

Abstract
In schizophrenia, genetic predisposition has been linked to chromosome 22q11 and myelin-specific genes are misexpressed in schizophrenia. Nogo-66 receptor 1 (NGR or RTN4R) has been considered to be a 22q11 candidate gene for schizophrenia susceptibility because it encodes an axonal protein that mediates myelin inhibition of axonal sprouting. Confirming previous studies, we found that variation at the NGR locus is associated with schizophrenia in a Caucasian case-control analysis, and this association is not attributed to population stratification. Within a limited set of schizophrenia-derived DNA samples, we identified several rare NGR nonconservative coding sequence variants. Neuronal cultures demonstrate that four different schizophrenia-derived NgR1 variants fail to transduce myelin signals into axon inhibition, and function as dominant negatives to disrupt endogenous NgR1. This provides the first evidence that certain disease-derived human NgR1 variants are dysfunctional proteins in vitro. Mice lacking NgR1 protein exhibit reduced working memory function, consistent with a potential endophenotype of schizophrenia. For a restricted subset of individuals diagnosed with schizophrenia, the expression of dysfunctional NGR variants may contribute to increased disease risk.
Keywords: schizophrenia, myelin, genetic linkage, axonal growth, Nogo, plasticity
Introduction
It is clear that genetic factors play a major role in the risk of schizophrenia. Gene expression analysis has demonstrated oligodendrocyte dysfunction in schizophrenia (Hakak et al., 2001; Hof et al., 2002; Davis and Haroutunian, 2003; Tkachev et al., 2003). Oligodendrocytes increase neuronal conduction velocity, provide trophic support (Marcus et al., 2002), and limit axonal sprouting and plasticity, through the Nogo/NgR1 pathway (Fournier et al., 2001; McGee and Strittmatter, 2003; Kim et al., 2004; Lee et al., 2004; Li et al., 2004, 2005; McGee et al., 2005; Cafferty and Strittmatter, 2006; Wang et al., 2006; Cafferty et al., 2007). Ultrastructural changes in oliogodendrocytes and myelin are present in schizophrenic brains (Uranova et al., 2001, 2004). Magnetic resonance imaging shows misaligned axons (Kubicki et al., 2003; Park et al., 2004). In some reports, white matter volumes are decreased in schizophrenia (Shenton et al., 2001; Hulshoff Pol et al., 2004; Park et al., 2004). Myelination of the prefrontal cortex occurs with late adolescence, the typical age of schizophrenia onset (Benes et al., 1986). It has been hypothesized that aberrant myelination contributes to the schizophrenia (Karoutzou et al., 2007; Segal et al., 2007).
Individuals with the DiGeorge syndrome [Mendelian Inheritance in Man (MIM) 188400] or the velo-cardio-facial syndrome (MIM 192430) (Scambler, 2000) are heterozygous for a 1.5 Mb deletion in 22q11 and exhibit a 20–30% prevalence of schizophrenia (Bassett and Chow, 1999, 2008; Bassett et al., 2003). There is an 80-fold increased prevalence of 22q11 deletion in adult schizophrenics, and a 22q11 deletion is present in 6% of childhood-onset schizophrenics (Karayiorgou et al., 1995; Cohen et al., 1999; Usiskin et al., 1999). Schizophrenia risk has been linked to a 22q11 locus (Baron, 2001).
Candidate genes within the 1.5 Mb critical 22q11 deletion interval include proline dehydrogenase (PRODH), ZDHHC8, and catechol-O-methyl transferase (COMT) (Gogos et al., 1998; Egan et al., 2001; Chakravarti, 2002; Jacquet et al., 2002; H. Liu et al., 2002a,b; Shifman et al., 2002; Akil et al., 2003). The region also encodes NgR1, an axonal receptor for myelin-derived growth inhibitors (Fournier et al., 2001; B. P. Liu et al., 2002, 2006; McGee and Strittmatter, 2003). Given the role of NgR1 in myelin-dependent regulation of plasticity (Fournier et al., 2001; McGee and Strittmatter, 2003; McGee et al., 2005), genetic variation at the NGR locus may contribute to schizophrenia risk.
An association of NGR single-nucleotide polymorphisms (SNPs) with schizophrenia is found in North Americans (H. Liu et al., 2002b), but not in Chinese (Meng et al., 2007), with a weak association in Afrikaners (Hsu et al., 2007). Rare coding variants in the NGR gene have been identified among schizophrenics (Sinibaldi et al., 2004; Hsu et al., 2007). Here, we confirm the association of NGR haplotypes with schizophrenia and show that several NgR1 variants are dysfunctional for signal transduction. Mice lacking NgR1 display impaired working memory. Thus, myelin-dependent restriction of CNS anatomical plasticity may reduce the risk of schizophrenia.
Materials and Methods
SNP genotyping
Case control DNA from Caucasians and African-Americans were obtained from the Coriell Institute and the Schizophrenia collection of the National Institute of Mental Health (NIMH) Center for Collaborative Genetic Studies on Mental Disorders. Our complete sample consists of 636 Caucasians (336 cases and 300 controls) and 296 African-American (196 cases and 100 controls). DNA from Chinese Trios was obtained from the NIMH Center for Collaborative Genetic Studies on Mental Disorders. This sample consists of 1122 trios (621 schizophrenic patients and 501 controls). Probands affected with schizophrenia were identified using Diagnostic and Statistical Manual of Mental Disorders IV criteria. Families were excluded if both parents were schizophrenic.
We included seven SNPs at the RTN4R locus: rs701421, rs701428, rs1567871, rs696880, rs854927, rs9606296, and rs701427. Automated high-throughput SNP genotyping was accomplished at The W. M. Keck Foundation Biotechnology Center at Yale using the TaqMan SNP Genotyping Assay (Applied Biosystems). The PCR was as follows: 2 min at 50°C, 10 min at 95°C, and 38 cycles at 95°C for 15 s and 60°C for 1 min. For each SNP, analysis of genotype distribution was performed to test deviation from Hardy–Weinberg equilibrium expectations using the Fisher's exact test. Linkage disequilibrium (LD) between pairs of SNPs was measured as D′ using Haploview (http://www.broad.mit.edu/mpg/haploview). Structures of haplotypes were analyzed from the parental genotypes based on LD pattern using the expectation–maximization algorithm (Slatkin and Excoffier, 1996). Conventional transmission/disequilibrium test (TDT) statistics were used to analyze transmission disequilibrium between the discrete trait schizophrenia or schizoaffective disorder and the SNPs (Spielman et al., 1993) using the trios from the NIMH with both available parents and one affected offspring. To adjust for multiple testing, the family-wise error rate was set at 0.05 significance level; for each test, the p value was adjusted using a threshold p = 1 − (1 − 0.05)^(1/n) for significance (where n is the number of SNPs).
In addition, single marker and haplotype association with schizophrenia were tested using the Family-Based Association Test (FBAT) software. When the phenotype is a single dichotomous trait and the number of a specific allele is counted to represent the offspring genotype, the test statistic is equivalent to the test statistics used in the conventional TDT (Spielman et al. 1993). FBAT also implements haplotype-based association tests for family-based studies when markers are tightly linked. When phase is unknown, haplotypes cannot be determined unambiguously. All markers were tested because they all had minor allele frequencies >5%.
Human DNA sequence analysis
DNA samples from 542 schizophrenic probands and NgR1variant pedigrees were obtained from the NIMH Center for Collaborative Genetic Studies on Mental Disorders. A group of 650 healthy control subject DNA samples were obtained from the Coriell Institute. The entire translated region plus ∼500 bp of 5′ and 3′ untranslated sequence was amplified in three PCRs, and the sequence was determined. The PCR amplification was performed in a 50 μl reaction [200 ng of genomic DNA with Platinium Pfx DNA polymerase (Invitrogen)] according to manufacturer's instruction. The cycles were performed using a iCycler (Bio-Rad) with the following cycles: 1 cycle of 5 min at 94°C; 35 cycles to 30 s at 94°C, 60 s at 60°C, and 60 s at 68°C; and 1 cycle of 10 min at 68°C.
Recombinant proteins
Alkaline phosphatase (AP)-Nogo-66, AP-myelin-associated glycoprotein (MAG), AP-oligodendrocyte myelin glycoprotein (OMgp), and AP-Lingo-1 constructs were described previously (Fournier et al., 2001; B. P. Liu et al., 2002; Barton et al., 2003; Park et al., 2006; Laurén et al., 2007).
NGR mutagenesis
NGR mutagenesis was accomplished using the QuickChange Multisite-Directed Mutagenesis kit (Stratagene). Human NGR was used as a template. All mutant sequences were confirmed by DNA sequencing.
COS-7 ligand binding assay
COS-7 binding assays were performed as described previously (Fournier et al., 2001). Conditioned media containing AP fused ligands were incubated with NgR1 transfected COS-7 cells for 1 h at room temperature before fixation. Bound AP was stained and measured using the NIH Image software.
