

Abstract
BACKGROUND & AIMS
Fibrosis is the hallmark of chronic liver diseases, yet many aspects of its mechanism remain to be defined. Chemokines are ubiquitous chemotactic molecules that mediate many acute and chronic inflammatory conditions, and CXC chemokine genes colocalize with a locus previously shown to include fibrogenic genes. We investigated the roles of the chemokine CXCL9 and its receptor CXCR3 in liver fibrosis.
METHODS
The effects of CXCL variants on fibrogenesis were analyzed using samples from patients with hepatitis C virus infection and by induction of fibrosis in CXCR3−/− and wild-type mice. In mice, intrahepatic immune cell subsets were investigated and interferon gamma messenger RNA levels were measured at baseline and after injury. Human serum CXCL9 levels were measured and correlated with CXCL9 variant and fibrosis severity. The effects of stimulation with CXCL9 were investigated on human hepatic stellate cells (LX-2).
RESULTS
Specific CXCL9 variants were associated with liver fibrosis in mice and humans; CXCL9 serum concentrations correlated with genotypes and levels of fibrosis in patients. In contrast to other chemokines, CXCL9 exerted antifibrotic effects in vitro, suppressing collagen production in LX-2 cells. CXCR3−/− mice had increased liver fibrosis; progression was associated with decreased numbers of intra-hepatic interferon gamma–positive T cells and reduced interferon gamma messenger RNA, indicating that CXCL9-CXCR3 regulates Th1-associated immune pathways.
CONCLUSIONS
This is the first description of a chemokine-based antifibrotic pathway in the liver; antifibrotic therapies might be developed to modulate CXC chemokine levels.
Liver fibrosis is a leading cause of morbidity and mortality worldwide. The main causes for advanced fibrosis are viral hepatitis, alcoholic liver disease, and metabolic conditions predisposing to nonalcoholic steatohepatitis.1 Among these, chronic hepatitis C virus (HCV) infection and nonalcoholic steatohepatitis are among the most prominent examples of the need to develop novel treatment options to halt progressive liver scarring that might otherwise result in liver cirrhosis and hepatocellular carcinoma. Notably, the reversibility of fibrosis in animal models has stimulated the enthusiasm for antifibrotic treatment strategies targeting critical regulatory pathways of liver fibrosis.2,3
The molecular mechanisms underlying liver fibrosis are complex and modulated by exogenous and endogenous factors.4 Although some of these factors (eg, sex, alcohol consumption, and presence of diabetes) are well recognized, the identification of genes that regulate liver fibrosis in humans is challenging.5,6 Most genetic studies lack reproducibility and do not provide functional data. Hence, we have proposed to identify modifier genes of polygenic diseases (such as liver fibrosis) in animal models first and validate these genes in human studies.7 The identification of such conserved key regulatory genes and molecular pathways might help to identify new antifibrotic strategies for the regression of tissue injury.
Chemokines are ubiquitous chemotactic molecules that play major roles in acute and chronic inflammatory conditions.8 They are divided into 4 families defined by the number of amino acids between the N-terminal cysteine residues (CC, CXC, CCX, and CX3C). The largest families are the CC and the CXC chemokines, members of which are expressed in acutely inflamed and fibrotic livers.9 The CXC chemokine family is a pleiotropic family of molecules that are involved in the trafficking of various leukocyte subsets, angiogenesis, and vascular remodeling. This class of chemokines has been shown to play a pivotal role in pulmonary fibrosis.10 Notably, the genes encoding CXC chemokines show an exceptional genomic organization, that is, the majority of them are clustered on the short arm of chromosome 4q12–21 in humans and chromosome 5 in mice, respectively. This CXC chemokine gene cluster drew our attention, because it colocalizes with a chromosomal region that we identified in earlier studies by in silico mapping to include fibrogenic genes.11
We show here that the gene encoding the interferon (IFN)-inducible chemokine CXCL9 (MIG) underlies this association in mice and humans, uncovering a new immunologic pathway of liver fibrosis by functional studies in vitro and in vivo.
Materials and Methods
Inbred Mice, Experimental Crosses, and Fibrosis Models
F1 hybrids of fibrosis-susceptible BALB/cJ and resistant A/J mice (Jackson Laboratory, Bar Harbor, ME) were crossed to obtain 358 F2 progeny ([A/J × BALB/cJ]F1 × [A/J × BALB/cJ]F1). Liver fibrosis in these mice was induced with CCl4.11 The generation of CXCR3−/− mice on the C57BL/6J background has been described previously.12 Male CXCR3−/− mice and wild-type litter-mates were challenged intraperitoneally with CCl4 (0.6 mL/kg twice weekly) or with thioacetamide (150 mg/kg body wt thrice a week) for 6 weeks. Staging of liver fibrosis was performed as described.7
Quantitative Trait Locus Analysis
A quantitative trait locus analysis of the Cxc chemokine cluster on murine chromosome 5 was performed in the F2 progeny. Single nucleotide polymorphisms (SNPs) that tag the proximal, middle, and distal part of the Cxc chemokine cluster were identified at http://snp.gnf.org/. The identified SNPs (Figure 1A) were genotyped using 5′-nuclease (TaqMan) assays on the ABI PRISM 7000 Sequence Detection System (Applera, Applied Biosystems, Foster City, CA). Supplementary Table 1 provides the primer and probe sequences for the allele-specific polymerase chain reactions. Regression analysis was calculated with Map Manager QTX (http://www.mapmanager.org/).
