

Large bowel cancer is one of the most common human malignancies in the Western world. The American Chemical Society estimated in 2007 that in the United States, there are 150,000 new cases and about 50,000 deaths due to colorectal cancer (CRC). Thus large bowel cancer is a major public health problem1. In the USA, CRC is the third most common type of cancer and the second most common cause of death due to cancer1,2. In general, colon cancer progresses from adenomatous polyps, a noncancerous growth in colon and rectum to malignant cancer in 10–15 years, hence providing a window of opportunity for effective treatment by intervention and prevention. However, despite intensive efforts, < 50 % of people aged 50 or older get colorectal cancer screening in this country.
Evidence in the literature supports the concept that dietary factors, smoking, alcohol and physical activities are key modulators of colorectal cancer. Epidemiological and animal model studies indicate that the etiology of colon cancer is complex3–5. Several studies have suggested that CRC may arise from the combined actions of environmental factors (carcinogens and co-carcinogens, formed through the preparation of food) and of promoting agents such as dietary fat6–8.
Particularly, chemoprevention, a science that has emerged during the last decade presents an alternative approach in reducing mortality of CRC as well as other cancers. Chemoprevention involves the long-term use of a variety of agents that can delay, block, or reverse the process of carcinogenesis or reduction of the underlying risk factors that can be applied across a continuum of the general population. Several naturally occurring and synthetic compounds including selenium compounds9–13 have been identified to exhibit chemopreventive activities both in vitro and in vivo. They are known to suppress cell growth, modulate cell differentiation, and induce apoptosis. Several agents have been identified which inhibit colon carcinogenesis in animal models11–23. These agents can protect against the development and progression of malignant cell and therefore proven plausible approach to inhibit or reverse carcinogenesis15, 24, 25. The role of arachidonic acid metabolism through lipooxygenase (LOX) and cyclooxygenase (COX) pathways, and involvement of inducible nitric oxide synthase (iNOS) activities in angiogenesis are well studied in the pathology of colon cancer26. Development of agents based on these studies led to few selective inhibitors that suppress these activities in prevention of colon cancer 27–30.
Nitric oxide (NO), an endogenous molecule, has been ascribed with many physiological and pathophysiological actions. Several L-arginine based selective iNOS inhibitors have been tested for chemoprevention of colon cancer. One such example is the use of iNOS inhibitor S,S′-1,4-phenylene-bis(1,2-ethanediyl)bis-isothiourea (PBIT, Figure 1), which was found to inhibit initiation and post-initiation stages of AOM-induced colonic ACF and iNOS activities in male F344 rat31. Stoner et al. found effective inhibition by PBIT in a diet against rat esophageal tumorigenesis32. Since, high concentrations of PBIT are needed for inhibitory potency, its utility in clinical settings is questionable. It is reported in the literature that incorporation of selenium in place of sulfur imparts new properties to the compound making it an effective inhibitor of diseases. For example, aminoethylisoselenourea (AE-SeU), which showed higher iNOS inhibitory potency than the corresponding isosteric sulfur analog in homogenized bovine endothelial cells33. However, selenium compound AE-SeU is unstable in aqueous solution above pH 6. Moreover, selenium is involved in the protection of tissue and membranes from oxidative stress. Additionally, low selenium intake and low plasma selenium levels are associated with increased cancer incidence34–36.
Figure 1.

Structures of PBIT, PBISe, PEIT, and PEISe
El-Bayoumy et al. have developed a synthetic organo-selenium compound that is a potent chemopreventive agent in animal models against colon37. Previously, we have reported that organoselenium compounds inhibit initiation and post-initiation chemical carcinogenesis38, 39. Selenium supplementation through diet or drinking water has been shown to inhibit neoplasm in the liver, skin, pancreas, colon, and mammary glands40–43. It has been reported that selenium-enriched broccoli protect against intestinal, chemically induced mammary, and colon cancers44–47. Selenium has also been shown to inhibit Akt activity in the PI3 kinase-signaling pathways thereby inhibiting its activity and that of the downstream signaling cascade48.
Normal colonic mucosa and hyperplastic polyps express very low levels of Akt, however, immuno reactivity to a pan-AKT antibody was detected in 57% of adenomas and also in similar number of cases in sporadic colorectal cancer49. In colon cancer Akt2 was found to be the predominant isoform. Parsons et al. reported that 40% of colorectal human tumors had PI3K pathways altered 50. Studies in chemical carcinogenesis and in genetic animal models confirm over expression of Akt in rat premalignanat colonocyte. In Apcmin/+ mice, total Akt and protein levels were significantly higher in colon tumors compared to normal cells51. Akt is activated by phosphatidylinositol 3-kinase (PI3K) which regulates multiple cellular functions, including survival, proliferation (increased cell numbers), growth (increased cell size), and various aspects of intermediary metabolism52.
