

Abstract
Human Rhinovirus (HRV) infection is the cause of about one-half of asthma and COPD exacerbations. With >100 serotypes in the HRV reference set an effort was undertaken to sequence their complete genomes so as to understand diversity, structural variation, and evolution of the virus. Analysis revealed conserved motifs, hypervariable regions, a potential fourth HRV species, within-serotype variation in field isolates, a non-scanning internal ribosome entry site, and evidence for HRV recombination. Techniques have now been developed using next generation sequencing to generate complete genomes from patient isolates with high throughput, deep coverage, and low costs. Thus relationships can now be sought between obstructive lung phenotypes and variation in HRV genomes in infected patients, and, potential novel therapeutic strategies developed based on HRV sequence.
Keywords: Asthma, COPD, inflammation, virus
The Human Rhinovirus (HRV) is the primary etiologic agent of the common cold, and HRV infection appears to trigger ~50% of asthma and chronic obstructive pulmonary disease (COPD) exacerbations.1–4 Early HRV infection also appears to be a major risk factor for developing asthma in later life.5 These lower airway effects represent significant morbidity and mortality associated with HRV, and were the impetus for us to sequence the full genomes of the archived reference set of 99 prototypic HRV serotypes. This provided an opportunity to ascertain the diversity of HRVs at the whole genome level, to define commonalties and differences, to begin to make refined structure/function predictions related to pathogenesis and novel therapies, and to develop tools for rapid full genome sequencing and analysis from patient isolates for epidemiologic studies. In this review, the salient findings from these endeavors6 are presented, with an emphasis on application to asthma and COPD exacerbations.
HRV TAXONOMY AND GENOMIC FEATURES
The taxonomic classification of HRV has been recently revised (http://www.ictvonline.org). Previously, HRV were placed in the Family Picornaviridae, Genus Rhinovirus, and each serotype was considered a species (such as hrv-16, hrv-28, etc.). The current taxonomy maintains HRV in the Picornaviridae family, but the Genus is Enterovirus, with three species: HRV-A, HRV-B, and HRV-C. Within each species there are multiple HRVs (variably designated as “serotypes,” “types,” or “strains”). Of note, the original ~100 HRVs collected from patient samples in the 1960’s and 1970’s (the reference or prototype set) which have been propagated and maintained by the American Type Culture Collection (ATCC), were in fact subjected to serotyping.7 Subsequently, as sequencing technology has improved, distinct HRVs are being discovered but are no longer serotyped. For the work discussed here, we utilized the collection of HRVs from ATCC since these are frequently utilized by many investigators in in vitro and in vivo experiments, and thus provides a collection relating phenotypes to an HRV with a known sequence. This reference set includes 74 HRV-A and 25 HRV-B serotypes, which have been assigned names by convention starting from “hrv-1.”8 The HRV-C species has only recently been recognized, and currently consists of at least 11 types whose complete genomes are known.6,9–11 To date, these HRV-Cs have not been successfully infected into host cells ex vivo, and thus there has been a limited amount of virus available for sequencing or biologic studies. However, there is a growing body of evidence suggesting that HRV-C infections may be associated with greater respiratory impairment or symptoms.12–14
HRV is a positive-sense single stranded RNA virus of ~7200 bp.15,16 The genome consists of a single gene, whose translated protein is cleaved by virally encoded proteases to yield 11 proteins (Fig 1). The 5′UTR is typically ~650 bases, the open reading frame ~6500 bases (~2100 encoded amino acids) and the 3′UTR consists of ~50 bases. At the 5′-terminal U, all HRVs are covalently linked to a small viral protein (VPg), which serves as a primer for genome replication.17 The 5′UTR contains a number of structural and sequence elements necessary for gene translation and other functions (see below). After cleavage of the single polypeptide product of the open reading frame, various proteins assemble to form the capsid, or, carry out other specific functions related to replication. The capsid is composed of the VP4, VP2, VP3 and VP1 proteins arranged together as a unit (termed the protomer). The protomers are organized into 12 pentamers, linked to each other along the 2-fold symmetry edges. Thus the entire capsid is composed of 60 copies each of the four capsid proteins. The full icosahedryl structure as determined by X-ray crystallography reveals a canyon in VP1 encircling each central plateau around the 5-fold axes of symmetry (see illustrations in references 15,16,18). Certain amino acids within these canyons provide the virion with attachment sites to cell surface ICAM-1 receptor,18–20 to which 88 of the HRV-A and -B bind. The other 11 (among the HRV-A) bind to the VLDL receptor. The cellular receptor for the HRV-Cs is not known. Within each VP1 protein, accessible through the canyon is a hydrophobic pocket, which is among the known binding sites for any of several anti-viral compounds.
Figure 1.
