

Abstract
A humanized IgG4 anti–HLA-DR monoclonal antibody (IMMU-114), engineered to avoid side effects associated with complement activation, was examined for binding and cytotoxicity on leukemia, lymphoma, and multiple myeloma cell lines and chronic lymphocytic leukemia (CLL) patient specimens, followed by evaluation of the effects of IMMU-114 on extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) signaling pathways. HLA-DR was expressed on the majority of these cells at markedly higher levels than CD20, CD22, and CD74. IMMU-114 was toxic to mantle cell lymphoma, CLL, acute lymphoblastic leukemia, hairy cell leukemia, non-Hodgkin lymphoma (including rituximab-resistant), and multiple myeloma cell lines, and also patient CLL cells. IMMU-114 induced disease-free survival in tumor-bearing SCID mice with early-stage disease and in models that are relatively resistant to anti-CD20 monoclonal antibodies. Despite positive staining, acute myelogenous leukemic cells were not killed by IMMU-114. The ability of IMMU-114 to induce activation of ERK and JNK signaling correlated with cytotoxicity and differentiates the mechanism of action of IMMU-114 from monoclonal antibodies against CD20 and CD74. Thus, antigen expression is not sufficient for cytotoxicity; antibody-induced hyperactivation of ERK and JNK mitogen activated protein kinase signaling pathways are also required.
Introduction
Monoclonal antibodies (mAbs) against many cell-surface differentiation antigens exert their in vivo effects largely through immunologic effector mechanisms, including complement-dependent cytotoxicity (CDC) and antibody-dependent cellular cytotoxicity (ADCC), although direct apoptosis also occurs.1,2 Thus, their efficacy is dependent mostly on intact immunologic mechanisms in the treated patient. Moreover, it is probable that these effector mechanisms mediate side effects observed on mAb administration, such as B-cell depletion3 and infusion-related toxicity. In contrast, hL243γ4P (IMMU-114) is an mAb with antitumor activity that is largely a consequence of direct cytotoxic (signaling) events. IMMU-114, a humanized IgG4 form of the murine anti–human leukocyte antigen (HLA-DR) mAb, L243, is a pan-HLA-DR mAb, recognizing a conformational epitope in the αchain of HLA-DR. It has been used against melanoma and in models for autoimmune disease, specifically rheumatoid arthritis.4,5 Because HLA-DR is expressed at high levels on a range of hematologic malignancies, there has been considerable interest in its role as a target for antibody-based therapy. However, safety concerns have been raised regarding the clinical use of HLA-DR–directed antibodies because the antigen is expressed on normal as well as tumor cells. In addition, anti–HLA-DR mAbs are potent inducers of complement activation,6 which plays a pivotal role in the pathogenesis of the side effects of mAb infusion.7 The IgG4 IMMU-114 antibody was specifically generated to derive an agent that is able to kill tumor cells without CDC or ADCC.8 Replacing the Fc region of hL243γ1, a humanized IgG1 anti–HLA-DR mAb, with the IgG4 isotype abrogated the effector cell functions of the antibody (ADCC and CDC), whereas the antigen-binding properties, antiproliferative capacity, and the ability to induce apoptosis concurrent with activation of the AKT survival pathway were retained. A point mutation, Ser241Pro (based on Kabat numbering), was introduced into the hinge region of the γ4 sequence to avoid formation of half molecules when the antibody is expressed and produced in mammalian cell cultures.9 Thus, IMMU-114 is indistinguishable from hL243γ1 and the parental murine mAb in assays dependent on antigen recognition, and the abrogation of ADCC and CDC may make this antibody an attractive clinical agent.
To define the potential use of IMMU-114 as a novel therapeutic, we examined its reactivity and cytotoxicity on a panel of leukemia, lymphoma, and multiple myeloma (MM) cell lines, and chronic lymphocytic leukemia (CLL) patient specimens. In addition, the signaling pathways activated by binding of IMMU-114 to the surface of malignant cells were studied. We found that antigen expression is not sufficient for cytotoxicity, but antibody-induced activation of extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) stress signaling pathways also are required. These data demonstrate that IMMU-114 may be useful in the treatment of mantle cell lymphoma (MCL), acute lymphoblastic leukemia (ALL), hairy cell leukemia (HCL), CLL, and MM, as well as non-Hodgkin lymphoma (NHL) shown in our earlier report.8
Methods
Cells
Cell lines were obtained as follows: the acute myelogenous leukemia (AML) lines, GDM-1, Kasumi-1, and Kasumi-3, from ATCC; MCL lines Jeko-1 and Granta-519, ALL lines RS4;11, REH, 697, and MN60, and the HCL line HC-1 from the German Collection of Microorganisms and Cell Cultures (DSMZ); CLL lines, MEC-1 and WAC, from Dr John Byrd (Ohio State University, Columbus, OH); NHL Burkitt lymphomas, Daudi, Raji, and Ramos, from ATCC; the non-Burkitt lymphoma cell line, RL, from Dr John Gribben (Dana-Farber Cancer Institute, Boston, MA); SU-DHL-6 from Dr Alan Epstein (University of Southern California, Los Angeles); WSU-FSCCL from Dr Mitchell Smith (Fox Chase Cancer Center, Philadelphia, PA); MM cell lines, KMS11, KMS-BM, and KMS12-PE, from Dr T. Otsuki (Kawasaki Medical School, Okayama, Japan); CAG from Dr Joshua Epstein (University of Arkansas, Fayetteville); OPM-2 from Dr Kenji Oritani (Osaka University, Japan); rituximab-resistant cell lines, Raji-2R, Raji-4RH, SU-DHL-4–2R, and SU-DHL-4–4RH (and the Raji and SU-DHL-4 parental cells from which these clones were derived) from Drs Myron Czuczman and Francisco J. Hernandez-Ilizaliturri (Roswell Park Cancer Institute, Buffalo, NY).
Patient CLL cells were collected from whole blood by Ficoll-Hypaque separation, and the percentage of CD5+/CD19+ cells was determined by 2-color flow cytometry. No further purification was performed because all specimens contained 85% or greater CD5+/CD19+ cells. The diagnosis of CLL was based on standard criteria, and patients were staged according to the Rai staging system.10 Cells were used fresh or viably frozen in freezing media consisting of 70% RPMI 1640, 20% fetal bovine serum, and 10% dimethyl sulfoxide at −135°C. All CLL patients gave written informed consent under an approved protocol of the Institutional Review Board of Weill Cornell Medical College.
Antibodies
The humanized mAbs milatuzumab (hLL1, anti-CD74 MAb),11 IMMU-114 (hL243γ4P),8 veltuzumab (hA20, anti-CD20),12 epratuzumab (hLL2, anti-CD22),13 and labetuzumab (hMN-14, anti-CD66e, anti–carcinoembryonic antigen)14 were provided by Immunomedics Inc. Rituximab was purchased from IDEC Pharmaceuticals Corp.
Immunophenotyping
Humanized anti-CD74, -HLA-DR, -CD20, -CD22, and isotype control (anti–carcinoembryonic antigen) antibodies were directly labeled with the AlexaFluor 488 mAb labeling kit (Invitrogen), according to the directions of the manufacturer for labeling the panel of cells described in Table 1. The degree of labeling was determined to be from 5 to 7 mol of dye per mole of IgG. Retention of immunoreactivity of the labeled antibodies was demonstrated by incubation with an excess of an anti-idiotype antibody developed against each IgG, and examining the shift to a higher molecular weight fraction by size-exclusion high-performance liquid chromatography. Flow cytometric analysis was performed using a FACSCalibur (BD Biosciences).
Table 1.
