

Abstract
Inflammatory regulators, including endogenous anti-inflammatory systems, can down-regulate inflammation thus providing negative feedback. Chronic inflammation can result from imbalance between levels of inflammatory mediators and regulators during immune responses. As a consequence, there are heightened inflammatory responses and irreversible tissue damage associated with many age-related chronic diseases. Alzheimer's disease (AD) brain is marked by prominent inflammatory features, in which microglial activation is the driving force for the elaboration of an inflammatory cascade. How the regulation of inflammation loses its effectiveness during AD pathogenesis remains largely unclear. In this article, we will first review current knowledge of microglial activation and its association with AD pathology. We then discuss four examples of anti-inflammatory systems that could play a role in regulating microglial activation: CD200/CD200 receptor, vitamin D receptor, peroxisome proliferator-activated receptors, and soluble receptor for advanced glycation end products. Through this, we hope to illustrate the diverse aspects of inflammatory regulatory systems in brain and neurodegenerative diseases such as AD. We also propose the importance of neuronal defense systems, because they are part of the integral inflammatory and anti-inflammatory systems. Augmenting the anti-inflammatory defenses of neurons can be included in the strategy for restoration of balanced immune responses during aging and neurodegenerative diseases.
Keywords: Aging, Alzheimer's disease, Anti-inflammation, CD200, CD200 receptor, Microglial activation, Peroxisome proliferator-activated receptor γ, Soluble receptor for advanced glycation end products, Vitamin D receptor, Vitamin D3
Introduction
Since the discovery of neuroinflammation as a consistent feature of pathology in Alzheimer's disease (AD) brains, many biochemical factors have been identified in AD brains that can exacerbate these activated microglia-mediated responses. These factors have been detected at elevated levels in AD brains and include activated complement proteins, cytokines, chemokines, free radicals, proteases, and arachidonic acid and its metabolites (reviews in [1–4]). In experimental systems using cultured microglia from human or animal sources, amyloid β peptide (Aβ), the major constituent of amyloid plaques, induced microglia to produce increased amounts of many of these factors; these data support the hypothesis that Aβ possesses significant activating effects on microglia [5–12].
Recent research has shown that chronic inflammation can result from an imbalance between the levels of inflammatory mediators and the levels of inflammatory regulators during immune responses. Inflammatory regulators include endogenous anti-inflammatory systems that can down-regulate inflammation, thus providing negative feedback. Under normal conditions, such systems regulate inflammatory responses so as to prevent uncontrolled inflammatory damage [13]. However, these can become deficient with aging and further defective under sustained inflammatory stimulation [14–16]. Restoration or enhancement of these systems may have potential to be therapeutic targets for disease modification.
Although a wide array of molecules has been identified to have inflammation regulatory functions, identification of molecules/systems contributing to such a deficiency in AD is still the subject of ongoing research. In this article, we will review current evidence for microglial activation in AD and provide four examples of anti-inflammatory systems that could be regulating this microglial activation, in order to illustrate the diverse aspects of inflammatory regulatory systems in brain and neurodegenerative diseases.
Microglial Activation
Microglia, as resident immune effectors cells of the central nervous system (CNS), have the principal function of managing brain homeostasis (housekeeping). They provide cellular surveillance using highly mobile filopodia-like processes as sensors and are usually described as “ramified” or “quiescent” microglia. The processes of quiescent microglia can contact adjacent neurons at a frequency of once every hour; this was shown by in vivo two-photon imaging of fluorescent-labeled neurons and microglia [17].
Microglia are the first responders against infectious, inflammatory, and pathophysiological stimuli. They react to these conditions by changing motility, shape of processes and cell bodies, phagocytic functions, releases of cytokines, chemokines, reactive oxygen species and prostaglandin metabolites, and expression of innate and adaptive immune-function molecules [13, 18–20]. These molecules include toll-like receptors (TLR), major histocompatability complex (MHC) II molecules, immunoglobulin Fc gamma receptors (FcγR), and complement receptors [21–24]. In response to stimuli, microglia can transform from a quiescent state to different activation states.
The magnitude of microglial activation depends on extrinsic and intrinsic conditions: for example, the type of insult, potency of the stimulus, distance from the stimulus, immediate microenvironment, and the “primed” (sensitized) state of microglia that have been exposed to prior and existing stimuli. Normally, primed microglia revert to a surveillance state when homeostasis is restored following removal of activation stimulant and cellular debris. However, sustained “priming” can occur when low levels of stimulation persist in the microenvironment; these can include factors associated with aging or chronic processes of disease. In fact, age-related priming of immune cell types, including CNS microglia, plays a role in the development of age-related inflammatory diseases [15, 25–28]. It was shown that there was a progressive increase in expression of MHCII by microglia with increasing age in rodents and primates [29]. This was associated with increased levels of certain pro-inflammatory cytokines [30, 31].
Priming can sensitize microglia to heightened activation responses when exposed to additional stimuli. In AD, this could come from progressively increasing amounts of soluble forms of Aβ or other associated neurodegenerative changes. This was demonstrated from a study of a group of subjects called high pathology controls, who had not met clinical and neuropathological criteria of AD. Their brains contained numerous amyloid plaques, but were low in neuritic pathology and activated microglia compared to AD brains. Studies of postmortem brain tissues from AD and high plaque non-demented control patients revealed that the severity of inflammation, indicated by numbers of MHCII positive microglia, inversely correlated with the concentration of synaptophysin (a synaptic vesicle protein) and positively correlated with neocortical concentrations of soluble (oligomeric) Aβ [32, 33]. Loss of synaptophysin is considered an index of synaptic and neuronal loss. Microglia pre-primed by age-related factors could be further activated (in high pathology controls) by increasing levels of oligomeric Aβ and synaptic loss, which eventually would become full-blown activation of microglia in AD brains.
Microglial Phagocytosis
Microglia are the major player in brain innate and adaptive immunity, and phagocytosis is a central housekeeping function. Microglia can phagocytose apoptotic cells in the absence of inflammation; this process is regulated by secretion of anti-inflammatory cytokines such as transforming growth factor-β [34, 35]. An array of phagocytic receptors for apoptotic cells have been identified that include phosphatidylserine receptors, scavenger receptor A and B, CD14, CD36, and triggering receptors expressed by myeloid cells [35, 36]. As a different process from phagocytosis of apoptotic cells, microglia can become activated to a neurotoxic phenotype when activated by adenosine triphosphate (ATP) released from injured/necrotic neurons. ATP can activate microglia through binding to purinergic receptors [37, 38]. Microglia phagocytose injured neurites and myelin debris in a slow process [39]. Activation of microglia increases the ability to phagocytose axonal and myelin debris [40, 41]. In the absence of priming and activation, slow removal of neuritic debris by microglia can impede the process of neuroregeneration.
How microglia interact with and phagocytose Aβ depends largely on the physical and biochemical properties of the Aβ. For example, soluble Aβ is internalized through macropinocytosis [42], while fibrillar Aβ interacts with the TLR receptors 2 and 4, and their co-receptor CD14; this results in activation of p38 MAP [43]. Aβ opsonized by antibodies can be phagocytosed by both FcγR-dependent and independent mechanisms [31, 44, 45]. This process is enhanced when complement proteins are also bound to antibodies or Aβ which can interact with different microglial complement receptors, including complement C1q receptor [46, 47].
Pathological conditions may cause sustained activation of phagocytic pathways that could overburden microglia, eventually resulting in an impairment of phagocytic function. It has been suggested that this may cause a deficiency in the clearance of amyloid by microglia or macrophages in AD brains [48, 49]. Autoantibodies against Aβ which can opsonize Aβ are present in serum at lower levels in AD patients than in controls [50]. Most amyloid plaques in AD brains are decorated with immunoglobulins (IgG). Moreover, AD patients with prominent IgG-labeled amyloid plaques are accompanied by increased CD68+ phagocytic microglia and reduced amyloid burden. This suggests that the binding of IgG to amyloid plaques enhances microglial ability to restrict amyloid load [51].
The ability of microglia to remove amyloid can be boosted by passive infusion of antibodies to the peptide or by active immunization with Aβ peptide so that the host produces specific antibodies. This has been shown in both amyloid precursor protein (APP) transgenic mouse model, and in human subjects [52–56]. However, this approach has resulted in significant autoimmune side effects; harnessing an already defective immune system in AD patients continues to be a key safety issue in using this strategy [57].
Microglial Activation in Relation to AD Pathology
Development of radioligands, [11C] (R)PK11195, [3H] (R) PK11195, and [(18)F]fluoroethyl-DAA1106, for peripheral benzodiazepine receptor(s) has provided a method for imaging activated microglia in AD animal models and human subjects with neurological disorders [56, 58–62]. Correlative analysis with the corresponding postmortem brain regions demonstrated that increased ligand retention was due to increased numbers of activated microglia [63]. This was observed in 50% of subjects with mild cognitive impairment (MCI) [60]. Microglia, detected with [3H] (R) PK11195, did not correlate with amyloid load, detected with the amyloid imaging Pittsburgh compound [11C] PIB, in AD patients. However, amyloid deposits are potent chemoattractants to the microglia; this could be illustrated using longitudinal in vivo multiphoton microscopy in the amyloid plaque-developing transgenic mouse strain APPswe/PS1d9xYFP. Activated microglia were shown to migrate to newly formed amyloid plaques within 1–2 days [64].
Morphology of Activated Microglia in AD Brains
Using an antibody to the monocytic cell-specific ionic binding adapter protein 1(Iba1), it has been reported recently that microglia in AD brains displayed degenerating features instead of the widely reported activation morphology [65, 66]. This disagreed with previously established concepts. The antibody against Iba1 is considered a pan-microglia marker and identifies both quiescent and activated microglia; its function is related to phagocytosis and migration [67, 68]. Using double immunohistochemistry with Iba1 and MHCII antibodies, we found in AD hippocampus that most MHCII immunoreactive microglia were Iba1 immunoreactive, indicating that the majority of microglia were activated. However, MHCII detected a smaller population of microglia when compared to Iba1. Therefore, MHCII is more specific than Iba1 for identifying activated microglia.
Here, we summarize three observations about microglial activation detected with MHCII immunohistochemistry. First, MHCII-immunoreactive microglia were more dense in gray matter areas enriched with amyloid pathology (Fig. 1a). Highly activated microglia were frequently associated with compact amyloid plaques. Second, there was a progressive increase in intensity of MHCII staining as it relates to the morphological features of microglia activation (Fig. 1b). Weak MHCII immunoreactivity was associated with small, round microglia with thin ramified processes, whereas intense immunoreactivity was associated with activated, hypertrophic microglia with thickened processes. Third, neurofibrillary tangle-bearing neurons were occasionally associated with MHCII positive microglia (Fig. 2a, c). The vast majority of the microglia showed intact and distinct nuclei and nucleolus when counterstained with neutral red dye, suggestive of an absence of degeneration. MHCII immunoreactive microglia were also found to associate with aberrant neurites detected with antibody to phosphorylated tau (Fig. 2b). Intensely immunoreactive microglia with ameboid shapes were associated with aberrant neuritic clusters (Fig. 2c).