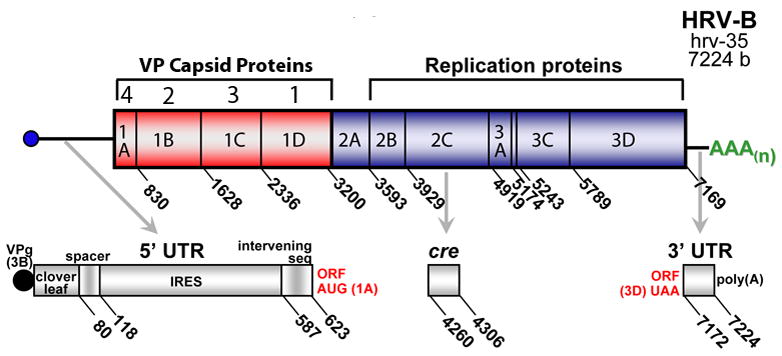
Organization of the HRV genome. The genome consists of 5′- and 3′UTRs, and a single open reading frame encoding one protein, which is subsequently cleaved to form 11 viral proteins which act to form the capsid (VP4-VP1) or are required for viral replication.
The remaining portion of the genome open reading frame encodes proteases, a polymerase, VPg and other proteins required for infection. Of note, the HRV 3D-polymerase, necessary for synthesis of new genome, has no proof-reading capacity, so mutations at this step are assumed to be frequent. If so, then like poliovirus a pool or “cloud” consisting of one (or more) high abundance HRVs and multiple low abundance mutated HRVs, of various related sequences, may be present during a given infection. For polio, this quasispeciation contributes to virulence.21 To date, the presence or relevance of non-clonal HRVs in a human infection is not known. Within the 3′UTR are structure elements with features that appear to act in the regulation of transcription.22
SEQUENCING THE REFERENCE SET OF HRVS
Our initial approach for sequencing the ATCC reference set of HRVs was to devise “universal primers” which might be suitable for PCR amplification along the genomes for all the HRVs from the ATCC as well as field samples. The original primers were designed from the eight full genome sequences that were available at that time and from limited sequence fragments of other HRVs. But this approach failed to give conditions that consistently provided amplicons for sequencing, so instead we utilized a random, sequence independent, single primer shotgun sequencing method.6,23 Briefly, the new primers for reverse transcription (to generate cDNA) consisted of all possible combinations of the 4 nucleotides as 7-mers, each of which had a unique 5′ tag. From a reverse transcriptase reaction with all ~4000 primers, then, there were matches to many sequences throughout each ~7200 bp genome, and a large number of products were generated. Polymerase chain reactions using the tag sequence for priming gave amplicons that could be cloned into a sequencing vector. After transformation ~300 colonies were picked for expansion, providing inserts of various sizes covering each genome, with substantial overlap in redundant sequence (Fig 2). The sequence of any areas in the genome where there were gaps or coverage was minimal was filled in after picking additional clones, or, by specific PCRs primed by adjacent sequence. This approach yielded ~6-fold coverage across each of the hrv genomes.24,25 Ultimately, we completed the full sequences of 88 HRV-A and HRV-B, including 10 HRVs from recent patient samples (here designated with an “fxx” along with the serotype. When our data were combined with that of two other groups who were also sequencing HRV24,25 we had the first description of the full genome set of the entire ATCC reference library, as well as the sequences of our additional field strains.
Figure 2.

Representative HRV sequence coverage by the sequence-independent random primer method. Each horizontal line represents contiguous sequence, ranging in size from ~50 to ~1000 bases. The vertical component represents the extent of redundancy for any given nucleotide or genome segment. The horizontal component represents the extent of the coverage of the genome.
STRUCTURE-BASED ALIGNMENT OF RNA AND AMINO ACID SEQUENCES
In order to begin to define the commonalities and differences between the serotypes, an accurate alignment of the RNA sequences, and the translated amino acid sequences, is necessary. The alignment is also critical for ascertaining phylogenetic relationships. We thus chose to utilize a molecular structure-based approach for the alignments so as to maximize potential structure/function relationships and phylogenetic inferences. For the proteins, the initial fits were by superimposition of the amino acids with known HRV and related virus structure maps derived from X-ray crystallography.26 In a stepwise manner, profile hidden Markov models (HMM) supplemented the founder set with remaining sequences. With these alignments the open reading frame was established for each HRV by reverse translation, and the insertion/deletion regions thereby identified. For the 5′ and 3′-UTRs, we utilized thermodynamically derived folding motifs as a template by which to progressively establish alignment. Here, minimal energy hydrogen bonding configurations were calculated for each HRV.27 Superimposition of these 5′ and 3′-motifs provided the core consensus profiles for RNA alignments. Complete genomes were then reconstructed by concatenating the aligned 5′, coding, and 3′ RNA sequences. These alignments which also include previously sequenced HRV-A and -B, and the recently sequenced HRV-C, can be found in the Supplementary Data of reference 6.