The existing iNOS inhibitor PBIT (1) (Figure 1) has limited effect on colon cancer and require high concentration for its inhibitory effects. Therefore, in the present communication, we investigated the efficacy of isosteric selenium analog of PBIT, namely Se,Se′-1,4-Phenylenebis(1,2-ethanediyl)bis-isoselenourea (PBISe, 2) (Figure 1) and evaluated in vitro inhibitory efficacy against colon cancer cells. Furthermore, the mechanism of action of PBISe and PBIT was also elucidated using in vitro studies.
Based on extensive mechanistic studies (in vitro and in vivo) and data from animal studies as well as investigation in CRC patients it has been well established that iNOS is over expressed in colon tumors but not in the normal adjacent mucosa and plays a pivotal role in the pathology of colon tumorigenesis. Previous reports have shown that selenium inhibit Akt activity. Although full explanation for causes, development, and control of colon cancer is awaiting further research, the growing knowledge about mechanisms by which various agents function defines opportunities to use specific agents or combination at critical stages of colon cancer initiation, promotion, or progression.
In the present study, we report the synthesis of PBIT (1), a known iNOS inhibitor and PBISe (2), its isosteric selenium analog. PBIT (1) and PBISe (2) were prepared by using similar procedure as reported in the literature with minor modification as shown (scheme I)53–56. In short, commercially available Methyl 1,4-phenylenediacetic acid (5) on reduction with LiAlH4 in THF yielded dihydrodiol 6 as white solid in quantitative yield. The diol 6 was converted to the dibromo derivative 7 as white powder by reacting with carbon tetrabromide and triphenyl phosphine. Dibromo derivative 7 was reacted with thiourea54,55 or selenourea56 to yield the desired product PBIT (1) and PBISe (2) as dihydrobromide salt, respectively. Monosubstituted Phenylethylisothiourea hydrobromide (PEIT, 3) was prepared by following literature method54 and isosteric selenium analog; Se-2-phenylethylisoselenourea hydrobromide (PEISe, 4) was prepared in a similar manner by reacting 2-bromoethyl benzene with selenourea to give the desired product PEISe, 4 in quantitative yield 57.
Scheme 1.

Reagents and condition: (a), LiAlH4, RT, 91%; (b) CBr4/PPh3, 74%; (c) NH2CSNH2 or NH2CSeNH2/EtOH, 78–69%.
To investigate the structural requirements and the effect of PBIT, PBISe, and PEIT, and PEISe on several colon adenocarcinoma cell viability58–59, In the present study we first compared increasing concentration of PBIT, PEIT and PEISe (10–100 μM) and PBISe (1–40 μM) against the viability of colon adenocarcinoma (Caco-2, HT 29, HCT 116, and SW 480) cells (Table 1). PBISe was highly potent at killing all four colon cancer cell lines with IC50 range of 2.4–5.55 μM59. PBIT showed an IC50 value range >59–100 μM. At the concentration range examined, monosubstituted PEIT and PEISe were less effective with IC50 value range >16–100 μM59. From dose response curves prepared for each compounds (data not shown), we concluded that di-substitution in the phenyl ring in the molecule was one of the important structural requirement for in vitro inhibitory efficacy against colon cancer. Based on these results for additional inhibitory efficacy studies, we have only compared PBIT (1) and its isosteric selenium analog; PBISe (2).
Table 1.
Inhibitory effect of PBISe, PBIT, PEIT, and PEISe on colon adenocarcinoma cell lines by MTS assay- IC50 value (μm).
| Cell line | PBIT | PBISe | PEIT | PEISe |
|---|---|---|---|---|
| Caco-2* | 59.4 ± 3.83 | 2.4 ± 0.43 | >100 | 16 ± 2 |
| HT-29 | >100 | 3.84 ± 0.32 | 21.92 ± 2.1 | >100 |
| HCT-116 | >100 | 4.72 ± 0.44 | >100 | >100 |
| SW-480 | >100 | 5.55 ± 1.28 | >100 | >100 |
=72 h
A representative example of in vitro cell viability on Caco-2 cells is shown in Figure 2. Treatment of Caco-2 cell lines with increasing concentrations of PBIT (0–120 μM) for 72 hours showed the predicted dose response curve with a resulting IC50 of above 60 μM (Figure 2)60. PBISe (0–10 μM) was dramatically more potent having an IC50 of only 2.4 μM on Caco-2 cell lines indicating a 25-fold increased inhibitory potency (Figure 2).
Figure 2.