Figure 1.

Murine quantitative trait locus and gene expression analysis. (A) Schematic diagram of the Cxc chemokine cluster on chromosome 5 (90,207,953 to 94,974,241 base pairs). The SNPs rs4225368, rs4225374, and rs3023048 tag the proximal, middle, and distal part of the gene cluster. Regression analysis in the F2 progeny showed that the SNPs tagging the middle and distal part of the cluster are significantly associated with the histologic stage of liver fibrosis induced by CCl4 (P = .026 and P = .036, respectively). (B –F) Quantitative expression of IFN-inducible chemokines and their receptor Cxcr3 in BALB/cJ and A/J mice after challenge with CCl4. Whereas the mRNA expression levels of (D) Cxcl10, E ( ) Cxcl11, and (F) Cxcr3 do not differ between the strains, the expression of Cxcl9 was 2.3-fold increased in the resistant strain (P = .002, B). (C) The increased mRNA expression of CXCL9 was confirmed by CXCL9 protein analysis (C).
Expression Analysis of Murine Chemokines and Cytokines
Total RNA was isolated from livers of mice and reversely transcribed using SuperScript (Invitrogen, Carlsbad, CA). Quantitative reverse-transcription polymerase chain reaction was performed for Cxcl9, Cxcl10 and Cxcl11, Cxcr3, and Ifn-γ with Assays on Demand from http://www.appliedbiosystems.com.
Fluorescence-Activated Cell Sorter Analysis of Intrahepatic IFN-γ–Positive Immune Cells
At baseline and after treatment of mice with CCl4, we performed flow cytometry analysis. Livers were flushed with phosphate-buffered saline and homogenized. Mononuclear cells were isolated by density gradient centrifugation. After 4-hour stimulation with phorbol myristate acetate (50 ng/mL) and ionomycin (500 ng/mL; both Sigma, Hamburg, Germany), 10 μg/mL brefeldin was added. The cells were stained with CD45-APC.Cy7 and CD3-APC (BD Biosciences, Heidelberg, Germany). After permeabilization, the cells were incubated with IFN-γ-PE or rat immunoglobulin G/PE (isotype specific control) and analyzed by flow cytometry using the FACS Canto II flow cytometer (BD Biosciences).
Human Study Populations
Two independent cohorts of white HCV-infected patients were enrolled in the study (together 441 individuals). The first cohort was from the university hospitals of Aachen, Bonn, and Regensburg, and the second cohort was recruited at Charité in Berlin. HCV infection was confirmed by a positive HCV RNA test result (Roche Diagnostics, Mannheim, Germany). Other chronic liver diseases were excluded by appropriate serologic tests. We also genotyped 232 healthy white individuals for the evaluation of CXC chemokine cluster haplotypes and Hardy–Weinberg equilibrium. The demographic data of the cohorts are given in Supplementary Table 2. All subjects were asked for the ethnicity of their second-degree relatives to minimize systematic differences in ancestry that might lead to population stratification.13 Informed consent was obtained from patients, and the protocol was approved by the local ethics committees. Liver biopsy specimens were obtained from all HCV-infected patients before antiviral treatment and were analyzed according to Desmet et al.14
Human Genetic Analysis
All haplotype-tagging SNPs (htSNPs) of the chromosomal region that harbors the genes for IFN-inducible chemokines (4q21: 77,380–77,420 kilobases) were selected from the International HapMap Project (Supplementary Figure 1). Using the HapMap data, we identified 8 SNPs with minor allele frequencies ≥0.05, which cover the genetic region of interest (Figure 2A). In our control cohort, 5 haplotypes with a frequency >5% were identified (Supplementary Table 3). These SNPs were genotyped in 404 white patients with mild (F0–F1) or severe (F2–F4) liver fibrosis. Thirty-seven patients of the total cohort were excluded due to insufficient amounts of DNA for genotyping all 8 SNPs. The haplotypes were named in order of frequency (the most prevalent haplotype was dCXC_1) (Figure 2B).
Figure 2.

Human genetic analyses. (A) Schematic diagram of the distal part of the human CXC chemokine cluster on chromosome 4q21 (77,380 –77,420 kilobases). Eight SNPs cover the genes for the chemokines CXCL9, CXCL10, and CXCL11. (B ) These SNPs were genotyped in 404 patients with mild (stage F0/F1, 2n = 474) or advanced (stage F2–F4, 2n = 334) liver fibrosis. The overall haplotype distribution is different between patients with mild fibrosis and subjects with severe fibrosis (P = .01). Specifically, the fourth most common haplotype, dCXC_4, is significantly more prevalent in individuals with advanced fibrosis (15.0%) compared with patients with mild fibrosis (6.4%; *P = .001). (C) dCXC_4 is tagged by rs3733236 in the 3′-untranslated region of CXCL9. The minor (risk) allele of this SNP is functionally associated with reduced serum concentrations in patients with HCV infection and mild fibrosis (n = 60, P = .02).