5′UTR SEQUENCE ANALYSIS
A number of intriguing findings came from comparative analysis of the 5′UTR sequences from the 99 HRV-A and -B and 7 HRV-C complete genomes. As was expected, we found the 5′ terminal portion of the 5′UTR modeled into a cloverleaf for each HRV (Fig 3A). The configuration of the cloverleaf appeared to be quite similar between species. In contrast, a pyrimidine-rich spacer tract just 3′ to the cloverleaf had a different nucleotide sequence for each HRV serotype from the reference set, and even within-serotype differences were observed when the reference samples were compared to recently obtained field isolates (Fig 3B). The analogous position in the poliovirus genome (denoted 10b) is critical for neurovirulence, while aphthoviruses and cardioviruses have poly(C) or poly(UC) tracts in this position which are also required for virulence. If this same region in HRV has a similar function, the marked sequence variation we found may indicate that serotypes have significant differences in virulence, and that virulence within serotype may be actively changing. Adjacent to the spacer tract (moving 3′) is the IRES which initiates translation for the open reading frame. Translation in enteroviruses via IRESs occurs by binding of the 40S ribosomal subunit to a 5′ triplet (such as AUG)28 with subsequent scanning through the intervening sequence to the initiator AUG. For all HRV, we found that the minimal energy model revealed that the initiator AUG was invariably paired to a 5′ upstream AUG (Fig 4). This pairing suggests a non-scanning IRES. That is, HRVs may use the proximity of the AUGs to orient the 40S ribosome to proceed with translation, in a bait and switch fashion, without the need for scanning. This mechanism may enhance HRV translation competitiveness. A similar pairing is not found with any other enterovirus.
Figure 3.

Variability of the 5′UTR pyrimidine-rich spacer tract in the HRV genome. Shown are representative modelings of the 5′UTR from the first base to ~130 bases, revealing a cloverleaf secondary structure motif followed by the spacer tract (A). This spacer tract, which is immediately 3′ to the cloverleaf, is highly variable between the serotypes (B). As indicated by the red boxes which compares the reference hrv-52 to the field sample hrv-52-f10, the tract is also variable within-serotype.
Figure 4.

Internal ribosome entry sites (IRES) for HRVs. Minimal energy secondary structure models were constructed for 5′UTRs of each HRV sequence. The 3′ region of each IRES (bases ~100 to ~625) forms an unbranched stem of varying length and composition. However, the statistically lowest energy conformation invariably pairs the last AUG of the IRES (light green box) with the ORF AUG (dark green box), as part of an HRV-specific mechanism to facilitate ribosome entry. Shown are representative stems from the three recognized species (HRV-A, -B, and -C) and clade D.
CODING SEQUENCE COMPARISONS
The amino acid identities between any two hrvs is shown in matrix form in Fig 5. Readily apparent are the high degrees of identity between members of a given species (e.g., orange and pink regions), and the lower identities when comparing across species (grey areas). It is also clear that there is heterogeneity within species, represented by “islands” of discontinuity in the color scheme. This suggested that certain HRVs may cluster together in terms of relatedness and perhaps function. Phylogenetic trees were constructed using the full genomes in order to assess relationships among HRVs. One such tree, constructed using a Neighbor-Joining algorithm, is shown in Fig 6. This approach may not be ideal for accurately defining the evolutionary path by which a given HRV arose (see below), but does provide an accurate model of the relationships between the various types. A poliovirus and two coxackie virus genomes were utilized as the outgroups to root the tree. As shown in Fig 6, HRV-A, -B, and -C each form distinct major clades. It also appears that HRV-A and HRV-C share a common ancestor which is a sister group to the ancestral HRV-B. The early divergence of HRV-C, and the presence of many serotypes within HRV-A and -B species, suggests evolutionary “space” within HRV-C, indicating that there may be many additional HRV-C yet to be discovered. We also note the distinct clustering and apparent early divergence of the small HRV-A clade denoted here as “clade-D.” This may be indicative that these represent a fourth, or newly emerging species (HRV-D). Within the larger HRV-A, there are clear mini-clades that may represent groupings of HRV with similar characteristics. The group starting with hrv-20 at the bottom of the figure, and moving counterclockwise to hrv-80, represents one such a mini-clade. Similarly, clusters of genotypes are noted elsewhere, such as hrv-89 through hrv-88 and hrv-81 through hrv-01. Furthermore, within the large group extending from hrv-09 through hrv-100, there are several additional clusters of >5 members with apparent early divergence from an ancestral HRV. Similar clustering is seen with HRV-B (Fig 6), and will likely be noted for HRV-C as more types are sequenced. Some of this clustering can be seen from the amino acid comparison matrix, such as the orange section encompassing hrv-12 through hrv-89 of Fig 5, which represents the first two mini-clades in Fig 6 discussed above. We compared the full genome tree with other trees constructed using only VP1 sequence, VP4+2 sequence, 3Dpol sequence, or the IRES, with the null-hypothesis being that these trees would not differ from the full genome tree. However, all four of these alternative trees differed statistically from the full-genome tree (P-values from ~10−6 to ~10−100). This suggested that HRVs may have evolved with mutations throughout the genome, rather than strictly at the most “immunologically exposed” regions.