Antigen expression on cell lines of B-cell malignancies
| Malignancy/cell line | Labetuzumab (isotype control) | IMMU-114 (HLA-DR) | Milatuzumab (CD74) | Epratuzumab (CD22) | Veltuzumab (CD20) |
|---|---|---|---|---|---|
| AML | |||||
| GDM-1 | 4.4 | 553.5 | 35.9 | 7.2 | 4.3 |
| Kasumi-1 | 6.6 | 8.9 | 9.5 | 8.5 | 7.5 |
| Kasumi-3 | 6.0 | 444.9 | 17.5 | 5.8 | 4.9 |
| MCL | |||||
| Jeko-1 | 5.6 | 539.0 | 29.0 | 5.8 | 250.1 |
| Granta-519 | 4.2 | 1797.2 | 41.4 | 21.6 | 805.4 |
| ALL | |||||
| RS4;11 | 3.3 | 252.2 | 35.2 | 59.5 | 3.0 |
| REH | 3.0 | 2583.1 | 70.6 | 50.8 | 15.0 |
| 697 | 4.0 | 1108.4 | 49.5 | 52.2 | 13.1 |
| MN60 | 8.4 | 2522.1 | 57.0 | 44.1 | 719.4 |
| HCL | |||||
| HC-1 | 8.1 | 5501.2 | 164.0 | 73.9 | 1079.4 |
| CLL | |||||
| MEC-1 | 5.4 | 2421.5 | 41.4 | 35.5 | 270.5 |
| WAC | 5.2 | 798.8 | 17.4 | 14.2 | 210.6 |
| NHL | |||||
| Daudi | 8.0 | 2092.5 | 49.8 | 130.2 | 499.8 |
| Namalwa | 5.4 | 554.5 | 24.9 | 17.4 | 24.9 |
| Raji | 6.2 | 2316.8 | 130.6 | 54.6 | 391.3 |
| Ramos | 3.8 | 373.3 | 45.2 | 38.7 | 516.0 |
| RL | 6.2 | 1663.6 | 56.0 | 34.2 | 264.5 |
| SU-DHL-6 | 3.3 | 1186.7 | 73.4 | 28.1 | 1466.4 |
| WSU-FSCCL | 5.3 | 2030.2 | 20.9 | 14.1 | 36.6 |
| MM | |||||
| CAG | 4.1 | 2803.9 | 86.9 | 4.1 | 4.1 |
| KMS-11 | 2.8 | 615.8 | 16.5 | 3.2 | 3.4 |
| KMS12-BM | 5.2 | 1501.8 | 17.9 | 10.5 | 318.0 |
| KMS12-PE | 3.1 | 939.7 | 16.3 | 3.2 | 3.0 |
| OPM-2 | 4.2 | 3.4 | 3.4 | 3.2 | 3.3 |
Values are mean fluorescence intensities.
Ex vivo assessment of mAb effects on PBCs
The effects of mAbs on peripheral blood cells (PBCs) from healthy volunteers were evaluated ex vivo using flow cytometry. Blood specimens were collected from healthy volunteers under a protocol approved by the New England Institutional Review Board (Wellesley, MA). Heparinized whole blood (150 μL) was incubated with test mAbs (50nM) for 2 days at 37°C and 5% CO2. Labetuzumab was used as a negative control mAb. The normal B and T cells were identified as CD19+ and CD3+ cells, respectively, in the lymphocyte gate. To evaluate the effects of the mAbs on additional cell types, blood samples were incubated with mAbs (33nM) or phosphate-buffered saline (PBS), in the presence of phytohemagglutinin (PHA, 2 μg/mL; Sigma-Aldrich), or lipopolysaccharide (LPS, 20 ng/mL, 0111:B4; Sigma-Aldrich), or PBS as nonactivation control for 2 days, and then stained with a mixture of mAbs containing anti–CD14/CD19-FITC (Invitrogen), anti–CD56-PE (Invitrogen), anti–CD3-PerCp (BD Biosciences), and anti–CD69-APC (BD Biosciences), followed by lysis of erythrocytes. The cells were fixed with 1.5% paraformadehyde before analysis by flow cytometry. All assays were done in triplicate. Student t test was used to evaluate statistical significance (P < .05).
In vitro MTT assay
The 3-(4,5-dimethyl thiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay, first described by Mosmann,15 was used to evaluate the in vitro cytotoxicity by quantifying viable cells after antibody treatments (5 μg/mL final concentration) for 4 days at 37°C in the absence or presence of goat anti–human (GAH) IgG (20 μg/mL final concentration).
Immunoblot analysis
Selected cell lines (1 × 107 cells) were treated with IMMU-114 (10nM), rituximab (20 μg/mL), veltuzumab (20 μg/mL), or milatuzumab (20 μg/mL) with or without a GAH IgG secondary antibody (10 μg/mL) for 24 hours. CLL patient samples (5 × 105 cells) were incubated with mAbs for 4 hours. Cells were treated with lower concentrations of IMMU-114 antibody because the antibody is potent and capable of inducing apoptosis at much lower concentrations. After incubation with mAbs, the cells were washed in PBS, centrifuged, and lysed in ice-cold radioimmunoprecipitation assay (RIPA) buffer. Equal amounts of protein samples (25 μg) were separated on 4% to 20% gradient Tris-glycine gels and transferred onto nitrocellulose membrane. Membranes were blocked in milk, incubated overnight with primary antibody, 1:1000 dilution in Tris-buffered saline/Tween 20 containing 5% bovine serum albumin at 4°C (Cell Signaling Technology), followed by probing with horseradish peroxidase-conjugated secondary antibody (1:5000 dilution). Blots were visualized with enhanced chemiluminescence (Thermo Scientific). Bands were quantified using a Kodak Image Station.
Annexin V binding assay
Cells were plated in 6-well plates (2 × 105 cells per well) and pretreated with various inhibitors for 2 hours. IMMU-114 (10nM) was added to the wells, and incubation continued for 24 hours. N-Acetylcysteine (NAC, 5mM) was used to block the reactive oxygen species (ROS).16 U0126 (10μM) blocks MEK phosophorylation and the ERK1/2 pathway.17 SP600125 (10μM) blocks the JNK pathway.18 Inhibitors were purchased from Calbiochem. After overnight incubation, cells were washed 3 times in PBS, centrifuged, and reconstituted in 100 μL of annexin binding buffer (10mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, 140mM NaCl, and 2.5mM CaCl2 in PBS). Cells were stained with 5 μL of annexin V conjugate for 20 minutes, followed by staining with propidium iodide (1 μg/mL) in 400 μL of annexin binding buffer, and then analyzed by flow cytometry.
Inhibition of ERK and JNK pathways by siRNA analysis
The specific siRNAs for the ERK and JNK MAPK pathways were bought from Cell Signaling Technology. Raji cells were transfected with either siERK, siJNK, or both siRNAs in the presence of transfection reagent DMRIE-C (Invitrogen) for 48 hours, as described by the manufacturers. IMMU-114 (10nM) was then added to the mixture, and the incubation continued for another 16 hours. Cells were washed with PBS, stained with annexin V conjugate for 20 minutes, and then analyzed by flow cytometry. Transfection reagent-treated cells and scrambled nonspecific siRNA-transfected cells were prepared as controls. To analyze the changes in expression profile of proteins, aliquots of the cells were washed in PBS, centrifuged, and lysed in ice-cold RIPA buffer. Equal amounts of protein samples (5 μg) were separated on 4% to 20% gradient Tris-glycine gels, transferred onto nitrocellulose membranes, and probed with anti-ERK1/2 or JNK antibodies.
Assessment of mitochondrial depolarization (Δψm) and ROS production
Mitochondrial membrane potential depolarization was determined using the fluorescent dye TMRE (Invitrogen), as described.19 Briefly, Raji cells (2 × 105 cells per well) in 6-well plates were treated with IMMU-114 for the indicated time points followed by incubation with TMRE (50nM final concentration) for 30 minutes at 37°C in the dark. Cells were washed 3 times with PBS and analyzed by flow cytometry. For ROS production, cells were incubated with the antibodies as for mitochondrial depolarization and stained with 2.5μM of dihydroethidium (Invitrogen) in PBS and incubated in the dark at 37° for 30 minutes as described.16 For the inhibition experiment, cells were pretreated with 5mM of NAC for 2 hours before IMMU-114 treatment.
In vivo studies in SCID mice bearing disseminated MM tumors
For studies on the therapeutic effect of the mAbs, 6- to 8-week-old female SCID mice were used (Taconic Farms). Myeloma cells were injected via the tail vein. Treatments were initiated after injection of tumor cells according to dose schedules described for each experiment. Mice were examined daily for signs of distress or hind-leg paralysis and weighed weekly. Paralysis of the hind legs or a weight loss of more than 20% was used as the survival endpoint, when animals were killed. Animal studies were performed under protocols approved by the Center for Molecular Medicine and Immunology Institutional Animal Care and Use Committee.
Results
Reactivity and cytotoxicity of IMMU-114 on a panel of malignant B-cell lines and CLL patient specimens compared with other anti–B-cell mAbs
We demonstrated previously that IMMU-114 is expressed at markedly higher levels and is much more potent in vitro than other naked mAbs tested in NHL cell lines.8 Expression of HLA-DR, CD74, CD22, and CD20 was compared on a panel of malignant B-cell leukemia, lymphoma, and myeloma cell lines (Table 1). HLA-DR was expressed on the cell surface of all MCL, ALL, HCL, CLL, and NHL cell lines tested, as well as 2 of 3 AML and 5 of 6 MM cell lines, with markedly greater binding of anti–HLA-DR than that of the anti-CD74, anti-CD22, and anti-CD20 mAbs. CD74 was expressed on the same cell lines as HLA-DR, but at substantially lower levels. There was little or no expression of CD22 and CD20 on the AML cell lines. CD22 expression was relatively low but positive on the ALL, HCL, CLL, and NHL cell lines, 1 of 2 MCL and 1 of 5 of the MM lines tested. CD20 was strongly positive on the MCL and CLL cell lines and variable on ALL, NHL, and MM.