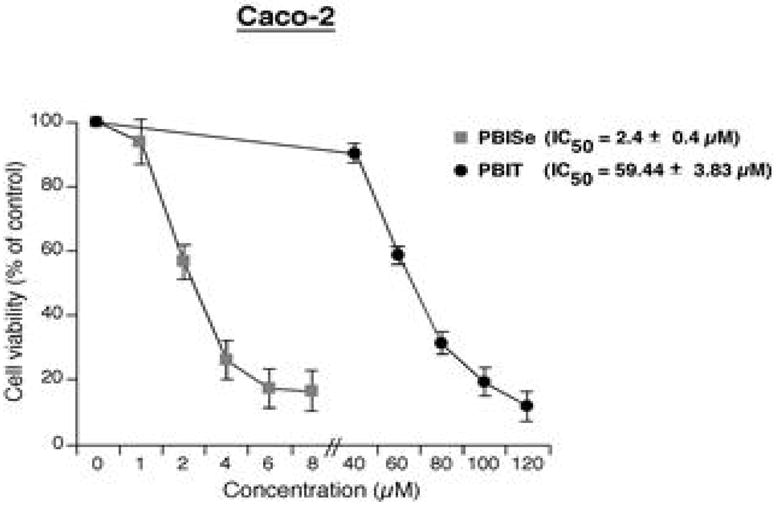
In vitro therapeutic efficacy of PBISe and PBIT in Caco-2 cancer cell viability study - IC50 value (μM)
Next, we examined the in vitro inhibitory efficacy of PBISe and PBIT in several cancer cell lines (Table 2)58,61. Cancer cell lines representing different cancer types were treated with increasing concentrations of PBIT and PBISe for 24 hours and cell viability measured using MTS assay. Dose response curves were prepared for growth inhibitory effects on normal fibroblast cells (FF2441) and with cultured cells from human vertical growth phase (VGP) melanoma cell line WM115, Lung adenocarcinoma (A549), fibrosarcoma (HT-1080), prostate adenocarcinoma (PC-3), ovarian carcinoma (OVCAR-3), and breast adenocarcinoma (MDA-MB-231) as shown in Figure 3. The cultured cancer cells were found to be 2–5 fold more sensitive to PBISe than normal fibroblast cells (Table 1). IC50 values calculated using GraphPad Prism software (Table 2) from dose response curves indicated that PBISe was more than >10 fold potent at decreasing cell viability of all the cancer cell lines tested (A549, HT-1080, PC-3, OVCAR-3, MDA-MB-231) compared to PBIT. PBISe inhibited the growth of cancer cell viability at concentrations as low as 2–10 μM (Table 2). However, PBIT was not effective at this concentration range and IC50 was observed to be more than 100 μM.
Table 2.
Inhibitory effect of PBISe and PBIT on various cancer cell lines by MTS assay- IC50 value (μm).
| Cell line | PBIT | PBISe |
|---|---|---|
| Melanoma (WM 115) | >100 | 9.7 ± 1.4 |
| Fibrosarcoma (HT-1080) | >100 | 3.22 ± 0.61 |
| Human lung carcinoma (A549) | >100 | 4.17 ± 0.58 |
| Human prostate adenocarcinoma (PC-3) | >100 | 6.64 ± 0.95 |
| Ovarian carcinoma (OVCAR3) | >100 | 5.33 ± 0.65 |
| Breast adenocarcinoma (MDA-MB-231) | >100 | 6.03 ± 0.54 |
Figure 3.

PBISe inhibit cancer cell survival more effectively compared to normal cells - IC50 value (μM)
We examine the iNOS inhibitory activity of PBISe in Caco-2 cells in terms of total nitrite (nitrate + nitrite) production using a colorimetric assay (Cayman chemical company) (Figure 4)62. Results showed that PBIT (40 μM) reduced the iNOS activity by 18% (One-way ANOVA) compared to control PBS treated cells. The PBISe reduced total nitrite production in Caco-2 cells (by ~ 25%) but at much lower concentrations (2 μM).
Figure 4.
