

Abstract
Using microcoil NMR technology, the uniformly 2H,15N-labeled integral membrane protein OmpX and the phosphocholine derivative detergent Fos-10 (n-decylphosphocholine), we investigated solutions of mixed protein–detergent micelles to determine the influence of the detergent concentration on the NMR spectra of the protein. In a first step, we identified key parameters that influence the composition of the micelle solutions, which resulted in a new protocol for the preparation of well-defined concentrated protein solutions. This led to the observation that high-quality 2D [15N,1H]-TROSY spectra of OmpX reconstituted in mixed micelles with Fos-10 were obtained only in a limited range of detergent concentrations. Outside of this range from about 90 mM to 180 mM, we observed a significant decrease of the average peak intensity. Relaxation-optimized NMR measurements of the rotational and translational diffusion coefficients of the OmpX/Fos-10 mixed micelles, Dr and Dt, respectively, then showed that the stoichiometry and the effective hydrodynamic radius of the protein-containing micelles are not significantly affected by high Fos-10 concentrations, and that the deterioration of NMR spectra is due to the increased viscosity at high detergent concentrations. The paper thus provides a basis for refined guidelines on the preparation of integral membrane proteins for structural studies.
Introduction
Recent publications show that atomic-resolution structures of integral membrane proteins (IMPs) can be determined either by X-ray diffraction in single crystals1 or by nuclear magnetic resonance spectroscopy (NMR) in solution2, provided that diffracting crystals or structure-quality solutions of IMPs incorporated into detergent or lipid micelles are available. In apparent contrast, the protein data bank3 contains only a small number of IMP structures when compared to soluble proteins, which is in no way correlated with the frequency at which the two classes of proteins are represented in the genomes of higher or lower organisms.4 The PDB thus documents a stringent bottleneck that slows down the structural biology of membrane proteins, i.e., the preparation of diffracting crystals or concentrated solutions of stable-isotope-labeled IMPs for high resolution structure determination. The present paper describes systematic studies of size and composition of mixed IMP–detergent micelles in solution under variable conditions of detergent concentration, protein concentration and experimental set-up. The results thus obtained should contribute to developing improved protocols for IMP preparation either for NMR or X-ray diffraction studies. The present experiments further bear on the solvation of IMP structures in aqueous solutions of mixed micelles with detergents, which may in turn also provide new insights into the behavior of IMPs in co-crystals with detergents and lipids, which are often obtained from such solutions.
The present study follows up on previous work by different authors, which showed that the physico-chemical properties of the detergent, in particular the CMC, the aggregation number, and the hydrophobicity and charge can play a key role in a successful sample preparation, and may strongly influence the quality of the NMR spectra recorded with solutions of micelle-solubilized IMPs.5–7 From the available data it is also readily apparent that appropriate choice of the detergent concentration is generally considered to be a key to successful solubilization and reconstitution of IMPs. For NMR structure determinations, detergent concentrations in the range from 200 mM to 600 mM have been reported,2,8 and it has also been suggested that the detergent micelle concentration should be much higher than the protein concentration,7 where the micelle concentration is estimated as (total detergent concentration − CMC)/Na, where Na is the aggregation number, which indicates the number of detergent molecules contained in a micelle.5
Using a previously presented microscale protocol for obtaining high-quality NMR data for the β-barrel outer membrane protein X (OmpX)9 as a starting platform, we optimized the procedure to obtain improved yields of OmpX under controlled solution conditions. Thereby it was of special concern that the detergent content in the concentrated protein solutions needed for NMR measurements or for crystallization attempts may greatly vary depending on the equipment and the conditions used for the concentration steps, and cannot in a straightforward way be monitored via the detergent concentration in the buffer solutions used. In this paper we tightly monitor the detergent concentration and the quality of the NMR spectra obtained at various stages of the OmpX preparation procedure (Fig 1), and thus derive rules on how the detergent concentration in the final NMR sample is affected by the concentration of the detergent in the NMR buffer, the type of centrifugal device used, the centrifugal device molecular weight cut-off (MWCO), the temperature and the protein concentration. Since this project required the screening of a large series of samples containing the uniformly 2H,15N-labeled IMP, the use of microscale NMR spectroscopy to monitor the composition of the buffer and the quality of the protein–detergent micelles during the sample preparation was key to making this study economically viable.