Neuronal culture
Embryonic day 7 (E7) chick retinal explants, E13 chick dorsal root ganglion (DRG) explants, dissociated E20 rat DRG cultures and herpes simplex virus (HSV) preparations have been described previously (Fournier et al., 2001; B. P. Liu et al., 2002). Briefly, permanox chamber slide were coated with poly-d-lysine (100 μg/ml) and laminin (5 μg/ml). For E7 retinal explants growth cone collapse experiments, HSV preparation was added 6 h after plating; the explants were allowed to grow for an additional 18–24 h. For E13 DRG explants, HSV preparations were added at the time of plating, and the protein allowed to express for 14–16 h. Growth cone collapse was allowed for 30 min, followed by fixation of the explants with 3.7% formaldehyde/20% sucrose. Cells were stained with a polyclonal anti-Flag antibody (Sigma-Aldrich) and rhodamine phalloidin. The growth cones of neurons expressing anti-Flag immunoreactive NgR1 were scored as spread or collapsed by an observer unaware of the treatment condition. Neurite outgrowth was measured after infecting dissociated E20 rat DRG neurons with virus for 4 h in suspension. Then infected cells were washed and plated on preformed monolayers of control or MAG-expressing CHO cells. A CHO line stably expressing mouse MAG was generated by transfection with a eukaryotic expression vector, selection with zeocin, and fluorescence-activated cell sorting of expressing cells. After fixation of cultures, neurites were visualized by anti-βIII tubulin staining and quantitated in an automated 96-well format with an ImageXpress apparatus (Molecular Devices). Under these conditions, >85% of the βIII-tubulin-positive neurons were also positive for recombinant protein expressing by anti-Flag immunohistochemistry or green fluorescent protein (GFP) visualization. Peptides were synthesized by the Keck Biotechnology Center at Yale. The sequence of the wild-type (WT) 377 peptide is biotin-PPGDSPPGNGSGPRHINDSPFGTLPGSAEP-amide, the R377Q peptide is biotin-PPGDSPPGNGSGPQHINDSPFGTLPGSAEP-amide, and the R377W peptide is biotin-PPGDSPPGNGSGPWHINDSPFGTLPGSAEP-amide.
Cognitive assessment in the spatial delayed alternation task
The spatial delayed alternation task has been shown to be sensitive to medial prefrontal cortex lesions in rats (Larsen and Divac, 1978). Mice were habituated to all testing procedures, and then assessed for spatial working memory abilities across 30 test sessions as described below.
Adaptation to testing procedures.
Cognitive testing requires extensive adaptation to the handling and training procedures to reduce stress-induced dysfunction and permit expression of spatial working memory performance. Animals were tested in a T maze of appropriate size for testing in mice (15 × 21 × 3“). The maze was constructed of wood and painted black, with a guillotine door separating the start box from the main stem of the maze. Testing occurred in a small room near the colony room under normal light conditions. All cognitive testing was performed between 8:00 A.M. and 5:00 P.M. during the animals' light cycle. Each mouse was tested by the same experimenter at the same time of day (e.g., 11:00 A.M.) everyday Monday through Friday. Before cognitive training, all animals were exposed to the food rewards (pieces of Fruit Loop cereal) in their home cage. All animals were fully habituated to the testing procedures by requiring them to pass criteria for three phases of training in the T maze: phase 1, the mouse had to be able to eat 10 rewards placed on the maze in 8 min or less for 2 consecutive days; phase 2, the mouse had to be able to eat 10 rewards placed on the maze in 5 min or less for 2 consecutive days while being picked up and placed back in the start box five times during the 5 min period; phase 3, the mouse was exposed to forced alternation, in which one of the two arms of the maze was blocked off on each trial. The mouse was rewarded only after traversing the entire length of the open arm, and was then picked up and put back in the start box. This phase was completed when the mouse was able to finish the forced alternation session in 8 min or less for 2 consecutive days.
Spatial delayed alternation testing.
After passing criteria for all three phases of training, mice were assessed for their performance of the spatial delayed alternation task. In this task, the animal was rewarded for choosing either the left or right arm on the first trial (not counted), but from then on had to choose the arm not entered on the previous trial. The animal was held in the start box between trials for a delay of ∼2.5 s while the choice point of the maze was wiped with alcohol to prevent olfactory cues from guiding behavior. The mice were tested for 30 test sessions; each daily test session consisted of 10 trials. The technician testing the animals was blind to mouse genotype and to the hypothesis under investigation. Performance (accuracy and response times) of the two groups was compared using a two-tailed independent t test.
Prepulse inhibition of acoustic startle
Testing was performed using an automated SRLab startle system (San Diego Instruments). The mice were placed in a 5-cm-diameter Plexiglas cylinders closed at both ends. During the sessions, the mice remained in the cylinders within a sound-attenuating cabinet in which a 73 dB white background noise was delivered. Stimuli were delivered and startle responses measured by the SRLab software (San Diego Instruments). For the habituation experiment, mice were subjected to 100 repetitions of a 50 ms startle stimulus of 110 dB. In a second set of experiments, the startle response of the animals to 50 ms broadband acoustic stimuli (75, 80, 90, 100, 110, 120 dB) was measured. For the prepulse inhibition (PPI) experiments, a total of 12 repetitions of six trials were delivered in a pseudorandom order with an interval of 20–30 s. Prepulse trials consisted of a single 110 dB pulse (S110) preceded by a 20 ms prepulse (PP) of 0, 2, 7, 12, 17, or 22 dB over baseline (i.e., 75, 80, 85, 90, or 95 dB). PPI was calculated according to the following formula: [1 − (S110 − PP)/S110]/100.
Results
SNP genotyping at the NGR locus
We examined seven SNPs at the NGR (RTN4R) locus spanning 19 kb in three different DNA samples, a Caucasian case–control sample (Fig. 1), an African-American case–control sample (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), and a Chinese Han trio sample (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). The SNPs used for each population were chosen to encompass genetic variation at the locus as predicted by SNPicker using HapMap data (http://www.hapmap.org/). In each case, the genotype frequency of the SNPs was consistent with Hardy–Weinberg equilibrium expectations. For the Caucasian population, rs701428 genotype is weakly associated with schizophrenia with the minor allele enriched in the schizophrenic group (Fig. 1B) (p = 0.03; but after correction for multiple testing, p = 0.1619). Two haplotypes, 01100 and 10001, have an association with disease status with p values of 0.0013 (adjusted for multiple comparisons, p = 0.012) and 0.034 (adjusted p = 0.27), respectively (Fig. 1C). Because rs701421 shows only weak linkage disequilibrium with the other SNPs (Fig. 1A), we performed an additional haplotype analysis without including this SNP. With rs701421 excluded, the haplotype 1001 (p = 0.0089 and adjusted p = 0.044) is associated with schizophrenia (data not shown).
Figure 1.

NGR locus is associated with schizophrenia in a Caucasian case–control study. A, Linkage disequilibrium plot. The relative position of the five SNPs relative to the NGR locus are depicted. B, Association of specific single markers with the disease. The five markers selected were in Hardy–Weinberg equilibrium (p > 0.05). HWE, Hardy–Weinberg p value; MA, minor allele; control, allele frequency in the control samples; case, allele frequency in the schizophrenic samples. C, Association of the haplotype obtained by analyzing the five SNPs with the disease. For each haplotype, 0 represents the major allele and 1 represents the minor allele. The order of the SNPs in haplotypes does not follow the order of the chromosomal position; it follows this order: rs9606296, rs701421, rs701428, rs1567871, rs696880. Uncorrected p values and values corrected for multiple testing are listed.
To ensure that this apparent association was not attributable to population stratification, we genotyped multiple markers informative for genetic admixture across all chromosomes in these populations (Pritchard and Rosenberg, 1999; Pritchard et al., 2000; Stein et al., 2004; Yang et al., 2005). No significant differences in genetic background of these Caucasian cases and controls were found (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
In the African-American case–control sample, we found one haplotype nominally associated with schizophrenia (p = 0.032, but adjusted p = 0.15) (supplemental Fig. 2D, available at www.jneurosci.org as supplemental material), and in Chinese Han, none (supplemental Fig. 3D, available at www.jneurosci.org as supplemental material). The positive association of NGR variation with schizophrenia in Caucasian but not in Chinese groups replicates the conclusions of previous studies (H. Liu et al., 2002b; Meng et al., 2007).
Specific human NGR genetic variants
The linkage of common intronic SNP haplotypes to schizophrenia in Caucasians does not provide insight into how NgR1 function might be altered. To explore whether any rare coding region variants might exist in the schizophrenic population, we examined DNA samples from 542 schizophrenic probands from NIMH Center for Collaborative Genetic Studies on Mental Disorders. The presence of nonsynonymous coding region variants was scored. None of the four previously described coding region variants, R119W, R196H, T134M, and L347R (Sinibaldi et al., 2004; Hsu et al., 2007), are present in any proband of the NIMH schizophrenia collection, but four novel missense variants were identified, R377W, R377Q, R227C, and R399W. In each of the individual DNA samples, a heterozygous substitution was identified (Fig. 2A,B). The altered residues are evolutionarily conserved among human, rat, and mouse. In previous studies of 600 healthy control subjects at the NGR locus, no nonsynonymous variants were reported (Sinibaldi et al., 2004; Hsu et al., 2007). In our study, an additional 650 control DNA samples were screened for nonsynonymous NGR variants, and 8 heterozygous variants were identified in single subjects, V53M, R68H, G141S, R227H, V263M, G314S, P329L, and V363M (Fig. 2A). The overall incidence of coding region variants in healthy and schizophrenic samples does not differ between groups.
Figure 2.