Fig. 1.
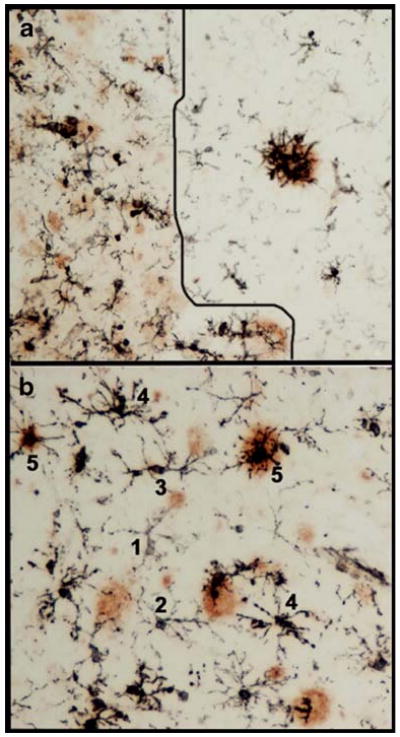
Microglial activation in amyloid plaque-enriched areas in Alzheimer's disease brains. Double immunohistochemistry was performed with an antibody against the microglial activation marker MHCII (LN3, MB Biomedical) and an anti-amyloid β peptide antibody (6E10, Covance). The black MHCII immunoreactive profiles are activated microglia, whereas amyloid deposits are shown in brown. a An artificial line was drawn to show less amyloid deposit area to the right and more amyloid deposits to the left; a clear difference between these two sides, demonstrating that the magnitude of microglial activation is greater in the area with more amyloid. b Microglia were assigned numbers 1 to 5 to represent graded intensity of MHCII immunoreactivites. Lower numbers denote lower level of MHCII expression and less activating morphology; whereas number 5 denotes higher level of MHCII expression and highly activated morphology
Fig. 2.
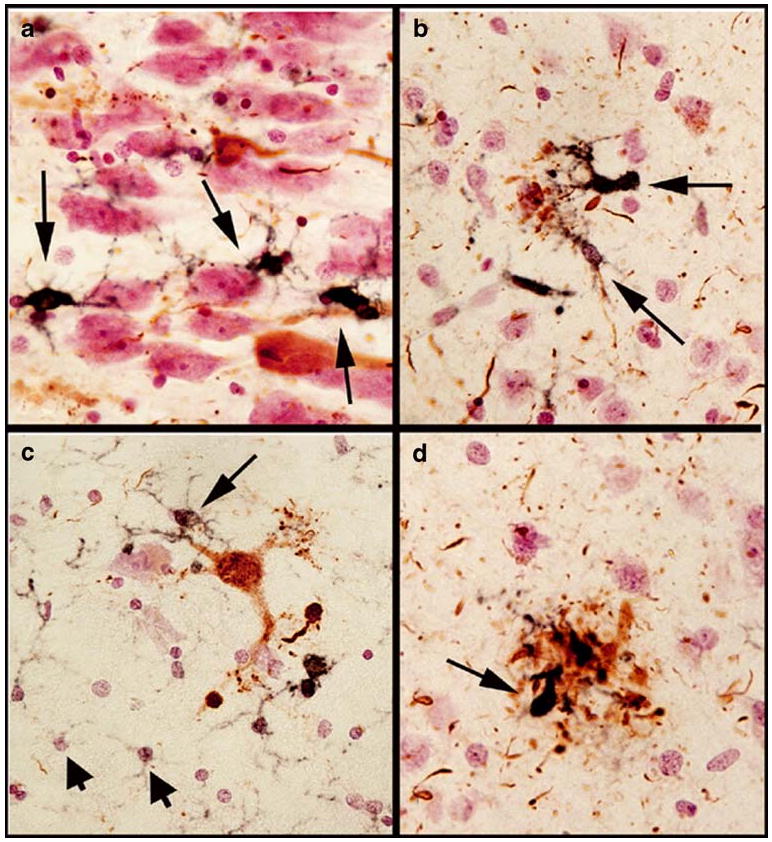
Microglial activation with tau pathology in Alzheimer's disease. Double immunohistochemistry was performed with an antibody against the microglial activation marker MHCII (LN3) and an anti-phospho tau antibody (Pierce). The black MHCII immunoreactive profiles are activated microglia, whereas phospho-tau containing neurons and neurites are shown in brown. Tissues were counterstained with neutral red to show the nuclei (in pink). Long arrows in a and c indicated MHCII immunoreactive microglia in association with neurons which are negative for tau-immunoreactivity (in a) or positive for tau-pathology (b). Short arrows in c indicate non-activated microglia expressing low amount of MHCII. MHCII immunoreactive microglia are associated with aberrant phospho-tau immunoreactive neurites (b) and neuritic clusters (d)
In the hippocampus, the most activated microglia were in corpus ammonus areas enriched with aberrant neurites, neurofibrillary tangles, and amyloid deposits. Only a few MHCII-immunoreactive microglia were observed in these regions in ND cases. Consistent with a low degree of microglial activation, these areas contained relatively few aberrant neuritic threads. The abundance of MHCII-immunoreactive microglia, aberrant neuritic threads, and neuritic plaques in the MCI cases was intermediate between the AD and ND cases. Thus, we have confirmed the association between the magnitude of microglial activation and the severity of AD pathological changes. Although microglia with degenerative morphology could be found within clusters of microglia localized at compact amyloid deposits, the predominant morphology was that of activated microglia.
Inflammatory Regulatory Systems—Do They Function in Human Brains?
Although substantial understanding of microglial activation and neuroinflammatory responses in AD has been achieved during the last two decades, knowledge of anti-inflammatory controls over the process of neuroinflammation is still emerging [69, 70]. In the following sections, we will review four examples of anti-inflammatory systems that have potential significance for understanding neuroinflammatory regulation in AD.
CD200 Receptor and CD200 System
One system that has become a subject of interest is CD200 receptor (CD200R) and CD200. The uniqueness of these molecules is that their only identified function to date is to interact with each other for the activation of anti-inflammatory signaling in CD200R-expressing mononuclear inflammatory cells. CD200, a highly glycosylated protein, is a member of the immunoglobulin superfamily of cell surface proteins [71]. Its expression can be prominently localized to neurons and oligodendrocytes in human brain, though astrocytes and brain endothelial cells have also been shown to express CD200 [72, 73]. It was demonstrated in rodents that there was a loss of CD200 messenger ribonucleic acid (mRNA) expression with increasing age [15]. In AD pathological brain regions, we have shown significantly lower CD200 expression when compared to these same brain regions in age-matched controls [73].
CD200R, also a member of the immunoglobulin superfamily, has cell-type and species-specific molecular weights ranging from 60 to 90 kDa [73–75]. Inflammatory cells including macrophages, neutrophils, microglia, and granulocytes, T lymphocytes, and non-immune associated cells in mice including astrocytes, oligodendrocyte, and epidermal keratinocytes and Langerhans cells, have been reported to express CD200R [76]. Our recent data indicate that human brain microglia express significantly lower levels of CD200R than blood-derived macrophages [73].
Function of CD200R Activation
CD200 has no attached signaling molecules and its sole apparent function is as a ligand for CD200R. Binding of CD200 to CD200R at the N-terminals of each of these molecules activates certain anti-inflammatory signaling pathways in CD200R expressing cells that down-regulate pro-inflammatory responses [77]. The activation of the extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and p38 mitogen-activated protein kinase (MAPK) pathways was inhibited by CD200R engagement with CD200 [78]. Mice deficient in expression of CD200 showed enhanced spontaneous inflammation, along with exacerbated inflammatory responses to injurious stimuli such as experimental arthritis, and experimental autoimmune encephalomyelitis (EAE) [79]. Mice lacking CD200R1 expression showed enhanced tumor necrosis factor-alpha (TNF-α) production in response to peripheral lipopolysaccharide (LPS) and a lack of ability of CD200 to suppress this inflammatory response [80].
Recent studies have demonstrated that CD200 and CD200R expression are both activated following stimulation with interleukin-4 (IL-4), along with interleukin-13 for CD200R [73, 78]. These anti-inflammatory cytokines bind to the same receptor complex and can activate the STAT-6 transcription factor [81]. Activation of STAT-6 occurs in IL-4-stimulated human brain microglia, and this correlates with increased expression of CD200R mRNA.
From these findings, there are two challenges for the CD200/CD200R anti-inflammatory system in aging human brains (both AD and non-demented controls): firstly, CD200 levels are reduced, and secondly, activated microglia express CD200R at levels that appear insufficient to effectively engage CD200 for anti-inflammatory signaling. However, our findings, and those of others, suggest both of these might be enhanced by increasing brain IL-4 levels [73, 82]. From this, we can suggest that the CD200R/CD200 might become therapeutic and functional if treatments can stimulate levels of IL-4 in brain without enhancing unwanted Th2 immune responses. Both statins and vitamin D(3) have been shown to enhance production of IL-4 with resulting anti-inflammatory effects [73, 83].
Vitamin D Receptor
Vitamin D3 has many roles in maintaining homeostasis throughout the body. It is essential for normal bone development and maintenance. It is involved in cellular neuronal signaling in the brain and has a central role in immunity [84]. Clinical data suggest that vitamin D3 insufficiency is associated with an increased risk of several CNS diseases, including multiple sclerosis, AD, Parkinson's disease (PD), seasonal affective disorder, and schizophrenia, though the results have not been consistent [reviewed in [85]]. AD patients have higher rates of bone catabolism with lower bone mass density and have been shown to be vitamin D deficient due to both nutritional causes and lack of exposure to sunlight [86]. Similar findings of reduced bone mass density and increased osteoporosis associated with vitamin D deficiency has been measured in PD [87]. Higher serum levels of 25-hydroxyvitamin D3 (the biologically active form of vitamin D3) correlated with higher mini-mental state exam scores in a cohort of patients with probable AD [88].
The cellular receptor for Vitamin D3, Vitamin D receptor (VDR), also known as nuclear receptor subfamily 1, group I, member 1 (NR1I1) and calcitriol receptor, is a member of the nuclear receptor family of transcription factors. Upon activation by vitamin D, VDR forms a heterodimer with the retinoid-X-receptors (RXRs), which binds to hormone responsive elements on deoxyribonucleic acid (DNA) resulting in increased expression or repression of specific genes. Our interest in VDR came from gene expression profiling experiments which showed that VDR mRNA expression was strongly upregulated in human brain microglia stimulated with aggregated Aβ 42 (2 μM) [12]. This finding was confirmed using real time polymerase chain reaction measurements of Aβ-stimulated microglia. Upregulation of VDR mRNA expression was stimulated by Aβ in a dose-dependent manner (Fig. 3a). These data, if replicated in vivo, suggest that activation of VDR, which should be increased on microglia around plaques, by use of vitamin D3 could be a therapeutic anti-inflammatory target.