Figure 5.

Amino acid identity heat map of HRVs. Shown are comparisons between all the reference HRV-A and -B, and five HRV-C. For clarity, the specific HRVs are not shown. The scale of identity ranges from ~45% (dark grey) to 100% (yellow). The diagonal set of yellow blocks represents the comparison of the same two HRVs. The dashed line shows the sequence identity comparison between hrv-32 and all other HRVs. Heterogeneity within-species, shown by blocks and islands of discontinuity in the identities, suggests that certain HRVs are clustered based on composition. See also the phylogenetic tree of Fig 6.
Figure 6.

Phylogeny of HRV as illustrated in a Neighbor-Joining tree. Three human enteroviruses (HEV-C: coxackie virus-a21 and -a13, and poliovirus-1m) were utilized for outgroup rooting of the tree. Individual serotypes with an “f” designation represent a field isolate obtained from a patient. The numbers at various branch points are bootstrap values indicating the confidence of the branching (100 is maximal).
RECOMBINATION BETWEEN HRVS
The nature of the diversity of HRVs was further explored by examining the potential for recombination. Unexpectedly, we found highly statistically significant evidence for recombination, as illustrated in Fig 7. In this example, hrv-53 and hrv-80 each contributed portions of their genomes to hrv-46. As can be seen, there is a high degree of identity between hrv-80 and hrv-46 in the 5′-halves of the genomes up to ~3600 b, which represents most of the 5′UTR, VP4, VP2, VP3 and VP1. The remaining ~3900 b of hrv-46 came from hrv-53, as indicated by the increased identity. Furthermore, the identities between the two parents (yellow line) is low, indicating that the recombination occurred between two relatively dissimilar hrvs (see location in the tree of Fig 5). Altogether, 23 HRVs in our total data set of full-length genomes, were found to originate through recombination events. Given that co-infection with two HRVs obviously occurs,29 these results provide compelling evidence for such conditions resulting in the creation of distinct new HRV. What is currently not known is the frequency of such events, when these events may have occurred, and whether additional newer recombinants will be found as more hrvs from patient samples are sequenced. Interestingly, within our virus cohort, the degree of identity between the donated parent segments and the analogous daughter segments never approached 100%. In the example shown, it peaked at ~65% and ~72% for each parental segment vs. the daughter segment. This may suggest that these recombination events were distant as opposed to recent, such that subsequent to the recombination, many additional mutations were fixed in the respective genomes. However, we don’t really know the within- or between-host mutation rate of HRV, so even the terms “recent” and “distant” are at best descriptive. An alternative hypothesis that cannot be excluded based on the data at hand is that there may be additional HRVs in circulation that we don’t know about which may have recently recombined. Under this scenario the lack of higher identities simply reflects an incomplete dataset of circulating HRVs. Sequencing genomes from large numbers of current patient samples will help to address these types of issues, and will further capture the extent of the diversity in the circulating pool of HRVs.
Figure 7.

HRV recombination has resulted in emergence of a third HRV. Shown is an example where hrv-53 and hrv-80 (parental strains) contributed sequence to generate the daughter strain hrv-46. The Y-axis represents the identity between a given pair of indicated hrvs at any nucleotide (X-axis) along the genome.
With the discovery of recombination, the use of phylogenetic methods that infer a strictly bifurcating tree becomes problematic if the goal of the analysis is to understand “how” a given hrv came to lie within a clade. There are techniques which allow for parallel or conflicting tracts of evolution reflecting recombination events or selection, essentially inferring relationships that represent for more complex models of evolution.30,31 The output from such algorithms appears as a network rather than a tree. We recently performed such an analysis with our full genome database,32 and the clusters found with the Neighbor-Joining method (Fig 6) are entirely consistent with the network model.