Haplotype distributions between the patient cohorts were compared by permutation testing, as implemented in PHASE 2.0.15 We reproduced our haplotype results with the Haploview 2.0 program, which supports linkage disequilibrium and haplotype block analysis; haplotype population frequency estimation; single SNP, global, and haplotype-specific association tests; and permutation testing for association significance.16 Subsequently, the frequencies of the most likely haplotypes as inferred by PHASE were compared between patients with mild and severe fibrosis. To further validate the robustness of the genetic data, the dCXC_4 htSNP rs3733236 C>T was analyzed independently in both cohorts of patients (Supplementary Table 4). Tests for association were performed by Fisher exact test for comparison of allele frequencies and with Cochran–Armitage trend test (http://ihg.gsf.de) for comparison of genotypes. We chose the stage of fibrosis as the main outcome variable in the analysis. However, the exact duration of infection was available in a subgroup of study subjects (n = 243). In these subjects, we also analyzed the association of the CXCL9 genotype with progression of fibrosis (fibrosis stage/duration in years).
Genotyping of Human SNPs and Direct Sequencing of CXCL9
Genotyping was performed with 5′-nuclease (Taq-Man) assays on the ABI PRISM 7000 System. Assays for genotyping were obtained from Applied Biosystems. Polymerase chain reaction amplification of the 4 exons and adjacent splice sites plus 300 base pairs upstream of the start codon of the human CXCL9 gene was performed using primers and annealing temperature conditions as listed in Supplementary Table 5.
Isolation and Stimulation of Stellate Cells
Culture of the human hepatic stellate cell (HSC) line LX-2 has previously been described.17 For chemo-kine stimulation, the serum content was reduced, and cells were stimulated with recombinant human IFN-γ, CXCL9, or CXCL4 (R&D Systems, Wiesbaden, Germany) for 48 hours. Total messenger RNA (mRNA) was isolated and collagen protein levels were determined by Western blot analysis after isolation of total protein from cultures. CXCR3 isoform mRNA was amplified with primers specific for the A and B splice variants.18 Quantification of mRNA was performed with SYBR green and β-actin as internal control. Crucial experiments performed in LX-2 cells were reproduced in primary myofibroblasts.
CXCL9 Expression Analysis in Human Fibrotic Liver
We isolated total RNA from formalin-fixed, paraffin-embedded liver biopsy specimens from 53 patients with HCV and 42 patients with nonalcoholic fatty liver disease (20 with fatty liver; 22 with steatohepatitis and fibrosis) as described.19 Quantitative reverse-transcription polymerase chain reaction for CXCL9, CXCR3-A, and CXCR3-B was performed as described previously.
CXCL9 and CXCL10 Serum Concentrations
Serum levels of CXCL9 and CXCL10 were determined in 180 HCV-infected patients. Serum was taken at the time of liver biopsy and analyzed by cytometric bead array (BD Pharmingen, Heidelberg, Germany). Serum levels of both chemokines were assessed with regard to stage of fibrosis by analysis of variance. For genotype-phenotype correlation, CXCL9 serum levels were measured in 60 patients with stage 0 and 1 fibrosis in which the CXCL9 rs3733236 genotype was determined as described.
Immunohistochemistry of CXCL9 in Human Liver Tissue
Immunohistochemical staining was performed with a mouse monoclonal anti-human CXCL9 antibody (R&D Systems).
Statistical Analysis
Statistical analysis was performed with GraphPad Prism and InStat (GraphPad Software, La Jolla, CA).19 Data are given as means and SEM. Categorical variables are compared with Fisher exact test and continuous variables with Student t test with Welsh correction in case of unequal variances. Multiple logistic regression analysis was performed with stage of fibrosis as the dependent variable and genotype, age, and sex as covariates. Continuous variables in more than 2 groups were compared by analysis of variance.
Results
The Distal Cxc Chemokine Cluster Determines the Severity of Liver Fibrosis in Experimental Crosses of Inbred Mice
We first performed a quantitative trait locus analysis in the F2 progeny from an experimental cross between the fibrosis-susceptible mouse strain BALB/cJ and the resistant A/J strain (Figure 1A). In this cross, regression analysis showed that the SNPs tagging the middle and distal parts of the cluster (rs4225374 - 0.3 ± 0.2 cM -rs3023048) are significantly associated with the histologic stage of liver fibrosis (P = .026 and P = .036, respectively; Figure 1A). Notably, the distal part of the cluster harbors the genes of the Ifn-inducible chemokines Cxcl9, Cxcl10, and Cxcl11, which are of particular interest because IFN-γ is suggested to exert antifibrotic effects.20,21
The Chemokine Cxcl9 Is Differentially Expressed in Fibrosis-Susceptible and Fibrosis-Resistant Mice
We therefore analyzed the hepatic mRNA expression of these genes and their receptor Cxcr3 in the parental strains after CCl4 treatment. As depicted in Figure 1B and C, we observed a significant difference in Cxcl9 mRNA and protein expression between BALB/cJ and A/J mice (P = .002 and P = .02, respectively), whereas mRNA levels of Cxcl10, Cxcl11, and Cxcr3 did not significantly differ between the strains (Figure 1D–F).