Cytotoxicity was examined in vitro using MTT assays. IMMU-114 was toxic to 2 of 2 MCL, 2 of 2 CLL, 2 of 4 ALL, 1 of 1 HCL, 2 of 2 NHL, and 2 of 2 MM cell lines without any added crosslinking agent (Table 2). RS4;11 and 697, the 2 ALL cell lines resistant to killing by IMMU-114, express 5- to 10-fold less HLA-DR than the 2 IMMU-114–sensitive ALL cell lines. Despite expression of HLA-DR, 3 of 3 AML cell lines were not killed by IMMU-114 in this assay. In the presence of GAH IgG second antibody, all cell lines except the AML lines and RS4;11 were killed by both IMMU-114 and milatuzumab. In the presence of GAH, veltuzumab was cytotoxic to the MCL, HCL, CLL, and NHL lines tested, but not the AML, 3 of 4 ALL, and the 2 MM lines cell lines. Cytotoxicity of IMMU-114 was compared with that of rituximab in rituximab-resistant lymphoma cell lines. These cell lines were generated from Raji and SU-DHL-4 by chronic exposure to rituximab with or without human serum.20 IMMU-114 was significantly more cytotoxic to both parental and rituximab-resistant NHL cell lines, compared with rituximab (P < .05), whether studied with or without crosslinking by GAH (Figure 1). Without crosslinking, rituximab yielded little or no inhibition relative to untreated cells in the parental or resistant cell lines, although GAH improved the efficacy of rituximab in these lines. As expected, rituximab was more effective in the Raji and SU-DHL-4 parental lines than in the corresponding rituximab-resistant cell lines (P < .05 for Raji-2R, Raji-4RH, and SU-DHL-4–4RH compared with corresponding parental lines).
Table 2.
Cytotoxicity of anti–B-cell mAbs
| Cell line | No second antibody |
With GAH IgG second antibody |
||||
|---|---|---|---|---|---|---|
| IMMU-114 (HLA-DR) | Milatuzumab (CD74) | Veltuzumab (CD20) | IMMU-114 (HLA-DR) | Milatuzumab (CD74) | Veltuzumab (CD20) | |
| AML | ||||||
| GDM-1 | 106 ± 14 | 102 ± 15 | 100 ± 11 | 104 ± 10 | 129 ± 10 | 112 ± 15 |
| Kasumi-1 | 92 ± 6 | 84 ± 5 | 88 ± 16 | 89 ± 11 | 88 ± 9 | 99 ± 5 |
| Kasumi-3 | 120 ± 8 | 116 ± 7 | 112 ± 8 | 114 ± 10 | 123 ± 9 | 112 ± 18 |
| MCL | ||||||
| Jeko-1 | 41 ± 5* | 117 ± 10 | 124 ± 8 | 26 ± 2* | 33 ± 1* | 67 ± 3* |
| Granta-519 | 79 ± 3* | 104 ± 6 | 95 ± 3 | 65 ± 2* | 65 ± 3* | 65 ± 2* |
| ALL | ||||||
| RS4;11 | 78 ± 4 | 102 ± 10 | 88 ± 11 | 91 ± 10 | 89 ± 5 | 107 ± 6 |
| REH | 50 ± 5* | 79 ± 10 | 88 ± 7 | 29 ± 3* | 29 ± 2* | 95 ± 10 |
| 697 | 91 ± 13 | 105 ± 8 | 115 ± 12 | 58 ± 4* | 59 ± 8* | 124 ± 8 |
| MN60 | 35 ± 3* | 88 ± 15 | 92 ± 10 | 27 ± 3* | 35 ± 3* | 56 ± 2* |
| HCL | ||||||
| HC-1 | 28 ± 2* | 121 ± 6 | 91 ± 11 | 17 ± 1* | 19 ± 1* | 49 ± 10* |
| CLL | ||||||
| MEC-1 | 69 ± 3* | 100 ± 8 | 99 ± 5 | 30 ± 2* | 52 ± 2* | 63 ± 6* |
| WAC | 54 ± 7* | 97 ± 11 | 102 ± 17 | 41 ± 4* | 51 ± 7* | 62 ± 8* |
| NHL | ||||||
| Raji | 63 ± 11* | 89 ± 4 | 95 ± 5 | 10 ± 28 | 36 ± 3* | 76 ± 10* |
| RL | 11 ± 1* | 126 ± 4 | 100 ± 16 | 20 ± 0* | 31 ± 3* | 56 ± 5* |
| MM | ||||||
| CAG | 69 ± 10* | 90 ± 21 | 82 ± 11 | 51 ± 11* | 67 ± 15* | 79 ± 8 |
| KMS-11 | 31 ± 4* | 89 ± 15 | 88 ± 3 | 21 ± 1* | 22 ± 5* | 63 ± 14 |
Values are percentage of untreated values in MTT assay.
Statistically significant (P < .05) decrease of 20% or more.
Figure 1.

Cytotoxicity of IMMU-114 and rituximab on rituximab-resistant cell lines. Percentage of untreated values in MTT assay is shown. 1IMMU-114 yields significantly more inhibition (P < .05) than rituximab when incubated without GAH. 2IMMU-114 yields significantly more inhibition (P < .05) than rituximab when incubated in the presence of GAH. 3Rituximab is significantly more effective (P < .05) in the parental lines than in the corresponding rituximab-resistant cell lines in Raji-2R, Raji-4RH, and SU-DHL-4–4RH.
Peripheral blood samples were obtained from 6 representative CLL patients, who ranged in age from 62 to 83 years and had received either no prior treatment or the various therapies shown in Table 3 but were not currently on active treatment. CD5+/CD19+ cells represented more than or equal to 85% of the total lymphocyte counts, indicating a high percentage of CLL cells. All patient specimens expressed HLA-DR. Four of the 6 samples expressed HLA-DR at markedly higher levels than CD20, with the remaining 2 showing similar HLA-DR and CD20 expression (Table 4). It is interesting to note that expression of HLA-DR was higher in patients with no prior therapy (mean fluorescence intensity, 505.8-919.0) than in the previously treated patients (mean fluorescence intensity, 23.2-148.6). Greater patient numbers will be needed to substantiate this trend. Cytotoxicity of anti–B-cell mAbs on 4 of the clinical CLL specimens was tested by incubating them with the mAbs, with and without second antibody crosslinking, for 2 days (Figure 2). Approximately 60% inhibition (range, 50.9% ± 7.6% to 61.9% ± 8.6% inhibition relative to untreated cells) was obtained after incubation with IMMU-114, regardless of the presence or absence of second antibody. In contrast, neither rituximab nor veltuzumab yielded relevant inhibition. Significant milatuzumab inhibition was obtained in 3 of 4 specimens; and consistent with previous data on milatuzumab cytotoxicity, crosslinking was necessary for activity.11
Table 3.
Clinical characteristics
| Patient ID | Age, y/sex | Rai stage | Previous therapy | WBC count, 1000/μL | CD5+/CD19+, percentage |
|---|---|---|---|---|---|
| CLL030 | 62/male | 1 | Id-KLH vaccine, IVIG | 28.3 | 91.4 |
| CLL037 | 72/female | 1 | No prior therapy | 137.3 | 84.5 |
| CLL078 | 75/female | 0 | No prior therapy | 118.4 | 97.4 |
| CLL107 | 83/male | 3 | Fludarabine, Cytoxan, IVIG | 52.2 | 97.0 |
| CLL113 | 63/female | 3 | No prior therapy | 214.5 | 95.8 |
All are Zap70+ and CD38−.
WBC indicates white blood cells; Id-KLH, idiotype conjugated to keyhole limpet hemocyanin; and IVIG, intravenous immunoglobulin G.
Table 4.
Antigen expression
| Patient ID | None | Labetuzumab (CEA, isotype control) | IMMU-114 (HLA-DR) | Milatuzumab (CD74) | Epratuzumab (CD22) | Veltuzumab (CD20) |
|---|---|---|---|---|---|---|
| CLL030 | 4.5 | 4.5 | 23.2 | 6.8 | 5.3 | 20.1 |
| CLL037 | 4.6 | 5.6 | 505.8 | 15.5 | 10.1 | 87.2 |
| CLL078 | 4.8 | 7.0 | 919.0 | 30.9 | 7.7 | 284.3 |
| CLL107 | 15.6 | 10.9 | 148.6 | 11.9 | 12.5 | 85.0 |
| CLL113 | 15.4 | 23.0 | 728.6 | 59.9 | 26.2 | 411.3 |
| CLL117 | 21.6 | 23.4 | 90.9 | 30.2 | 25.2 | 111.0 |
Values are mean fluorescence intensities.
Figure 2.

Cytotoxicity of anti–B-cell mAbs on clinical CLL specimens. Percentage of untreated values in MTT assay is shown. *P < .05 versus isotype control. □ represent no second antibody;  , GAH IgG included in the incubation. ND indicates not done.
, GAH IgG included in the incubation. ND indicates not done.