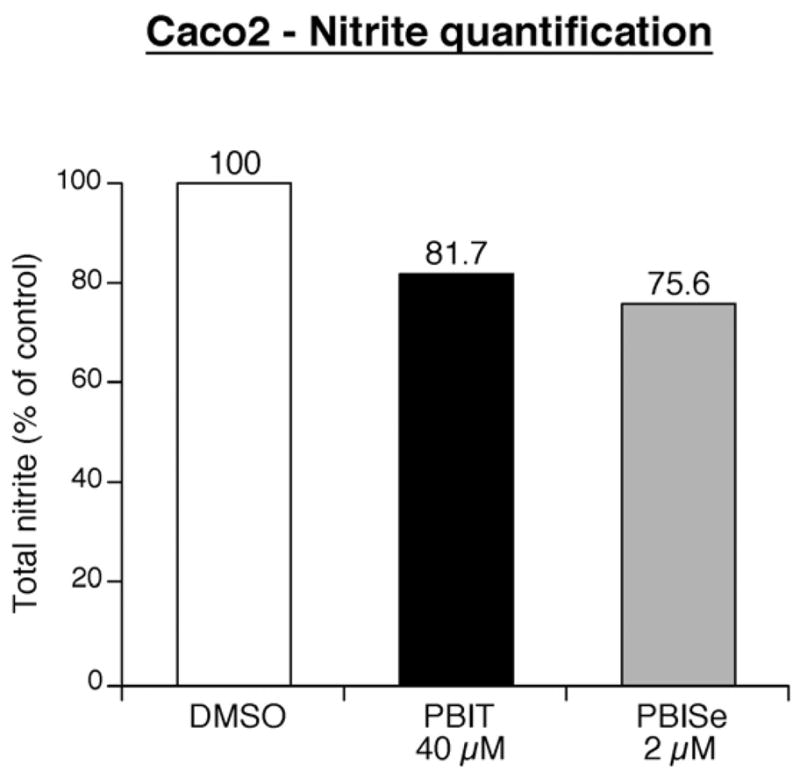
Effect of PBIT and PBISe on total nitrite production by Caco-2 cells
To elucidate the effects of PBISe and PBIT on molecular targets that are critical in the development of colon cancer, we focused on PI3 kinase and downstream targets63. Effect of PBISe on MAP and PI3 kinase signaling using Western blot analysis is shown in Figure 3A and 3B. Figure 3A shows decreased pAkt, and Akt2 levels that are relatively specific for colon cancer, and downstream pPRAS40 levels accompanied by an increase in cleaved PARP, an apoptosis marker demonstrating decreased PI3 kinase activity upon exposure to PBISe but not PBIT. Figure 3B shows decreased cyclin D1 levels at higher concentration with a simultaneous increase in cell cycle inhibitor p27, indicating inhibition of MAP kinase activity following exposure to PBISe but not PBIT.
In summary, the present article shows that newly developed novel selenium compound PBISe is clearly a more potent agent than its isosteric sulfur analog, PBIT. These preliminary data are significant, demonstrating that PBISe significantly inhibits iNOS, PI3 kinase signaling and pAkt signal in colon cancer cells to dramatically increase apoptosis and decrease proliferation. These are critical observations needed to support the utility of PBISe in future animal studies. Mechanisms of action of PBISe and in vivo animal study with various cancer types are under investigation.
Acknowledgments
The authors would like to thank Dr. Jyh-Ming Lin from the Penn State Hershey Cancer Institute Instrumentation Facility for NMR spectra and Jenny Dai for performing the MS analysis. This study was supported by NIH R03 CA1378763 (DD), NCI contract NCI-CB-56603 (SA), and funds from Penn State Cancer Institute.
LITERATURE CITED
- 1.Boursi B, Arber N. Curr Pharm Des. 2007;13:2274. doi: 10.2174/138161207781368783. [DOI] [PubMed] [Google Scholar]
- 2.American Cancer Society. Colorectal Cancer Fact and Figures. Special. 2008. p. 1. [Google Scholar]
- 3.Potter JD. Cancer Causes Control. 1996;7:127. doi: 10.1007/BF00115644. [DOI] [PubMed] [Google Scholar]
- 4.Correa P, Haenszel W. Adv Cancer Res. 1978;26:1. doi: 10.1016/s0065-230x(08)60086-x. [DOI] [PubMed] [Google Scholar]
- 5.Wynder EL, Kajitani T, Ishikawa S, Dodo H, Takano A. Cancer. 1969;23:1210. doi: 10.1002/1097-0142(196905)23:5<1210::aid-cncr2820230530>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 6.National Research Council. Diet, Nutrition and Cancer, Assembly of Life Sciences. National Academy Press; Washington, D.C.: 1982. [Google Scholar]
- 7.Reddy BS. In: Diet, Nutrition and Cancer. A critical Evaluation. Reddy BS, Cohen LA, editors. CRC Press; Boca Raton, FL: 1986. pp. 47–65. [Google Scholar]
- 8.Sugimura T, Wakabayashi K, Ohgaki H, Takayama S, Nagao MG, Esumi H. In: Xenobiotics and Cancer. Ernster L, Esumi H, Fujii Y, Gelboin HV, Kato R, Sugimura T, editors. Japan Sci. Soc. Press; 1991. pp. 279–288. [Google Scholar]
- 9.Kelloff GJ, Crowell JA, Steele VE, Malone WE, Boone CW, Kopelovich L, Hawk ET, Lieberman R, Lawrence JA, Ali I, Viner JL, Sigman CC. Progress in cancer chemoprevention: development of diet-derived chemopreventive agents. 2000;130:467s–471s. doi: 10.1093/jn/130.2.467S. [DOI] [PubMed] [Google Scholar]