The Distal Part of the Orthologous Human CXC Chemokine Cluster Is Associated With the Severity of Liver Fibrosis in Patients With Chronic Hepatitis C
We next performed a haplotype cluster analysis of 8 SNPs in 404 HCV-infected patients with mild (F0/F1) and severe liver fibrosis (F2–F4). As in our control population (Supplementary Table 1), 5 common haplotypes (frequency >0.05) of the distal CXC (dCXC) cluster were identified in HCV-infected patients. The overall haplo-type distribution did not differ between our total patients’ cohort and the control cohort (data not shown). However, the distribution of dCXC haplotypes between patients with mild fibrosis and individuals with severe fibrosis differed significantly (P = .01) by permutation testing with 2 independent programs.15,16 Specifically, the fourth most frequent haplotype (dCXC_4) is significantly more prevalent in subjects with severe fibrosis (15.0%) compared with patients with mild fibrosis (6.4%, P = .001, Figure 2B). This haplotype is tagged by an SNP in the 3′-untranslated region of CXCL9 (rs3733236). Genetic association of this htSNP with the severity of liver fibrosis was evident in both investigated cohorts of HCV-infected patients (Supplementary Table 1). The association of the CXCL9 htSNP with liver fibrosis held true after correction for sex and age in multiple logistic regression analysis. The duration of HCV infection was only available in a subgroup of patients (n = 243). Despite the resulting loss of statistical power, progression of fibrosis was markedly higher in carriers of rs3733236 T alleles (0.38 fibrosis stages/year) compared with individuals homozygous for the C allele (0.27 fibrosis stages/year). The association of rs3733236 with liver fibrosis is not due to linkage with common coding variants in our patients upon sequencing of CXCL9 exons (data not shown). Notably, when analyzing the serum concentrations of CXCL9 in relation to rs3733236 alleles, we found significantly reduced CXCL9 levels in patients carrying the T allele (Figure 2C), suggesting a functional significance of the htSNP. This analysis was performed in patients with only mild fibrosis to exclude independent effects of the fibrosis stage on CXCL9 serum levels.
CXCL9 Increases With More Advanced Stages of Fibrosis in Humans
We next assessed the intrahepatic expression of CXCL9 at different stages of liver fibrosis. As shown in Figure 3A, the mRNA expression of CXCL9 significantly increases with advanced stages of HCV-induced liver fibrosis (P = .04). An increase in CXCL9 mRNA levels with regard to fibrosis was confirmed in liver biopsy specimens from patients with nonalcoholic steatohepatitis with and without liver fibrosis (P = .002, Figure 3B) and by immunohistochemistry in advanced HCV-induced fibrosis (Figure 3C). We also assessed the mRNA expression of the splice variants A and B of the CXCL9 receptor CXCR3 in HCV-infected liver. As shown in Figure 3D, both splice variants were equally expressed. The elevated intrahepatic levels of CXCL9 mRNA were also reflected by elevated serum concentrations at higher stages of liver fibrosis (P = .01, Figure 3E). This association is not specific for CXCL9 but is also found for serum levels of CXCL10, another CXCR3 agonist (P = .001, Figure 3F).
Figure 3.

Analyses of CXCL9 in human liver fibrosis. (A) CXCL9 mRNA expression is significantly associated with the degree of liver fibrosis in HCV (P = .04). (B) Association of CXCL9 mRNA expression with liver fibrosis is confirmed in patients with fibrosis due to nonalcoholic steatohepatitis (P = .002). (C) Immunohistochemistry of CXCL9 confirms a strong up-regulation in chronic liver damage (normal liver, left; HCV-induced cirrhosis, right). (D ) The mRNA of both splice variants of the CXCL9 receptor CXCR3 are present in HCV-infected liver. (E) CXCL9 and (F) CXCL10 serum concentrations are also associated with the severity of liver fibrosis in HCV infection (P = .01 and P = .001, respectively).
CXCL9 Exerts Antifibrotic Effects In Vitro
We next assessed the functional relevance of CXCL9 with regard to activated stellate cells. Isolated primary HSCs display low basal CXCL9 mRNA expression, which significantly (P = .02) increases after IFN-γ stimulation of the cells (Figure 4A). CXCL9 specifically binds to the chemokine receptor CXCR3, which is expressed on HSCs.22 Interestingly, stellate cells express equal amounts of both splice variants of the receptor (Figure 4E). We tested the direct effects of CXCL9 on profibrogenic markers of LX-2 and primary stellate cells. As shown in Figure 4B and C, increasing concentrations of CXCL9 lead to dose-dependent down-regulation of transforming growth factor β1 (P = .01) and collagen 1α1 mRNA (P = .001). Importantly, collagen protein expression of stellate cells was strongly suppressed by CXCL9 (Figure 4D and Supplementary Figure 2). We also treated LX-2 cells with CXCL4, an agonist for CXCR3-B,18 to determine specific effects of the CXCR3-B splice variant. As depicted in Figure 4F, CXCL4 had only minor effects on collagen mRNA expression in LX-2 cells. This was also true for TIMP-1 and TGF-β1 mRNA (data not shown).
Figure 4.

Functional analysis of CXCL9 in stellate cells. (A) CXCL9 mRNA is expressed in HSCs and is significantly increased (P = .02) after treatment of the cells with IFN-γ. (B and C) Stimulation of LX-2 cells with CXCL9 leads to a dose-dependent down-regulation of TGFB (P = .01) and COLA1A mRNA expression (P = .001), respectively. The columns represent mean values ± SEM of 3 independent experiments. (D) The down-regulation of collagen by CXCL9 in HSCs is confirmed by Western blot with an antibody detecting the immature (175 kilodaltons) and mature (120 kilodaltons) chain of collagen. (E) Both splice variants of the CXCL9 receptor CXCR3 are present in LX-2 cells. (F) However, stimulation of the CXCR3-B isoform with CXCL4 leads only to a modest increase in COLA1A mRNA.