Fig. 3.

Detection of vitamin D receptor (VDR) mRNA by quantitative polymerase chain reaction. a Microglia isolated from human postmortem brains expressed VDR mRNA and the level was increased significantly by treating with 2 and 5 μM of aggregated Aβ1-42. b The levels of VDR mRNA were detected in mRNA samples of parietal brain tissues of neuropathologically confirmed autopsy cases of 9 ND, 10 MCI, and 13 AD
VDR is widely expressed in the brain and immune system. We have detected a significant upregulation of VDR mRNA expression in AD neocortex when comparing to MCI and ND neocortex (Fig. 3b). Vitamin D can upregulate expression of several neurotrophins; increase secretion of the anti-inflammatory cytokine IL-4; reduce secretion of pro-inflammatory cytokines TNF-α and interleukin-1 beta (IL-1β); and inhibit differentiation of dendritic cells [89–91]. In the brain, VDR expression has been localized to oligodendrocytes and neurons in rodents; rat oligodendrocytes respond to vitamin D3 by increased expression of VDR and the p75 neurotrophin receptor, while chronic exposure to Vitamin D3 protected rat cortical neurons from glutamate neurotoxicity. Similarly, vitamin D3 was effective in protecting rodent dopaminergic neurons, in vivo and in vitro, from the toxic effects of 6-hydroxydopamine. In human brain tissues, VDR expression has been demonstrated in neurons and glia [92]. We have identified VDR expression in microglia and neurons (Fig. 4a, b).
Fig. 4.
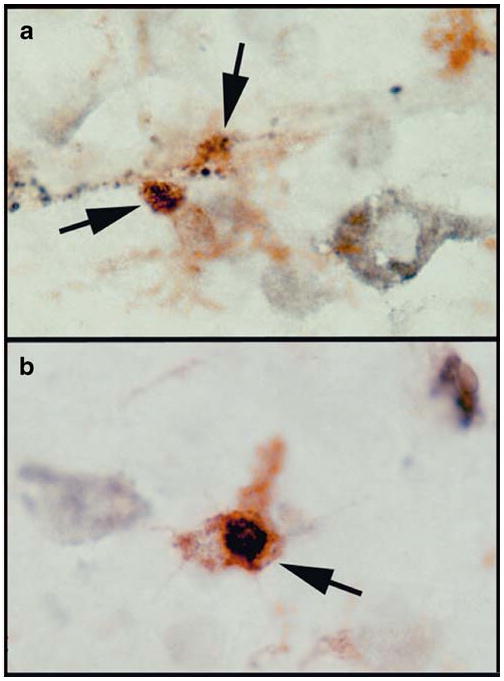
The VDR protein expression in human brains. VDR was detected in microglia in neocortical tissues using double immunohistochemistry of a VDR antibody (Epitomics) and the MHCII antibody (LN3). VDR immunoreactivity is in black and the MHCII immunoreactivity is in brown (indicated by arrows). VDR immunoreactivities exhibit in granular profiles (a), and intracellular profile resembling nuclear localization (b)
Higher occurrence of polymorphisms of the VDR gene has been shown in AD [93]. The Aa VDR genotype was associated with 2.3-fold increased risk of AD, while the AT VDR genotype was significantly higher in controls suggesting a protective effect.
Expression of VDR was found in different immune-effector cells of the myeloid and lymphoid lineage under resting and activating conditions. An anti-inflammatory role for vitamin D(3) has been shown; 1,25-dihydroxyvitamin D3 (1,25-(OH)2D3), the hormonally active vitamin D metabolite, was effective in blocking the progression of experimental allergic encephalomyelitis (EAE), an animal model for multiple sclerosis, through its immunosuppressive effects [94]. Studies have shown that 1, 25-dihydroxyvitamin D3 (1, 25-(OH)2D3) can induce expression of the anti-inflammatory cytokines IL-4 and transforming growth factor β-1 [95]. Treatment of EAE affected mice with (1,25-(OH)2D3) resulted in significant reduction in the accumulation of macrophages in affected spinal cord, possibly due to their increased apoptosis [94]. With respect to neuroinflammation and AD, there is limited evidence for a protective role for VDR or vitamin D3. However, clinical studies have indicated that AD patients are deficient in vitamin D3 and have lower bone mass density [96]. A deficiency of vitamin D3 necessary for healthy bone function might also indicate a deficiency in the other homeostatic functions of vitamin D3, including immunity.
Peroxisome Proliferator-Activated Receptor γ
Peroxisome proliferator-activated receptors (PPARs) belong to the superfamily of nuclear hormone receptors, which comprise steroid, thyroid, retinoid receptors, and PPAR. The main function of PPARs is to regulate glucose and lipid metabolism and their subsequent storage [97, 98]. In association with RXRs, the PPARs/RXRs heterodimers regulate gene transcription by binding to peroxisome-proliferator response elements (PPREs). Ligands binding to either PPAR or RXR activate PPAR, and their effects can be additive. In the absence of ligand, these heterodimers binds to co-repressor complexes blocking gene transcription [97].
There are three different PPAR isotypes (α, β/δ, and γ), and they exhibit distinct tissue distributions and ligand specificities [97, 98]. In the adult CNS, PPARβ/δ are ubiquitously expressed; whereas PPARα is only sparsely expressed in astrocytes. PPARγ is the dominant isoform in microglia [99].
Recently, PPARγ has been recognized as a prime target to modulate inflammation. PPARγ are capable of inhibiting pro-inflammatory gene expression independently of binding to PPREs, a process termed PPARγ-mediated transcriptional transrepression [98]. Although the underlying mechanism remains to be clarified, it is believed that PPARγ agonist complexes stabilize the co-repressor complexes residing on the promoters of NFκB, thus preventing inflammatory gene expression [100, 101]. Polyunsaturated fatty acids, eicosanoids, and the prostaglandin metabolite 15-deoxy-delta-12,14-prostaglandin J2 (15d-PGJ2) are naturally occurring PPARγ ligands [102]; whereas synthetic thiazolidinediones are selective PPARγ agonists [103].
Suppression of immune responses by PPARγ agonists was initially demonstrated in monocytes/macrophages [104, 105]. In these two studies, PPARγ agonists were found to inhibit the activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB), activator protein-1 (AP-1), and signal transducer and activator of transcription-1 (STAT-1), and the expression of TNF-α, IL-1β, and interleukin-6 by activated monocyte/macrophage cells. Along the same line, the effect of PPARγ agonists on microglia activation was also studied. In the BV-2 mouse microglial cell line, 15d-PGJ2 suppressed LPS-induced inducible nitric oxide synthase (iNOS) and subsequent nitric oxide production, yet had no effect on the nuclear translocation or DNA binding of NFκB [102]. Furthermore, LPS-induced nitric oxide production was not affected by a synthetic PPARγ agonist troglitazone, suggesting that 15d-PGJ2 may inhibit iNOS gene expression through a receptor-independent mechanism [102]. However, in a later study using rat primary microglial cultures, the effects of 15d-PGJ2 on LPS-induced NO and TNF-α were found to be PPARγ-dependent [106]. Such discrepancy may be derived from the different cell types used in these two studies; the BV-2 microglial cell line does not express PPARγ, while primary microglia do.
The observations that PPARγ are able to suppress inflammatory responses in macrophages and microglia led to the idea that PPARγ might be beneficial for CNS disorders possessing an inflammatory component, such as AD and PD. The actions of PPARγ in AD have been investigated epidemiologically, empirically, and clinically. A number of epidemiological studies have demonstrated that non-steroidal anti-inflammatory drugs (NSAIDs) known to activate PPARγ and inhibit inflammatory response were associated with a reduced risk of cognitive impairment and AD [107–109]. Such observations are supported by the finding that PPARγ activation in microglia suppressed iNOS expression and NO production, which was associated with reduced neuronal cell death. Furthermore, PPARγ agonists have been shown to suppress Aβ-mediated activation of microglia in vitro, and prevent cortical or hippocampal neuron cell death [110, 111]. However, APP transgenic (Tg2576) mice that received pioglitazone orally for six months did not have altered amyloid plaque loads or inflammatory markers in the cortex [112]. The poor effect of pioglitazone may be explained by the limited blood brain barrier (BBB) penetration of this drug. In a similar study, APP transgenic (APPV717I) mice that received a larger dose of pioglitazone exhibited significant reduction in amyloid plaque load, and reduced microglial and astroglial activation [113]. The phenomenon of PPARγ agonists reducing amyloid plaque burden has been attributed to reduced β-secretase 1 (BACE1) transcription and/or enhanced Aβ clearance [113–115]. In clinical trials, PPARγ agonist rosiglitazone was found to improve cognition in a subset of AD patients without apolipoprotein E allele 4 (APOE ε4) [116, 117]. Because rosiglitazone does not pass the BBB and hyperinsulinemia, insulin resistance and type II diabetes have been associated with increased risk for AD and memory impairment; the beneficial effect of rosiglitazone was interpreted as being due to enhancement of peripheral insulin sensitivity [118–120]. Whether rosiglitazone has significant benefit in APOE ε4-stratified subjects with mild to moderate AD still needs to be confirmed [117, 121].
Rosiglitazone was also shown to reverse the age-related decrease of IL-4 in hippocampus during aging. After LPS stimulation, rosiglitazone attenuated the expression of MHCII and IL-1β in glia prepared from wild-type mice, but not in glia prepared from IL-4-/- mice. This suggested that the anti-inflammatory actions of rosiglitazone may be mediated by modulating IL-4 expression [122].
Soluble Receptor for Advanced Glycation End Products
The discovery of soluble receptor for advanced glycation end products (sRAGE) endogenously present under physiological conditions has provided insight into the protective function of soluble RAGE molecules. These molecules can be generated by alternative splicing (called endogenous secretory RAGE, esRAGE) or by enzymatic cleavage of RAGE (called ecRAGE) [123–125]. The carboxyl terminal sequences are different between ecRAGE and esRAGE. These soluble forms of RAGE can act as decoy receptors and compete for the ligands of full-length RAGE (fl-RAGE). Ligand binding with esRAGE and ecRAGE avoids the adverse consequences of activating signal transduction pathways that occurs when cell-associated RAGE is activated. Many studies have documented that RAGE activation results in increased inflammation and cellular perturbation. Interaction of cell-surface fl-RAGE with activates transcription factor NFκB, resulting in increased signaling of neuronal oxidative stress and microglial inflammatory responses [126]. Evidence of this was established from studies in human postmortem brain tissues, primary cell cultures, and transgenic mouse models that overexpressed human RAGE in neurons and microglia [127].