NGR locus and genetic variation in schizophrenia. A, The NGR coding sequence from 542 schizophrenic individuals and 650 healthy controls was determined. The nonsynonymous coding sequence variants listed were present heterozygously in 12 individuals. Those variants listed in red are predicted to be “probably damaging” by the PolyPhen program. In addition, the four nonsynonymous coding sequence variants from two previous studies containing 328 schizophrenic individuals and 600 healthy controls are cited. B, 5′–3′ DNA sequence traces from a control individual with two WT NGR alleles, an affected individual heterozygous for a G>A transition at nucleotide 1130 resulting in the R377Q substitution, or an affected individual heterozygous for a C>T transition at nucleotide 1129 resulting in the R377W substitution. C, The observed numbers of coding variants predicted to be “probably damaging” or not by PolyPhen (http://genetics.bwh.harvard.edu/pph/) (Sunyaev et al., 2001) in the two groups is tabulated and analyzed statistically for the existing study. The metaanalysis table includes the individuals from previous studies and assesses the overall incidence of “probably damaging” variants in the two populations. D, E, Pedigree for R377W and R377Q families. Schizophrenic probands are indicated by the arrows. The filled symbols represent individuals with major psychiatric disease: schizophrenia, major depression with psychotic features, mild mental retardation with schizoid personality, or manic-depressive illness. F, The occurrence of R377Q variant or the transheterozygous R377Q variant in the presence of the 5′ noncoding change is tabulated for the family in D.
We noted that amino acid variants in the schizophrenic group were nonconservative changes, whereas those in the control group were conservative. For example, four NGR variants that altered the electrostatic charge of an amino acid side chain were present only in the schizophrenic group, but none were found in 1250 control subjects (Fig. 2A, red). A computational algorithm predicts the likelihood of disrupted protein function based on protein structure, phylogenetic conservation, and amino acid similarity (PolyPhen; http://genetics.bwh.harvard.edu/pph/) (Sunyaev et al., 2001). In our study, three of four variants in the schizophrenic samples are predicted to disrupt protein function (R377W, R227C, and R399W), whereas zero of eight variants in the control samples are predicted to be disruptive (Fig. 2C) (p = 0.0047, Fisher's exact test). In a metaanalysis that includes all samples in the two previous studies, four variants in 870 schizophrenic samples are predicted to disrupt protein function (R119W, R377W, R227C, and R399W), whereas 0 variants in 1250 control samples are predicted to be disruptive (Fig. 2C) (p = 0.016, χ2 test). Thus, there is a higher incidence of rare nonconservative NGR variants in the schizophrenia population.
Next, we were able to obtain DNA samples from the R377Q and R377W pedigrees. The R377W pedigree is small and most members carry one copy of the variant sequence (Fig. 2D,E). It is not possible to draw any conclusion regarding the association of the variant with psychiatric disease in this family. The R377Q family is larger, and some individuals have a C>T substitution at position −130 in the 5′-untranslated region of the NGR gene (Fig. 2E). The untranslated C>T substitution is on the opposite allele from the R377Q variant and was not present in any other probands or controls. Within this family, some members exhibit schizophrenia, whereas one displays mild mental retardation plus schizoid personality and another depression with psychotic features. There is a weakly significant correlation between the presence of both NGR alleles and major psychiatric disease within this one relatively small R377Q pedigree (Fig. 2F).
Functional analysis of human NgR1 variants: signaling domain variants
Although the sequencing studies suggest that rare nonconservative NGR protein variants are present in a subset of schizophrenics, the small numbers prevent stringent statistical proof of altered function NGR function in schizophrenia. Therefore, we analyzed the function of four of the NGR variants in vitro to determine whether the schizophrenia-associated variants are in fact dysfunctional. The R377Q and R377W substitutions lie outside of the NgR1 ligand-binding domain, but are centered in the region of the protein (amino acids 311–450) that is required for signal transduction (Fournier et al., 2002). Within this signaling domain, R377 lies within a most highly conserved cluster of amino acids. We hypothesized that these sequence variations might disrupt myelin ligand signaling through NgR1.
As a first step toward characterizing the function of R377Q-NgR1 and R377W-NgR1, binding to myelin ligands was tested (Fig. 3). The binding affinity of AP-Nogo-66, AP-MAG, and AP-OMgp for NgR1 is unaffected by either of these amino acid substitutions (Fig. 3A). At least three proteins may mediate NgR1 signaling: p75-NTR, Taj/Troy, and Lingo-1 (K. C. Wang et al., 2002; Wong et al., 2002; Mi et al., 2004; Park et al., 2005; Shao et al., 2005). Sequence variants that disrupt signaling may alter NgR1 interaction with these coreceptors. In coimmunoprecipitation experiments, NgR1 association with p75NTR or Troy or Lingo-1 is unaltered in the R377Q and R377W NgR1 variants (Fig. 3D) (data not shown). This result demonstrates that these amino acid variants do not abolish binding to putative coreceptors, but cannot rule out the possibility that R377Q and R377W disrupt signaling, because transduction might be altered without a change in physical association.
Figure 3.

Schizophrenia-derived NgR1 signaling domain variants bind myelin ligands and interact with known coreceptors. A, Schematic structure of the NgR1 protein. The ligand-binding domain is composed of eight leucine-rich repeats. The segment from amino acid 311 to 450 is required for signaling through coreceptors and is anchored to the membrane via a GPI (glycosylphosphatidylinositol) moiety (7, 35). The site of amino acid 377 in the signaling domain is indicated. B, COS-7 cells were transfected with control vector, WT-NgR-expressing expression vector, R377Q-NgR1 vector, or R377W-NgR1 vector. The Kd for the binding of Nogo-66, MAG, and OMgp is reported. The binding of 10 nm AP-Nogo-66 ligand is detected as a dark reaction product on NgR1-expressing cells (right). C, AP-Nogo-66 binding assays were conducted over a range of ligand concentrations, and bound AP was measured. Error bars indicate SEM. D, The data from B are replotted. E, HEK293T cells were transfected with the indicated expression vectors (WT-NgR, WT; R377Q-NgR, Q; R377W-NgR, W). Lysates were immunoblotted directly or subjected to immunoprecipitation with the indicated antibodies and then immunoblotted. NgR1 amino acid variants do not significantly affect interaction with p75 or Lingo-1.
To study the signaling function of the R377 variants, WT-NgR1 or NgR1 variants were expressed in various chick neurons, and growth cone collapse was tested in response to the myelin ligand Nogo-66, which requires NgR1 expression (Fournier et al., 2001; Chivatakarn et al., 2007). Expression levels and transport to the axonal growth cone are indistinguishable for the NgR1 variants (Fig. 4A). In young E7 retinal neurons, NgR1 is not detectable and Nogo-66 does not induce growth cone collapse (Fournier et al., 2001). E7 retinal explants transfected with a virus expressing WT-NgR1 sensitizes these neurons to Nogo-66, whereas the R377Q or R377W variants do not (Fig. 4A,B). Similarly, MAG-mediated inhibition of neurite outgrowth in E20 rat DRG neurons is supported by WT-NgR1 but not by R377 substituted NgR1 (Fig. 4C,D). Thus, the schizophrenia-associated NgR1 variants are incapable of transducing inhibitory myelin signals. We considered whether the variant NgR1 proteins might disrupt endogenous WT-NgR1 function. E13 chick DRG neurons express NgR1 and are responsive to Nogo-66 (Fournier et al., 2001). Viral overexpression of WT-NgR1 does not alter responsiveness in DRGs at this stage. In contrast, the R377Q and R377W substituted proteins blocks the function of the endogenous NgR1 and function as dominant negatives (Fig. 4E). Accordingly, individuals bearing alleles containing the R377Q or R377W substitutions may have severe disruption of NgR1 signaling.
Figure 4.

The R377Q and R377W-NgR1 variants are dominant-negative disruptors of myelin signaling. A, Chick E7 retinal neurons were cultured with or without herpes simplex virus directing the expression of WT-NgR, R377Q-NgR1, or R377W-NgR1 as indicated. The cultures were exposed to 100 nm GST (glutathione S-transferase)-Nogo-66, for 30 min, and then fixed and stained with phalloidin for F-actin and with anti-NgR1 antibody. Note that the viruses drive the expression of NgR1 protein. WT-NgR1 allows growth cones to collapse in response to Nogo-66, but R-377Q-NgR1 does not. B, The fraction of collapsed E7 retinal growth cones expressing WT-NgR1 is significantly greater than that for growth cones expressing GFP after exposure to 100 nm Nogo-66 (*p < 0.05, one-way ANOVA). C, Neurite outgrowth over 16 h from E20 rat DRG was assessed on a monolayer of control or MAG-expressing CHO cells after infection of neurons with virus expressing GFP, WT-NgR, R377Q-NgR1, or R377W-NgR. Neurons are visualized with anti-βIII-tubulin immunohistology. D, Mean neurite outgrowth from neurons expressing WT-NgR1 is significantly decreased on MAG cells relative to control monolayers (*p < 0.05, one-way ANOVA). Outgrowth from neurons expressing variant NgR1 is not reduced by MAG. E, The percentage of collapsed E13 chick DRG growth cones expressing the indicated proteins is reported from cultures similar to those in A. The values with R377Q-NgR1 and R377W-NgR1 virus are significantly different from those with control GFP virus (*p < 0.05, one-way ANOVA). F, Growth cone collapse of E13 chick DRG growth cones in the presence of 100 nm Nogo-66, 100 nm MAG-Fc, 3 μg/ml myelin protein, or 10 nm Sema3A. Collapse was assessed without peptide, with 1 μm WT peptide, with 1 μm R377Q peptide, or with 1 μm R377W peptide. Note that the R377W peptide suppresses collapse by Nogo-66, MAG, and myelin but not by Sema3A. Values with the variant NgR1 peptides are significantly different from those with the WT NgR1 peptide (*p < 0.05, one-way ANOVA). G, E13 chick DRG growth cone collapse in presence of 100 nm MAG-Fc and various concentrations of R377W peptide. All data are mean ± SEM from n = 3–7 experiments.