Genetic Deletion of the CXCL9 Receptor CXCR3 Leads to Increased Fibrosis In Vivo
Based on these results, we hypothesized that genetic deletion of the specific CXCL9 receptor CXCR3 should lead to increased liver damage in vivo. We thus challenged CXCR3−/− mice and wild-type littermates for 6 weeks with CCl4. Upon histologic examination, CXC3−/− animals indeed displayed significantly higher stages of fibrosis (Figure 5A and C). This exaggerated fibrogenic response in CXCR3−/− mice was confirmed by increased concentrations of the collagen-specific amino acid hydroxyproline in knockout animals (P = .03, Figure 5B). Next we extended the findings of an exaggerated fibrogenic response in CXCR3−/− mice to a second fibrosis model, thioacetamide, which was chronically administered for 6 weeks. As shown in Figure 5D–F, lack of CXCR3 resulted in increased stages of fibrosis (P = .02) and significantly higher hydroxyproline concentrations in knockout mice compared with their wild-type littermates (P = .004), confirming the results in the CCl4 model.
Figure 5.
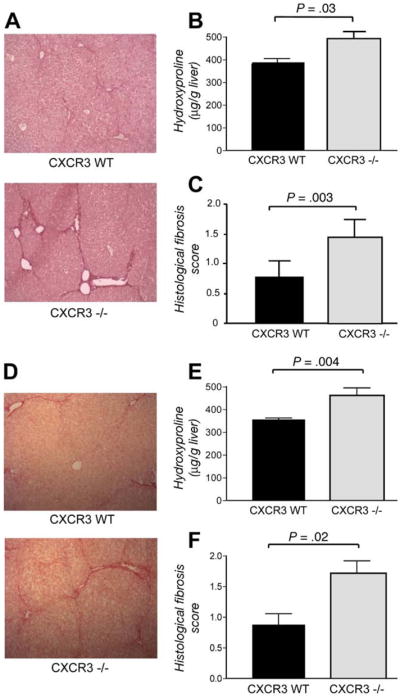
In vivo evidence for a role of CXCR3 for liver fibrosis. A ( ) Representative Sirius red stainings of wild-type and CXCR3−/− mice after challenge with CCl4. (B) Augmented fibrosis in CXCR3−/− mice is validated by increased concentrations of hydroxyproline (P = .03) and (C ) significantly higher semiquantitative fibrosis scores (P = .003). D ( ) The exaggerated fibrogenic response in CXCR3−/− mice is confirmed in the thioacetamide model of liver fibrosis as shown by representative Sirius red stains. Increased deposition of collagen in CXCR3−/− animals is shown by (E) hydroxyproline concentrations (P = .004) and (F) increased fibrosis scores (P = .02).
Increased Fibrosis in CXCR3−/− Mice Is Associated With a Blunted Intrahepatic IFN-γ Response
Because the CXCL9/CXCR3 system mediates the recruitment of immune cells upon liver injury, we next analyzed the hepatic mRNA expression of Ifn-γ at baseline and after treatment with CCl4. As depicted in Figure 6A, CXCR3−/− mice had significantly decreased Ifn-γ mRNA levels compared with wild-type mice after induction of fibrosis (P = .04). To validate these findings, we next performed fluorescence-activated cell sorter analysis for intrahepatic Ifn-γ–positive T cells before and after CCl4 treatment. As shown in Figure 6B and C, the recruitment of Ifn-γ–positive T cells into the liver after CCl4 challenge is reduced in CXCR3−/− mice compared with their wild-type littermates (P = .01). In contrast to the fibrotic features, the necroinflammatory response is not significantly altered in CXCR3−/− mice as shown by histology (Figure 6D) and aspartate aminotransferase/ala-nine aminotransferase levels (Figure 6E).
Figure 6.

Immunologic characterization of CXCR3−/− mice. (A ) Deletion of CXCR3 significantly alters the intrahepatic Ifn-γ mRNA expression after induction of fibrosis (P = .04). (B ) Representative fluorescence-activated cell sorter analysis of intracellular Ifn-γ–positive T cells in fibrotic livers of CXCR3−/− mice and wild-type littermates. (C ) Abundance of CXCR3 leads to a significant reduction of Ifn-γ–positive T cells after CCl4 administration for 6 weeks (P = .01). Despite changes in fibrotic features, the necroinflammatory response is not significantly altered in CXCR3−/− mice as demonstrated by (D) histology and (E) aspartate aminotrans-ferase/alanine aminotransferase levels.
Discussion
We here identify the CXCL9/CXCR3 system as an antifibrotic pathway of liver fibrosis in vivo and in vitro. The concordant results in murine models and the data obtained in patients with chronic liver diseases suggest that the CXCL9/CXCR3 pathway is conserved between species and might therefore be an attractive target for the evaluation of antifibrotic treatment strategies.