Effects of IMMU-114 on PBCs from healthy blood donors
The toxicity of IMMU-114 on normal PBCs was examined using an ex vivo flow cytometry assay on whole blood from healthy volunteer donors. Data shown in Figure 3 are representative of those obtained from experiments with 5 healthy volunteer blood donors. Two-day incubation of heparinized whole blood with IMMU-114 or rituximab yielded significant decreases in B cells relative to control incubations in all cases (P < .05). IMMU-114 yielded decreases in B-cell counts that ranged from 43.6% plus or minus 2.9% to 70.0% plus or minus 1.0%. Incubation with rituximab yielded significantly greater killing than IMMU-114 in 4 cases (range, 76.2% ± 2.4% to 90.1% ± 2.73%, P < .04, relative to IMMU-114) and similar killing (36.4% ± 10.1%, P = .315) in the fifth. None of the treatments decreased T cells significantly.
Figure 3.
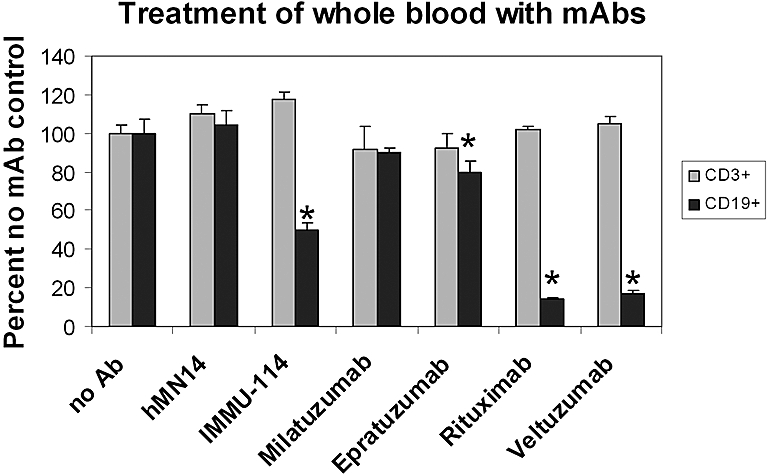
The effect of IMMU-114 on peripheral blood lymphocytes from healthy volunteers. Decreases in CD19+ and CD3+ cells present in the lymphocyte gate after a 2-day incubation of heparinized whole blood of healthy volunteers with mAbs were measured. Data are shown as percentage of untreated control. Error bars represent SD of 3 replicates. *Significant decrease (P < .05) in positive cell count relative to no mAb control.
A second study was designed to test the effects of IMMU-114 and rituximab on the absolute number of monocytes, B cells, T cells, NK cells (CD56high and CD56medium), and NKT cells. The percentage of activated T cells, NK, and NKT cells was measured by CD69 staining. The blood cells were evaluated without stimulation and after PHA or LPS stimulation. PHA is a T-cell mitogen, with only a marginal ability to activate B cells, whereas LPS is a B-cell mitogen.21 Consistent with the published effects of PHA and LPS, we observed that only 1% to 2% of B cells were activated (CD86+) after one day of PHA stimulation, compared with more than 10% after LPS stimulation (data not shown). The gating strategy for monocytes and lymphocyte subpopulations is shown in supplemental Figure 1A (available on the Blood Web site; see the Supplemental Materials link at the top of the online article). As shown in Table 5 and supplemental Figure 1B, these experiments confirmed that, in unstimulated and PHA-stimulated blood, B-cell count decreases caused by IMMU-114 were less than those caused by rituximab. However, in LPS-stimulated blood, B-cell depletion by IMMU-114 exceeded that of rituximab (P < .05 for IMMU-114 vs rituximab). This suggests that IMMU-114 preferentially eliminates activated B cells.
Table 5.
Effects of mAbs on monocytes and lymphocyte subsets in human whole blood
| Stimulation/mAb | B cells | Monocytes | CD56high NK | CD56medium NK | NKT cells | T cells | CD69+CD56high NK cells | CD69+CD56medium NK cells | CD69+ NKT cells | CD69+ T cells |
|---|---|---|---|---|---|---|---|---|---|---|
| PBS | ||||||||||
| None | 4.72 ± 0.30 | 0.63 ± 0.06 | 0.21 ± 0.06 | 1.88 ± 0.06 | 3.20 ± 0.79 | 39.3 ± 3.4 | 1.51 ± 0.75 | 2.47 ± 0.34 | 1.99 ± 0.12 | 0.98 ± 0.12 |
| Labetuzumab | 3.92 ± 0.53 | 0.69 ± 0.16 | 0.34 ± 0.21 | 1.69 ± 0.05 | 3.52 ± 0.68 | 37.0 ± 2.1 | 4.21 ± 2.67 | 8.83 ± 4.15 | 3.16 ± 0.71 | 1.15 ± 0.52 |
| IMMU-114 | 2.11 ± 0.77* | 0.41 ± 0.08* | 0.18 ± 0.05 | 1.90 ± 0.09 | 3.18 ± 0.38 | 38.4 ± 1.9 | 1.96 ± 1.00 | 2.55 ± 0.52 | 1.83 ± 0.13 | 0.76 ± 0.16 |
| Rituximab | 1.45 ± 0.32* | 0.95 ± 0.04 | 0.19 ± 0.02 | 1.84 ± 0.06 | 3.21 ± 0.18 | 37.3 ± 1.2 | 2.83 ± 0.76 | 4.20 ± 0.82 | 2.14 ± 0.55 | 0.83 ± 0.27 |
| PHA | ||||||||||
| None | 3.82 ± 0.56 | 1.03 ± 0.15 | 0.19 ± 0.05 | 1.76 ± 0.02 | 3.02 ± 0.25 | 34.9 ± 0.7 | 2.26 ± 1.14 | 2.38 ± 0.65 | 1.84 ± 0.42 | 0.79 ± 0.09 |
| Labetuzumab | 3.51 ± 0.62 | 1.13 ± 0.07 | 0.15 ± 0.03 | 1.60 ± 0.09 | 2.83 ± 0.30 | 33.7 ± 0.9 | 2.14 ± 1.05 | 4.92 ± 0.47 | 1.93 ± 0.17 | 0.83 ± 0.03 |
| IMMU-114 | 1.94 ± 0.57* | 0.46 ± 0.13* | 0.22 ± 0.09 | 1.82 ± 0.02 | 3.29 ± 0.61 | 36.8 ± 1.8 | 3.07 ± 0.64 | 3.75 ± 0.34 | 2.56 ± 0.45 | 1.04 ± 0.40 |
| Rituximab | 1.23 ± 0.17* | 0.94 ± 0.36 | 0.22 ± 0.10 | 1.72 ± 0.09 | 3.20 ± 0.29 | 34.5 ± 0.2 | 3.59 ± 1.45 | 6.00 ± 2.64 | 3.21 ± 1.58 | 1.38 ± 0.80 |
| LPS | ||||||||||
| None | 3.13 ± 0.48 | 0.26 ± 0.05 | 0.41 ± 0.09 | 1.63 ± 0.12 | 2.88 ± 0.15 | 34.1 ± 0.4 | 30.75 ± 9.56 | 42.58 ± 7.36 | 10.91 ± 2.65 | 2.47 ± 0.24 |
| Labetuzumab | 3.09 ± 0.21 | 0.33 ± 0.06 | 0.30 ± 0.11 | 1.62 ± 0.03 | 2.92 ± 0.25 | 34.9 ± 0.6 | 14.79 ± 8.96 | 27.94 ± 11.15 | 5.57 ± 2.48 | 1.43 ± 0.36 |
| IMMU-114 | 0.92 ± 0.17*† | 0.18 ± 0.06 | 0.38 ± 0.22 | 1.67 ± 0.26 | 2.81 ± 0.40 | 34.3 ± 2.2 | 24.08 ± 22.77 | 37.02 ± 22.37 | 11.22 ± 7.94 | 2.26 ± 0.76 |
| Rituximab | 2.47 ± 0.30* | 0.32 ± 0.10 | 0.36 ± 0.07 | 1.66 ± 0.24 | 2.90 ± 0.40 | 35.5 ± 0.6 | 26.15 ± 12.03 | 40.79 ± 9.35 | 10.11 ± 2.16 | 2.38 ± 0.45 |
B cells, monocytes, CD56high and CD56medium NK cells, NKT cells, and T cells were gated according to the strategy described in supplemental Figure 1. The percentages of these cell subsets in nucleated cells are shown. Data are representative of 4 blood donors. Activated CD56high NK cells, CD56medium NK cells, NKT cells, and T cells are the percentages of CD69+ cells in each of these cell subsets.
Statistically significant (P < .05) decreases of more than 20% vs both untreated and control mAb (labetuzumab) treatment.
Statistically significant (P < .05) difference between IMMU-114 and rituximab.
IMMU-114, but not rituximab, reduced the number of monocytes (33%–59% reduction vs control mAb). T cells were unaffected under all conditions. Compared with the control mAb, neither IMMU-114 nor rituximab significantly decreased NK cells (including CD56medium and CD56high) or NKT cells. In 3 of 4 blood donors, IMMU-114 did not significantly affect activated (CD69+) T cells, NK, or NKT cells compared with the control mAb, either in unstimulated blood or after PHA or LPS stimulation. However, significant decreases (P < .05) were observed in 1 of the 4 donors in PHA-stimulated CD69+CD56medium NK and CD69+ NKT cells, and in unstimulated CD69+CD56medium NK cells.