Figure 1.
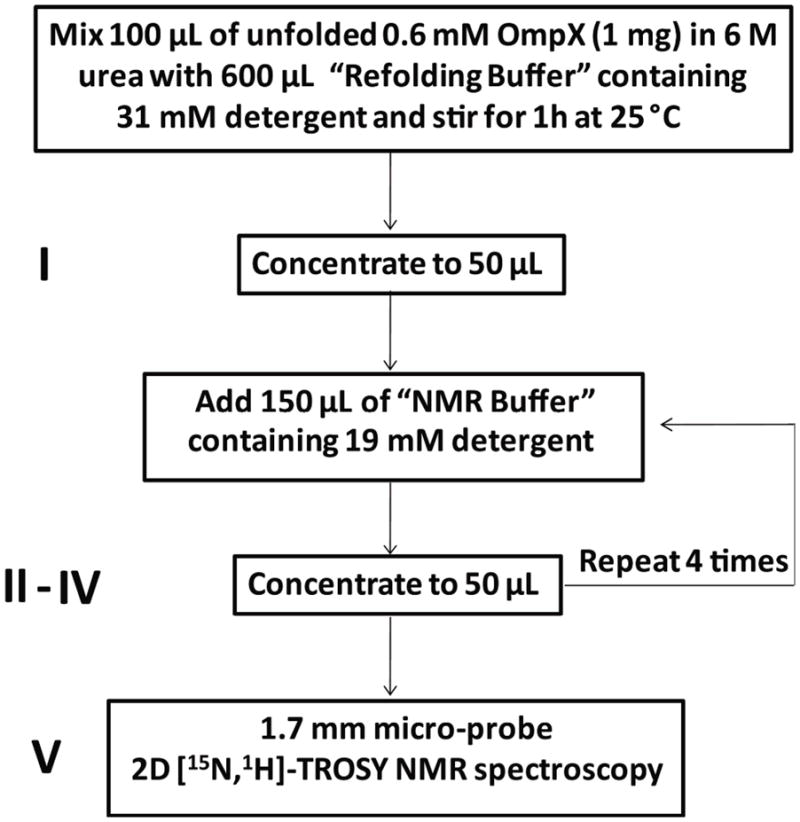
Standard protocol used for microscale preparation of NMR samples of OmpX reconstituted with the detergent Fos-10. In the first step, urea-unfolded OmpX is reconstituted in Fos-10 micelles. In the second step the volume of the solution of the mixed micelles in the refolding buffer is reduced to 50 μl, using centrifugal concentration devices with membranes of MWCO = 10 kDa. In each of four subsequent steps the concentrated protein solution is diluted with a three-fold excess of NMR buffer, and with the aforementioned centrifugal device the sample volume is again reduced to 50 μl. The protein and detergent concentrations, the MWCO of the centrifugal device, the reaction temperature, the reaction times and the repeats of individual steps indicated in the figure are the result of a procedure optimization described in the first part of this paper (the buffers are described in the Materials and Methods section). They are different from the conditions used previously with the same overall set-up9. The optimization was pursued by monitoring the detergent and protein concentrations in the solutions I–V. The solution V corresponds to the final sample, for which a 2D [15N,1H]-correlation NMR experiment was also recorded (see Materials and Methods).
The aforementioned preparative and analytical work sets the stage for investigations on the influence of various solution parameters on the size of the mixed micelles in solution, the quality of the high-resolution NMR spectra obtained, and other parameters. This part of the project is pursued with a suite of NMR experiments for investigations of the rotational and translational diffusion of biological macromolecules and macromolecular assemblies.
Materials and Methods
Unlabeled and uniformly [2H,15N]-labeled OmpX was expressed as inclusion bodies in E. coli and purified to yield a solution of the unfolded protein in 6 M urea.9 The detergent selected for OmpX reconstitution in this study is the phosphocholine derivative n-decylphosphocholine (Fos-10, Anatrace, Maumee, OH), which is a representative of a family of detergents that have previously been shown to yield high-quality NMR spectra of the protein OmpX9 and other β-barrel membrane proteins (unpublished data).
