10.1016/s0896-6273(00)81047-8. [DOI] [PubMed] [Google Scholar]
- Sutula T, Koch J, Golarai G, Watanabe Y, McNamara JO. NMDA receptor dependence of kindling and mossy fiber sprouting: evidence that the NMDA receptor regulates patterning of hippocampal circuits in the adult brain. J Neurosci. 1996;16:7398–7406. doi: 10.1523/JNEUROSCI.16-22-07398.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swann JW, Hablitz JJ. Cellular abnormalities and synaptic plasticity in seizure disorders of the immature nervous system. Ment Retard Dev Disabil Res Rev. 2000;6:258–267. doi: 10.1002/1098-2779(2000)6:4<258::AID-MRDD5>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Swann JW, Smith KL, Lee CL. Neuronal activity and the establishment of normal and epileptic circuits during brain development. Int Rev Neurobiol. 2001;45:89–118. doi: 10.1016/s0074-7742(01)45007-0. [DOI] [PubMed] [Google Scholar]
- Tancredi V, Avoli M, Hwa GG. Low-magnesium epilepsy in rat hippocampal slices: inhibitory postsynaptic potentials in the CA1 subfield. Neurosci Lett. 1988;89:293–298. doi: 10.1016/0304-3940(88)90542-3. [DOI] [PubMed] [Google Scholar]
- Tancredi V, Hwa GG, Zona C, Brancati A, Avoli M. Low magnesium epileptogenesis in the rat hippocampal slice: electrophysiological and pharmacological features. Brain Res. 1990;511:280–290. doi: 10.1016/0006-8993(90)90173-9. [DOI] [PubMed] [Google Scholar]
- Traub RD, Colling SB, Jefferys JG. Cellular mechanisms of 4-amino-pyridine-induced synchronized after-discharges in the rat hippocampal slice. J Physiol. 1995;489:127–140. doi: 10.1113/jphysiol.1995.sp021036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub RD, Knowles WD, Miles R, Wong RK. Synchronized afterdischarges in the hippocampus: simulation studies of the cellular mechanism. Neuroscience. 1984;12:1191–1200. doi: 10.1016/0306-4522(84)90013-7. [DOI] [PubMed] [Google Scholar]
- Traub RD, Knowles WD, Miles R, Wong RK. Models of the cellular mechanism underlying propagation of epileptiform activity in the CA2-CA3 region of the hippocampal slice. Neuroscience. 1987;21:457–470. doi: 10.1016/0306-4522(87)90135-7. [DOI] [PubMed] [Google Scholar]
- Traub RD, Wong RK. Synchronized burst discharge in disinhibited hippocampal slice. II. Model of cellular mechanism. J Neurophysiol. 1983;49:459–471. doi: 10.1152/jn.1983.49.2.459. [DOI] [PubMed] [Google Scholar]
- Traub RD, Wong RK, Miles R, Michelson H. A model of a CA3 hippocampal pyramidal neuron incorporating voltage-clamp data on intrinsic conductances. J Neurophysiol. 1991;66:635–650. doi: 10.1152/jn.1991.66.2.635. [DOI] [PubMed] [Google Scholar]
- Walther H, Lambert JD, Jones RS, Heinemann U, Hamon B. Epileptiform activity in combined slices of the hippocampus, subiculum and entorhinal cortex during perfusion with low magnesium medium. Neurosci Lett. 1986;69:156–161. doi: 10.1016/0304-3940(86)90595-1. [DOI] [PubMed] [Google Scholar]
- Westbrook GL. Glutamate receptor update. Curr Opin Neurobiol. 1994;4:337–346. doi: 10.1016/0959-4388(94)90094-9. [DOI] [PubMed] [Google Scholar]
- Wilcox KS, Buchhalter J, Dichter MA. Properties of inhibitory and excitatory synapses between hippocampal neurons in very low density cultures. Synapse. 1994;18:128–151. doi: 10.1002/syn.890180206. [DOI] [PubMed] [Google Scholar]
- Wuarin JP, Dudek FE. Electrographic seizures and new recurrent excitatory circuits in the dentate gyrus of hippocampal slices from kainate-treated epileptic rats. J Neurosci. 1996;16:4438–4448. doi: 10.1523/JNEUROSCI.16-14-04438.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong ZQ, Stringer JL. Prolonged bursts occur in normal calcium in hippocampal slices after raising excitability and blocking synaptic transmission. J Neurophysiol. 2001;86:2625–2628. doi: 10.1152/jn.2001.86.5.2625. [DOI] [PubMed] [Google Scholar]
- Zhang A, Fan SH, Cheng TP, Altura BT, Wong RK, Altura BM. Extracellular Mg2+ modulates intracellular Ca2+ in acutely isolated hippocampal CA1 pyramidal cells of the guinea-pig. Brain Res. 1996;728:204–220. doi: 10.1016/0006-8993(96)00401-5. [DOI] [PubMed] [Google Scholar]