FIELD ISOLATES
Although we had full genome sequences from only 10 field samples recently obtained from patients (nasal secretions), it was useful to see the variations between these genomes and the analogous HRV represented by the ATCC samples, given that they differed by >30 years in the collection times. From our phylogenetic analysis (Fig 6), it was readily apparent that each field isolate was identifiably close to one of the reference serotypes (i.e., see hrv-89-f09, hrv-89-f08 and hrv-89 at the bottom of the tree). So, these fit readily into the scaffold as minor variants in all cases. Nevertheless, up to ~800 base changes in RNA sequence were sometimes found between some of these recent isolates and their reference counterparts, representing ~10% of the genome. In terms of nonsynonymous variations (those that change the amino acid), ~50 such variations were noted between some recent isolates and their analogous ATCC sample. We found no readily apparent pattern regarding regions of the HRV genome that were hypervariable or conserved within-serotype, but the sample size is inadequate for such an analysis. We sequenced 2 field isolates each of hrv-09, -81, and -89. These were all collected in 2005–2006, and interestingly, there were far fewer variations between the most recently collected isolates (within serotype) compared to their analogous, older, ATCC isolates.6 While it is tempting to calculate a “mutation rate,” realistically, our sample size is simply too limiting to consider this analysis, and, of course, one cannot exclude that the variant strains, albeit recently collected, were not also in circulation when the ATCC samples were collected. It is important to note that one should not expect that all future patient isolates will necessarily be variants (however so defined) of one of the fully sequenced HRVs from the ATCC set. Indeed, within GenBank one can find HRV-like sequences (typically far shorter than full-length) that may represent novel types. As an example, we recently reported32 the full genome sequence of an HRV (denoted hrv-A101) that fell within the HRV-A species, but had only 76% nucleotide identity with its nearest neighbor (a lower value than virtually all other HRVs compared to their closest match), and, was distinctly localized within the HRV phylogenetic network. While specific definitions of an HRV type or strain have not yet been articulated by the ICTV (in progress), the totality of the evidence from this HRV genome is indicative that it is unique from those previously sequenced.32 GenBank searches showed partial sequences with high identity to hrv-A101 reported from isolates from China, Germany, Spain, Australia, Belgium and the U.S. Thus even within the “packed” HRV-A species, there are likely to be other types awaiting discovery. Given that recombination is prevalent, it will be important to have full genome sequence of new HRV isolates in order to place them into context within the group of known isolates.
IMPLICATIONS FOR OBSTRUCTIVE LUNG DISEASE AND FUTURE DIRECTIONS
The results discussed in this review provide opportunities to explore basic questions about HRV biology, and the link between HRV infection and asthma or COPD pathophysiology. First, we now have a scaffold (the alignments and phylogeny) to integrate additional strains as they are identified into the three (or four) well-defined HRV species. Furthermore, the pipeline methods have been developed to rapidly sequence the complete genomes of HRVs obtained from patient samples. So, the diversity of HRV at the full genome level can now be ascertained within a community during a single cold season. Similarly, the mutation rate of an HRV, from onset to recovery, within a single individual could be determined, as well as how the same virus, when naturally transmitted to another person, may mutate in response to different host factors. The effects of concomitant bacterial or other viral infection, or the upper airway microbiome, could also be addressed.
With proper study design, it should be possible to begin to relate a clinical phenotype to an HRV genomic sequence. Among possible approaches, for example, consider a hypothetical study which might reveal that out of 30 HRVs identified from a group of asthmatics with symptomatic upper respiratory tract infection, only those who had an HRV belonging to a certain mini-clade had an asthmatic exacerbation. Such a finding would lead to a scrutiny of those HRV genomes to ascertain which features promote the “pro-exacerbation” phenotype. Alternatively, it might be found that the most robust association is with a particular genome segment, which does not necessarily track with whole genome phylogeny. Again, such a finding would prompt study of that region of the genome and the unique properties of the group that was associated with exacerbation. On an even finer scale, it may be found that certain variations (“polymorphisms”) within one or more parts of HRV genomes are the real determinants of exacerbation. Possibly, these HRVs could be found scattered throughout the species tree, even when a tree is constructed with small genome segments.
Some of the future studies outlined above, such as those examining mutation rates, obviously require the availability of full genome sequences. In some of the other studies, our bias is to “start big.” That is, use all of the genetic information from a complete genome in these association studies with asthma phenotypes. This bias is based on our assumption that the HRV evolve within the context of the full genome, rather than as isolated segments without dependence on other parts of the genome. As more data are gathered, we may well be able to utilize shorter HRV sequences to identify those asthmatics who will exacerbate, but given the advances in high-throughput sequencing technology there is little reason (now) to compromise a study by using limited HRV sequence information. Regardless, many samples are going to be required to carry out these analyses, and an effort by the International Rhinovirus Consortium (http://www.international-rhinovirus-consortium.org) to collect and sequence HRVs from adequately phenotyped subjects is underway.
The full set of genome sequence data, RNA structural predictions, phylogeny and recombination results have multiple implications for development of anti-HRV therapeutics. Many attempts have been made to devise broad-based efficacious drugs which act at one or more events in the HRV infection and replication cycles. Thus far, these efforts have taken a “one drug fits all” approach. Drugs targeting viral attachment (soluble ICAM 1, pleconaril), viral fusion to the cell membrane (arbidol), capsid uncoating (pleconaril), capsid stabilizers (Ro-09-0410), viral replication (enviroxime), translation (IRES-based oligomers), and post-translational processing (inhibition of proteases by agents such as rupintrivir) have been developed and studied in vitro and in clinical trials. Typically these experiments show excellent antiviral effects in cell culture systems, but minimally significant or non-significant effects in human clinical trials. Even when a realized prophylactic effect to experimental HRV infection can be observed (such as decrease of ~1–2 days of clinical symptoms, nasal viral titers, or secretions), further studies assessing prophylaxis, or, acute treatment of naturally occurring colds from HRV, often show no significant effect.