We considered whether the dominant-negative effect of the R377Q and R377W proteins might be mimicked by 30 aa peptides composed of the region flanking R377. These peptides do not induce growth cone collapse (Fig. 4F). However, the two variant peptides block Nogo-66-induced growth cone collapse. The dominant-negative effect of the R377W peptide extends to both MAG and myelin. Its potency and specificity as an inhibitor of MAG-induced collapse are high; the EC50 is 150 nm (Fig. 4G) and semaphorin-induced growth cone collapse is unaffected (Fig. 4F). These data confirm that the R377 amino acid substitutions inhibit NgR1 signaling.
Functional analysis of human NgR1 variants: binding domain variants
In contrast to the R377 variants, the two Italian NgR1 variants, R119W and R196H (Sinibaldi et al., 2004), are located in the ligand-binding domain of NgR1 (amino acids 27–310). Previously, the surface residues required for NgR1 ligand binding have been mapped using a mutagenesis strategy based on the crystal structure of NgR1 (Barton et al., 2003; He et al., 2003; Park et al., 2006; Laurén et al., 2007). The core of the binding domain (in red) is centered across the concave surface of NgR1 with residues differentially used by Nogo versus MAG versus OMgp surrounding this core. Much of the convex surface appears dispensable for ligand binding (Fig. 5A). The Italian variants map to the edge of the principal ligand binding domain (R119W) and on the convex surface of the protein (R196H) (Fig. 5B). This suggests that the R119W substitution may have specific effects on one NgR1 ligand but not on another, and that the R196H substitution would be inconsequential for binding.
Figure 5.

Ligand-binding domain in NgR1 and the effect of schizophrenia-derived sequence variants. A, The human NgR1 surface required for ligand binding is summarized from previous analysis of 74 Ala substitution variants (Park et al., 2006; Laurén et al., 2007). Residues required for Nogo-66, MAG, and OMgp are highlighted in red, and residues required for one ligand but not all are shown in yellow. B, Location of the two separate coding region variants identified in an Italian population (green) (Sinibaldi et al., 2004). The R119 residue is at the edge of the region required for ligand binding. The R196 residue is centered on the convex surface not implicated in ligand binding. C, Binding of AP-tagged myelin ligands to WT-NgR1, NgR-R119W, and NgR-R196H variants. Pictures shown are at the Kd for each ligand for WT-NgR. Note the absence of binding of AP-MAG and AP-OMgp to R119W-NgR. D, AP-Nogo-66, AP-MAG, and AP-OMgp binding assays were conducted over a range of ligand concentrations, and bound AP was measured.
To examine the functional consequence of these aa substitutions, we measured the affinity of myelin ligands for the R119W-NgR1 and R196H-NgR1 proteins expressed in COS-7 cells. The R196H substitution has no effect on ligand binding, as predicted from the ligand mapping study. In contrast, the R119W substitution has selective effects on binding of some myelin ligands. MAG and OMgp bind with substantially reduced affinity to the R119W-NgR, whereas Nogo-66 binding is similar to control values (Fig. 5C,D). Because the R196H variant resides in the NgR1 surface opposite the ligand binding, we considered whether it might alter coreceptor interactions. Physical associations with p75NTR, Lingo-1, and Troy are identical for WT-NgR, R119W-NgR1, and R196H-NgR1 (Fig. 6A) (data not shown). Thus, the R119W substitution selectively reduces MAG and OMgp interaction, but other known protein associations are detected at normal levels with the Italian NgR1 variant proteins.
Figure 6.

NgR1 ligand-binding domain variants interact with known coreceptors but do not mediate myelin signaling. A, Interaction of NgR1 Italian variants with putative NgR1 coreceptors. HEK293T cells were transfected with the indicated expression vectors (WT-NgR, WT; R119W-NgR, W; R196H-NgR, H). Lysates were immunoblotted directly or subjected to immunoprecipitation and then immunoblot, as indicated. These NgR1 amino acid variations do not significantly alter interaction with p75 or Lingo-1. B, Growth cone collapse induced by myelin ligands in E7 retinal explants expressing WT-NgR1, R119W-NgR1, or R196H-NgR1. Values with variant NgR1-expressing virus are significantly different from those with wild-type NgR1 expression (*p < 0.05, one-way ANOVA). C, Dominant-negative effect of NgR1 variants in E13 cDRG expressing indicated constructs. Values with variant NgR1-expressing virus are significantly different from those with wild-type NgR1 expression (*p < 0.05, one-way ANOVA). All data are mean ± SEM from n = 3–6 experiments.
The reduced R119W-NgR1 binding of MAG and OMgp predicts that the growth cone collapse function of these ligands will be reduced in neurons containing the variant R119W-NgR. Indeed, MAG and OMgp do not induce collapse of growth cones of E7 retinal neurons expressing R119W-NgR1 despite normal NgR1 surface expression levels (Fig. 6B). Interestingly, although COS-7-expressed R119W-NgR1 protein binds Nogo-66, neuronal R119W-NgR1 fails to mediate Nogo-66-induced growth cone collapse (Fig. 5B). This implies that the R119W-NgR1 binds Nogo-66 in a nonproductive signaling complex. Similarly, the R196H substitution does not alter ligand binding or coreceptor association, but when expressed in E7 chick retinal neurons, the R196H-NgR1 variant is incapable of mediating growth cone collapse by myelin ligands. This amino acid substitution may abolish signal transduction by altering ligand-induced conformational changes, without modulating the extent of protein–protein association. Because both Italian NgR1 variants fail to signal, we assessed any potential dominant-negative activity. The R119W-NgR1 and R196W-NgR1 proteins suppress endogenous NgR-mediated responsiveness to myelin ligands (Fig. 6C). Together, four human NgR1 variants from schizophrenic individuals are functionally inactive in myelin-induced growth cone collapse assay and possess dominant-negative function in vitro.
Behavioral and cognitive assessments: impaired spatial working memory in the absence of NgR1
If human inactivating sequence variants in NGR contribute to increased schizophrenic risk, then mice lacking NgR1 might exhibit a parallel propensity to what are considered to be schizophrenic-associated behaviors or “endophenotypes.” We find that mice lacking NgR1 have impaired working memory with little change in motor or emotional behavior. In previous studies of motor activity, we noted that mice lacking the NgR1 protein have slightly impaired performance on the Rotarod test (Kim et al., 2004). In open-field tests, the NGR−/− mice have a tendency to avoid the center of the arena and are mildly hypoactive (Kim et al., 2004). Here, we considered whether a more specific measure of anxiety might be altered in this strain. No differences in light/dark preference were detected, arguing against a generalized anxiety disorder (Fig. 7 A,B).
Figure 7.