Reconstitution of OmpX in Fos-10 micelles for NMR spectroscopy
The protocol for the preparation of NMR samples containing OmpX in mixed micelles with Fos-10 starts with the purified protein in 6 M urea solution. The protein is reconstituted by the addition of an excess of “refolding buffer” containing 20 mM Tris–HCl at pH 8.0, 5 mM EDTA, 600 mM L-Arg and the detergent, and by stirring of the resulting new solution (Figure 1). Next, the solution of the refolded protein is concentrated to 50 μl using centrifugal concentration devices with a given molecular weight cut-off (MWCO). This concentrated solution is then repeatedly diluted with “NMR buffer” containing 5 mM sodium phosphate at pH 6.8, 10 mM NaCl, 0.3 % NaN3 and the detergent, and the volume is again reduced to 50 μl by centrifugation. After a sufficient number of dilution/concentration cycles for complete replacement of the refolding buffer with NMR buffer, a sample for microcoil NMR spectroscopy is obtained. The “standard conditions” indicated in Figure 1 were selected based on optimization of this procedure in the present paper, and differ from the previously reported conditions used with the same set-up.9 The final NMR sample contained 45 μl of the concentrated OmpX solution supplemented with 5 μl of D2O and 1 μl of a 100 mM solution of 2,2-dimethyl-2-silapentane-3,3,4,4,5,5-d6-5-sulfonate sodium salt (DSS), which was added as an internal reference for the 1H chemical shifts as well as the peak integrals in the 1D 1H NMR spectra.
NMR Spectroscopy
All NMR experiments were recorded at 25 °C on a Bruker DRX-700 spectrometer equipped with a 1.7 mm TXI microcoil probehead (Bruker, Billerica, MA). 1D 1H NMR spectra were collected with the following parameters: complex data size = 16 k, acquisition time = 1.38 s; number of scans = 128, sweep width = 11900 Hz. 2D [15N,1H]-TROSY correlation experiments were recorded as described previously,9,10 and details of the TROSY parameter settings are given in the legend of Figure 4. The NMR data were processed using the software TOPSPIN 1.3 (Bruker). The analysis of the [15N,1H]-TROSY correlation spectra was performed using XEASY,11 and the TRACT and REST data (see below) were analyzed using in-house TOPSPIN macros in combination with the program XMGRACE (http://plasma-gate.weizmann.ac.il).
Figure 4.

(A) – (C) 2D [15N,1H]-TROSY correlation NMR spectra of OmpX/Fos-10 mixed micelles at 53 mM, 102 mM and 881 mM Fos-10 concentrations and 0.84, 1.04 and 0.77 mM [2H,15N]-OmpX concentrations in the NMR sample (solution V in Fig. 1). The arrow points at the well-separated cross-peak of G143, for which the peak intensities, Irel, in the cross sections along ω1(15N) are shown in the panels (D) – (F). Identical acquisition and processing parameters were used to record the three data sets: data size 100 (t1) × 1024 (t2) complex points; t1max = 35.24 ms, t2max = 86.11 ms; 64 scans per t1 increment, overall measurement time 4 h per experiment. Before Fourier transformation the data matrices were multiplied with an exponential window function in the acquisition dimension and with a 75°-shifted sine bell window21 in the indirect dimension.
Monitoring the sample composition at discrete points of the preparation protocol
The detergent-concentration was determined in the solutions I–V (Fig. 1) by comparing the integral of the Fos-10 trimethylamino signal at 3.22 ppm in the 1D 1H NMR spectra with the DSS signal intensity at 0 ppm (Fig. 2). To monitor the quality of the NMR spectra obtained in the different experiments, 2D [15N,1H]-TROSY correlation spectra were recorded in the solution V.
Figure 2.