Comparison of signaling pathways in IMMU-114–sensitive and -resistant cell lines and CLL patient specimens
Signaling pathways were studied to elucidate why cytotoxicity does not always correlate with antigen expression in the malignant B-cell lines examined in Tables 1 and 2. Various pathways were compared in IMMU-114–sensitive and –resistant HLA-DR–expressing cell lines. The AML lines, Kasumi-3 and GDM-1, were used as examples of HLA-DR+ cell lines resistant to IMMU-114 cytotoxicity. IMMU-114–sensitive cells included NHL (Raji), MCL (Jeko-1 and Granta-519), CLL (WAC and MEC-1), and ALL (REH and MN60). Results of Western blot analyses of these cell lines revealed that IMMU-114 induces phosphorylation and activation of ERK and JNK mitogen activated protein (MAP) kinases in all the cells defined as IMMU-114–sensitive by the cytotoxicity assays, but not the IMMU-114–resistant cell lines, Kasumi-3 and GDM-1 (Figure 4A). p38 MAP kinase was found to be constitutively active in these cell lines, and no further activation beyond basal levels was noted (data not shown).
Figure 4.

Comparison of signaling pathways in IMMU-114–sensitive and –resistant cells. (A) Western blot analyses of ERK1/2 and JNK1/2 phosphorylation on cell extracts of IMMU-114–sensitive and –resistant cell lines after mAb treatment. (B) Effect of ERK, JNK, and ROS inhibitors on IMMU-114–mediated apoptosis in Raji. NAC (5mM) was used to block the ROS. U0126 (10μM) blocks MEK phosophorylation and the ERK1/2 pathway. SP600125 (10μM) blocks the JNK pathway. (C) Effect of siERK and siJNK RNAs on the expression of ERK and JNK proteins. Error bars represent SD of 3 replicates. (D) Western blot analyses of ERK and JNK pathways in CLL patient samples.
Two methods were used to confirm the importance of the ERK and JNK signaling pathways in the IMMU-114 mechanism of action, specific chemical inhibitors of these pathways and siRNA. ERK, JNK, and ROS inhibitors used were as follows: NAC (5mM) blocks ROS,16 U0126 (10μM) blocks MEK phosphorylation and the ERK1/2 pathway,17 and SP600125 (10μM) blocks the JNK pathway.18 As shown in Figure 4B, inhibition of ERK, JNK, or ROS by their respective inhibitors decreased apoptosis in Raji cells, although the inhibition was not complete when any single inhibitor was used. This may have been the result of activation of multiple pathways because inhibition of 2 or more pathways by specific inhibitors abolished the IMMU-114–induced apoptosis. Transfection of Raji cells with siERK and siJNK RNAs effectively lowered the expression of ERK and JNK proteins (supplemental Figure 1), and significantly inhibited IMMU-114–induced apoptosis (Figure 4C) validating the role of these pathways in IMMU-114 cell killing.
Consistent with the cytotoxicity data of Table 2, the AML lines, Kasumi-3 and GDM-1, were resistant to apoptosis mediated by IMMU-114 (as measured by annexin V, data not shown). Significant changes in mitochondrial membrane potential and generation of ROS also were not observed on treatment of these AML cell lines with IMMU-114 (not shown). Sensitive lines, such as Raji, show a greater degree of homotypic aggregation on treatment with IMMU-114, whereas aggregation was not observed in AML lines, such as Kasumi-3 (data not shown).
Activation of ERK1/2 and JNK signaling pathways was also assessed in CLL patient samples (Figure 4D). Patient cells were incubated with IMMU-114 for 4 hours because the cells in these samples were much smaller than those of the established cell lines. Moreover, the shorter incubation time avoids the risk of higher apoptosis and cell death. Similar to our observations in the IMMU-114–sensitive cell lines, activation and phosphorylation of the ERK1/2 and JNK pathways were observed in the CLL patient cells, indicating the generation of stress in these samples. Almost 4- to 5-fold activation of ERK and JNK pathways was observed on incubation with IMMU-114 over untreated controls, although no such activation was seen on treatment with rituximab or milatuzumab.
IMMU-114 induces ROS production and changes in mitochondrial membrane potential
To further investigate the molecular mechanism whereby IMMU-114 induces cell death, we investigated the effect of IMMU-114 on changes in mitochondrial membrane potential and production of ROS. Treatment with IMMU-114 induced a time-dependent mitochondrial membrane depolarization that could be detected in Raji cells as well as in other sensitive lines. A time-course analysis in Raji cells indicated a change in mitochondrial membrane depolarization of 46% in as little as 30 minutes of treatment, and a further increase to 66% in 24 hours (Figure 5A). Similar changes in ROS levels were observed (Figure 5A). A 30-minute incubation with IMMU-114 induced a 24% change in ROS levels that increased to 33% to 44% on overnight incubation (Figure 5A-B). Preincubation of Raji cells with the ROS inhibitor NAC blocked the generation of ROS on treatment with IMMU-114; only 8% ROS was observed in IMMU-114 plus NAC-treated cells (Figure 5B). Changes in mitochondrial membrane potential were also abrogated by the ROS inhibitor (Figure 5B).
Figure 5.

IMMU-114 induces changes in mitochondrial membrane potential, generation of ROS, and release of AIF. (A) At the indicated time points (top panel), changes in mitochondrial membrane potential were determined by shift in the emission spectrum toward the left on staining with the dye, TMRE. At the indicated points (bottom panel), generation of ROS was measured by staining with dihydroethidium dye. Dihydroethidium becomes oxidized in the presence of ROS and intercalates within double-stranded DNA and fluoresces. Increased fluorescence measured by the shift to the right indicates increased production of ROS. (B) ROS scavenging by NAC inhibits IMMU-114–induced mitochondrial membrane depolarization and generation of ROS. Cells were pretreated with NAC (5mM) for 2 hours, followed by treatment with IMMU-114 overnight. (C) IMMU-114 induces phosphorylation of pERK1/2, JNK, and release of AIF in a time-dependent manner. Raji cells were incubated with IMMU-114 for the indicated times, and expression levels of various proteins were determined by Western blot analysis of cytosolic extracts on probing with specific antibodies. IMMU-114–mediated apoptosis is caspase-independent, as observed by unchanged levels of caspase-3 and -9.
ROS has been reported to play a crucial role in caspase-independent apoptosis by rituximab.22 We therefore investigated the effects of IMMU-114 on cleavage of caspases-3 and -9 in a time-dependent manner. We did not observe any change or cleavage of these caspases (Figure 5C), although phosphorylation of ERK1/2 and the JNK MAP kinases was observed in as little as 15 minutes. It is probable that the increase in the ROS levels creates intracellular stress in the cell, inducing activation of MAP kinases. The MAP kinases further induce changes in mitochondrial membrane potential, causing the release of mitochondrial apoptosis-inducing factor (AIF) to the cytosol. Time-dependent analysis of the release of AIF revealed that IMMU-114 caused the release of AIF in as little as 15 minutes, with a further increase to steady-state levels in approximately 1 hour (Figure 5C). These data suggest that ROS generation plays a crucial role in IMMU-114–induced cell death and are consistent with the action of IMMU-114 on ROS being an early effect occurring before apoptosis.
In vivo activity of IMMU-114 on tumor-bearing SCID mice
The therapeutic activity of IMMU-114 was evaluated in 3 NHL models: WSU-FSCCL, Namalwa, and Raji. IMMU-114 was more effective than the other anti–B-cell mAbs tested in all models. In the WSU-FSCCL study (Figure 6A), where the mAbs were administered at 12.5 mg/kg (250 μg/injection) beginning the day after injection of tumor cells, IMMU-114 treatment yielded 100% long-term survival. Milatuzumab was also effective, yielding 7 of 10 long-term survivors. Although the other anti–B-cell mAbs also induced significant responses compared with untreated mice, median survival times were markedly less, and no long-term survivors were seen. The dose of IMMU-114 was lowered to 1.75 mg/kg (35 μg/injection) in the Namalwa model, and compared with 25 mg/kg (500 μg/injection) doses of milatuzumab, epratuzumab, and veltuzumab. IMMU-114 yielded 4 of 6 long-term survivors in the Namalwa-bearing SCID mice, compared with none in the other groups (Figure 6B). Because Namalwa and WSU-FSCCL express relatively low levels of CD20, it was not surprising that veltuzumab and rituximab had relatively low activity. In the Raji model (Figure 6C), IMMU-114 was compared with veltuzumab, each at 1.75 mg/kg (35 μg/injection), with initiation of therapy delayed until day 5 after injection of tumor cells. Both antibodies significantly improved survival above the control animals (P < .001). Median survival times were 28 days and 26 days, respectively, for IMMU-114 and veltuzumab; hence, a statistically significance difference was not reached between the 2 mAbs (P = .0608). The survival data obtained in these 3 models demonstrate the in vivo potency of IMMU-114.