- 10.Ip C, Dong Y, Ganther HE. Cancer Metastasis Rev. 2002;21:281. doi: 10.1023/a:1021263027659. [DOI] [PubMed] [Google Scholar]
- 11.El-Bayoumy KE, Sinha R, Pinto JT, Rivlin RS. J Nutri. 2006:864S. doi: 10.1093/jn/136.3.864S. [DOI] [PubMed] [Google Scholar]
- 12.Clark LC, Combs GF, Jr, Tumbull BW, Slate EH, Chalker DK, Chow J, Davis LS, Glover RA, Graham GF, Gross EG, Krongrad A, Lesher JL, Jr, Park HK, Sanders BB, Jr, Smith CL, Taylor JR. J Am Med Assoc. 1996;276:1957. [PubMed] [Google Scholar]
- 13.Clark LC, Dalkin B, Krongrad GF, Combs GF, Jr, Tumbull BW, Slate EH, Witherington R, Herlong JH, Janosko E, Carpenter D, Borosso C, Falk S, Rounder J. Br J Urol. 1998;81:730. doi: 10.1046/j.1464-410x.1998.00630.x. [DOI] [PubMed] [Google Scholar]
- 14.Narisawa T, Sato M, Tani M, Kudo T, Takahashi T, Goto A. Cancer Res. 1981;41:1954. [PubMed] [Google Scholar]
- 15.Wattenberg LW. Cancer Res. 1983;43:2448s. [PubMed] [Google Scholar]
- 16.Rao CV, Tokumo K, Kelloff G, Reddy BS. Carcinogenesis. 1991;12:1051. doi: 10.1093/carcin/12.6.1051. [DOI] [PubMed] [Google Scholar]
- 17.Kudo T, Narisawa T, Abo S. Gann. 1980;71:260. [PubMed] [Google Scholar]
- 18.Reddy BS, Maruyama H, Kelloff GJ. Cancer Res. 1987;47:5340. [PubMed] [Google Scholar]
- 19.Reddy BS, Rao CV, Rivenson A, Kelloff GJ. Cancer Res. 1993;53:3493. [PubMed] [Google Scholar]
- 20.Pollard M, Luckert PH. J Natl Cancer Inst. 1983;70:1103. [PubMed] [Google Scholar]
- 21.Nigro N, Bull AW, Boyd ME. J natl Cancer Inst. 1986;77:1309. [PubMed] [Google Scholar]
- 22.Kingsworth AN, King WK, Diekema KA, McCann PP, Ross JS, Malt RA. Cancer Res. 1983;43:2545. [PubMed] [Google Scholar]
- 23.Nayini JR, Sugie S, El-Bayoumy KE, Rao CV, Rigotty J, Sohn OS, Reddy BS. Nutr Cancer. 1991;15:129. doi: 10.1080/01635589109514120. [DOI] [PubMed] [Google Scholar]
- 24.Wattenberg LW. In: Cancer Chemoprevention. Wattenberg LW, Lipkin M, Boone CW, Kelloff GJ, editors. CRC Press; Boca Raton, FL: 1990. pp. 19–39. [Google Scholar]
- 25.Newmark HL. Nutr Cancer. 1984;6:58. doi: 10.1080/01635588509513807. [DOI] [PubMed] [Google Scholar]
- 26.Sporn MB. Cancer Res. 1976;36:2699. [PubMed] [Google Scholar]
- 27.Reddy BS, Rao CV, Seibert K. Cancer Res. 1996;56:4566. [PubMed] [Google Scholar]
- 28.Kawamori T, Rao CV, Seibert K, Reddy BS. Carcinogenesis. 1998;58:409. [PubMed] [Google Scholar]
- 29.Rao CV, Kawamori T, Hamid R, Reddy BS. Carcinogenesis. 1999;20:641. doi: 10.1093/carcin/20.4.641. [DOI] [PubMed] [Google Scholar]
- 30.Reddy BS, Hirsoe Y, Lubet R, Steele VE, Kelloff GJ, Paulson S, Seibert K, Rao CV. Cancer Res. 2000;60:293. [PubMed] [Google Scholar]
- 31.Rao CV, Indranie C, Simi B, Manning PT, Connor JR, Reddy BS. Cancer Res. 2002;62:165. [PubMed] [Google Scholar]
- 32.Chen T, Nines RG, Peschke SM, Kresty LA, Stoner GD. Cancer Res. 2004;64:3714. doi: 10.1158/0008-5472.CAN-04-0302. [DOI] [PubMed] [Google Scholar]
- 33.Southan GJ, Salzman AL, Szabo C. Life Sci. 1996;58:1139. doi: 10.1016/0024-3205(96)00072-0. [DOI] [PubMed] [Google Scholar]
- 34.Reinhold U, Biltz H, Bayer W, Schmidt KH. Acta Derm Venereol. 1989;69:132. [PubMed] [Google Scholar]
- 35.Reid ME, Duffield-Lillico AJ, Slate E, Natarajan N, Turnbull B, Jacobs E, Combs GF, Jr, Alberts DS, Clark LC, Marshall JR. Nutr Cancer. 2008;60:155. doi: 10.1080/01635580701684856. [DOI] [PubMed] [Google Scholar]