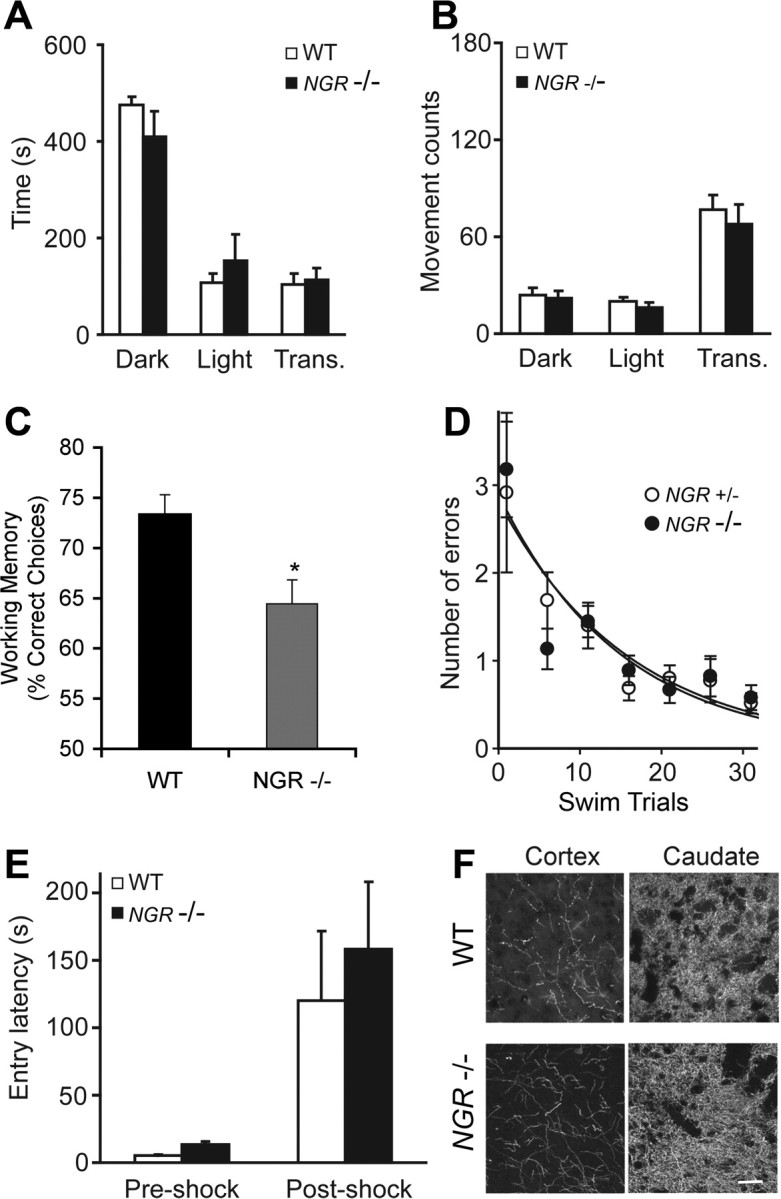
Selectively impaired working memory in NGR−/− mice. Mice were tested on a battery of tests to assess affective and cognitive responses. A, B, The light/dark exploration test was performed by placing the mouse in a cage (44 × 21 × 21 cm) that has one dark chamber (Dark) and one light chamber (Light). The animal was initially placed in the lighted side, and transitions between sides (trans) and the time spent in each chamber were recorded for 10 min using computer-monitored photocell beams. Mice were from a mixed 129 × C57BL/6 strain background. No significant difference of anxiety-like behavior was observed between WT and NGR−/− animals (n = 21, 10 WT, 11 −/−). C, The importance of NgR1 signaling to the spatial working memory functions of the prefrontal cortex in mice. NGR-deficient mice (−/−; n = 8) performed significantly worse on the spatial delayed alternation task than WT mice (+/+; n = 5). All mice were of a pure C57BL/6J genetic background (>10 backcrosses for the NGR mutation). Data represent mean ± SEM percentage correct over the 30 daily test sessions; *p = 0.017. D, NGR heterozygote and null littermate mice from a mixed strain background were subjected to a 2 d six-arm radial-arm water-maze paradigm as described previously (Morgan et al., 2000). To minimize odor cues, the goal arm was randomly assigned for each mouse. The start arm was varied for each trial, with the goal arm remaining constant. The mice were tested in the same manner on day 2. The number of incorrect arm entries (errors) was measured for 1 min. Mice made an arm choice within 20 s in every experiment. Each mouse's errors for five consecutive trials were averaged. E, Passive avoidance testing was performed in a mouse passive avoidance chamber (Ugo Basile). Mice used were from mixed 129 × C57BL/6 background. Testing occurred on 3 consecutive days; for all trials, the mouse was initially placed in the light chamber. On day 1, the mouse was allowed to move freely between the two compartments for 5 min. On day 2 (preshock), the entry latency into the dark chamber was measured; after entry, the door between the light and dark compartments closed, and a 2 s electric shock (0.2 mA) was administered through the grid floor. On day 3 (postshock), entry latency into the dark chamber was recorded. No differences in passive avoidance learning were observed in the WT and NGR−/− mice (n = 34, 17 WT, 17 −/−). F, Tyrosine hydroxylase-immunoreactive fibers in the prefrontal cerebral cortex and the caudate nucleus were visualized in sections from WT and NGR−/− adult mice. Scale bar, 100 μm. All data are mean ± SEM.
The spatial delayed alternation task depends on the integrity of the medial (prelimbic and infralimbic) prefrontal cortex (Divac and Larsen, 1978) and may be most relevant as a schizophrenic-associated endophenotype. Mice tested on this task are sensitive to changes in noradrenergic α-2A or dopamine D1 receptor stimulation, much like rats and primates (Franowicz et al., 2002; Lidow et al., 2003). Wild-type (+/+) and NGR-deficient (−/−) mice were required to achieve criteria during habituation and training to ensure that all mice were equally responsive to the maze, food rewards, handling, and trial procedures before cognitive assessment. NGR-deficient mice tended to take slightly longer to achieve criterion to handling in the maze (WT average of 4.0 ± 1.3 trials; NGR deficient average of 20.8 ± 8.3 trials), but this did not reach significance because of the large variability in responses of the NGR-deficient mice (p = 0.12). The NGR-deficient mice also took longer to habituate to the testing procedures assessed by the forced alternation criterion (WT average of 4.8 ± trials; NGR-deficient mice average of 9.4 ± 1.6 trials; p = 0.05). These stringent training criteria were used to ensure that the delayed alternation testing measured cognitive changes rather than affective responses to the testing procedures. After this strict training protocol, mice were assessed for 30 sessions on the delayed alternation task. NGR-deficient mice did not differ in their response times, but had impaired accuracy compared with wild-type controls (Fig. 7C). Wild-type (+/+) mice performed at an average of 73.3 ± 1.96% correct over the 30 test sessions, whereas NGR-deficient (−/−) mice performed an average of only 64.4 ± 2.39% correct (p = 0.017). In this test, 50% correct reflects random choices and the absence of working memory. There were no significant differences in response times between the groups; wild-type (+/+) mice responded in an average of 211.5 ± 3.8 s per test session, whereas NGR deficient (−/−) mice responded in an average of 232.0 ± 14.1 s (p > 0.25).
The specificity of impaired working memory was considered by examining the mice in other learning and memory tasks that required longer-term memory consolidation. Spatial learning and memory consolidation were examined in the radial arm water maze. The NGR−/− mice were indistinguishable from control mice in this examination (Fig. 7D). Passive avoidance learning was also normal in NGR−/− mice (Fig. 7E). Thus, NGR−/− mice exhibited impaired spatial working memory without widespread cognitive dysfunction.
Because brain dopamine levels and dopamine antagonist medications are intensely studied in schizophrenia and play a role in prefrontal cortex function, we considered whether there were any gross alterations in these fiber systems in mice lacking NgR. Although NgR1 is expressed in tyrosine hydroxylase positive neurons (data not shown), there was no obvious change in tyrosine hydroxylase immunopositive axons in the cerebral cortex or caudate nucleus NGR−/− mice (Fig. 7F). Quantitative measurements of DA, DOPAC, and homovanillic acid levels in NGR−/− brain revealed no significant difference from wild-type mouse brain (data not shown). Thus, NgR1-dependent alterations in prefrontal cortex itself, or in other ascending neuromodulators, may account for the decreased working memory function.
A recent report has demonstrated that another schizophrenia endophenotype, PPI of the acoustic startle response, is not altered in NGR−/− animals (Hsu et al., 2007). Similarly, we find that PPI shows no consistent change across strain backgrounds in mice lacking NgR1 (Fig. 8B,E). However, we noted that a statistically significant subset of NGR−/− mice exhibits an absence of PPI response (Fig. 8C,F). This may reflect a stochastic interaction of the NGR genotype with other processes controlling the development of this endophenotype.
Figure 8.

A small subset of mice lacking NgR1 exhibit decreased prepulse inhibition. Experiments used mice of a mixed background 129 × C57BL/6J with littermate controls (A–C) or a pure C57BL/6J genetic background (D–F). A, Acoustic startle response was measured as a function of stimulus intensity from mice of the indicated genotypes. B, Acoustic startle was measured with a stimulus of 110 dB stimulus preceded 100 ms by a pretone of the indicted sound level in NGR+/+ (n = 24), NGR+/− (n = 41), or NGR−/− (n = 38) mice. The trend to reduced NGR−/− PPI is not significant. C, Percentage of mice with absent PPI from the data set in B. Absent PPI is defined as <3% reduction in startle magnitude by prepulses of ≥85 dB. The incidence of absent PPI is greater in the NGR−/− population (*p < 0.05, χ2 test). D, Acoustic startle response was similar in NGR+/+ and NGR−/− mice (n = 57, 28 WT, and 29 NGR null). E, Prepulse inhibition of the acoustic startle was not significantly different in WT versus NGR−/− mice in this strain background, as opposed to the mixed background (n = 73, 34 WT, and 39 NGR null). F, The percentage of mice with absent PPI is reported. There is a significant increase in this percentage in the NGR−/− group (*p < 0.05, χ2). Error bars indicate SEM.
Discussion
A range of evidence supports the notion that lack of myelin-based restriction of axonal plasticity in the postnatal brain contributes to the risk of schizophrenia (Karoutzou et al., 2007; Segal et al., 2007). Previous studies have revealed altered myelin gene expression in schizophrenic brain and genetic linkage of schizophrenic risk to the 22q11 genomic region that includes NGR. The statistical correlation of NGR variation with schizophrenia reaches statistical significance in our Caucasian study, but is not strong. Here, we describe additional rare NgR1 sequence variants in schizophrenic individuals, and note that variants predicted to alter protein function are more common in the schizophrenic population. Because these variants are rare, biological tests of function, rather than genetic association statistics, provide an alternate and essential means to assess their role. Critically, we show that four of these variant proteins are loss-of-function mutants in tissue culture, incapable of mediating myelin inhibition of axonal growth. The schizophrenia-derived NgR1 variants disrupt WT-NgR1 signaling in a dominant-negative pattern. In addition, mice lacking NgR1 protein exhibit impaired spatial working memory. These findings support the hypothesis that one mechanism for increased schizophrenic risk is a failure to restrict anatomical plasticity in the brain.