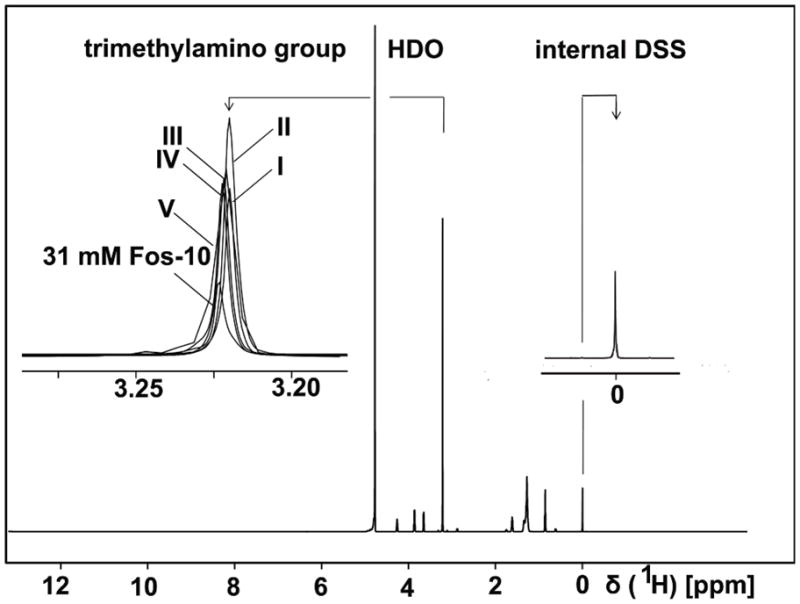
1D 1H NMR spectra used to monitor the Fos-10 concentration of the solutions I to V identified in Fig. 1 for the following experiment: detergent concentration in the NMR buffer = 19 mM, OmpX concentration = 1.0 mM. The spectra were recorded at 700 MHz, using a 1.7 mm microprobe. The singlet resonance at 3.22 ppm corresponds to the trimethylamino group of Fos-10. The DSS signal at 0 ppm was used as an internal reference for both the 1H chemical shifts and the peak integrals in the 1D 1H NMR spectra. Expanded plots of the Fos-10 trimethylamino signal, which shows small chemical shift differences in the various buffer compositions, and the DSS signal in the solutions I to V are superimposed in the two inserts, as indicated in the Figure.
The OmpX concentration was checked by UV spectroscopy, as follows: 1 μl of the concentrated solution of refolded OmpX was diluted 25-fold in 20 mM Tris-HCl at pH 8.5 that contained 6 M urea, and this solution was then stirred at 25°C for 12 h to ensure complete unfolding of the protein. Then the absorption at 280 nm was measured with a nano-drop instrument (ND -1000 Spectrophotometer).
NMR characterization of the OmpX/Fos-10 mixed micelles
The effective rotational correlation time of the OmpX/Fos-10 micelles, τc, was determined using the TRACT experiment.12 A total of 128 relaxation delays ranging from 1 ms to 129 ms were used, with 512 scans per relaxation delay. The relaxation rates Rα and Rβ were obtained by fitting the integrals over all the peaks between 10.0 and 7.0 ppm to a one-parameter exponential decay, and were then used to calculate τc.12
The translational diffusion constant, Dt, was measured using a relaxation-optimized 15N-edited stimulated echo (STE) experiment (REST).13 A series of 16 diffusion-weighted one-dimensional spectra were recorded in a two-dimensional manner, using a pair of gradient pulses of 4.5 ms duration separated by 100 ms, with gradient strengths ranging from 3 to 55 Gcm−1. The pulsed field gradient strengths were calibrated with the residual 1H signal in 99.9 % D2O, using a self-diffusion coefficient for HDO at 25 °C of 1.902 ± 0.002 × 10−9 m2s−1.14 Dt was then obtained by fitting the signal volume with Eq. (1):
| (1) |
γ is the 1H gyromagnetic ratio, and G, δ and Δ are the amplitude, the duration and the separation of the gradient pulses, respectively. To determine the translational diffusion constant of OmpX, the integral over the NMR signals in the interval 7.0 to 10.0 ppm was measured to obtain values for I and I0.
Analysis of the rotational and translational diffusion constants
We use the Stokes–Einstein (SE)-Model to describe the dependence of the rotational and translational diffusion constants on the solution viscosity (η) and the hydrodynamic radius (“Stokes radius”, Rh) for spherical, non-interacting particles:
| (2) |
| (3) |
kB is the Boltzmann constant, T the absolute temperature, and Dt and Dr are the translational and rotational diffusion constants, respectively. Dr is related to the effective rotational correlation time, τc, by
| (4) |
The equations (2) and (3) lead to an expression for the effective hydrodynamic radius of an equivalent sphere, Rh, which is independent of η:
| (5) |
The experimental translational and rotational diffusion constants can thus be used to determine Rh of macromolecular assemblies, such as protein–detergent mixed micelles, within the validity of the Stokes–Einstein model for an equivalent sphere representing the macromolecular structure.