- 36.Camargo MC, Burk RF, Bravo LE, Piazuelo MB, Hill KE, Fontham ET, Motley AK, Yepez MC, Mora Y, Schneider BG, Correa P. Arch Med Res. 2008;39:443. doi: 10.1016/j.arcmed.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reddy BS, Rivenson A, El-Bayoumy KE, Upadhyaya P, Pittman B, Rao CV. J Natl Cancer Inst. 1997;89:506. doi: 10.1093/jnci/89.7.506. [DOI] [PubMed] [Google Scholar]
- 38.El-Bayoumy KE. Nutr Cancer. 2001;40:4. doi: 10.1207/S15327914NC401_3. [DOI] [PubMed] [Google Scholar]
- 39.El-Bayoumy KE, Sinha R, Pinto JT, Rivlin RS. J Nutr. 2006;136:864S. doi: 10.1093/jn/136.3.864S. [DOI] [PubMed] [Google Scholar]
- 40.Rayman MP. Proc Nutr Soc. 2005;64:527. doi: 10.1079/pns2005467. [DOI] [PubMed] [Google Scholar]
- 41.Letavayova L, Vlckova V, Brozmanova J. Toxicology. 2006;227:1. doi: 10.1016/j.tox.2006.07.017. [DOI] [PubMed] [Google Scholar]
- 42.Aboul-Fadl T. Curr Med Chem Anticancer Agents. 2005;5:637. doi: 10.2174/156801105774574676. [DOI] [PubMed] [Google Scholar]
- 43.Yan L, Yee JA, McGuire MH, Graef GL. Nutr Cancer. 1997;28:165. doi: 10.1080/01635589709514570. [DOI] [PubMed] [Google Scholar]
- 44.Davis CD, Zeng H, Finley JW. J Nutr. 2002;132:307. doi: 10.1093/jn/132.2.307. [DOI] [PubMed] [Google Scholar]
- 45.Higdon JV, Delage B, Williams DE, Dashwood RH. Pharmacol Res. 2007;55:224. doi: 10.1016/j.phrs.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Finley JW, Davis CD. Biofactors. 2001;14:191. doi: 10.1002/biof.5520140124. [DOI] [PubMed] [Google Scholar]
- 47.Zeng H, Davis CD, Finley JW. J Nutr Biochem. 2003;14:227. doi: 10.1016/s0955-2863(03)00005-6. [DOI] [PubMed] [Google Scholar]
- 48.Wu Y, Zu K, Warren MA, Wallace PK, Ip C. Mol Cancer Ther. 2006;5:246. doi: 10.1158/1535-7163.MCT-05-0376. [DOI] [PubMed] [Google Scholar]
- 49.Roy HK, Olusola BF, Clemens DL, Karolski WJ, Ratashak A, Lynch HT, Smyrk TC. Carcinogenesis. 2002;23:201. doi: 10.1093/carcin/23.1.201. [DOI] [PubMed] [Google Scholar]
- 50.Parsons DW, Wang TL, Samuels Y. Nature. 2005;436:792. doi: 10.1038/436792a. [DOI] [PubMed] [Google Scholar]
- 51.Ju J, Hong J, Zhou J-N, Pan Z, Bose M, Liao J, Yang G-Y, Liu YY, Hou Z, Lin Y, Ma J, Shih WJ, Carothers AM, Yang CS. Cancer Res. 2005;65:10623. doi: 10.1158/0008-5472.CAN-05-1949. [DOI] [PubMed] [Google Scholar]
- 52.Altomare DA, Testa JR. Oncogene. 2005;24:7455. doi: 10.1038/sj.onc.1209085. [DOI] [PubMed] [Google Scholar]
- 53.General synthesis: Melting points were recorded on a Fisher-Johnson melting point apparatus and are uncorrected. Unless stated otherwise, proton NMR spectra were recorded in CDCl3 using a Bruker 500 MHz instruments. The chemical shifts are reported in ppm downfield from TMS. MS were run on 4000 Q trap hybrid triple quadrupole/linear ion trap instrument (Applied Biosystems/MDS Sciex) at the proteomic facility in Penn State Cancer Institute at Penn State College of Medicine, Hershey, PA. High-resolution MS were determined at the Instrument Center, University of Buffalo, Buffalo, NY. Thin-layer chromatography (TLC) was on aluminum-supported, pre-coated silica gel plates (EM Industries, Gibbstown, NJ). All starting materials and reagents were obtained from Aldrich Chemical Co. (Milwaukee, WI) and used without further purification.