As a first approach to identify novel mediators of fibrogenesis, we performed extensive genetic analysis in inbred mice and patients with HCV infection. In both species, we identified variants in the genetic region of IFN-inducible chemokines that are statistically associated with the degree of liver fibrosis. In mice, the associated gene variants were fine mapped within the distal part of the Cxc chemokine cluster, a region that has been linked to experimental fibrosis in an earlier study.11 However, identification of new genes and related pathways has been hampered by the lack of reproducibility and scarce functional data. To circumvent these limitations, the Complex Trait Consortium23 has proposed that new candidate genes identified in experimental crosses should be validated by functional analyses.
We therefore performed expression analysis in the parental strains of our experimental cross and found that Cxcl9 is differentially expressed on the mRNA and protein level between BALB/cJ and A/J mice. Interestingly, protein and mRNA expression of Cxcl9 was higher in the resistant strain, suggesting a potential antifibrotic action of this chemokine, which is expressed by hepatocytes,24 endothelial cells,25 and HSCs.26 In infectious murine models, Cxcl9 is strongly up-regulated in the liver during viral infection, thereby enhancing T-cell infiltration.27 Notably, in accordance with our results, Cxcl9 has already been shown to have protective roles in viral-induced encephalitis and hepatitis.28,29
To further dissect the functional role of Cxcl9, we performed experiments in CXCR3−/− mice in which the specific receptor for Cxcl9 is deleted.12 Consistent with our genetic and expression results, these mice displayed pronounced liver fibrosis in 2 independent models of chronic liver injury. Enhanced fibrosis in CXCR3−/− mice was associated with reduced mRNA expression levels of Ifn-γ and a reduction of Ifn-γ–positive intrahepatic T cells. Importantly, such a Th2-dominated cytokine environment has already also been associated with enhanced pulmonary and renal fibrosis in CXCR3−/− mice.30,31
Interestingly, we were able to validate the genetic findings in mice by haplotype analysis in patients with HCV infection. In this cohort, an htSNP in the 3′-untranslated region of CXCL9 was associated with severe fibrosis with odds ratios that are expected for genetically complex diseases.32 We did not analyze mRNA stability, but the risk allele of this SNP was functionally associated with reduced serum levels of CXCL9 in our patients. Because the risk allele is overrepresented in subjects with severe fibrosis, the reduced serum concentrations of the IFN-inducible CXCL9 are a first hint for a potential antifibrotic role of this chemokine in humans as well. Whether genetic variation in the CXCL9 receptor CXCR3 also contributes to the findings in our cohort needs to be investigated, but only one SNP has yet been identified in CXCR3 in white subjects (www.hapmap.org). Nevertheless, while the antifibrotic effects of IFN and related pathways are well proven in mice,33 the evidence is less convincing in humans. In fact, the largest intervention trial with IFN-γ for reversion of HCV-induced liver fibrosis did not show an overall beneficial effect of this cytokine.34
However, subgroup analysis indicated that individuals with a strong up-regulation of IFN-inducible chemokines do benefit from IFN-γ.34 We therefore further investigated the role of CXCL9 in humans by measuring intra-hepatic and peripheral concentrations. In line with recent studies,35,36 intrahepatic CXCL9 mRNA and protein increased with higher stages of fibrosis. We extend these results by showing higher CXCL9 mRNA levels in non-alcoholic steatohepatitis and a correlation between CXCL9 serum levels and liver fibrosis. At this stage, it is difficult to explain why a potentially antifibrotic chemokine is elevated at higher stages of fibrosis. This might be due to differences in IFN responsiveness between species and the known differences in CXCR3 isoform expression between mice and humans,18,37 but more experiments are clearly warranted to mechanistically resolve these aspects.
Interestingly, HSCs are known to express CXCR3.22 Thus, we assessed the direct effects of CXCL9 on HSCs and LX-2 cells17 in vitro, revealing reproducible repression of collagen mRNA and protein. However, the anti-fibrotic action of CXCL9 on HSCs could be counterbalanced by a greater inflammatory infiltrate associated with this chemokine, leading to increased fibrosis, despite the direct effects of CXCL9 on stellate cells. We also acknowledge that the antifibrotic action of CXCL9 on stellate cells might not be strong enough to ultimately cause a reduction in fibrosis in human diseases. Notably, HSCs expressed both splice variants of CXCR3, which can mediate opposite effects.38 We therefore also stimulated LX-2 cells with CXCL4, a selective ligand for CXCR3-B.18 As shown in Figure 4F, CXCL4 had only minor effects on collagen mRNA expression. Therefore, the observed effects of CXCL9 on stellate cells do not seem to be mediated by the CXCR3-B isoform. However, CXCL4 has been shown to interact with other chemokines and might thus mediate effects via other receptors on HSCs, for example, by interacting with the CCR5 ligand CCL5.39
In summary, we here identify CXCL9 and its receptor CXCR3 as regulators of liver fibrosis in mice and provide first data for a role of this chemokine system in patients with HCV infection. These findings reinforce the involvement of mediators classically associated with the innate immune system in liver regeneration and chronic injury40 and set the stage for further evaluation of this chemokine system as a potential antifibrotic target.
Supplementary Material
Acknowledgments
The authors thank all patients for providing serum and DNA samples for this study.