Figure 6.

Effect of B-cell mAbs on survival of tumor-bearing SCID mice. (A) SCID mice (10 per group) were injected intravenously with WSU-FSCCL (2.5 × 106 cells/mouse). mAbs were given twice weekly for 4 weeks starting 1 day after injection of cells. (B) SCID mice (6 per group) were injected intravenously with Namalwa (5 × 106 cells). Therapy began 1 day after injection of cells. Mice received 1.75 mg/kg (35 μg) subcutaneously of IMMU-114 3 times per week for 2 weeks. Other groups received 25 mg/kg (500 μg) intraperitoneal injections of veltuzumab, milatuzumab, or epratuzumab every 4 days for a total of 8 injections. (C) SCID mice (10 per group) were injected intravenously with Raji (2.5 × 106 cells/mouse). Therapy began 5 days after injection of cells. Mice received 1.75 mg/kg (35 μg) subcutaneously of veltuzumab or IMMU-114 3 times per week for 2 weeks.
Discussion
The chimeric anti-CD20 mAb, rituximab, was approved by the U.S. Food and Drug Administration in 1997 for treatment of B-cell non-Hodgkin lymphoma after failing other chemotherapy regimens.23 Rituximab induces responses in approximately 50% of patients with previously treated, low-grade, follicular lymphoma, with approximately 10% having a complete response.24 In patients with more aggressive histologic lymphoma subtypes, rituximab therapy has an approximately 30% overall response rate,25 but combined with CHOP chemotherapy, improved objective responses and durations compared with CHOP alone, making R-CHOP a standard protocol for the treatment of diffuse large B-cell lymphoma.26–29 Rituximab's success has led to an expansion of its use in a variety of diseases30–36 and has generated an ongoing effort to improve on the efficacy of rituximab treatments for B-cell malignancies. Efforts include the development of humanized anti-CD20 mAbs,12,37 combining mAbs with other biologic agents, eg, interleukin-226 and interferon,38 combining mAb therapy with chemotherapy, as was done with CHOP,39,40 and the use of mAbs that target antigens other than CD20.41,42
Here we evaluated the antitumor effects and mechanism of action of the humanized anti–HLA-DR IgG4 mAb, IMMU-114. Several factors combine to generate considerable interest in the development of HLA-DR as a target for antibody-based lymphoma therapy. These include the substantial effect of anti–HLA-DR mAbs on lymphoma growth, the lack of antigen modulation after mAb binding, observations of signaling mechanisms playing a major role in antibody-induced toxicity, and the expression of HLA-DR at high levels on a range of hematologic malignancies. Because IMMU-114 does not induce ADCC and CDC,8 this antibody has the potential to avoid the complications associated with complement activation that may have limited the use of another anti–HLA-DR antibody, Hu1D10.6,43 Evaluation of the cytotoxicity of IMMU-114 on a panel of leukemia and lymphoma cell lines showed that HLA-DR is expressed on all but a few of the B-lymphoma, leukemia, and MM cell lines tested at markedly higher levels than CD20, CD22, and CD74. Consistent with our earlier observations in NHL,8 neither crosslinking nor effector-mediated mechanisms (ADCC and CDC) were needed for in vitro cytotoxicity in the cell line panel and on primary B-CLL cells. Interestingly, despite expression of HLA-DR on AML, MCL, ALL, HCL, CLL, NHL, and MM, the AML cell lines were distinguished from the others because they were not killed by IMMU-114 even when HLA-DR is highly expressed.
Direct in vitro and in vivo comparisons between the efficacy of IMMU-114 and the anti-CD20 mAbs, rituximab and veltuzumab, demonstrated remarkable efficacy of IMMU-114 in disease models in which the anti-CD20 mAbs have relatively low activity. In vitro, IMMU-114 was consistently more cytotoxic than rituximab and veltuzumab when tested on established leukemia, lymphoma, and MM cell lines, on the rituximab-resistant cell lines generated by Czuczman et al,20 and on CLL patient specimens. In vivo, IMMU-114 induced long-term, disease-free survival in tumor-bearing SCID mice treated at an early stage of disease and in models that are relatively resistant to anti-CD20 mAbs, WSU-FSCCL and Namalwa. Because IMMU-114-treatment was initiated only 1 day after administration of tumor cells in these studies, these studies may be measuring inhibition of engraftment rather than treatment of established disease. However, mice bearing the anti–CD20-sensitive cell line, Raji, treated with low mAb doses at an advanced stage of disease yielded significant, but not-long term, prolongation of survival with either IMMU-114 or veltuzumab. Future work will include dose-escalation studies in established tumors in additional models to demonstrate the potency of IMMU-114 under a variety of conditions.
Despite higher toxicity than anti-CD20 mAbs on malignant B cells, both in vitro and in vivo, IMMU-114 resulted in significantly less normal B-cell depletion than rituximab and veltuzumab in unstimulated blood. However, when whole blood from healthy donors was stimulated with LPS, IMMU-114 yielded more B-cell depletion than rituximab. These data are consistent with previous reports of HLA-DR mAbs killing activated, but not resting, normal B cells in addition to tumor cells.44 A dual requirement for both MHC-II expression and cell activation for antibody-induced death has been suggested; and because most peripheral B cells are resting, the potential side effect resulting from killing of normal B cells may be minimal. Differing stages of B-cell differentiation may also lead to different outcomes after engagement of HLA-DR with mAbs or natural ligands.4 IMMU-114 also caused a significant reduction of the monocyte population in whole blood, ranging from 33% to 59%, depending on the status of stimulation. In view of the normal functions of circulating monocytes in antigen processing and presentation, cytokine production, and as precursors of other antigen-presenting cells, such as macrophages and dendritic cells, possible impairment of immune functions and/or inflammatory responses must be considered during clinical evaluation of IMMU-114.
Past studies of signaling through anti–HLA-DR mAbs have yielded inconsistent results. There have been reports of anti–HLA-DR inducing apoptosis directly45 or through CD95 (Fas)-mediated mechanisms.46 Other studies describe caspase-dependent or caspase-independent pathways.19,47–49 However, there is agreement that the mechanisms of action of anti–HLA-DR mAbs include induction of apoptosis, generation of ROS, and loss of mitochondrial membrane potential.19 To characterize the mechanism of IMMU-114, we examined the activation profiles of ERK, JNK, and p38 MAP kinases. MAP kinases are a group of protein serine/threonine kinases that are activated in response to a variety of extracellular stimuli, and induce signaling cascades in the cellular microenvironment culminating in cell growth, differentiation, and apoptosis.50 Three prominent groups of MAP kinases have been established, although recent reports describe additions to the family of MAP kinases. The ERK group of MAP kinases is activated primarily by mitogenic stimuli, such as growth factors,51 and are known to play an important role in inducing cell growth, whereas JNK and the p38 group of MAP kinases respond to cellular stress signals, such as ultraviolet irradiation, hydrogen peroxide, DNA damage, heat, and osmotic shock.52 Each of the 3 types of MAP kinases, ERKs, JNKs, and p38, phosphorylates a distinct set of transcription factors; and as a result, activation of different combinations of MAP kinases could lead to distinct biologic responses. Thus, differential activation of MAP kinases by extracellular stimuli could lead to distinct outcomes in terms of survival and apoptosis.53,54
We compared the activation of ERK, JNK, and p38 MAP kinases in our panel of cell lines and CLL patient cells. We found that JNK1/2 and ERK1/2 phosphorylation was up-regulated after exposure of cells to IMMU-114 in sensitive cell lines, such as the CLL patient cells, and the Raji and Jeko-1 cell lines, but not in the IMMU-114–resistant AML cell lines, such as Kasumi-3 and GDM-1. We observed up to 5-fold activation of the ERK and JNK signaling pathways on treatment with IMMU-114 at a modest 10-nM concentration. p38 MAP kinase was found to be constitutively active in these cell lines, and no further activation beyond basal levels was noted. Inhibition of the ERK and JNK signaling cascades by their respective inhibitors could modestly inhibit the apoptosis induced by IMMU-114; however, apoptosis was completely inhibited when 2 inhibitors were used together, indicating the activation of multiple MAP kinases by IMMU-114. IMMU-114–induced apoptosis was also significantly inhibited by siERK and siJNK RNAs. Thus, IMMU-114 cytotoxicity correlates with activation of ERK and JNK signaling. In addition, the results of these studies differentiate the mechanism of action of IMMU-114 cytotoxicity from that of the anti-CD74 (milatuzumab) and anti-CD20 mAbs (exemplified by comparison with rituximab and the humanized anti-CD20 mAb, veltuzumab). These findings highlight the importance of stress generation in the cells that further induces apoptosis.