Because myelin-mediated inhibition of neuritic sprouting is likely to be one of the last steps in neuronal development (McGee et al., 2005; Cafferty et al., 2008), this model fits with the recognition that schizophrenia is a neurodevelopmental condition with onset typically in late adolescence to early adulthood. Neural stem cell proliferation, neuronal differentiation, cell migration, axon guidance, and synapse formation are mostly completed before myelin-dependent restriction of axonal plasticity via the NgR1 pathway. Although NgR1 is not expressed in all neuronal subtypes (Fournier et al., 2001; Hunt et al., 2002; Josephson et al., 2002; X. Wang et al., 2002), its distribution is wide enough in the adult brain to explain the involvement of multiple neurotransmitter systems in schizophrenia. Although the data presented here support the hypothesis that myelin restriction of neuritic plasticity via the NgR1 is necessary to stabilize CNS circuitry and limit the risk of schizophrenia, it is clear that multiple pathways regulate connectivity in the brain. It is plausible that these multiple influences on axonal plasticity in the adult brain account for complex genetic influences on schizophrenic risk. Of note, NGR coding region genetic variants appear to be present in only a small fraction (1%) of schizophrenic cases. Furthermore, the mouse NGR−/− phenotype of absent sensorimotor gating is of low penetrance and is strain-background dependent.
Recently, it has been noted that NgR1 expression plays a role in regulating glutaminergic synaptic transmission (Lee et al., 2008). This provides an additional pathway by which NgR1 variants might contribute to schizophrenia without frank alteration in axonal sprouting or circuitry per se.
Patients with schizophrenia have profound impairments in prefrontal cortical cognitive abilities (Keefe et al., 2006), including deficits in spatial working memory performance (Park and Holzman, 1992; Keedy et al., 2006). The current study thus assessed the spatial working abilities of the NgR1 knock-out mice to observe whether loss of Nogo signaling altered prefrontal abilities in mice. The mice were assessed using an established working memory paradigm (Franowicz et al., 2002; Lidow et al., 2003). As with patients with schizophrenia, NGR−/− mice showed deficits in spatial working memory performance.
Acute reduction of Nogo/NgR1 function in mature animals has the beneficial effect of improving recovery from stroke and spinal cord injury via axonal sprouting and regeneration (GrandPré et al., 2002; Li and Strittmatter, 2003; Kim et al., 2004; Lee et al., 2004; Li et al., 2004). Constitutive absence of NgR1 throughout neurodevelopment and adulthood may possibly increase schizophrenic risk. Improved axon regeneration and impaired sensorimotor gating may be opposite sides of the same coin and be inextricably linked. Alternatively, increased risk of schizophrenia may require absent NgR1 function specifically during the late adolescent completion of myelination when neuronal networks are stabilized. If the latter scenario is valid, then relatively transient NgR1 loss of function in the injured adult CNS may be achieved without enhanced risk of neuropsychiatric disease. Future studies inactivating the NGR gene during different developmental epochs should clarify this issue. It is conceivable that pharmacological NgR1 agonism may either reduce psychiatric risk before overt manifestations or reverse schizophrenic symptomatology after onset. In conclusion, the presence of dominant-negative NgR1 sequence variants in schizophrenic individuals supports a disease model of failed stabilization in brain wiring during the last phases of neuronal development.
Footnotes
This work was supported in part by grants from the National Institutes of Health and the Falk Medical Research Trust (S.M.S.) and a Scottish Rite fellowship (S.B.). S.M.S. and A.F.T.A. are members of the Kavli Institute of Neuroscience at Yale. We acknowledge that data and biomaterials were collected in three projects that participated in the National Institute of Mental Health Schizophrenia Genetics Initiative. From 1991 to 1997, the principal investigators and coinvestigators were as follows: Harvard University (Boston, MA) (Grant U01 MH46318), Ming T. Tsuang, MD, PhD, DSc, Stephen Faraone, PhD, and John Pepple, PhD; Washington University (St. Louis, MO) (Grant U01 MH46276), C. Robert Cloninger, MD, Theodore Reich, MD, and Dragan Svrakic, MD; and Columbia University (New York, NY) (Grant U01 MH46289), Charles Kaufmann, MD, Dolores Malaspina, MD, and Jill Harkavy Friedman, PhD.
References
- Akil M, Kolachana BS, Rothmond DA, Hyde TM, Weinberger DR, Kleinman JE. Catechol-O-methyltransferase genotype and dopamine regulation in the human brain. J Neurosci. 2003;23:2008–2013. doi: 10.1523/JNEUROSCI.23-06-02008.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron M. Genetics of schizophrenia and the new millennium: progress and pitfalls. Am J Hum Genet. 2001;68:299–312. doi: 10.1086/318212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton WA, Liu BP, Tzvetkova D, Jeffrey PD, Fournier AE, Sah D, Cate R, Strittmatter SM, Nikolov DB. Structure and axon outgrowth inhibitor binding of the Nogo-66 receptor and related proteins. EMBO J. 2003;22:3291–3302. doi: 10.1093/emboj/cdg325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett AS, Chow EW. 22q11 deletion syndrome: a genetic subtype of schizophrenia. Biol Psychiatry. 1999;46:882–891. doi: 10.1016/s0006-3223(99)00114-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett AS, Chow EW. Schizophrenia and 22q11.2 deletion syndrome. Curr Psychiatry Rep. 2008;10:148–157. doi: 10.1007/s11920-008-0026-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett AS, Chow EW, AbdelMalik P, Gheorghiu M, Husted J, Weksberg R. The schizophrenia phenotype in 22q11 deletion syndrome. Am J Psychiatry. 2003;160:1580–1586. doi: 10.1176/appi.ajp.160.9.1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benes FM, Davidson J, Bird ED. Quantitative cytoarchitectural studies of the cerebral cortex of schizophrenics. Arch Gen Psychiatry. 1986;43:31–35. doi: 10.1001/archpsyc.1986.01800010033004. [DOI] [PubMed] [Google Scholar]
- Cafferty WB, Strittmatter SM. The Nogo-Nogo receptor pathway limits a spectrum of adult CNS axonal growth. J Neurosci. 2006;26:12242–12250. doi: 10.1523/JNEUROSCI.3827-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cafferty WB, Kim JE, Lee JK, Strittmatter SM. Response to correspondence: Kim et al., “axon regeneration in young adult mice lacking Nogo-A/B.” Neuron 38, 187–199. Neuron. 2007;54:195–199. doi: 10.1016/j.neuron.2007.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cafferty WB, McGee AW, Strittmatter SM. Axonal growth therapeutics: regeneration or sprouting or plasticity? Trends Neurosci. 2008;31:215–220. doi: 10.1016/j.tins.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarti A. A compelling genetic hypothesis for a complex disease: PRODH2/DGCR6 variation leads to schizophrenia susceptibility. Proc Natl Acad Sci U S A. 2002;99:4755–4756. doi: 10.1073/pnas.092158299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chivatakarn O, Kaneko S, He Z, Tessier-Lavigne M, Giger RJ. The Nogo-66 receptor NgR1 is required only for the acute growth cone-collapsing but not the chronic growth-inhibitory actions of myelin inhibitors. J Neurosci. 2007;27:7117–7124. doi: 10.1523/JNEUROSCI.1541-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen E, Chow EW, Weksberg R, Bassett AS. Phenotype of adults with the 22q11 deletion syndrome: a review. Am J Med Genet. 1999;86:359–365. doi: 10.1002/(sici)1096-8628(19991008)86:4<359::aid-ajmg10>3.0.co;2-v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis KL, Haroutunian V. Global expression-profiling studies and oligodendrocyte dysfunction in schizophrenia and bipolar disorder. Lancet. 2003;362:758. doi: 10.1016/S0140-6736(03)14297-3. [DOI] [PubMed] [Google Scholar]