Supported by grants from the Deutsche Forschungsgemeinschaft WA 2557/1-1, LA 997/4-1, and SFB-TRR57; National Institutes of Health grant R01DK56621 (to S.L.F.); Aachen University (START grants to H.E.W. and R.W.); and the Interdisciplinary Centre for Clinical Research “BIOMAT” within the Faculty of Medicine at the RWTH Aachen University (to H.E.W.).
Abbreviations in this paper
- HCV
hepatitis C virus
- HSC
hepatic stellate cell
- htSNP
haplotype-tagging single nucleotide polymorphism
- IFN
interferon
- SNP
single nucleotide polymorphism
Footnotes
Supplementary Data: Note: To access the supplementary material accompanying this article, visit the online version of Gastroenterology at www.gastrojournal.org, and at doi: 10.1053/j.gastro.2009.03.053.
Conflicts of interest
The authors disclose no conflicts.
References
- 1.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wynn T. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655–1669. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Missiha SB, Ostrowski M, Heathcote EJ. Disease progression in chronic hepatitis C: modifiable and nonmodifiable factors. Gastroenterology. 2008;134:1699–1714. doi: 10.1053/j.gastro.2008.02.069. [DOI] [PubMed] [Google Scholar]
- 5.Juran BD, Lazaridis KN. Applying genomics to the study of complex disease. Semin Liver Dis. 2007;27:3–12. doi: 10.1055/s-2006-960167. [DOI] [PubMed] [Google Scholar]
- 6.Huang H, Shiffman ML, Friedman S, et al. A 7 gene signature identifies the risk of developing cirrhosis in patients with chronic hepatitis C. Hepatology. 2007;46:297–306. doi: 10.1002/hep.21695. [DOI] [PubMed] [Google Scholar]
- 7.Hillebrandt S, Wasmuth HE, Weiskirchen R, et al. Complement factor 5 is a quantitative trait gene that modifies liver fibrogenesis in mice and humans. Nat Genet. 2005;37:835–843. doi: 10.1038/ng1599. [DOI] [PubMed] [Google Scholar]
- 8.Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med. 2006;354:610–621. doi: 10.1056/NEJMra052723. [DOI] [PubMed] [Google Scholar]
- 9.Heydtmann M, Adams DH. Chemokines in the immunopathogenesis of hepatitis C infection. Hepatology. 2009;49:676–688. doi: 10.1002/hep.22763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Strieter RM, Gomperts BN, Keane MP. The role of CXC chemokines in pulmonary fibrosis. J Clin Invest. 2007;117:549–556. doi: 10.1172/JCI30562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hillebrandt S, Goos C, Matern S, et al. Genome-wide analysis of hepatic fibrosis in inbred mice identifies the susceptibility locus Hfib1 on chromosome 15. Gastroenterology. 2002;123:2041–2051. doi: 10.1053/gast.2002.37069. [DOI] [PubMed] [Google Scholar]
- 12.Hancock WW, Lu B, Gao W, et al. Requirement of the chemokine receptor CXCR3 for acute allograft rejection. J Exp Med. 2000;192:1515–1520. doi: 10.1084/jem.192.10.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wacholder S, Rothman N, Caporaso N. Counterpoint: bias from population stratification is not a major threat to the validity of conclusions from epidemiological studies of common polymorphisms and cancer. Cancer Epidemiol Biomarkers Prev. 2002;11:513–520. [PubMed] [Google Scholar]
- 14.Desmet VJ, Gerber M, Hoofnagle JH, et al. Classification of chronic hepatitis: diagnosis, grading and staging. Hepatology. 1994;19:1513–1520. [PubMed] [Google Scholar]
- 15.Stephens M, Donnelly P. A comparison of Bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet. 2003;73:1162–1169. doi: 10.1086/379378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barrett JC, Fry B, Maller J, et al. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 17.Xu L, Hui AY, Albanis E, et al. Human hepatic stellate cell lines, LX-1 and LX-2: new tools for analysis of hepatic fibrosis. Gut. 2005;54:142–151. doi: 10.1136/gut.2004.042127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lasagni L, Francalanci M, Annunziato F, et al. An alternatively spliced variant of CXCR3 mediates the inhibition of endothelial cell growth induced by IP-10, Mig, and I-TAC, and acts as functional receptor for platelet factor 4. J Exp Med. 2003;197:1537–1549. doi: 10.1084/jem.20021897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wiederholt T, von Westernhagen M, Zaldivar MM, et al. Genetic variations of the chemokine scavenger receptor D6 are associated with liver inflammation in chronic hepatitis C. Hum Immunol. 2008;69:861–866. doi: 10.1016/j.humimm.2008.08.275. [DOI] [PubMed] [Google Scholar]
- 20.Jeong WI, Park O, Radaeva S, et al. STAT1 inhibits liver fibrosis in mice by inhibiting stellate cell proliferation and stimulating NK cell cytotoxicity. Hepatology. 2006;44:1441–1451. doi: 10.1002/hep.21419. [DOI] [PubMed] [Google Scholar]
- 21.Weng H, Mertens PR, Gressner AM, et al. IFN-gamma abrogates profibrogenic TGF-beta signaling in liver by targeting expression of inhibitory and receptor Smads. J Hepatol. 2007;46:295–303. doi: 10.1016/j.jhep.2006.09.014. [DOI] [PubMed] [Google Scholar]