In conclusion, contemporary cancer drug development is focused on “targeted” therapies, which includes agents that selectively attack a survival pathway for cancer cells; thus, antibodies that can perform this function also are of great interest. The anti–HLA-DR mAb, IMMU-114, an agent that reacts with a variety of hematologic malignancies, is one of the most effective therapeutic mAbs that we have examined, including showing cytotoxicity in rituximab-resistant NHL cell lines. The efficacy is apparently related to high antigen expression and effects on signaling. Having engineered a humanized IgG4 version of this antibody to avoid the side effects associated with complement activation7 and with impressive evidence of enhanced antitumor activity over that of an anti-CD20 IgG, clinical evaluation of this new agent is planned. In addition, variation in expression and cytotoxicity profiles between the mAbs suggests that combination therapies may yield greater effects in these various malignancies than the mAbs given singly, as reported previously in NHL cell lines.8
Supplementary Material
Acknowledgments
The authors thank Rosana Michel for providing the technical assistance in the ROS assays.
This work was supported in part by the National Institutes of Health (USPHS grant 1P01-CA103985; D.M.G., R.S.), New Jersey Commission on Cancer Research (grant 09-1976-CCR-EO), and the Thomas and Agnes Carvel Foundation.
Footnotes
An Inside Blood analysis of this article appears at the front of this issue.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: R.S., P.G., and X.C. designed and performed research, analyzed data, and wrote the paper; T.M.C. designed and performed research and analyzed data; R.R.F. provided clinical specimens and analyzed data; S.C. performed research; C.-H.C. designed research; and D.M.G. designed research and wrote the paper.
Conflict-of-interest disclosure: P.G., T.M.C., C.-H.C., and D.M.G. have employment, stock, and/or stock options with Immunomedics Inc. The remaining authors declare no competing financial interests.
Correspondence: Rhona Stein, Center for Molecular Medicine and Immunology, 520 Belleville Ave, Belleville, NJ 07109; e-mail: rstein@gscancer.org.
References
- 1.Cartron G, Watier H, Golay J, Solal-Celigny P. From the bench to the bedside: ways to improve rituximab efficacy. Blood. 2004;104(9):2635–2642. doi: 10.1182/blood-2004-03-1110. [DOI] [PubMed] [Google Scholar]
- 2.Keating MJ, Flinn I, Jain V, et al. Therapeutic role of alemtuzumab (Campath-1H) in patients who have failed fludarabine: results of a large international study. Blood. 2002;99(10):3554–3561. doi: 10.1182/blood.v99.10.3554. [DOI] [PubMed] [Google Scholar]
- 3.Reff ME, Carner K, Chambers KS, et al. Depletion of B cells in vivo by a chimeric mouse human monoclonal antibody to CD20. Blood. 1994;83(2):435–445. [PubMed] [Google Scholar]
- 4.Altomonte M, Visintin A, Tecce R, et al. Targeting of HLA-DR molecules transduces agonistic functional signals in cutaneous melanoma. J Cell Physiol. 2004;200(2):272–276. doi: 10.1002/jcp.20015. [DOI] [PubMed] [Google Scholar]
- 5.Brozek CM, Savage SM, Marnell LL, Searles RP. Anti-DR antibodies inhibit in vitro production of human rheumatoid factor. J Clin Lab Immunol. 1990;31(3):105–109. [PubMed] [Google Scholar]
- 6.Rech J, Repp R, Rech D, et al. A humanized HLA-DR antibody (hu1D10, apolizumab) in combination with granulocyte colony-stimulating factor (filgrastim) for the treatment of non-Hodgkin's lymphoma: a pilot study. Leuk Lymphoma. 2006;47(10):2147–2154. doi: 10.1080/10428190600757944. [DOI] [PubMed] [Google Scholar]
- 7.van der Kolk LE, Grillo-Lopez AJ, Baars JW, Hack CE, van Oers MH. Complement activation plays a key role in the side effects of rituximab treatment. Br J Haematol. 2001;115(4):807–811. doi: 10.1046/j.1365-2141.2001.03166.x. [DOI] [PubMed] [Google Scholar]
- 8.Stein R, Qu Z, Chen S, Solis D, Hansen HJ, Goldenberg DM. Characterization of a humanized IgG4 anti–HLA-DR monoclonal antibody that lacks effector cell functions but retains direct antilymphoma activity and increases the potency of rituximab. Blood. 2006;108(8):2736–2744. doi: 10.1182/blood-2006-04-017921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schuurman J, Perdok GJ, Gorter AD, Aalberse RC. The inter-heavy chain disulfide bonds of IgG4 are in equilibrium with intra-chain disulfide bonds. Mol Immunol. 2001;38(1):1–8. doi: 10.1016/s0161-5890(01)00050-5. [DOI] [PubMed] [Google Scholar]
- 10.Cheson BD, Bennett JM, Grever M, et al. National Cancer Institute-sponsored Working Group guidelines for chronic lymphocytic leukemia: revised guidelines for diagnosis and treatment. Blood. 1996;87(12):4990–4997. [PubMed] [Google Scholar]
- 11.Stein R, Qu Z, Cardillo TM, et al. Antiproliferative activity of a humanized anti-CD74 monoclonal antibody, hLL1, on B-cell malignancies. Blood. 2004;104(12):3705–3711. doi: 10.1182/blood-2004-03-0890. [DOI] [PubMed] [Google Scholar]
- 12.Stein R, Qu Z, Chen S, et al. Characterization of a new humanized anti-CD20 monoclonal antibody, IMMU-106, and its use in combination with the humanized anti-CD22 antibody, epratuzumab, for the therapy of non-Hodgkin's lymphoma. Clin Cancer Res. 2004;10(8):2868–2878. doi: 10.1158/1078-0432.ccr-03-0493. [DOI] [PubMed] [Google Scholar]
- 13.Leung SO, Goldenberg DM, Dion AS, et al. Construction and characterization of a humanized, internalizing, B-cell (CD22)-specific, leukemia/lymphoma antibody, LL2. Mol Immunol. 1995;32(17):1416–1427. doi: 10.1016/0161-5890(95)00080-1. [DOI] [PubMed] [Google Scholar]
- 14.Sharkey RM, Juweid M, Shevitz J, et al. Evaluation of a complementarity-determining region-grafted (humanized) anti-carcinoembryonic antigen monoclonal antibody in preclinical and clinical studies. Cancer Res. 1995;55(suppl):5935S–5945S. [PubMed] [Google Scholar]
- 15.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65(1):55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 16.Lebedeva IV, Su ZZ, Sarkar D, et al. Melanoma differentiation associated gene-7, mda-7/interleukin-24, induces apoptosis in prostate cancer cells by promoting mitochondrial dysfunction and inducing reactive oxygen species. Cancer Res. 2003;63(23):8138–8144. [PubMed] [Google Scholar]
- 17.Favata MF, Horiuchi KY, Manos EJ, et al. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem. 1998;273(29):18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- 18.Shin M, Yan C, Boyd D. An inhibitor of c-jun aminoterminal kinase (SP600125) represses c-Jun activation, DNA-binding and PMA-inducible 92-kDa type IV collagenase expression. Biochim Biophys Acta. 2002;1589(3):311–316. doi: 10.1016/s0167-4889(02)00195-7. [DOI] [PubMed] [Google Scholar]
- 19.Carlo-Stella C, Di Nicola M, Turco MC, et al. The anti-human leukocyte antigen-DR monoclonal antibody 1D09C3 activates the mitochondrial cell death pathway and exerts a potent antitumor activity in lymphoma-bearing nonobese diabetic/severe combined immunodeficient mice. Cancer Res. 2006;66(3):1799–1808. doi: 10.1158/0008-5472.CAN-05-1200. [DOI] [PubMed] [Google Scholar]
- 20.Czuczman MS, Olejniczak S, Gowda A, et al. Acquirement of rituximab resistance in lymphoma cell lines is associated with both global CD20 gene and protein down-regulation regulated at the pretranscriptional and posttranscriptional levels. Clin Cancer Res. 2008;14(5):1561–1570. doi: 10.1158/1078-0432.CCR-07-1254. [DOI] [PubMed] [Google Scholar]
- 21.Andersson LC, Nordling S, Häyry P. Proliferation of B and T cells in mixed lymphocyte cultures. J Exp Med. 1973;138(1):324–329. doi: 10.1084/jem.138.1.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bellosillo B, Villamor N, López-Guillermo A, et al. Complement-mediated cell death induced by rituximab in B-cell lymphoproliferative disorders is mediated in vitro by a caspase-independent mechanism involving the generation of reactive oxygen species. Blood. 2001;98(9):2771–2777. doi: 10.1182/blood.v98.9.2771. [DOI] [PubMed] [Google Scholar]