Computational evaluation of the micelle radius Rm
A hypothetical value for the radius of OmpX/Fos-10 mixed micelles was estimated for the situation that the Fos-10 molecules in the solution would either be bound to mixed micelles or monomeric, i.e., with the assumption that there would be no empty Fos-10 micelles in the solution. For this situation, the volume of the mixed micelles, Vm, was computed as
| (6) |
where VOmpX is the volume of the protein (18.2 × 103 Å3),15 VFos10 is the Fos-10 monomer volume (494 Å3)16, Na is the aggregation number, CMCFOS10 = 10.8 mM is the critical micelle concentration for Fos-10, and cFos10 and cOmpX are the experimentally determined total concentrations of Fos-10 and OmpX in the solution in molar units. If the mixed micelles are represented by an equivalent sphere, the radius of this sphere, Rm, is given by
| (7) |
The Rm value obtained with Eq. (7) is a lower bound for the effective hydrodynamic radius Rh, since the equations (6) and (7) do not take into account the hydration shell of the mixed micelle, and deviations from spherical shape always lead to larger effective Rh values for the equivalent sphere.17
Results and Discussion
It has been well established that the nature and concentration of the detergents used for integral membrane protein (IMP) solubilization and reconstitution in aqueous solvents are critical factors in successful IMP preparations for structural biology. However, although it is straightforward to establish well-defined detergent concentrations in the starting buffer solutions, variation of the detergent concentration during dialysis and sample concentration results in uncertainty about the composition of the highly concentrated protein solutions needed either for NMR spectroscopy or for crystallization trials.18 In a first part of the present project we therefore introduce an experimental set-up that enables to monitor the amount of detergent present at discrete stages of the sample preparation and in the final, concentrated protein solution. We then use this procedure to investigate the influence of different parameters on the final sample composition, such as the buffers used, the protein concentration, the type of membrane used in the centrifugal concentration devices and the temperature during the concentration steps. The resulting concentrated protein solutions with known detergent content were then used to study the size and composition of the protein–detergent micelles at variable conditions.
Preparation of concentrated membrane protein solutions with defined detergent concentration
In order to analyze the evolution of the detergent concentration during the reconstitution protocol (Fig. 1), the Fos-10 concentration in the protein solution was determined by 1D 1H NMR spectroscopy (Fig. 2), as described in the Materials and Methods section. The figure 3A shows the results for an experiment performed with 19 mM Fos-10 in the NMR buffer. The comparison of the concentration steps in I and II shows a small increase of detergent content in the sample, which we rationalize by the markedly different buffer compositions in the solutions I and II. The amounts of Fos-10 at the end of steps II and V are similar, with only a slight trend toward lower concentrations arising from the fact that the NMR buffer contains lower amount of detergent than the refolding buffer (see Fig. 1 and the Materials and Methods section for details). We then varied the detergent concentration in the NMR buffer and measured the resulting variation of the detergent concentration in the NMR sample (Fig. 3B). A linear relationship was thus found between the detergent concentrations in the NMR buffer and in the NMR sample, which provided a calibration curve for the preparation of OmpX/Fos-10 samples with specified detergent concentrations. In view of the pronounced up-concentration of the detergent in the NMR sample V, when compared to the NMR buffer (Fig. 1), we further investigated the impact of other experimental parameters on this process.
Figure 3.

Up-concentration of Fos-10 during OmpX refolding monitored by microscale 1D 1H NMR. (A) Histogram presentation of the detergent concentrations in the solutions I to V of the sample preparation protocol (Fig. 1) when using 19 mM Fos-10 in the NMR buffer. The OmpX concentration is given for the NMR sample volume of 50 μl. (B) Relation between the Fos-10 concentrations in the NMR buffer and the NMR sample (solution V in Fig. 1). The solid line represents a linear regression. (C). Dependence of the detergent concentration in the NMR sample (solution V in Fig. 1) on the total amount of OmpX added to the sample. Measurements were performed with 19 mM and 31 mM Fos-10 concentration in the NMR buffer. The broken lines represent control experiments in which no protein was added to the solution. Each data point in (B) and (C) represents an OmpX reconstitution experiment with the protocol of Figure 1.