- Egan MF, Goldberg TE, Kolachana BS, Callicott JH, Mazzanti CM, Straub RE, Goldman D, Weinberger DR. Effect of COMT Val108/158 Met genotype on frontal lobe function and risk for schizophrenia. Proc Natl Acad Sci U S A. 2001;98:6917–6922. doi: 10.1073/pnas.111134598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier AE, GrandPré T, Strittmatter SM. Identification of a receptor mediating Nogo-66 inhibition of axonal regeneration. Nature. 2001;409:341–346. doi: 10.1038/35053072. [DOI] [PubMed] [Google Scholar]
- Fournier AE, Gould GC, Liu BP, Strittmatter SM. Truncated soluble Nogo receptor binds Nogo-66 and blocks inhibition of axon growth by myelin. J Neurosci. 2002;22:8876–8883. doi: 10.1523/JNEUROSCI.22-20-08876.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franowicz JS, Kessler LE, Borja CM, Kobilka BK, Limbird LE, Arnsten AF. Mutation of the α2A-adrenoceptor impairs working memory performance and annuls cognitive enhancement by guanfacine. J Neurosci. 2002;22:8771–8777. doi: 10.1523/JNEUROSCI.22-19-08771.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogos JA, Morgan M, Luine V, Santha M, Ogawa S, Pfaff D, Karayiorgou M. Catechol-O-methyltransferase-deficient mice exhibit sexually dimorphic changes in catecholamine levels and behavior. Proc Natl Acad Sci U S A. 1998;95:9991–9996. doi: 10.1073/pnas.95.17.9991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GrandPré T, Li S, Strittmatter SM. Nogo-66 receptor antagonist peptide promotes axonal regeneration. Nature. 2002;417:547–551. doi: 10.1038/417547a. [DOI] [PubMed] [Google Scholar]
- Hakak Y, Walker JR, Li C, Wong WH, Davis KL, Buxbaum JD, Haroutunian V, Fienberg AA. Genome-wide expression analysis reveals dysregulation of myelination-related genes in chronic schizophrenia. Proc Natl Acad Sci U S A. 2001;98:4746–4751. doi: 10.1073/pnas.081071198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He XL, Bazan JF, McDermott G, Park JB, Wang K, Tessier-Lavigne M, He Z, Garcia KC. Structure of the Nogo receptor ectodomain: a recognition module implicated in myelin inhibition. Neuron. 2003;38:177–185. doi: 10.1016/s0896-6273(03)00232-0. [DOI] [PubMed] [Google Scholar]
- Hof PR, Haroutunian V, Copland C, Davis KL, Buxbaum JD. Molecular and cellular evidence for an oligodendrocyte abnormality in schizophrenia. Neurochem Res. 2002;27:1193–1200. doi: 10.1023/a:1020981510759. [DOI] [PubMed] [Google Scholar]
- Hsu R, Woodroffe A, Lai WS, Cook MN, Mukai J, Dunning JP, Swanson DJ, Roos JL, Abecasis GR, Karayiorgou M, Gogos JA. Nogo receptor 1 (RTN4R) as a candidate gene for schizophrenia: analysis using human and mouse genetic approaches. PLoS ONE. 2007;2:e1234. doi: 10.1371/journal.pone.0001234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulshoff Pol HE, Schnack HG, Mandl RC, Cahn W, Collins DL, Evans AC, Kahn RS. Focal white matter density changes in schizophrenia: reduced inter-hemispheric connectivity. Neuroimage. 2004;21:27–35. doi: 10.1016/j.neuroimage.2003.09.026. [DOI] [PubMed] [Google Scholar]
- Hunt D, Mason MR, Campbell G, Coffin R, Anderson PN. Nogo receptor mRNA expression in intact and regenerating CNS neurons. Mol Cell Neurosci. 2002;20:537–552. doi: 10.1006/mcne.2002.1153. [DOI] [PubMed] [Google Scholar]
- Jacquet H, Raux G, Thibaut F, Hecketsweiler B, Houy E, Demilly C, Haouzir S, Allio G, Fouldrin G, Drouin V, Bou J, Petit M, Campion D, Frébourg T. PRODH mutations and hyperprolinemia in a subset of schizophrenic patients. Hum Mol Genet. 2002;11:2243–2249. doi: 10.1093/hmg/11.19.2243. [DOI] [PubMed] [Google Scholar]
- Josephson A, Trifunovski A, Widmer HR, Widenfalk J, Olson L, Spenger C. Nogo-receptor gene activity: cellular localization and developmental regulation of mRNA in mice and humans. J Comp Neurol. 2002;453:292–304. doi: 10.1002/cne.10408. [DOI] [PubMed] [Google Scholar]
- Karayiorgou M, Morris MA, Morrow B, Shprintzen RJ, Goldberg R, Borrow J, Gos A, Nestadt G, Wolyniec PS, Lasseter VK. Schizophrenia susceptibility associated with interstitial deletions of chromosome 22q11. Proc Natl Acad Sci U S A. 1995;92:7612–7616. doi: 10.1073/pnas.92.17.7612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karoutzou G, Emrich HM, Dietrich DE. The myelin-pathogenesis puzzle in schizophrenia: a literature review. Mol Psychiatry. 2007;13:245–260. doi: 10.1038/sj.mp.4002096. [DOI] [PubMed] [Google Scholar]
- Keedy SK, Ebens CL, Keshavan MS, Sweeney JA. Functional magnetic resonance imaging studies of eye movements in first episode schizophrenia: smooth pursuit, visually guided saccades and the oculomotor delayed response task. Psychiatry Res. 2006;146:199–211. doi: 10.1016/j.pscychresns.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Keefe RSE, Bilder RM, Harvey PD, Davis SM, Palmer BW, Gold JM, Meltzer HY, Green MF, Miller DD, Canive JM, Adler LW, Manschreck TC, Swartz M, Rosenheck R, Perkins DO, Walker TM, Stroup TS, McEvoy JP, Lieberman JA. Baseline neurocognitive deficits in the CATIE schizophrenia trial. Neuropsychopharmacology. 2006;31:2033–2046. doi: 10.1038/sj.npp.1301072. [DOI] [PubMed] [Google Scholar]
- Kim JE, Liu BP, Park JH, Strittmatter SM. Nogo-66 receptor prevents raphespinal and rubrospinal axon regeneration and limits functional recovery from spinal cord injury. Neuron. 2004;44:439–451. doi: 10.1016/j.neuron.2004.10.015. [DOI] [PubMed] [Google Scholar]
- Kubicki M, Westin CF, Nestor PG, Wible CG, Frumin M, Maier SE, Kikinis R, Jolesz FA, McCarley RW, Shenton ME. Cingulate fasciculus integrity disruption in schizophrenia: a magnetic resonance diffusion tensor imaging study. Biol Psychiatry. 2003;54:1171–1180. doi: 10.1016/s0006-3223(03)00419-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen JK, Divac I. Selective ablations within the prefrontal cortex of the rat and performance of delayed alternation. Physiol Psychol. 1978;6:15–17. [Google Scholar]
- Laurén J, Hu F, Chin J, Liao J, Airaksinen MS, Strittmatter SM. Characterization of myelin ligand complexes with neuronal Nogo-66 receptor family members. J Biol Chem. 2007;282:5715–5725. doi: 10.1074/jbc.M609797200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Raiker SJ, Venkatesh K, Geary R, Robak LA, Zhang Y, Yeh HH, Shrager P, Giger RJ. Synaptic function for the Nogo-66 receptor NgR1: regulation of dendritic spine morphology and activity-dependent synaptic strength. J Neurosci. 2008;28:2753–2765. doi: 10.1523/JNEUROSCI.5586-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JK, Kim JE, Sivula M, Strittmatter SM. Nogo receptor antagonism promotes stroke recovery by enhancing axonal plasticity. J Neurosci. 2004;24:6209–6217. doi: 10.1523/JNEUROSCI.1643-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Strittmatter SM. Delayed systemic Nogo-66 receptor antagonist promotes recovery from spinal cord injury. J Neurosci. 2003;23:4219–4227. doi: 10.1523/JNEUROSCI.23-10-04219.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Liu BP, Budel S, Li M, Ji B, Walus L, Li W, Jirik A, Rabacchi S, Choi E, Worley D, Sah DW, Pepinsky B, Lee D, Relton J, Strittmatter SM. Blockade of nogo-66, myelin-associated glycoprotein, and oligodendrocyte myelin glycoprotein by soluble nogo-66 receptor promotes axonal sprouting and recovery after spinal injury. J Neurosci. 2004;24:10511–10520. doi: 10.1523/JNEUROSCI.2828-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Kim JE, Budel S, Hampton TG, Strittmatter SM. Transgenic inhibition of Nogo-66 receptor function allows axonal sprouting and improved locomotion after spinal injury. Mol Cell Neurosci. 2005;29:26–39. doi: 10.1016/j.mcn.2004.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lidow MS, Koh PO, Arnsten AF. D1 dopamine receptors in the mouse prefrontal cortex: immunocytochemical and cognitive neuropharmacological analyses. Synapse. 2003;47:101–108. doi: 10.1002/syn.10143. [DOI] [PubMed] [Google Scholar]
- Liu BP, Fournier A, GrandPré T, Strittmatter SM. Myelin-associated glycoprotein as a functional ligand for the Nogo-66 receptor. Science. 2002;297:1190–1193. doi: 10.1126/science.1073031. [DOI] [PubMed] [Google Scholar]