The basis of these prior failures is not altogether clear. In some cases, the route of administration (or the formulation) may have been sub-optimal. However, many of these failures might be attributable to our overall lack of understanding of the basic biology of HRV infection in the human airway, and, the genetic diversity and real-time evolution of natural HRV strains. The clinical experiences with pleconaril exemplify such issues.33,34 This agent showed inhibition of ~90% of HRV serotypes in vitro, and in two placebo-controlled double-blind clinical studies showed some degree of efficacy for treating naturally occurring colds. Observations included a decrease in time to illness resolution (7.9 days vs. 6.8 days for placebo), illness severity scores, and nasal mucous viral RNA at Day 3 (97.7% decrease vs. 90.3% decrease for placebo). Viral cultures and in vitro sensitivities of the isolated HRVs were also carried out at baseline and at Day 3. Fewer pleconaril-treated patients had positive cultures at Day 3 compared to placebo-treated patients (53% vs. 72%, respectively). The clinical benefit appeared to correlate somewhat with the in vitro sensitivities at Day 1: EC50 values of ≤0.38 μg/ml had reductions in symptom duration, while those with higher EC50 values showed no benefit. This latter group represented 60% of the subjects. So, while there was an overall benefit generically, clearly the drug was ineffective for a substantial subset of subjects. Moreover, in 10.7% of the pleconaril-treated subjects, the Day 3 in vitro susceptibility was actually lower, and in 2.7% the virus was fully resistant to pleconaril. These latter data suggested the development of pleconaril-induced resistance during treatment in up to ~13% of patients. It would be highly informative to know the full genome sequences (not just the serotype) of the HRVs that showed low sensitivity to pleconaril at Day 1, and, of those that developed drug resistance. Potentially, pleconaril might be highly efficacious for a subset of HRVs, based on a particular genomic sequence. A rapid diagnostic test could identify patients most likely to benefit based on HRV sequence at the onset of symptoms. In essence, this is a form of “personalized medicine,” where the genome of the pathogen could be used to help guide treatment.
The potential for a successful vaccine seem remote at this time. Unfortunately, the very nature of HRV genetic diversity, fueled by dual-infection recombination and a high genome-specific mutation rate, are exactly the same parameters which work against effective vaccine development. The constant co-circulation of the 99 (100+?) defined serotypes of HRV-A and HRV-B, let alone the added immunogenetic assortment of the numerous HRV-C, make it difficult to envision how a monovalent or even polyvalent formulation could cover this diversity. The sequences tell us there are no common epitopes conserved on the various capsid surfaces. The receptor binding-canyons are thought to be too deep and narrow for antibody recognition, and even if these regions could be targeted artificially, protective immunity against HRV is likely to require an IgA (mucosal) response, not the more common IgG (circulating) response, as elicited by other efficacious (e.g., polio) picornavirus vaccines.
CONCLUDING REMARKS
In summary, the complete reference set of HRVs has now been sequenced at the full genome level. The results provide new insights into HRV evolution and diversity, and suggest levels of relatedness that may provide for signatures for asthma or COPD exacerbations. Structural predictions from the genomes may provide for novel therapeutics targeted to subsets of HRVs based on specific sequences.
Acknowledgments
The authors thank Esther Moses for manuscript preparation.
Supported by National Institutes of Health grants HL091490 and AI070503, and NIAID contract HHSN272200900009C.
Abbreviations
- HRV
human rhinovirus
- IRES
internal ribosome entry site
- UTR
untranslated region
- ATCC
American Type Culture Collection
Glossary
- Arbidol
An antiviral compound that has been used to treat influenza, rhinovirus, and hepatitis C
- Capsid
The protein shell of the virus that protects the genome and allows entry into the host cells. The capsid is usually composed of redundant proteins so that if one subunit is damaged the virus can continue to be infective using its other subunits. H1N1 of the Influenza virus is an example of a capsid protein that determines virulence
- Enviroxime
An antiviral agent that inhibits viral replication by inhibiting the viral protein 3A
- Hidden Markov Models
A statistical model system that can be applied to bioinformatics for predicting and testing relationships. In this model, the state is not visible but the output dependent on the state is visible. Based on this, one can predict the likelihood that any given observation reflects the hidden state
- ICAM-1
ICAM-1 (intracellular adhesion molecule-1), is expressed on numerous cell-types. Many rhinovirus types bind ICAM-1 expressed on nasal epithelial cells
- Neighbor-Joining Algorithm
A methodology used in bioinformatics that uses DNA or protein sequences to construct phylogenetic trees
- Open Reading Frame
The sequence of bases that could encode a protein. An ORF is located between the start codon (ATG, methionine) and the stop codon; any ORF can be read in 6 reading frames (in double stranded DNA).