- 54.Methyl 1,4-phenylenediacetic acid (5), 1,4-phenylene diethanol (6), and 1,4-di(2-bromoethyl)benzene (7) were prepared as reported by Garvey EP, Oplinger JA, Tanoury GJ, Sherman PA, Fowler M, Marshall S, Harmon MF, Paith JE, Furfine ES. J Biol Chem. 1994;269:26669.
- 55.Synthesis of S,S′-1,4-phenylene-bis(1,2-ethanediyl)bis-isothiourea dihydrobromide (PBIT, 1): To a mixture of 1,4-di(2-bromoethyl)benzene (7) (1.0 g, 3.43 mmol) in ethanol (50 mL) was added thiourea (0.52 g, 6.86 mmol). The reaction mixture was heated to reflux for 2h. After concentration of ethanol (20 mL), the precipitated solid was filtered, washed with hexane, methylene chloride, and dried to yield 1 (1.19 g, 78%) as white powder, mp, 234–236 °C54; 1H NMR (D2O): 7.25 (s, 4H, aromatic), 3.37 (t, 4H, Ph-CH2, J=7.2 Hz), 3.00 (t, 4H, C-CH2, J = 6.86 Hz); MS m/z (intensity) 283 (M+1, 100), 266 (40), 207 (30), 142 (70), 131 (100), 104 (10), 77 (30).
- 56.Synthesis of Se,Se′-1,4-phenylene-bis(1,2-ethanediyl)bis-isoselenourea dihydrobromide (PBISe, 2): Compound 2 was prepared in a similar manner as reported for compound 1 by reacting 1,4-di(2-bromoethyl)benzene (7)(1.0 g, 3.43 mmol) with selenourea (0.84 g, 6.86 mmol) to yield 2 (1.3 g, 69%), mp, 247–249 °C; 1H NMR (D2O): 7.25 (s, 4H, aromatic), 3.37 (t, 4H, Ph-CH2, J=7.2 Hz), 3.21 (t, 4H, C-CH2, J=6.86 Hz); MS m/z; ion intensity 377 & 379 (M+, 50), 335 & 337 (80), 253 and 255 (70), 188 and 190 (100), 131 (50), 73 (10). ESI-MS: m/z, calcd: 378.9934 for C12H19N4Se2; found: 378.9935.
- 57.Synthesis of Se-2-phenylethylisoselenourea hydrobromide (PEISe, 4): Compound 4 was prepared in similar manner as reported above for compound 2 by reacting 2-bromoethyl benzene (1.0g, 5.4 mmol) with selenourea (0.67g, 5.4 mmol) to yield 4 (1.26g, 80%); mp, 140 –142°C; 1H NMR (D2O): 7.29–7.20 (m, 5H, aromatic), 3.02 (t, 2H, CH2-Ph, J=7.0 Hz), 3.48 (t, 2H, CH2-Se, J=7.0 Hz); EMS (m/z, intensity): 229 (M+, 100), 181 (80), 159 (10), 105 (10).
- 58.Cell lines and culture conditions: Colon adenocarcinoma cell line (Caco-2; ATCC No. HTB-37, HT-29; ATCC No. HTB-38, HCT-116; ATCC No. CCL-247, and SW-480; CCL-228) were grown in Advanced DMEM (2Mm) supplemented with 10% heat treated (56°C for 30 minutes) FBS (Hyclone, Logan, UT) and L-glutamine or RPMI-1640 containing 10% FBS. Lung adenocarcinoma (A549; ATCC No. CCL-185), fibrosarcoma (HT-1080; ATCC No’ CCL-121), prostate adenocarcinoma (PC-3; ATCC No.CRL-1435), ovarian adenocarcinoma (NIH: OVCAR-3; ATCC No. HTB-161), and a breast adenocarcinoma cell line (MDA-MB-231; ATCC No. HTB-26) were grown in DMEM supplemented with 10% FBS. The human vertical growth phase (VGP) melanoma cell line WM115 was maintained in Tu2% medium lacking calcium chloride, but supplemented with 2% heat treated (56°C for 30 minutes) FBS and L-glutamine as described previously.