The effect of the total amount of protein present in the solution (some of which may not be properly reconstituted, depending on the detergent concentration) on the Fos-10 up-concentration was evaluated by two series of refolding experiments using Fos-10 concentrations in the NMR buffer of 19 mM and 31 mM, respectively. The amount of OmpX added to the sample was 0.0, 0.2, 1.0 and 1.7 mg. For both sets of experiments, the final detergent concentration of about 95 mM and 185 mM, respectively, was independent of the amount of OmpX added (Fig. 3C).
The molecular weight cut-off (MWCO) and the type of the centrifugal devices are known to largely affect the resulting detergent concentration.18 Here, we compared Vivaspin polyethersulfone membranes (PES) and Amicon regenerated cellulose membranes (C) with MWCO-values of 10, 30, 50 and 100 kDa (Table 1). The results show that the MWCO-value plays an important role, whereas the type of membrane has only a negligibly small influence. Reduced detergent up-concentration at higher temperature was observed for all the tested concentration devices,19 as illustrated in Table 1 for Vivaspin PES membranes. Larger MWCO values resulted in increased loss of protein from the concentrated solutions (I – V in Fig. 1), both by absorption in the membrane and by flow-through. For example, at 25°C only about 70 % and 50 % of the protein were retained in the solution I (Fig. 1) when using 50 kDa and 100 kDa MWCO, respectively. For the OmpX reconstitution we therefore used 10 kDa MWCO devices at 25°C, which is a reasonable compromise regarding loss of OmpX, up-concentration of the detergent during the concentration steps, and the duration of the sample preparation.
Table 1.
Fos-10 up-concentration in the NMR sample (V in Fig. 1) observed when using different centrifugal devices and different temperatures for the sample preparation.a
| Device\MWCOb | 10 kDa | 30 kDa | 50 kDa | 100 kDa | |
|---|---|---|---|---|---|
| Vivaspin (polyethersulfone, PES) | 4 °C 25 °C |
5.5 4.9 |
4.2 3.2 |
3.6 1.5 |
1 1 |
| Amicon (regenerated cellulose, C) | 4 °C | 5.9 | 4.6 | 3.9 | 1 |
The numbers in the Table represent the ratio of the Fos-10 concentrations in the solution V of Fig. 1 and in the NMR buffer.
MWCO is the molecular weight cut-off of the centrifugal filter device
Optimizing the NMR spectra of OmpX/Fos-10 micelles
Using samples of [2H,15N]-labeled OmpX reconstituted in mixed micelles with Fos-10 at defined detergent concentrations, the quality of the 2D [15N,1H]-TROSY correlation spectra of OmpX was evaluated for a wide range of detergent concentrations (Fig. 4). The pattern of cross-peaks in these spectra was found to be very similar for all the Fos-10 concentrations used, indicating that the conformation of the NMR-observable protein in these solutions is independent of the detergent content. In contrast, the cross-peak intensities depend strongly on the detergent content. A survey of six experiments of the type of Figure 4 shows that the average peak intensity, Irel, varies over nearly an order of magnitude among different detergent concentrations (Fig. 5A). These differences in the signal intensities cannot be explained by variation of the protein concentration, which is closely similar in all the different samples (Fig. 5A). While the NMR sensitivity at the lowest detergent concentration of 53 mM indicates incomplete reconstitution of OmpX (see also text below), the signal variations at higher detergent concentrations called for additional investigations.
Figure 5.