The function of fl-RAGE in human cerebral vessels has emerged as an important subject for understanding how Aβ is transported in and out of the brain [128, 129]. Fl-RAGE can mediate influx of circulating Aβ across the BBB, whereas lipoprotein receptor-related protein-1, which is expressed on the abluminal surface of cerebral endothelial cells, transports Aβ from the brain to the blood. RAGE protein expression in blood vessels increases with severity of AD pathology, which could lead to enhanced passage of Aβ into the brain from the circulation. Using the Tg 2576 strain of amyloid plaque-developing transgenic mice, it was shown that infusion of Aβ into the blood resulted in its RAGE-mediated transport into the brain. Uptake of Aβ was blocked using an antibody to RAGE infused into the mice along with the Aβ; the antibody bound to cerebral vessels expressing RAGE, thus preventing endothelial-RAGE-Aβ interactions [128]. The same blocking effect was observed if mice were infused with recombinant soluble RAGE protein along with the Aβ. This was the first indication that soluble forms of RAGE protein might have therapeutic use in AD. This same study showed that administration of recombinant soluble RAGE to plaque developing mice for 6 months significantly reduced their accumulation of Aβ in the brain [128]. Aβ bound to recombinant soluble RAGE could be detected in the circulation of these treated mice. This study was of particular interest as it established that Aβ from the circulation can significantly contribute to Aβ accumulation in the brain.
Reduced sRAGE levels in plasma have been detected in mild cognitive impaired (MCI) and AD subjects [130]. Other neurodegenerative disease such as multiple sclerosis, amytrophic lateral sclerosis, and peripheral inflammatory vascular or metabolic diseases (rheumatoid arthritis, atherosclerosis, coronary artery disease, diabetes) are also marked by reduced circulating sRAGE [131–137]. These findings suggested that the protective role of sRAGE is vulnerable to age-related disease processes.
Measurements of plasma of healthy adults showed that esRAGE levels represented only one fifth of the total soluble RAGE in the circulation. This suggests that additional mechanisms besides alternative splicing are involved in the generation of the soluble forms of RAGE present in the circulation. Recent studies identified enzymes capable of cleaving fl-RAGE, resulting its release from the plasma membrane. These enzymes are matrix metalloproteinase 9, and/or “a disintegrin and metalloprotease 10” [124, 125, 138]. It appears that RAGE shedding can occur under constitutive and inducible conditions. Therefore, elevating endogenous sRAGE levels by increasing enzymatic cleavage could have significant therapeutic potential.
RAGE Ligands, the S100/Calgranulin Proteins
RAGE ligands play a key role in activating RAGE-mediated inflammatory responses. Three members of the S100 protein family identified as RAGE ligands are linked to peripheral innate immune responses, including anti-infection, phagocytosis, and inflammatory responses. These are S100A8, S100A9, and S100A12. They are constitutively expressed, but can be translocated to the cytoskeleton, membrane structures and be actively secreted via an alternative pathway when activated by pro-inflammatory signals [139]. Extracellular S100 proteins have autocrine and paracrine effects and exert different effects on different cell types. These S100 proteins can act in concert to achieve enhanced activation at sites of inflammation.
Using immunohistochemical analysis, S100A8, S100A9, S100A12, and S100B were shown associated with vascular structures in brains of control and sporadic AD cases, but in AD cases only S100A9, S100A12, and S100B were closely associated with amyloid plaques and neurofibrillary tangles [140]. S100B was specifically associated with astrocytes, whereas S100A9 and S100A12 were associated with neurons in AD cases [140–142]. We have detected that S100A8 mRNA levels in AD hippocampus were 2-fold those of ND cases. Moreover, S100A8 expression can be induced approximately 30-fold in cultured human microglia by chronic treatment with oligomeric Aβ1-42 [12].
Taken together, evidence supports an important adverse role for fl-RAGE and ligands, including Aβ and S100 A8, A9, and A12, in neuroinflammation and neurodegeneration in AD. However, the mechanisms leading to progressive deficiency of circulating soluble RAGE during the development of AD are currently unknown. As sRAGE is a protective decoy receptor for these inflammatory ligands, avenues that can restore circulating sRAGE to normal levels may facilitate regulation of inflammation from the periphery.
Conclusion
As AD contains features of autoimmune reactions, but is complicated by immune senescence and regulatory deficiency, it has been a challenge to tackle with currently available immunosuppressant or anti-inflammatory drugs. Clinical trials using NSAIDs for treatment or prevention have been hampered by issues of safety, choice of drugs, and criteria of when and whom to treat. Immune systems have both aspects of Yang (inflammation) and Ying (anti-inflammation). The latter keeps inflammatory responses under control (Fig. 5). Extensive studies have been done from the aspect of “Yang”. Better knowledge of the Ying—the anti-inflammatory systems, however, is needed to obtain a full picture of the dysregulation occurring in the immune responses of AD patients. We have reviewed four of anti-inflammatory systems that are deficient in AD. One important point is that neurons are part of the integral inflammatory and anti-inflammatory systems. Neuronal anti-inflammatory roles need to be included in the strategy of restoration of healthy immune response during aging and neurodegenerative diseases.
Fig. 5.
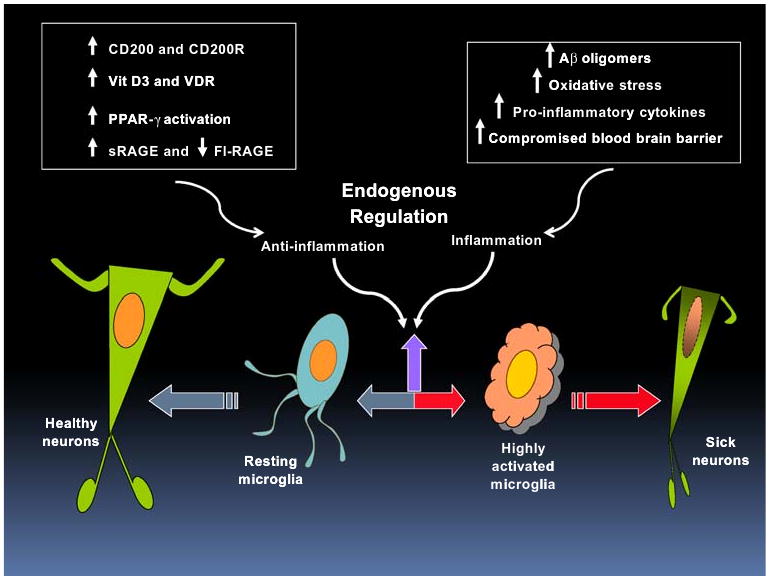
Enhancing endogenous anti-inflammatory regulatory systems in Alzheimer's disease (AD). AD is characterized by heightened microglial activation and inflammatory responses which can be caused by increasing oligomeric amyloid β peptide (Aβ), oxidative stress, pro-inflammatory cytokines, and compromised blood brain barrier. These responses driven by activated microglia have been shown to cause neuronal distress and injury (right-pointing red arrow). Emerging evidence indicated that some of the endogenous anti-inflammatory systems might not be functioning properly. Resting microglia contribute to keeping neurons healthy (left-pointing gray arrow). This could be accomplished by proper expression and function of several anti-inflammatory systems, including CD200/CD200R, vitamin D3 (Vit D3)/Vitamin D receptor (VDR), PPAR-γ, and soluble and full-length receptor for advanced glycation end-products (sRAGE and fl-RAGE). Achieving a fine balance of anti-inflammatory regulation of inflammatory responses (up-pointing purple arrow) might be of potential therapeutic value
Acknowledgments
We thank the following supports: NINDS (RO1 NS049075 to LFL) and Alzheimer's Association (IIRG-09-91996 to LFL; IIRG-06-26125 to DGW). We are grateful to the Banner Sun Health Research Institute Brain Donation Program of Sun City, Arizona for the provision of human biological materials for the study of human cells and postmortem tissues. The Brain Donation Program is supported by the National Institute on Aging (P30 AG19610 Arizona Alzheimer's Disease Core Center), the Arizona Department of Health Services (contract 211002, Arizona Alzheimer's Research Center), the Arizona Biomedical Research Commission (contracts 4001, 0011, 05-901 and 1001 to the Arizona Parkinson's Disease Consortium) and the Prescott Family Initiative of the Michael J. Fox Foundation for Parkinson's Research.
Contributor Information
Lih-Fen Lue, Email: lihfen.lue@bannerhealth.com, Laboratory of Neuroregeneration, Banner Sun Health Research Institute, 10515 West Santa Fe Drive, Sun City, AZ 85351, USA.
Yu-Min Kuo, Department of Cell Biology and Anatomy, National Cheng Kung University Medical College, Tainan, Taiwan.
Thomas Beach, Civin Laboratory of Neuropathology, Banner Sun Health Reesearch Institute, Sun City, AZ, U.S.A..