- 59.To investigate the structural requirements, we first compared the cell viability of colon adenocarcinoma cells (Caco-2, HT-29, HCT-116, SW-480) upon treating with PBIT (1), PBISe (2), and their corresponding monosubstituted compounds PEIT (3), and PEISe (4) (Table 1). The IC50 values were calculated by treating with increasing concentrations of PBIT, PEIT, PEISe (10–100 μM) or PBISe (1.6 – 50 μM) for 72 hours and the number of viable cells quantified using 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) (Promega, WI).
- 60.Representative example of inhibitory effect of PBISe and PBIT against Caco-2 cells is shown in Figure 2. In brief, the assay was carried out by plating 5×103 cells per well in 100 μL media for 24h followed by treatment with either vehicle control PBS or with increasing concentrations of PBIT (0–120μM) or PBISe (0–10μM) for 72h. The percentages of viable cells compared to control PBS treated cells were determined using MTS assay and IC50 values calculated using GraphPad Prism version 4.01 (GraphPad software, San Diego, CA). IC50 value for each compound was determined from at least three independent experiments and represented with a standard error (Figure 2).
- 61.In vitro inhibitory effect of PBIT and PBISe on various cancer cell lines:By following similar procedure as reported above against colon cancer cells60; growth inhibitory effects on normal fibroblast cells (FF2441), and cultured cancer cells from human vertical growth phase (VGP) melanoma (WM115), Lung adenocarcinoma (A549), fibrosarcoma (HT-1080), prostate adenocarcinoma (PC-3), ovarian carcinoma (OVCAR-3), and breast adenocarcinoma (MDA-MB-231) were tested with increasing concentrations of PBIT (10–100μM) or PBISe (1–40μM) (Table 2, Figure 3).
- 62.Inhibition of nitrite production by PBISe in Caco-2 cells:To demonstrate that PBISe and PBIT similarly inhibited nitrite production in Caco-2 cells, the iNOS activity in terms of total nitrite (nitrate + nitrite) produced was measured using a colorimetric assay (Cayman chemical company, Ann Arbor, MI) (Figure 4). Caco-2 cells (1×106) were plated in 60 mm culture dishes in advanced DMEM containing 10% heat treated FBS. Twelve hours later, growing cells were conditioned (~12 h) with phenol-red free DMEM containing 0.5% FBS and then treated with increasing concentrations of PBIT (40 μM) or PBISe (2μM) dissolved in phenol-red free DMEM (2 ml) containing 0.5% FBS for 72 hours. Culture supernatants were collected, centrifuged (500 × g) and total nitrite (nitrate + nitrite) measured by incubating 80 μL supernatant with enzyme cofactor mixture (10μL) and nitrate reductase (10μL) for 2 hours. The addition of Griess reagent-I and II (50μL each) produced characteristic color that was measured at 540 nm using a SPECTRAmax M2 plate reader (Molecular Devices, Sunnyvale, CA). A nitrate standard curve (5–35μM) was simultaneously prepared and total nitrite present in samples was measured.
- 63.Western blot analysis:cell lysates were harvested by addition of lyses buffer containing 50 mM HEPES (pH 7.5), 150 mM NaCl, 10 mM EDTA, 10% glycerol, 1% Triton X-100, 1 mM sodium orthovanadate, 0.1 mM sodium molybdate, 1 mM phenylmethylsulfonyl fluoride, 20 μg/ml aprotinin, and 5 μg/ml leupeptin. Whole cell lysates were centrifuged (≥10,000 × g) for 10 minutes at 4°C to remove cell debris. Protein concentrations were quantitated using the BCA assay (Pierce; Rockford, IL), and 30 μg of lysate loaded per lane onto NuPAGE Gels (Life Technologies, Inc. Carlsbad, CA). Following electrophoresis, samples were transferred to polyvinylidene difluoride (PVDF) membrane (Pall Corporation, Pensacola, FL) and the blots probed with antibodies according to each supplier’s recommendations: antibodies to PRAS40 and phosphorylated PRAS40 from Invitrogen (Invitrogen Corporation, Carlsbad, CA); antibodies to cyclin D1, p27, and Erk2 from Santa Cruz Biotechnology (Santa Cruz, CA); and antibodies to Akt3, phosphorylated-Akt (Ser473), phosphorylated-Erk 1/2, and cleaved PARP from Cell Signaling Technology (Danvers, MA). Secondary antibodies conjugated with horseradish peroxidase were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Immunoblots were developed using the enhanced chemiluminescence (ECL) detection system (Pierce Biotechnology, Rockford, IL) (Figure 5).
Figure 5.

Effect of PBISe on vital pathways regulating colon cancer development.