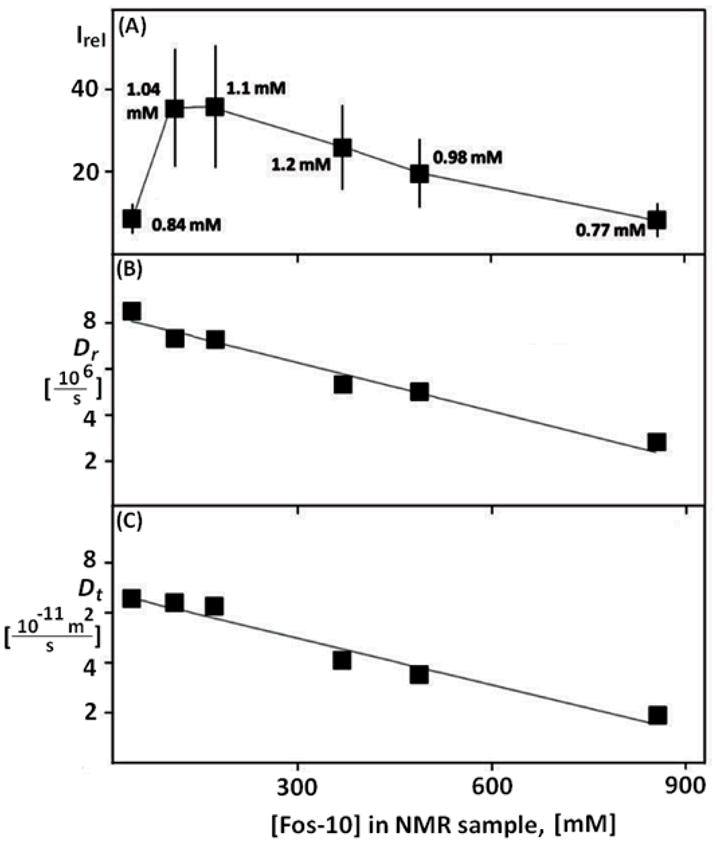
(A) Mean intensity of the cross-peaks in the 2D [15N,1H]-TROSY correlation spectra of OmpX, Irel, at variable Fos-10 concentrations in the NMR sample (solution V in Fig. 1). The data at 53 mM, 102 mM and 881 mM Fos-10 correspond to the spectra of Fig. 4, A–C, respectively. The error bars represent the standard deviation to the mean taken over all the backbone 15N–1H cross peaks. For each data point the OmpX concentration is indicated, as determined using UV spectroscopy at 280 nm. (B) Rotational diffusion constant of OmpX, Dr, at variable Fos-10 concentrations, as determined using the TRACT NMR experiment.12 (C) Translational diffusion coefficient, Dt, at variable Fos-10 concentrations as determined using the REST NMR experiment.13 The data presented in (A) to (C) were recorded with the same OmpX/Fos-10 solutions. For further details on the NMR experiments used in (B) and (C) see the Materials and Methods section.
Size and composition of OmpX/Fos-10 micelles from studies of rotational and translational diffusion
The rotational diffusion coefficient, Dr, was evaluated from the overall rotational correlation time, τc, measured with the TRACT experiment (see Materials and Methods). The experimental Dr values were found to depend linearly on the detergent concentration, with
| (8) |
where Dr,o and κr are fit parameters. A linear regression of the data gave Dr,o and κr values of 7.93 ± 0.20 × 106 s−1 and 7.10 ± 0.03 × 102 M−1, respectively (Fig. 5B), showing that the rotational tumbling of the mixed micelles slows down at high detergent concentrations. This variation of τc with the detergent concentration could be caused either by a change in the size of the protein–detergent mixed micelles or by an increase of the viscosity due to the formation of empty micelles at high Fos-10 concentrations. To discriminate between these two possible explanations, we determined the dependence of the mixed-micelle hydrodynamic radii, Rh, on the detergent concentration by combining the information provided by the rotational and translational diffusion constants, Dr and Dt, respectively. Dt values for the same range of detergent concentrations as for Dr were determined using the REST experiment. We found that Dt depends linearly on the detergent concentration,
| (9) |
with the following values for the two fit parameters: 6.93 ± 0.19 × 10−11 m2 s−1 for Dt,0 and 8.34 ± 0.06 × 102 M−1 for κt (Fig. 6C).
Figure 6.