Douglas G. Walker, Laboratory of Neuroinflammation, Banner Sun Health Research Institute, Sun City, AZ, USA
References
- 1.Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O'Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wilkinson BL, Landreth GE. The microglial NADPH oxidase complex as a source of oxidative stress in Alzheimer's disease. J Neuroinflammation. 2006;3:30–41. doi: 10.1186/1742-2094-3-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walter S, Letiembre M, Liu Y, Heine H, Penke B, Hao W, Bode B, Manietta N, Walter J, Schulz-Schuffer W, Fassbender K. Role of the toll-like receptor 4 in neuroinflammation in Alzheimer's disease. Cell Physiol Biochem. 2007;20:947–956. doi: 10.1159/000110455. [DOI] [PubMed] [Google Scholar]
- 4.Hoozemans JJ, Rozemuller JM, van Haastert ES, Veerhuis R, Eikelenboom P. Cyclooxygenase-1 and -2 in the different stages of Alzheimer's disease pathology. Curr Pharm Des. 2008;14:1419–1427. doi: 10.2174/138161208784480171. [DOI] [PubMed] [Google Scholar]
- 5.Korotzer AR, Watt J, Cribbs D, Tenner AJ, Burdick D, Glabe C, Cotman CW. Cultured rat microglia express C1q and receptor for C1q: implications for amyloid effects on microglia. Exp Neurol. 1995;134:214–221. doi: 10.1006/exnr.1995.1051. [DOI] [PubMed] [Google Scholar]
- 6.O'Barr S, Cooper NR. The C5a complement activation peptide increases IL-1 beta and IL-6 release from amyloid-beta primed human monocytes: implications for Alzheimer's disease. J Neuroimmunol. 2000;109:87–94. doi: 10.1016/s0165-5728(00)00291-5. [DOI] [PubMed] [Google Scholar]
- 7.Lue LF, Rydel R, Brigham EF, Yang LB, Hampel H, Murphy GM, Jr, Brachova L, Yan SD, Walker DG, Shen Y, Rogers J. Inflammatory repertoire of Alzheimer's disease and nondemented elderly microglia in vitro. Glia. 2001;35:72–79. doi: 10.1002/glia.1072. [DOI] [PubMed] [Google Scholar]
- 8.Hoozemans JJ, Veerhuis R, Janssen I, van Elk EJ, Rozemuller AJ, Eikelenboom P. The role of cyclo-oxygenase 1 and 2 activity in prostaglandin E(2) secretion by cultured human adult microglia: implications for Alzheimer's disease. Brain Res. 2002;951:218–226. doi: 10.1016/s0006-8993(02)03164-5. [DOI] [PubMed] [Google Scholar]
- 9.Veerhuis R, Van Breemen MJ, Hoozemans JM, Morbin M, Ouladhadj J, Tagliavini F, Eikelenboom P. Amyloid beta plaque-associated proteins C1q and SAP enhance the Abeta1-42 peptide-induced cytokine secretion by adult human microglia in vitro. Acta Neuropathol. 2003;105:135–144. doi: 10.1007/s00401-002-0624-7. [DOI] [PubMed] [Google Scholar]
- 10.Gan L, Ye S, Chu A, Anton K, Yi S, Vincent VA, von Schack D, Chin D, Murray J, Lohr S, Patthy L, Gonzalez-Zulueta M, Nikolich K, Urfer R. Identification of cathepsin B as a mediator of neuronal death induced by Abeta-activated microglial cells using a functional genomics approach. J Biol Chem. 2004;279:5565–5572. doi: 10.1074/jbc.M306183200. [DOI] [PubMed] [Google Scholar]
- 11.Lindberg C, Selenica ML, Westlind-Danielsson A, Schultzberg M. Beta-amyloid protein structure determines the nature of cytokine release from rat microglia. J Mol Neurosci. 2005;27:1–12. doi: 10.1385/JMN:27:1:001. [DOI] [PubMed] [Google Scholar]
- 12.Walker DG, Link J, Lue LF, sing-Hernandez JE, Boyes BE. Gene expression changes by amyloid beta peptide-stimulated human postmortem brain microglia identify activation of multiple inflammatory processes. J Leukoc Biol. 2006;79:596–610. doi: 10.1189/jlb.0705377. [DOI] [PubMed] [Google Scholar]
- 13.Griffiths M, Neal JW, Gasque P. Innate immunity and protective neuroinflammation: new emphasis on the role of neuroimmune regulatory proteins. Int Rev Neurobiol. 2007;82:29–55. doi: 10.1016/S0074-7742(07)82002-2. [DOI] [PubMed] [Google Scholar]
- 14.Ye SM, Johnson RW. An age-related decline in interleukin-10 may contribute to the increased expression of interleukin-6 in brain of aged mice. Neuroimmunomodulation. 2001;9:183–192. doi: 10.1159/000049025. [DOI] [PubMed] [Google Scholar]
- 15.Frank MG, Barrientos RM, Biedenkapp JC, Rudy JW, Watkins LR, Maier SF. mRNA up-regulation of MHC II and pivotal pro-inflammatory genes in normal brain aging. Neurobiol Aging. 2006;27:717–722. doi: 10.1016/j.neurobiolaging.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 16.Stahel PF, Flierl MA, Morgan BP, Persigehl I, Stoll C, Conrad C, Touban BM, Smith WR, Beauchamp K, Schmidt OI, Ertel W, Leinhase I. Absence of the complement regulatory molecule CD59a leads to exacerbated neuropathology after traumatic brain injury in mice. J Neuroinflammation. 2009;6:2. doi: 10.1186/1742-2094-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wake H, Moorhouse AJ, Jinno S, Kohsaka S, Nabekura J. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J Neurosci. 2009;29:3974–3980. doi: 10.1523/JNEUROSCI.4363-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jack CS, Arbour N, Manusow J, Montgrain V, Blain M, McCrea E, Shapiro A, Antel JP. TLR signaling tailors innate immune responses in human microglia and astrocytes. J Immunol. 2005;175:4320–4330. doi: 10.4049/jimmunol.175.7.4320. [DOI] [PubMed] [Google Scholar]
- 19.Town T, Nikolic V, Tan J. The microglial “activation” continuum: from innate to adaptive responses. J Neuroinflammation. 2005;2:2. doi: 10.1186/1742-2094-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garden GA, Moller T. Microglia biology in health and disease. J Neuroimmune Pharmacol. 2006;1:127–137. doi: 10.1007/s11481-006-9015-5. [DOI] [PubMed] [Google Scholar]
- 21.Woodroofe MN, Hayes GM, Cuzner ML. Fc receptor density, MHC antigen expression and superoxide production are increased in interferon-gamma-treated microglia isolated from adult rat brain. Immunology. 1989;68:421–426. [PMC free article] [PubMed] [Google Scholar]
- 22.Peress NS, Fleit HB, Perillo E, Kuljis R, Pezzullo C. Identification of Fc gamma RI, II and III on normal human brain ramified microglia and on microglia in senile plaques in Alzheimer's disease. J Neuroimmunol. 1993;48:71–79. doi: 10.1016/0165-5728(93)90060-c. [DOI] [PubMed] [Google Scholar]
- 23.Bsibsi M, Ravid R, Gveric D, van Noort JM. Broad expression of toll-like receptors in the human central nervous system. J Neuropathol Exp Neurol. 2002;61:1013–1021. doi: 10.1093/jnen/61.11.1013. [DOI] [PubMed] [Google Scholar]
- 24.Webster SD, Park M, Fonseca MI, Tenner AJ. Structural and functional evidence for microglial expression of C1qR(P), the C1q receptor that enhances phagocytosis. J Leukoc Biol. 2000;67:109–116. doi: 10.1002/jlb.67.1.109. [DOI] [PubMed] [Google Scholar]
- 25.Franceschi C, Bonafe M, Valensin S, Olivieri F, De LM, Ottaviani E, De BG. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann NY Acad Sci. 2000;908:244–254. doi: 10.1111/j.1749-6632.2000.tb06651.x. [DOI] [PubMed] [Google Scholar]
- 26.Blalock EM, Chen KC, Stromberg AJ, Norris CM, Kadish I, Kraner SD, Porter NM, Landfield PW. Harnessing the power of gene microarrays for the study of brain aging and Alzheimer's disease: statistical reliability and functional correlation. Ageing Res Rev. 2005;4:481–512. doi: 10.1016/j.arr.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 27.Maher FO, Nolan Y, Lynch MA. Downregulation of IL-4-induced signalling in hippocampus contributes to deficits in LTP in the aged rat. Neurobiol Aging. 2005;26:717–728. doi: 10.1016/j.neurobiolaging.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 28.Nolan Y, Maher FO, Martin DS, Clarke RM, Brady MT, Bolton AE, Mills KH, Lynch MA. Role of interleukin-4 in regulation of age-related inflammatory changes in the hippocampus. J Biol Chem. 2005;280:9354–9362. doi: 10.1074/jbc.M412170200. [DOI] [PubMed] [Google Scholar]
- 29.Sheffield LG, Berman NE. Microglial expression of MHC class II increases in normal aging of nonhuman primates. Neurobiol Aging. 1998;19:47–55. doi: 10.1016/s0197-4580(97)00168-1. [DOI] [PubMed] [Google Scholar]
- 30.Ye SM, Johnson RW. Increased interleukin-6 expression by microglia from brain of aged mice. J Neuroimmunol. 1999;93:139–148. doi: 10.1016/s0165-5728(98)00217-3. [DOI] [PubMed] [Google Scholar]
- 31.Henry CJ, Huang Y, Wynne AM, Godbout JP. Peripheral lipopolysaccharide (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both pro-inflammatory IL-1beta and anti-inflammatory IL-10 cytokines. Brain Behav Immun. 2009;23:309–317. doi: 10.1016/j.bbi.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lue LF, Brachova L, Civin WH, Rogers J. Inflammation, A beta deposition, and neurofibrillary tangle formation as correlates of Alzheimer's disease neurodegeneration. J Neuropathol Exp Neurol. 1996;55:1083–1088. [PubMed] [Google Scholar]
- 33.Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol. 1999;155:853–862. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.von Zahn J, Moller T, Kettenmann H, Nolte C. Microglial phagocytosis is modulated by pro- and anti-inflammatory cytokines. Neuroreport. 1997;8:3851–3856. doi: 10.1097/00001756-199712220-00003. [DOI] [PubMed] [Google Scholar]
- 35.Stolzing A, Grune T. Neuronal apoptotic bodies: phagocytosis and degradation by primary microglial cells. FASEB J. 2004;18:743–745. doi: 10.1096/fj.03-0374fje. [DOI] [PubMed] [Google Scholar]
- 36.Hirt UA, Leist M. Rapid, noninflammatory and PS-dependent phagocytic clearance of necrotic cells. Cell Death Differ. 2003;10:1156–1164. doi: 10.1038/sj.cdd.4401286. [DOI] [PubMed] [Google Scholar]
- 37.Haynes SE, Hollopeter G, Yang G, Kurpius D, Dailey ME, Gan WB, Julius D. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat Neurosci. 2006;9:1512–1519. doi: 10.1038/nn1805. [DOI] [PubMed] [Google Scholar]
- 38.Wu LJ, Vadakkan KI, Zhuo M. ATP-induced chemotaxis of microglial processes requires P2Y receptor-activated initiation of outward potassium currents. Glia. 2007;55:810–821. doi: 10.1002/glia.20500. [DOI] [PubMed] [Google Scholar]