- 23.Scott SD. Rituximab: a new therapeutic monoclonal antibody for non-Hodgkin's lymphoma. Cancer Practice. 2002;6(3):195–197. doi: 10.1046/j.1523-5394.1998.006003195.x. [DOI] [PubMed] [Google Scholar]
- 24.McLaughlin P, Grillo-Lopez AJ, Link BK, et al. Rituximab chimeric anti-CD20 monoclonal antibody therapy for relapsed indolent lymphoma: half of patients respond to a four dose treatment program. J Clin Oncol. 1998;16(8):2825–2833. doi: 10.1200/JCO.1998.16.8.2825. [DOI] [PubMed] [Google Scholar]
- 25.Coiffier B, Haioun C, Ketterer N, et al. Rituximab (anti-CD20 monoclonal antibody) for the treatment of patients with relapsing or refractory aggressive lymphoma: a multicenter phase II study. Blood. 1998;92(6):1927–1932. [PubMed] [Google Scholar]
- 26.Friedberg JW, Neuberg D, Gribben JG, et al. Combination immunotherapy with rituximab and interleukin 2 in patients with relapsed or refractory follicular non-Hodgkin's lymphoma. Br J Haematol. 2002;117(4):828–834. doi: 10.1046/j.1365-2141.2002.03535.x. [DOI] [PubMed] [Google Scholar]
- 27.Gisselbrecht C. Use of rituximab in diffuse large B-cell lymphoma in the salvage setting. Br J Haematol. 2008;143(5):607–621. doi: 10.1111/j.1365-2141.2008.07383.x. [DOI] [PubMed] [Google Scholar]
- 28.Leonard JP, Martin P, Barrientos J, Elstrom R. Targeted treatment and new agents in diffuse large B-cell lymphoma. Semin Hematol. 2008;45(suppl 2):S11–S16. doi: 10.1053/j.seminhematol.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 29.Mounier N, Briere J, Gisselbrecht C, et al. Rituximab plus CHOP (R-CHOP) overcomes bcl-2–associated resistance to chemotherapy in elderly patients with diffuse large B-cell lymphoma (DLBCL). Blood. 2003;101(11):4279–4284. doi: 10.1182/blood-2002-11-3442. [DOI] [PubMed] [Google Scholar]
- 30.Dimopoulos MA, Gertz MA, Kastritis E, et al. Update on treatment recommendations from the Fourth International Workshop on Waldenstrom's Macroglobulinemia. J Clin Oncol. 2009;27(1):120–126. doi: 10.1200/JCO.2008.17.7865. [DOI] [PubMed] [Google Scholar]
- 31.Furst DE, Keystone EC, Kirkham B, et al. Updated consensus statement on biological agents for the treatment of rheumatic diseases, 2008. Ann Rheum Dis. 2008;67(suppl 3):2–25. doi: 10.1136/ard.2008.100834. [DOI] [PubMed] [Google Scholar]
- 32.Isaksen K, Jonsson R, Omdal R. Anti-CD20 treatment in primary Sjögren's syndrome. Scand J Immunol. 2008;68(6):554–564. doi: 10.1111/j.1365-3083.2008.02185.x. [DOI] [PubMed] [Google Scholar]
- 33.Mariette X. Therapeutic potential for B-cell modulation in Sjögren's syndrome. Rheum Dis Clin North Am. 2008;34(4):1025–1033. doi: 10.1016/j.rdc.2008.08.014. [DOI] [PubMed] [Google Scholar]
- 34.Ramos-Casals M, Brito-Zeron P, Munoz S, Soto MJ. A systematic review of the off-label use of biological therapies in systemic autoimmune diseases. Medicine (Baltimore) 2008;87(6):345–364. doi: 10.1097/MD.0b013e318190f170. [DOI] [PubMed] [Google Scholar]
- 35.Roccatello D, Baldovino S, Alpa M, et al. Effects of anti-CD20 monoclonal antibody as a rescue treatment for ANCA-associated idiopathic systemic vasculitis with or without overt renal involvement. Clin Exp Rheumatol. 2008;26(suppl 49):S67–S71. [PubMed] [Google Scholar]
- 36.Venkateshan SP, Sidhu S, Malhotra S, Pandhi P. Efficacy of biologicals in the treatment of rheumatoid arthritis: a meta-analysis. Pharmacology. 2009;83(1):1–9. doi: 10.1159/000165777. [DOI] [PubMed] [Google Scholar]
- 37.Vugmeyster Y, Howell K, Bakshl A, Flores C, Canova-Davis E. Effect of anti-CD20 monoclonal antibody, Rituxan, on cynomolgus monkey and human B cells in a whole blood matrix. Cytometry. 2003;52(2):101–109. doi: 10.1002/cyto.a.10030. [DOI] [PubMed] [Google Scholar]
- 38.Salles G, Mounier N, de Guibert S, et al. Rituximab combined with chemotherapy and interferon in follicular lymphoma patients: results of the GELA-GOELAMS FL2000 study. Blood. 2008;112(13):4824–4831. doi: 10.1182/blood-2008-04-153189. [DOI] [PubMed] [Google Scholar]
- 39.Czuczman M. CHOP plus rituximab chemoimmunotherapy of indolent B-cell lymphoma. Semin Oncol. 1999;26(5) suppl 14:88–96. [PubMed] [Google Scholar]
- 40.Coiffier B, Lepage E, Briere J, et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med. 2002;346(4):235–242. doi: 10.1056/NEJMoa011795. [DOI] [PubMed] [Google Scholar]
- 41.Carnahan J, Stein R, Qu Z, et al. Epratuzumab, a CD22-targeting recombinant humanized antibody with a different mode of action from rituximab. Mol Immunol. 2007;44(6):1331–1341. doi: 10.1016/j.molimm.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 42.Stein R, Mattes MJ, Cardillo TM, et al. CD74: A new candidate target for the immunotherapy of hematological malignancies. Clin Cancer Res. 2007;13(18) suppl:5556S–5563S. doi: 10.1158/1078-0432.CCR-07-1167. [DOI] [PubMed] [Google Scholar]
- 43.Shi JD, Bullock C, Hall WC, et al. In vivo pharmacodynamic effects of Hu1D10 (remitogen), a humanized antibody reactive against a polymorphic determinant of HLA-DR expressed on B cells. Leuk Lymphoma. 2002;43(6):1303–1312. doi: 10.1080/10428190290026376. [DOI] [PubMed] [Google Scholar]
- 44.Nagy Z, Hubner B, Lohning C, et al. Fully human, HLA-DR-specific monoclonal antibodies efficiently induce programmed death of malignant lymphoid cells. Nat Med. 2002;8(8):801–807. doi: 10.1038/nm736. [DOI] [PubMed] [Google Scholar]
- 45.Newell MK, VanderWall J, Beard KS, Freed JH. Ligation of major histocompatibility complex class II molecules mediates apoptotic cell death in resting B lymphocytes. Proc Natl Acad Sci U S A. 1993;90(22):10459–10463. doi: 10.1073/pnas.90.22.10459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Truman JP, Choqueux C, Tschopp J, et al. HLA class II-mediated death is induced via Fas/Fas ligand interactions in human splenic B lymphocytes. Blood. 1997;89(6):1996–2007. [PubMed] [Google Scholar]
- 47.Blancheteau V, Charron D, Mooney N. HLA class II signals sensitize B lymphocytes to apoptosis via Fas/CD95 by increasing FADD recruitment to activated Fas and activation of caspases. Hum Immunol. 2002;63(5):375–383. doi: 10.1016/s0198-8859(02)00384-1. [DOI] [PubMed] [Google Scholar]
- 48.Drenou B, Blancheteau V, Burgess DH, Fauchet R, Charron DJ, Mooney NA. A caspase-independent pathway of MHC class II antigen-mediated apoptosis of human B lymphocytes. J Immunol. 1999;163(8):4115–4124. [PubMed] [Google Scholar]
- 49.Mone AP, Huang P, Pelicano H, et al. Hu1D10 induces apoptosis concurrent with activation of the AKT survival pathway in human chronic lymphocytic leukemia cells. Blood. 2004;103(5):1846–1854. doi: 10.1182/blood-2003-08-2836. [DOI] [PubMed] [Google Scholar]
- 50.Chan-Hui PY, Weaver R. Human mitogen-activated protein kinase kinase kinase mediates the stress-induced activation of mitogen-activated protein kinase cascades. Biochem J. 1998;336(3):599–609. doi: 10.1042/bj3360599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Minden A, Karin M. Regulation and function of the JNK subgroup of MAP kinases. Biochim Biophys Acta. 1997;1333(2):F85–F104. doi: 10.1016/s0304-419x(97)00018-8. [DOI] [PubMed] [Google Scholar]
- 52.Ichijo H. From receptors to stress-activated MAP kinases. Oncogene. 1999;18(45):6087–6093. doi: 10.1038/sj.onc.1203129. [DOI] [PubMed] [Google Scholar]
- 53.Sutherland CL, Heath AW, Pelech SL, Young PR, Gold MR. Differential activation of the ERK, JNK, and p38 mitogen-activated protein kinases by CD40 and the B cell antigen receptor. J Immunol. 1996;157(8):3381–3390. [PubMed] [Google Scholar]
- 54.Roovers K, Assoian RK. Integrating the MAP kinase signal into the G1 phase cell cycle machinery. Bioessays. 2000;22(9):818–826. doi: 10.1002/1521-1878(200009)22:9<818::AID-BIES7>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}