- 22.Bonacchi A, Romagnani P, Romanelli RG, et al. Signal transduction by the chemokine receptor CXCR3: activation of Ras/ERK, Src, and phosphatidylinositol 3-kinase/Akt controls cell migration and proliferation in human vascular pericytes. J Biol Chem. 2001;276:9945–9954. doi: 10.1074/jbc.M010303200. [DOI] [PubMed] [Google Scholar]
- 23.Abiola O, Angel JM, Avner P, et al. The nature and identification of quantitative trait loci: a community’s view. Nat Rev Genet. 2003;4:911–916. doi: 10.1038/nrg1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ren X, Kennedy A, Colletti LM. CXC chemokine expression after stimulation with interferon-gamma in primary rat hepatocytes in culture. Shock. 2002;17:513–520. doi: 10.1097/00024382-200206000-00013. [DOI] [PubMed] [Google Scholar]
- 25.Schrage A, Wechsung K, Neumann K, et al. Enhanced T cell transmigration across the murine liver sinusoidal endothelium is mediated by transcytosis and surface presentation of chemokines. Hepatology. 2008;48:1262–1272. doi: 10.1002/hep.22443. [DOI] [PubMed] [Google Scholar]
- 26.Holt AP, Haughton EL, Lalor PF, et al. Liver myofibroblasts regulate infiltration and positioning of lymphocytes in human liver. Gastroenterology. 2009;136:705–714. doi: 10.1053/j.gastro.2008.10.020. [DOI] [PubMed] [Google Scholar]
- 27.Lang KS, Georgiev P, Recher M, et al. Immunoprivileged status of the liver is controlled by Toll-like receptor 3 signaling. J Clin Invest. 2006;116:2456–63. doi: 10.1172/JCI28349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hokeness KL, Deweerd ES, Munks MW, et al. CXCR3-dependent recruitment of antigen-specific T lymphocytes to the liver during murine cytomegalovirus infection. J Virol. 2007;81:1241–1250. doi: 10.1128/JVI.01937-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muse M, Kane JA, Carr DJ, et al. Insertion of the CXC chemokine ligand 9 (CXCL9) into the mouse hepatitis virus genome results in protection from viral-induced encephalitis and hepatitis. Virology. 2008;382:132–144. doi: 10.1016/j.virol.2008.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jiang D, Liang J, Hodge J, et al. Regulation of pulmonary fibrosis by chemokine receptor CXCR3. J Clin Invest. 2004;114:291–299. doi: 10.1172/JCI16861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakaya I, Wada T, Furuichi K, et al. Blockade of IP-10/CXCR3 promotes progressive renal fibrosis. Nephron Exp Nephrol. 2007;107:e12–e21. doi: 10.1159/000106505. [DOI] [PubMed] [Google Scholar]
- 32.Bodmer W, Bonilla C. Common and rare variants in multifactorial susceptibility to common diseases. Nat Genet. 2008;40:695–701. doi: 10.1038/ng.f.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shi Z, Wakil AE, Rockey DC. Strain-specific differences in mouse hepatic wound healing are mediated by divergent T helper cytokine responses. Proc Natl Acad Sci U S A. 1997;94:10663–10668. doi: 10.1073/pnas.94.20.10663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pockros PJ, Jeffers L, Afdhal N, et al. Final results of a double-blind, placebo-controlled trial of the antifibrotic efficacy of interferon-gamma1b in chronic hepatitis C patients with advanced fibrosis or cirrhosis. Hepatology. 2007;45:569–578. doi: 10.1002/hep.21561. [DOI] [PubMed] [Google Scholar]
- 35.Zeremski M, Petrovic LM, Chiriboga L, et al. Intrahepatic levels of CXCR3-associated chemokines correlate with liver inflammation and fibrosis in chronic hepatitis C. Hepatology. 2008;48:1440–1450. doi: 10.1002/hep.22500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bieche I, Asselah T, Laurendeau I, et al. Molecular profiling of early stage liver fibrosis in patients with chronic hepatitis C virus infection. Virology. 2005;332:130–144. doi: 10.1016/j.virol.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 37.Petrai I, Rombouts K, Lasagni L, et al. Activation of p38(MAPK) mediates the angiostatic effect of the chemokine receptor CXCR3-B. Int J Biochem Cell Biol. 2008;40:1764–1774. doi: 10.1016/j.biocel.2008.01.008. [DOI] [PubMed] [Google Scholar]
- 38.Datta D, Flaxenburg JA, Laxmanan S, et al. Ras-induced modulation of CXCL10 and its receptor splice variant CXCR3-B in MDA-MB-435 and MCF-7 cells: relevance for the development of human breast cancer. Cancer Res. 2006;66:9509–9518. doi: 10.1158/0008-5472.CAN-05-4345. [DOI] [PubMed] [Google Scholar]
- 39.von Hundelshausen P, Koenen RR, Sack M, et al. Heterophilic interactions of platelet factor 4 and RANTES promote monocyte arrest on endothelium. Blood. 2005;105:924–930. doi: 10.1182/blood-2004-06-2475. [DOI] [PubMed] [Google Scholar]
- 40.Gao B, Jeong WI, Tian Z. Liver: an organ with predominant innate immunity. Hepatology. 2008;47:729–736. doi: 10.1002/hep.22034. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.