- 39.Bechmann I, Nitsch R. Astrocytes and microglial cells incorporate degenerating fibers following entorhinal lesion: a light, confocal, and electron microscopical study using a phagocytosis-dependent labeling technique. Glia. 1997;20:145–154. doi: 10.1002/(sici)1098-1136(199706)20:2<145::aid-glia6>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 40.Vallieres N, Berard JL, David S, Lacroix S. Systemic injections of lipopolysaccharide accelerates myelin phagocytosis during Wallerian degeneration in the injured mouse spinal cord. Glia. 2006;53:103–113. doi: 10.1002/glia.20266. [DOI] [PubMed] [Google Scholar]
- 41.Tanaka T, Ueno M, Yamashita T. Engulfment of axon debris by microglia requires p38 MAPK activity. J Biol Chem. 2009;284:21626–21636. doi: 10.1074/jbc.M109.005603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mandrekar S, Jiang Q, Lee CY, Koenigsknecht-Talboo J, Holtzman DM, Landreth GE. Microglia mediate the clearance of soluble Abeta through fluid phase macropinocytosis. J Neurosci. 2009;29:4252–4262. doi: 10.1523/JNEUROSCI.5572-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reed-Geaghan EG, Savage JC, Hise AG, Landreth GE. CD14 and toll-like receptors 2 and 4 are required for fibrillar A {beta}-stimulated microglial activation. J Neurosci. 2009;29:11982–11992. doi: 10.1523/JNEUROSCI.3158-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bacskai BJ, Kajdasz ST, McLellan ME, Games D, Seubert P, Schenk D, Hyman BT. Non-Fc-mediated mechanisms are involved in clearance of amyloid-beta in vivo by immunotherapy. J Neurosci. 2002;22:7873–7878. doi: 10.1523/JNEUROSCI.22-18-07873.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lue LF, Walker DG. Modeling Alzheimer's disease immune therapy mechanisms: interactions of human postmortem microglia with antibody-opsonized amyloid beta peptide. J Neurosci Res. 2002;70:599–610. doi: 10.1002/jnr.10422. [DOI] [PubMed] [Google Scholar]
- 46.Brazil MI, Chung H, Maxfield FR. Effects of incorporation of immunoglobulin G and complement component C1q on uptake and degradation of Alzheimer's disease amyloid fibrils by microglia. J Biol Chem. 2000;275:16941–16947. doi: 10.1074/jbc.M000937200. [DOI] [PubMed] [Google Scholar]
- 47.Webster SD, Galvan MD, Ferran E, Garzon-Rodriguez W, Glabe CG, Tenner AJ. Antibody-mediated phagocytosis of the amyloid beta-peptide in microglia is differentially modulated by C1q. J Immunol. 2001;166:7496–7503. doi: 10.4049/jimmunol.166.12.7496. [DOI] [PubMed] [Google Scholar]
- 48.Fiala M, Lin J, Ringman J, Kermani-Arab V, Tsao G, Patel A, Lossinsky AS, Graves MC, Gustavson A, Sayre J, Sofroni E, Suarez T, Chiappelli F, Bernard G. Ineffective phagocytosis of amyloid-beta by macrophages of Alzheimer's disease patients. J Alzheimer's Dis. 2005;7:221–232. doi: 10.3233/jad-2005-7304. [DOI] [PubMed] [Google Scholar]
- 49.Hickman SE, Allison EK, El KJ. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer's disease mice. J Neurosci. 2008;28:8354–8360. doi: 10.1523/JNEUROSCI.0616-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sohn JH, So JO, Hong HJ, Kim JW, Na DR, Kim M, Kim H, Nam E, Ha HJ, Kim YH, Mook-Jung I. Identification of autoantibody against beta-amyloid peptide in the serum of elderly. Front Biosci. 2009;14(3879–83):3879–3883. doi: 10.2741/3496. [DOI] [PubMed] [Google Scholar]
- 51.Kellner A, Matschke J, Bernreuther C, Moch H, Ferrer I, Glatzel M. Autoantibodies against beta-amyloid are common in Alzheimer's disease and help control plaque burden. Ann Neurol. 2009;65:24–31. doi: 10.1002/ana.21475. [DOI] [PubMed] [Google Scholar]
- 52.Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, Seubert P, Schenk D, Yednock T. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- 53.Monsonego A, Maron R, Zota V, Selkoe DJ, Weiner HL. Immune hyporesponsiveness to amyloid beta-peptide in amyloid precursor protein transgenic mice: implications for the pathogenesis and treatment of Alzheimer's disease. Proc Natl Acad Sci USA. 2001;98:10273–10278. doi: 10.1073/pnas.191118298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wilcock DM, Rojiani A, Rosenthal A, Levkowitz G, Subbarao S, Alamed J, Wilson D, Wilson N, Freeman MJ, Gordon MN, Morgan D. Passive amyloid immunotherapy clears amyloid and transiently activates microglia in a transgenic mouse model of amyloid deposition. J Neurosci. 2004;24:6144–6151. doi: 10.1523/JNEUROSCI.1090-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Morgan D. Modulation of microglial activation state following passive immunization in amyloid depositing transgenic mice. Neurochem Int. 2006;49:190–194. doi: 10.1016/j.neuint.2006.03.017. [DOI] [PubMed] [Google Scholar]
- 56.Nicoll JA, Barton E, Boche D, Neal JW, Ferrer I, Thompson P, Vlachouli C, Wilkinson D, Bayer A, Games D, Seubert P, Schenk D, Holmes C. Abeta species removal after abeta42 immunization. J Neuropathol Exp Neurol. 2006;65:1040–1048. doi: 10.1097/01.jnen.0000240466.10758.ce. [DOI] [PubMed] [Google Scholar]
- 57.Lemere CA. Developing novel immunogens for a safe and effective Alzheimer's disease vaccine. Prog Brain Res. 2009;175(83–93):83–93. doi: 10.1016/S0079-6123(09)17506-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Edison P, Archer HA, Gerhard A, Hinz R, Pavese N, Turkheimer FE, Hammers A, Tai YF, Fox N, Kennedy A, Rossor M, Brooks DJ. Microglia, amyloid, and cognition in Alzheimer's disease: an [11C] (R)PK11195-PET and [11C]PIB-PET study. Neurobiol Dis. 2008;32:412–419. doi: 10.1016/j.nbd.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 59.Higuchi M. Visualization of brain amyloid and microglial activation in mouse models of Alzheimer's disease. Curr Alzheimer Res. 2009;6:137–143. doi: 10.2174/156720509787602906. [DOI] [PubMed] [Google Scholar]
- 60.Okello A, Edison P, Archer HA, Turkheimer FE, Kennedy J, Bullock R, Walker Z, Kennedy A, Fox N, Rossor M, Brooks DJ. Microglial activation and amyloid deposition in mild cognitive impairment: a PET study. Neurology. 2009;72:56–62. doi: 10.1212/01.wnl.0000338622.27876.0d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Venneti S, Lopresti BJ, Wang G, Slagel SL, Mason NS, Mathis CA, Fischer ML, Larsen NJ, Mortimer AD, Hastings TG, Smith AD, Zigmond MJ, Suhara T, Higuchi M, Wiley CA. A comparison of the high-affinity peripheral benzodiazepine receptor ligands DAA1106 and (R)-PK11195 in rat models of neuroinflammation: implications for PET imaging of microglial activation. J Neurochem. 2007;102:2118–2131. doi: 10.1111/j.1471-4159.2007.04690.x. [DOI] [PubMed] [Google Scholar]
- 62.Venneti S, Lopresti BJ, Wang G, Hamilton RL, Mathis CA, Klunk WE, Apte UM, Wiley CA. PK11195 labels activated microglia in Alzheimer's disease and in vivo in a mouse model using PET. Neurobiol Aging. 2009;30:1217–1226. doi: 10.1016/j.neurobiolaging.2007.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Venneti S, Wang G, Nguyen J, Wiley CA. The positron emission tomography ligand DAA1106 binds with high affinity to activated microglia in human neurological disorders. J Neuropathol Exp Neurol. 2008;67:1001–1010. doi: 10.1097/NEN.0b013e318188b204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Meyer-Luehmann M, Spires-Jones TL, Prada C, Garcia-Alloza M, de Calignon A, Rozkalne A, Koenigsknecht-Talboo J, Holtzman DM, Bacskai BJ, Hyman BT. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer's disease. Nature. 2008;451:720–724. doi: 10.1038/nature06616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Streit WJ. Microglia and neuroprotection: implications for Alzheimer's disease. Brain Res Brain Res Rev. 2005;48:234–9. doi: 10.1016/j.brainresrev.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 66.Streit WJ, Braak H, Xue QS, Bechmann I. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer's disease. Acta Neuropathol. 2009;118:475–485. doi: 10.1007/s00401-009-0556-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ito D, Tanaka K, Suzuki S, Dembo T, Fukuuchi Y. Enhanced expression of Iba1, ionized calcium-binding adapter molecule 1, after transient focal cerebral ischemia in rat brain. Stroke. 2001;32:1208–1215. doi: 10.1161/01.str.32.5.1208. [DOI] [PubMed] [Google Scholar]
- 68.Kanazawa H, Ohsawa K, Sasaki Y, Kohsaka S, Imai Y. Macrophage/microglia-specific protein Iba1 enhances membrane ruffling and Rac activation via phospholipase C-gamma-dependent pathway. J Biol Chem. 2002;277:20026–20032. doi: 10.1074/jbc.M109218200. [DOI] [PubMed] [Google Scholar]
- 69.Walker D, Lue LF. Anti-inflammatory and immune therapy for Alzheimer's disease: current status and future directions. Curr Neuropharmacol. 2007;5:232–243. doi: 10.2174/157015907782793667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McGeer EG, McGeer PL. Neuroinflammation in Alzheimer's disease and mild cognitive impairment: a field in its infancy. J Alzheimers, Dis. 2009 doi: 10.3233/JAD-2010-1219. [DOI] [PubMed] [Google Scholar]
- 71.McCaughan GW, Clark MJ, Hurst J, Grosveld F, Barclay AN. The gene for MRC OX-2 membrane glycoprotein is localized on human chromosome 3. Immunogenetics. 1987;25:133–135. doi: 10.1007/BF00364281. [DOI] [PubMed] [Google Scholar]
- 72.Koning N, Bo L, Hoek RM, Huitinga I. Downregulation of macrophage inhibitory molecules in multiple sclerosis lesions. Ann Neurol. 2007;62:504–514. doi: 10.1002/ana.21220. [DOI] [PubMed] [Google Scholar]