- Liu BP, Cafferty WB, Budel SO, Strittmatter SM. Extracellular regulators of axonal growth in the adult central nervous system. Philos Trans R Soc Lond B Biol Sci. 2006;361:1593–1610. doi: 10.1098/rstb.2006.1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Heath SC, Sobin C, Roos JL, Galke BL, Blundell ML, Lenane M, Robertson B, Wijsman EM, Rapoport JL, Gogos JA, Karayiorgou M. Genetic variation at the 22q11 PRODH2/DGCR6 locus presents an unusual pattern and increases susceptibility to schizophrenia. Proc Natl Acad Sci U S A. 2002a;99:3717–3722. doi: 10.1073/pnas.042700699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Abecasis GR, Heath SC, Knowles A, Demars S, Chen YJ, Roos JL, Rapoport JL, Gogos JA, Karayiorgou M. Genetic variation in the 22q11 locus and susceptibility to schizophrenia. Proc Natl Acad Sci U S A. 2002b;99:16859–16864. doi: 10.1073/pnas.232186099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus J, Dupree JL, Popko B. Myelin-associated glycoprotein and myelin galactolipids stabilize developing axo-glial interactions. J Cell Biol. 2002;156:567–577. doi: 10.1083/jcb.200111047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGee AW, Strittmatter SM. The Nogo-66 receptor: focusing myelin inhibition of axon regeneration. Trends Neurosci. 2003;26:193–198. doi: 10.1016/S0166-2236(03)00062-6. [DOI] [PubMed] [Google Scholar]
- McGee AW, Yang Y, Fischer QS, Daw NW, Strittmatter SM. Experience-driven plasticity of visual cortex limited by myelin and Nogo receptor. Science. 2005;309:2222–2226. doi: 10.1126/science.1114362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng J, Shi Y, Zhao X, Guo S, Wang H, Zheng Y, Tang R, Feng G, Gu N, Liu H, Zhu S, He L. No association between the genetic polymorphisms in the RTN4R gene and schizophrenia in the Chinese population. J Neural Transm. 2007;114:249–254. doi: 10.1007/s00702-006-0538-y. [DOI] [PubMed] [Google Scholar]
- Mi S, Lee X, Shao Z, Thill G, Ji B, Relton J, Levesque M, Allaire N, Perrin S, Sands B, Crowell T, Cate RL, McCoy JM, Pepinsky RB. LINGO-1 is a component of the Nogo-66 receptor/p75 signaling complex. Nat Neurosci. 2004;7:221–228. doi: 10.1038/nn1188. [DOI] [PubMed] [Google Scholar]
- Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, Duff K, Jantzen P, DiCarlo G, Wilcock D, Connor K, Hatcher J, Hope C, Gordon M, Arendash GW. A beta peptide vaccination prevents memory loss in an animal model of Alzheimer's disease. Nature. 2000;408:982–985. doi: 10.1038/35050116. [DOI] [PubMed] [Google Scholar]
- Park S, Holzman PS. Schizophrenics show spatial working memory deficits. Arch Gen Psychiatry. 1992;49:975–982. doi: 10.1001/archpsyc.1992.01820120063009. [DOI] [PubMed] [Google Scholar]
- Park HJ, Westin CF, Kubicki M, Maier SE, Niznikiewicz M, Baer A, Frumin M, Kikinis R, Jolesz FA, McCarley RW, Shenton ME. White matter hemisphere asymmetries in healthy subjects and in schizophrenia: a diffusion tensor MRI study. Neuroimage. 2004;23:213–223. doi: 10.1016/j.neuroimage.2004.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JB, Yiu G, Kaneko S, Wang J, Chang J, He XL, Garcia KC, He Z. A TNF receptor family member, TROY, is a coreceptor with Nogo receptor in mediating the inhibitory activity of myelin inhibitors. Neuron. 2005;45:345–351. doi: 10.1016/j.neuron.2004.12.040. [DOI] [PubMed] [Google Scholar]
- Park JH, Widi GA, Gimbel DA, Harel NY, Lee DH, Strittmatter SM. Subcutaneous Nogo receptor removes brain amyloid-β and improves spatial memory in Alzheimer's transgenic mice. J Neurosci. 2006;26:13279–13286. doi: 10.1523/JNEUROSCI.4504-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Rosenberg NA. Use of unlinked genetic markers to detect population stratification in association studies. Am J Hum Genet. 1999;65:220–228. doi: 10.1086/302449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scambler PJ. The 22q11 deletion syndromes. Hum Mol Genet. 2000;9:2421–2426. doi: 10.1093/hmg/9.16.2421. [DOI] [PubMed] [Google Scholar]
- Segal D, Koschnick JR, Slegers LH, Hof PR. Oligodendrocyte pathophysiology: a new view of schizophrenia. Int J Neuropsychopharmacol. 2007;10:503–511. doi: 10.1017/S146114570600722X. [DOI] [PubMed] [Google Scholar]
- Shao Z, Browning JL, Lee X, Scott ML, Shulga-Morskaya S, Allaire N, Thill G, Levesque M, Sah D, McCoy JM, Murray B, Jung V, Pepinsky RB, Mi S. TAJ/TROY, an orphan TNF receptor family member, binds Nogo-66 receptor 1 and regulates axonal regeneration. Neuron. 2005;45:353–359. doi: 10.1016/j.neuron.2004.12.050. [DOI] [PubMed] [Google Scholar]
- Shenton ME, Dickey CC, Frumin M, McCarley RW. A review of MRI findings in schizophrenia. Schizophr Res. 2001;49:1–52. doi: 10.1016/s0920-9964(01)00163-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shifman S, Bronstein M, Sternfeld M, Pisanté-Shalom A, Lev-Lehman E, Weizman A, Reznik I, Spivak B, Grisaru N, Karp L, Schiffer R, Kotler M, Strous RD, Swartz-Vanetik M, Knobler HY, Shinar E, Beckmann JS, Yakir B, Risch N, Zak NB. A highly significant association between a COMT haplotype and schizophrenia. Am J Hum Genet. 2002;71:1296–1302. doi: 10.1086/344514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinibaldi L, De Luca A, Bellacchio E, Conti E, Pasini A, Paloscia C, Spalletta G, Caltagirone C, Pizzuti A, Dallapiccola B. Mutations of the Nogo-66 receptor (RTN4R) gene in schizophrenia. Hum Mutat. 2004;24:534–535. doi: 10.1002/humu.9292. [DOI] [PubMed] [Google Scholar]
- Slatkin M, Excoffier L. Testing for linkage disequilibrium in genotypic data using the expectation-maximization algorithm. Heredity. 1996;76:377–383. doi: 10.1038/hdy.1996.55. [DOI] [PubMed] [Google Scholar]
- Spielman RS, McGinnis RE, Ewens WJ. Transmission test for linkage disequilibrium: the insulin gene region and insulin-dependent diabetes mellitus (IDDM) Am J Hum Genet. 1993;52:506–516. [PMC free article] [PubMed] [Google Scholar]
- Stein MB, Schork NJ, Gelernter J. A polymorphism of the beta1-adrenergic receptor is associated with low extraversion. Biol Psychiatry. 2004;56:217–224. doi: 10.1016/j.biopsych.2004.05.020. [DOI] [PubMed] [Google Scholar]
- Sunyaev S, Ramensky V, Koch I, Lathe W, 3rd, Kondrashov AS, Bork P. Prediction of deleterious human alleles. Hum Mol Genet. 2001;10:591–597. doi: 10.1093/hmg/10.6.591. [DOI] [PubMed] [Google Scholar]
- Tkachev D, Mimmack ML, Ryan MM, Wayland M, Freeman T, Jones PB, Starkey M, Webster MJ, Yolken RH, Bahn S. Oligodendrocyte dysfunction in schizophrenia and bipolar disorder. Lancet. 2003;362:798–805. doi: 10.1016/S0140-6736(03)14289-4. [DOI] [PubMed] [Google Scholar]
- Uranova N, Orlovskaya D, Vikhreva O, Zimina I, Kolomeets N, Vostrikov V, Rachmanova V. Electron microscopy of oligodendroglia in severe mental illness. Brain Res Bull. 2001;55:597–610. doi: 10.1016/s0361-9230(01)00528-7. [DOI] [PubMed] [Google Scholar]
- Uranova NA, Vostrikov VM, Orlovskaya DD, Rachmanova VI. Oligodendroglial density in the prefrontal cortex in schizophrenia and mood disorders: a study from the Stanley Neuropathology Consortium. Schizophr Res. 2004;67:269–275. doi: 10.1016/S0920-9964(03)00181-6. [DOI] [PubMed] [Google Scholar]
- Usiskin SI, Nicolson R, Krasnewich DM, Yan W, Lenane M, Wudarsky M, Hamburger SD, Rapoport JL. Velocardiofacial syndrome in childhood-onset schizophrenia. J Am Acad Child Adolesc Psychiatry. 1999;38:1536–1543. doi: 10.1097/00004583-199912000-00015. [DOI] [PubMed] [Google Scholar]
- Wang KC, Kim JA, Sivasankaran R, Segal R, He Z. P75 interacts with the Nogo receptor as a co-receptor for Nogo, MAG and OMgp. Nature. 2002;420:74–78. doi: 10.1038/nature01176. [DOI] [PubMed] [Google Scholar]
- Wang X, Chun SJ, Treloar H, Vartanian T, Greer CA, Strittmatter SM. Localization of Nogo-A and Nogo-66 receptor proteins at sites of axon-myelin and synaptic contact. J Neurosci. 2002;22:5505–5515. doi: 10.1523/JNEUROSCI.22-13-05505.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Baughman KW, Basso DM, Strittmatter SM. Delayed Nogo receptor therapy improves recovery from spinal cord contusion. Ann Neurol. 2006;60:540–549. doi: 10.1002/ana.20953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong ST, Henley JR, Kanning KC, Huang KH, Bothwell M, Poo MM. A p75(NTR) and Nogo receptor complex mediates repulsive signaling by myelin-associated glycoprotein. Nat Neurosci. 2002;5:1302–1308. doi: 10.1038/nn975. [DOI] [PubMed] [Google Scholar]
- Yang BZ, Zhao H, Kranzler HR, Gelernter J. Practical population group assignment with selected informative markers: characteristics and properties of Bayesian clustering via STRUCTURE. Genet Epidemiol. 2005;28:302–312. doi: 10.1002/gepi.20070. [DOI] [PubMed] [Google Scholar]