- Phylogenetics
Phylogenetics uses sequencing data to evaluate the evolutionary relatedness between organisms
- Pleconaril
An oral antiviral that integrates into the hydrophobic core of the picornavirus capsid
- Reverse translation
Refers to the process of entering a protein sequence into a program that then turns the amino acid sequence back into a DNA nucleotide sequence
- Rupintrivir
An antiviral agent that inhibits viral replication by inhibiting the rhinovirus 3C protease
- Universal primers
Primers used in PCR (polymerase chain reaction) and DNA sequencing that recognize the multiple cloning sites found in plasmid vectors. These plasmid regions are used as the starting site for sequencing the specific cloned DNA fragment of interest
- 5′ UTR (untranslated region)
3′ UTR, The untranslated region of mRNA located upstream and downstream of the coding sequence respectively
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gwaltney JM, Jr, Hendley JO, Simon G, Jordan WS., Jr Rhinovirus infections in an industrial population. I. The occurrence of illness. N Engl J Med. 1966;275:1261–8. doi: 10.1056/NEJM196612082752301. [DOI] [PubMed] [Google Scholar]
- 2.Nicholson KG, Kent J, Ireland DC. Respiratory viruses and exacerbations of asthma in adults. BMJ. 1993;307:982–6. doi: 10.1136/bmj.307.6910.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johnston SL, Pattemore PK, Sanderson G, Smith S, Campbell MJ, Josephs LK, et al. The relationship between upper respiratory infections and hospital admissions for asthma: a time-trend analysis. Am J Respir Crit Care Med. 1996;154:654–60. doi: 10.1164/ajrccm.154.3.8810601. [DOI] [PubMed] [Google Scholar]
- 4.Varkey JB, Varkey B. Viral infections in patients with chronic obstructive pulmonary disease. Curr Opin Pulm Med. 2008;14:89–94. doi: 10.1097/MCP.0b013e3282f4a99f. [DOI] [PubMed] [Google Scholar]
- 5.Jackson DJ, Gangnon RE, Evans MD, Roberg KA, Anderson EL, Pappas TE, et al. Wheezing rhinovirus illnesses in early life predict asthma development in high risk children. Am J Respir Crit Care Med. 2008;178:667–72. doi: 10.1164/rccm.200802-309OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Palmenberg AC, Spiro D, Kuzmickas R, Wang S, Djikeng A, Rathe JA, et al. Sequencing and analyses of all known human rhinovirus genomes reveals structure and evolution. Science. 2009;324:55–9. doi: 10.1126/science.1165557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Conant RM, Hamparian VV. Rhinoviruses: basis for a numbering system. II. Serologic characterization of prototype strains. J Immunol. 1968;100:114–9. [PubMed] [Google Scholar]
- 8.Rhinoviruses: a numbering system. Nature. 1967;213:761–2. doi: 10.1038/213761a0. [DOI] [PubMed] [Google Scholar]
- 9.Dominguez SR, Briese T, Palacios G, Hui J, Villari J, Kapoor V, et al. Multiplex MassTag-PCR for respiratory pathogens in pediatric nasopharyngeal washes negative by conventional diagnostic testing shows a high prevalence of viruses belonging to a newly recognized rhinovirus clade. J Clin Virol. 2008;43:219–22. doi: 10.1016/j.jcv.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McErlean P, Shackelton LA, Andrews E, Webster DR, Lambert SB, Nissen MD, et al. Distinguishing molecular features and clinical characteristics of a putative new rhinovirus species, human rhinovirus C (HRV C) PLoS ONE. 2008;3:e1847. doi: 10.1371/journal.pone.0001847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lau SK, Yip CC, Tsoi HW, Lee RA, So LY, Lau YL, et al. Clinical features and complete genome characterization of a distinct human rhinovirus (HRV) genetic cluster, probably representing a previously undetected HRV species, HRV-C, associated with acute respiratory illness in children. J Clin Microbiol. 2007;45:3655–64. doi: 10.1128/JCM.01254-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang T, Wang W, Bessaud M, Ren P, Sheng J, Yan H, et al. Evidence of recombination and genetic diversity in human rhinoviruses in children with acute respiratory infection. PLoS ONE. 2009;4:e6355. doi: 10.1371/journal.pone.0006355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jin Y, Yuan XH, Xie ZP, Gao HC, Song JR, Zhang RF, et al. Prevalence and clinical characterization of a newly identified human rhinovirus C species in children with acute respiratory tract infections. J Clin Microbiol. 2009;47:2895–900. doi: 10.1128/JCM.00745-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Piralla A, Rovida F, Campanini G, Rognoni V, Marchi A, Locatelli F, et al. Clinical severity and molecular typing of human rhinovirus C strains during a fall outbreak affecting hospitalized patients. J Clin Virol. 2009;45:311–7. doi: 10.1016/j.jcv.2009.04.016. [DOI] [PubMed] [Google Scholar]