- 73.Walker DG, sing-Hernandez JE, Campbell NA, Lue LF. Decreased expression of CD200 and CD200 receptor in Alzheimer's disease: a potential mechanism leading to chronic inflammation. Exp Neurol. 2009;215:5–19. doi: 10.1016/j.expneurol.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wright GJ, Jones M, Puklavec MJ, Brown MH, Barclay AN. The unusual distribution of the neuronal/lymphoid cell surface CD200 (OX2) glycoprotein is conserved in humans. Immunology. 2001;102:173–179. doi: 10.1046/j.1365-2567.2001.01163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gorczynski R, Chen Z, Kai Y, Lee L, Wong S, Marsden PA. CD200 is a ligand for all members of the CD200R family of immunoregulatory molecules. J Immunol. 2004;172:7744–7749. doi: 10.4049/jimmunol.172.12.7744. [DOI] [PubMed] [Google Scholar]
- 76.Rijkers ES, de Ruiter T, Baridi A, Veninga H, Hoek RM, Meyaard L. The inhibitory CD200R is differentially expressed on human and mouse T and B lymphocytes. Mol Immunol. 2008;45:1126–1135. doi: 10.1016/j.molimm.2007.07.013. [DOI] [PubMed] [Google Scholar]
- 77.Hatherley D, Barclay AN. The CD200 and CD200 receptor cell surface proteins interact through their N-terminal immunoglobulin-like domains. Eur J Immunol. 2004;34:1688–1694. doi: 10.1002/eji.200425080. [DOI] [PubMed] [Google Scholar]
- 78.Zhang S, Cherwinski H, Sedgwick JD, Phillips JH. Molecular mechanisms of CD200 inhibition of mast cell activation. J Immunol. 2004;173:6786–6793. doi: 10.4049/jimmunol.173.11.6786. [DOI] [PubMed] [Google Scholar]
- 79.Hoek RM, Ruuls SR, Murphy CA, Wright GJ, Goddard R, Zurawski SM, Blom B, Homola ME, Streit WJ, Brown MH, Barclay AN, Sedgwick JD. Down-regulation of the macrophage lineage through interaction with OX2 (CD200) Science. 2000;290:1768–1771. doi: 10.1126/science.290.5497.1768. [DOI] [PubMed] [Google Scholar]
- 80.Boudakov I, Liu J, Fan N, Gulay P, Wong K, Gorczynski RM. Mice lacking CD200R1 show absence of suppression of lipopolysaccharide-induced tumor necrosis factor-alpha and mixed leukocyte culture responses by CD200. Transplantation. 2007;84:251–257. doi: 10.1097/01.tp.0000269795.04592.cc. [DOI] [PubMed] [Google Scholar]
- 81.Hebenstreit D, Wirnsberger G, Horejs-Hoeck J, Duschl A. Signaling mechanisms, interaction partners, and target genes of STAT6. Cytokine Growth Factor Rev. 2006;17:173–188. doi: 10.1016/j.cytogfr.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 82.Lyons A, Downer EJ, Crotty S, Nolan YM, Mills KH, Lynch MA. CD200 ligand receptor interaction modulates microglial activation in vivo and in vitro: a role for IL-4. J Neurosci. 2007;27:8309–8313. doi: 10.1523/JNEUROSCI.1781-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Deluca HF, Cantorna MT. Vitamin D: its role and uses in immunology. FASEB J. 2001;15:2579–2585. doi: 10.1096/fj.01-0433rev. [DOI] [PubMed] [Google Scholar]
- 84.Fernandes de Abreu DA, Eyles D, Feron F. Vitamin D, a neuro-immunomodulator: implications for neurodegenerative and autoimmune diseases. Psychoneuroendocrinology. 2009;34(Suppl 1):S265–277. doi: 10.1016/j.psyneuen.2009.05.023. [DOI] [PubMed] [Google Scholar]
- 85.Annweiler C, Allali G, Allain P, Bridenbaugh S, Schott AM, Kressig RW, Beauchet O. Vitamin D and cognitive performance in adults: a systematic review. Eur J Neurol. 2009;16:1083–1089. doi: 10.1111/j.1468-1331.2009.02755.x. [DOI] [PubMed] [Google Scholar]
- 86.Sato Y, Iwamoto J, Kanoko T, Satoh K. Amelioration of osteoporosis and hypovitaminosis D by sunlight exposure in hospitalized, elderly women with Alzheimer's disease: a randomized controlled trial. J Bone Miner Res. 2005;20:1327–1333. doi: 10.1359/JBMR.050402. [DOI] [PubMed] [Google Scholar]
- 87.Evatt ML, Delong MR, Khazai N, Rosen A, Triche S, Tangpricha V. Prevalence of vitamin D insufficiency in patients with Parkinson disease and Alzheimer disease. Arch Neurol. 2008;65:1348–1352. doi: 10.1001/archneur.65.10.1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Oudshoorn C, Mattace-Raso FU, van der Veldi N, Colin EM, van der Cammen TJ. Higher serum vitamin D3 levels are associated with better cognitive test performance in patients with Alzheimer's disease. Dement Geriatr Cogn Disord. 2008;25:539–543. doi: 10.1159/000134382. [DOI] [PubMed] [Google Scholar]
- 89.Neveu I, Naveilhan P, Baudet C, Brachet P, Metsis M. 1, 25-dihydroxyvitamin D3 regulates NT-3, NT-4 but not BDNF mRNA in astrocytes. NeuroReport. 1994;6:124–126. doi: 10.1097/00001756-199412300-00032. [DOI] [PubMed] [Google Scholar]
- 90.Naveilhan P, Neveu I, Baudet C, Funakoshi H, Wion D, Brachet P, Metsis M. 1, 25-Dihydroxyvitamin D3 regulates the expression of the low-affinity neurotrophin receptor. Brain Res Mol Brain Res. 1996;41:259–268. doi: 10.1016/0169-328x(96)00103-9. [DOI] [PubMed] [Google Scholar]
- 91.Hewison M, Freeman L, Hughes SV, Evans KN, Bland R, Eliopoulos AG, Kilby MD, Moss PA, Chakraverty R. Differential regulation of vitamin D receptor and its ligand in human monocyte-derived dendritic cells. J Immunol. 2003;170:5382–5390. doi: 10.4049/jimmunol.170.11.5382. [DOI] [PubMed] [Google Scholar]
- 92.Eyles DW, Smith S, Kinobe R, Hewison M, McGrath JJ. Distribution of the vitamin D receptor and 1 alpha-hydroxylase in human brain. J Chem Neuroanat. 2005;29:21–30. doi: 10.1016/j.jchemneu.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 93.Gezen-Ak D, Dursun E, Ertan T, Hanagasi H, Gurvit H, Emre M, Eker E, Ozturk M, Engin F, Yilmazer S. Association between vitamin D receptor gene polymorphism and Alzheimer's disease. Tohoku J Exp Med. 2007;212:275–282. doi: 10.1620/tjem.212.275. [DOI] [PubMed] [Google Scholar]
- 94.Jiao ZM, Fu YH, Fu J, Zhang F, Wang WZ. 1, 25-dihydroxyvitamin D3 promotes the apoptosis of inflammatory cells in acute experimental autoimmune encephalomyelitis: experiment with rats. Zhonghua Yixue Zazhi. 2008;88:2350–2354. [PubMed] [Google Scholar]
- 95.Cantorna MT, Woodward WD, Hayes CE, Deluca HF. 1, 25-dihydroxyvitamin D3 is a positive regulator for the two anti-encephalitogenic cytokines TGF-beta 1 and IL-4. J Immunol. 1998;160:5314–5319. [PubMed] [Google Scholar]
- 96.Wilkins CH, Birge SJ, Sheline YI, Morris JC. Vitamin D deficiency is associated with worse cognitive performance and lower bone density in older African Americans. J Natl Med Assoc. 2009;101:349–354. doi: 10.1016/s0027-9684(15)30883-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr Rev. 1999;20:649–688. doi: 10.1210/edrv.20.5.0380. [DOI] [PubMed] [Google Scholar]
- 98.Kielian T, Drew PD. Effects of peroxisome proliferator-activated receptor-gamma agonists on central nervous system inflammation. J Neurosci Res. 2003;71:315–325. doi: 10.1002/jnr.10501. [DOI] [PubMed] [Google Scholar]
- 99.Basu-Modak S, Braissant O, Escher P, Desvergne B, Honegger P, Wahli W. Peroxisome proliferator-activated receptor beta regulates acyl-CoA synthetase 2 in reaggregated rat brain cell cultures. J Biol Chem. 1999;274:35881–35888. doi: 10.1074/jbc.274.50.35881. [DOI] [PubMed] [Google Scholar]
- 100.Ogawa S, Lozach J, Benner C, Pascual G, Tangirala RK, Westin S, Hoffmann A, Subramaniam S, David M, Rosenfeld MG, Glass CK. Molecular determinants of crosstalk between nuclear receptors and toll-like receptors. Cell. 2005;122:707–721. doi: 10.1016/j.cell.2005.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ghisletti S, Huang W, Ogawa S, Pascual G, Lin ME, Willson TM, Rosenfeld MG, Glass CK. Parallel SUMOylation-dependent pathways mediate gene- and signal-specific transrepression by LXRs and PPARgamma. Mol Cell. 2007;25:57–70. doi: 10.1016/j.molcel.2006.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Petrova TV, Akama KT, Van Eldik LJ. Cyclopentenone prostaglandins suppress activation of microglia: down-regulation of inducible nitric-oxide synthase by 15-deoxy-delta12, 14-prostaglandin J2. Proc Natl Acad Sci USA. 1999;96:4668–4673. doi: 10.1073/pnas.96.8.4668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma) J Biol Chem. 1995;270:12953–12956. doi: 10.1074/jbc.270.22.12953. [DOI] [PubMed] [Google Scholar]
- 104.Jiang C, Ting AT, Seed B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature. 1998;391:82–86. doi: 10.1038/34184. [DOI] [PubMed] [Google Scholar]
- 105.Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. 1998;391:79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- 106.Bernardo A, Levi G, Minghetti L. Role of the peroxisome proliferator-activated receptor-gamma (PPAR-gamma) and its natural ligand 15-deoxy-delta12, 14-prostaglandin J2 in the regulation of microglial functions. Eur J NeuroSci. 2000;12:2215–2223. doi: 10.1046/j.1460-9568.2000.00110.x. [DOI] [PubMed] [Google Scholar]
- 107.Heneka MT, Landreth GE, Feinstein DL. Role for peroxisome proliferator-activated receptor-gamma in Alzheimer's disease. Ann Neurol. 2001;49:276. doi: 10.1002/1531-8249(20010201)49:2<276::aid-ana53>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 108.Sastre M, Dewachter I, Rossner S, Bogdanovic N, Rosen E, Borghgraef P, Evert BO, Dumitrescu-Ozimek L, Thal DR, Landreth G, Walter J, Klockgether T, van Leuven F, Heneka MT. Nonsteroidal anti-inflammatory drugs repress beta-secretase gene promoter activity by the activation of PPAR-gamma. Proc Natl Acad Sci USA. 2006;103:443–448. doi: 10.1073/pnas.0503839103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kummer MP, Heneka MT. PPARs in Alzheimer's disease. PPAR Res. 2008;2008:403896. doi: 10.1155/2008/403896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Combs CK, Johnson DE, Karlo JC, Cannady SB, Landreth GE. Inflammatory mechanisms in Alzheimer's disease: inhibition of beta-amyloid-stimulated proinflammatory responses and neurotoxicity by PPARgamma agonists. J Neurosci. 2000;20:558–567. doi: 10.1523/JNEUROSCI.20-02-00558.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Heneka MT, Klockgether T, Feinstein DL. Peroxisome proliferator-activated receptor-gamma ligands reduce neuronal inducible nitric oxide synthase expression and cell death in vivo. J Neurosci. 2000;20:6862–6867. doi: 10.1523/JNEUROSCI.20-18-06862.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yan Q, Zhang J, Liu H, Babu-Khan S, Vassar R, Biere AL, Citron M, Landreth G. Anti-inflammatory drug therapy alters beta-amyloid processing and deposition in an animal model of Alzheimer's disease. J Neurosci. 2003;23:7504–7509. doi: 10.1523/JNEUROSCI.23-20-07504.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Heneka MT, Sastre M, Dumitrescu-Ozimek L, Hanke A, Dewachter I, Kuiperi C, O'Banion K, Klockgether T, van Leuven F, Landreth GE. Acute treatment with the PPARgamma agonist pioglitazone and ibuprofen reduces glial inflammation and Abeta1-42 levels in APPV717I transgenic mice. Brain. 2005;128:1442–1453. doi: 10.1093/brain/awh452. [DOI] [PubMed] [Google Scholar]
- 114.Sastre M, Dewachter I, Landreth GE, Willson TM, Klockgether T, van Leuven F, Heneka MT. Nonsteroidal anti-inflammatory drugs and peroxisome proliferator-activated receptor-gamma agonists modulate immunostimulated processing of amyloid precursor protein through regulation of beta-secretase. J Neurosci. 2003;23:9796–9804. doi: 10.1523/JNEUROSCI.23-30-09796.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Camacho IE, Serneels L, Spittaels K, Merchiers P, Dominguez D, De SB. Peroxisome-proliferator-activated receptor gamma induces a clearance mechanism for the amyloid-beta peptide. J Neurosci. 2004;24:10908–10917. doi: 10.1523/JNEUROSCI.3987-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Watson GS, Cholerton BA, Reger MA, Baker LD, Plymate SR, Asthana S, Fishel MA, Kulstad JJ, Green PS, Cook DG, Kahn SE, Keeling ML, Craft S. Preserved cognition in patients with early Alzheimer disease and amnestic mild cognitive impairment during treatment with rosiglitazone: a preliminary study. Am J Geriatr Psychiatry. 2005;13:950–958. doi: 10.1176/appi.ajgp.13.11.950. [DOI] [PubMed] [Google Scholar]
- 117.Risner ME, Saunders AM, Altman JF, Ormandy GC, Craft S, Foley IM, Zvartau-Hind ME, Hosford DA, Roses AD. Efficacy of rosiglitazone in a genetically defined population with mild-to-moderate Alzheimer's disease. Pharmacogenomics J. 2006;6:246–254. doi: 10.1038/sj.tpj.6500369. [DOI] [PubMed] [Google Scholar]
- 118.Luchsinger JA, Tang MX, Stern Y, Shea S, Mayeux R. Diabetes mellitus and risk of Alzheimer's disease and dementia with stroke in a multiethnic cohort. Am J Epidemiol. 2001;154:635–641. doi: 10.1093/aje/154.7.635. [DOI] [PubMed] [Google Scholar]
- 119.Messier C. Impact of impaired glucose tolerance and type 2 diabetes on cognitive aging. Neurobiol Aging. 2005;26(Suppl 1):26–30. 26–30. doi: 10.1016/j.neurobiolaging.2005.09.014. Epub;%2005 Oct;%19. [DOI] [PubMed] [Google Scholar]
- 120.Ronnemaa E, Zethelius B, Sundelof J, Sundstrom J, german-Gunnarsson M, Berne C, Lannfelt L, Kilander L. Impaired insulin secretion increases the risk of Alzheimer disease. Neurology. 2008;71:1065–1071. doi: 10.1212/01.wnl.0000310646.32212.3a. [DOI] [PubMed] [Google Scholar]
- 121.Combarros O, Rodriguez-Rodriguez E, Mateo I, Vazquez-Higuera JL, Infante J, Berciano J, Sanchez-Juan P. APOE dependent-association of PPAR-gamma genetic variants with Alzheimer's disease risk. Neurobiol Aging. 2010 doi: 10.1016/j.neurobiolaging.2009.07.004. in press. [DOI] [PubMed] [Google Scholar]
- 122.Loane DJ, Deighan BF, Clarke RM, Griffin RJ, Lynch AM, Lynch MA. Interleukin-4 mediates the neuroprotective effects of rosiglitazone in the aged brain. Neurobiol Aging. 2009;30:920–931. doi: 10.1016/j.neurobiolaging.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 123.Yonekura H, Yamamoto Y, Sakurai S, Watanabe T, Yamamoto H. Roles of the receptor for advanced glycation endproducts in diabetes-induced vascular injury. J Pharm Sci. 2005;97:305–311. doi: 10.1254/jphs.cpj04005x. [DOI] [PubMed] [Google Scholar]
- 124.Galichet A, Weibel M, Heizmann CW. Calcium-regulated intramembrane proteolysis of the RAGE receptor. Biochem Biophys Res Commun. 2008;370:1–5. doi: 10.1016/j.bbrc.2008.02.163. [DOI] [PubMed] [Google Scholar]
- 125.Zhang L, Bukulin M, Kojro E, Roth A, Metz VV, Fahrenholz F, Nawroth PP, Bierhaus A, Postina R. Receptor for advanced glycation end products is subjected to protein ectodomain shedding by metalloproteinases. J Biol Chem. 2008;283(51):35507–35516. doi: 10.1074/jbc.M806948200. [DOI] [PubMed] [Google Scholar]
- 126.Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, Slattery T, Zhao L, Nagashima M, Morser J, Migheli A, Nawroth P, Stern D, Schmidt AM. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer's disease. Nature. 1996;382:685–691. doi: 10.1038/382685a0. [DOI] [PubMed] [Google Scholar]
- 127.Arancio O, Zhang HP, Chen X, Lin C, Trinchese F, Puzzo D, Liu S, Hegde A, Yan SF, Stern A, Luddy JS, Lue LF, Walker DG, Roher A, Buttini M, Mucke L, Li W, Schmidt AM, Kindy M, Hyslop PA, Stern DM, Du Yan SS. RAGE potentiates Abeta-induced perturbation of neuronal function in transgenic mice. EMBO J. 2004;23:4096–4105. doi: 10.1038/sj.emboj.7600415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Deane R, Du YS, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, Welch D, Manness L, Lin C, Yu J, Zhu H, Ghiso J, Frangione B, Stern A, Schmidt AM, Armstrong DL, Arnold B, Liliensiek B, Nawroth P, Hofman F, Kindy M, Stern D, Zlokovic B. RAGE mediates amyloid-beta peptide transport across the blood–brain barrier and accumulation in brain. Nat Med. 2003;9:907–913. doi: 10.1038/nm890. [DOI] [PubMed] [Google Scholar]
- 129.Deane R, Sagare A, Zlokovic BV. The role of the cell surface LRP and soluble LRP in blood–brain barrier Abeta clearance in Alzheimer's disease. Curr Pharm Des. 2008;14:1601–1605. doi: 10.2174/138161208784705487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hernanz A, De la Fuente M, Navarro M, Frank A. Plasma aminothiol compounds, but not serum tumor necrosis factor receptor II and soluble receptor for advanced glycation end products, are related to the cognitive impairment in Alzheimer's disease and mild cognitive impairment patients. Neuroimmunomodulation. 2007;14:163–167. doi: 10.1159/000110641. [DOI] [PubMed] [Google Scholar]
- 131.Humpert PM, Djuric Z, Kopf S, Rudofsky G, Morcos M, Nawroth PP, Bierhaus A. Soluble RAGE but not endogenous secretory RAGE is associated with albuminuria in patients with type 2 diabetes. Cardiovasc Diabetol. 2007;6:9. doi: 10.1186/1475-2840-6-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Koyama H, Yamamoto H, Nishizawa Y. RAGE and soluble RAGE: potential therapeutic targets for cardiovascular diseases. Mol Med. 2007;13:625–635. doi: 10.2119/2007-00087.Koyama. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Pullerits R, Bokarewa M, Dahlberg L, Tarkowski A. Synovial fluid expression of autoantibodies specific for RAGE relates to less erosive course of rheumatoid arthritis. Rheumatology (Oxford) 2007;46:1367–1371. doi: 10.1093/rheumatology/kem141. [DOI] [PubMed] [Google Scholar]
- 134.Santilli F, Bucciarelli L, Noto D, Cefalu AB, Davi V, Ferrante E, Pettinella C, Averna MR, Ciabattoni G, Davi G. Decreased plasma soluble RAGE in patients with hypercholesterolemia: effects of statins. Free Radic Biol Med. 2007;43:1255–1262. doi: 10.1016/j.freeradbiomed.2007.06.017. [DOI] [PubMed] [Google Scholar]
- 135.Sternberg Z, Weinstock-Guttman B, Hojnacki D, Zamboni P, Zivadinov R, Chadha K, Lieberman A, Kazim L, Drake A, Rocco P, Grazioli E, Munschauer F. Soluble receptor for advanced glycation end products in multiple sclerosis: a potential marker of disease severity. Mult Scler. 2008;14:759–763. doi: 10.1177/1352458507088105. [DOI] [PubMed] [Google Scholar]
- 136.Chiang KH, Huang PH, Huang SS, Wu TC, Chen JW, Lin SJ. Plasma levels of soluble receptor for advanced glycation end products are associated with endothelial function and predict cardiovascular events in nondiabetic patients. Coron Artery Dis. 2009;20:267–273. doi: 10.1097/MCA.0b013e32832c459c. [DOI] [PubMed] [Google Scholar]
- 137.Ilzecka J. Serum-soluble receptor for advanced glycation end product levels in patients with amyotrophic lateral sclerosis. Acta Neurol Scand. 2009;120:119–122. doi: 10.1111/j.1600-0404.2008.01133.x. [DOI] [PubMed] [Google Scholar]
- 138.Raucci A, Cugusi S, Antonelli A, Barabino SM, Monti L, Bierhaus A, Reiss K, Saftig P, Bianchi ME. A soluble form of the receptor for advanced glycation endproducts (RAGE) is produced by proteolytic cleavage of the membrane-bound form by the sheddase a disintegrin and metalloprotease 10 (ADAM10) FASEB J. 2008;22:3716–3727. doi: 10.1096/fj.08-109033. [DOI] [PubMed] [Google Scholar]
- 139.Hsieh HL, Schafer BW, Weigle B, Heizmann CW. S100 protein translocation in response to extracellular S100 is mediated by receptor for advanced glycation endproducts in human endothelial cells. Biochem Biophys Res Commun. 2004;316:949–959. doi: 10.1016/j.bbrc.2004.02.135. [DOI] [PubMed] [Google Scholar]
- 140.Shepherd CE, Goyette J, Utter V, Rahimi F, Yang Z, Geczy CL, Halliday GM. Inflammatory S100A9 and S100A12 proteins in Alzheimer's disease. Neurobiol Aging. 2006;27:1554–1563. doi: 10.1016/j.neurobiolaging.2005.09.033. [DOI] [PubMed] [Google Scholar]
- 141.Mrak RE, Griffinbc WS. The role of activated astrocytes and of the neurotrophic cytokine S100B in the pathogenesis of Alzheimer's disease. Neurobiol Aging. 2001;22:915–922. doi: 10.1016/s0197-4580(01)00293-7. [DOI] [PubMed] [Google Scholar]
- 142.Simpson JE, Ince PG, Lace G, Forster G, Shaw PJ, Matthews F, Savva G, Brayne C, Wharton SB. Astrocyte phenotype in relation to Alzheimer-type pathology in the ageing brain. Neurobiol Aging. 2010;31:578–590. doi: 10.1016/j.neurobiolaging.2008.05.015. [DOI] [PubMed] [Google Scholar]