- 15.Turner RB, Lee W-M. Rhinovirus. In: Richman DD, Whitley RJ, Hayden FG, editors. Clinical Virology. Washington, DC: ASM Press; 2009. pp. 1063–82. [Google Scholar]
- 16.Couch RB. Rhinoviruses. In: Knipe DM, Howley PM, editors. Fields Virology. Philadelphia: Lippincott Williams & Wilkins; 2001. pp. 777–98. [Google Scholar]
- 17.Paul AV, van Boom JH, Filippov D, Wimmer E. Protein-primed RNA synthesis by purified poliovirus RNA polymerase. Nature. 1998;393:280–4. doi: 10.1038/30529. [DOI] [PubMed] [Google Scholar]
- 18.Colonno RJ, Condra JH, Mizutani S, Callahan PL, Davies ME, Murcko MA. Evidence for the direct involvement of the rhinovirus canyon in receptor binding. Proc Natl Acad Sci U S A. 1988;85:5449–53. doi: 10.1073/pnas.85.15.5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Olson NH, Kolatkar PR, Oliveira MA, Cheng RH, Greve JM, McClelland A, et al. Structure of a human rhinovirus complexed with its receptor molecule. Proc Natl Acad Sci U S A. 1993;90:507–11. doi: 10.1073/pnas.90.2.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vlasak M, Roivainen M, Reithmayer M, Goesler I, Laine P, Snyers L, et al. The minor receptor group of human rhinovirus (HRV) includes HRV23 and HRV25, but the presence of a lysine in the VP1 HI loop is not sufficient for receptor binding. J Virol. 2005;79:7389–95. doi: 10.1128/JVI.79.12.7389-7395.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vignuzzi M, Stone JK, Arnold JJ, Cameron CE, Andino R. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature. 2006;439:344–8. doi: 10.1038/nature04388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Herold J, Andino R. Poliovirus RNA replication requires genome circularization through a protein-protein bridge. Mol Cell. 2001;7:581–91. doi: 10.1016/S1097-2765(01)00205-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Djikeng A, Halpin R, Kuzmickas R, Depasse J, Feldblyum J, Sengamalay N, et al. Viral genome sequencing by random priming methods. BMC Genomics. 2008;9:5. doi: 10.1186/1471-2164-9-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kistler AL, Webster DR, Rouskin S, Magrini V, Credle JJ, Schnurr DP, et al. Genome-wide diversity and selective pressure in the human rhinovirus. Virol J. 2007;4:40. doi: 10.1186/1743-422X-4-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tapparel C, Junier T, Gerlach D, Cordey S, Van Belle S, Perrin L, et al. New complete genome sequences of human rhinoviruses shed light on their phylogeny and genomic features. BMC Genomics. 2007;8:224. doi: 10.1186/1471-2164-8-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Palmenberg AC, Sgro J-Y. Alignments and comparative profiles of picornavirus genera. In: Semler BL, Wimmer E, editors. Molecular Biology of Picornaviruses. New York: ASM Press; 2001. pp. 149–58. [Google Scholar]
- 27.Palmenberg AC, Sgro J-Y. Topological organization of picornaviral genomes: statistical prediction of RNA structural signals. Seminars in Virology. 1997;8:231–41. [Google Scholar]
- 28.Jackson RJ. Initiation site selection mechanisms. In: Hershey WB, Mathews MB, Sonenberg N, editors. Translational Control. Cold Spring Harbor Laboratory Press; 1996. pp. 71–112. [Google Scholar]
- 29.Savolainen C, Mulders MN, Hovi T. Phylogenetic analysis of rhinovirus isolates collected during successive epidemic seasons. Virus Res. 2002;85:41–6. doi: 10.1016/s0168-1702(02)00016-3. [DOI] [PubMed] [Google Scholar]
- 30.Huson DH. SplitsTree: analyzing and visualizing evolutionary data. Bioinformatics. 1998;14:68–73. doi: 10.1093/bioinformatics/14.1.68. [DOI] [PubMed] [Google Scholar]
- 31.Huson DH, Bryant D. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol. 2006;23:254–67. doi: 10.1093/molbev/msj030. [DOI] [PubMed] [Google Scholar]
- 32.Rathe JA, Liu X, Tallon LJ, Gern JE, Liggett SB. Full-genome sequence and analysis of a novel human Rhinovirus strain within a divergent HRV-A clade. Arch Virol. 2010;155:83–7. doi: 10.1007/s00705-009-0549-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hayden FG, Herrington DT, Coats TL, Kim K, Cooper EC, Villano SA, et al. Efficacy and safety of oral pleconaril for treatment of colds due to picornaviruses in adults: results of 2 double-blind, randomized, placebo-controlled trials. Clin Infect Dis. 2003;36:1523–32. doi: 10.1086/375069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pevear DC, Hayden FG, Demenczuk TM, Barone LR, McKinlay MA, Collett MS. Relationship of pleconaril susceptibility and clinical outcomes in treatment of common colds caused by rhinoviruses. Antimicrob Agents Chemother. 2005;49:4492–9. doi: 10.1128/AAC.49.11.4492-4499.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]