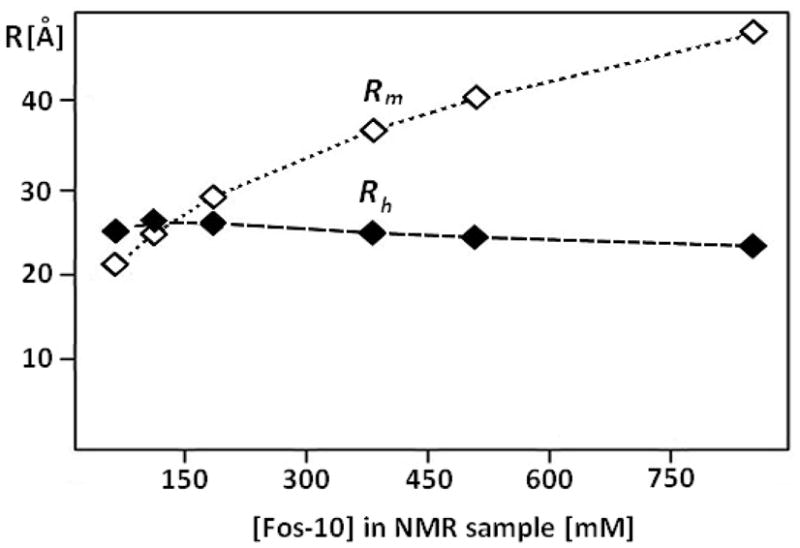
Dependence of the radius of an equivalent sphere representing the volume of the OmpX/Fos-10 mixed-micelles on the Fos-10 concentration. The open diamonds represent the results of a model calculation (Rm) based on the assumptions that the OmpX concentration is 1mM and detergent molecules are present exclusively either in mixed micelles with OmpX or as monomers (see Materials and Methods). The black diamonds represent Stokes radii, Rh, of the mixed micelles calculated with Eq. (5) from the translational and rotational diffusion coefficients Dt and Dr experimentally determined at an OmpX concentration of 0.1 ± 0.2 mM (Fig. 5). The broken lines connect points of the same data set, and are drawn to guide the eye.
We then determined the value of the hydrodynamic radius Rh for each detergent concentration from the measured values of Dr and Dt, using Equation (5), and compared them to the values for the radius of an equivalent sphere representing the micelles, Rm, calculated using different model assumptions on the influence of variable detergent concentrations on the size of the mixed micelles (see Materials and Methods). A plot of the experimental Rh values versus the detergent concentration shows only a weak dependence, with a maximum of 26.0 Å at 102 mM Fos-10, and a minimum of 23.3 Å at 881 mM Fos-10. In contrast, a model in which all the detergent molecules that are not present as monomers in the solution would be incorporated into the mixed micelles predicts that the Rm value would increase from 25 Å at 102 mM Fos-10 to 47 Å at 881 mM Fos-10 (Fig. 6). Since Rm values calculated with Eq. (7) represent a lower bound of Rh, the results of Figure 6 indicate that the deterioration of the NMR spectrum at high detergent concentrations is not due to an increase of the size of the mixed micelles. An increase in the viscosity of the solution due to the excess detergent, which leads to concentrations far above the CMC and must therefore be present in the form of empty micelles, thus seems to be responsible for the decrease of Dt and Dr, and hence of Irel (Figs. 4 and 5A).
The linear dependence of Dt and Dr is in agreement with earlier theoretical and experimental studies, which report a linear concentration dependence of Dr in suspensions of particles with constant sizes, and rationalize this behavior by the increased motional restrictions due to crowding effects at higher particle concentrations.20 In this context, the fit parameters Dt,0 and Dr,0 can be interpreted as single-particle diffusion coefficients at infinite dilution which satisfy the Stokes–Einstein-relations of the equations (2)–(4), with the corresponding Stokes radius, Rh,0, and viscosity, η0:
| (10) |
| (11) |
| (12) |
The value of Rh,0 for OmpX/Fos-10 mixed micelles is thus found to be 25.9 ± 0.3 Å. The Dr,o value of 7.93 × 106 s−1 corresponds to an effective rotational correlation time of 21 ns, which coincides closely with the experimental values of 21–25 ns, where τc = 21 ns was measured in samples with low detergent concentrations.12 These values indicate that the OmpX/Fos-10 mixed micelles are approximately 50 kDa in size, with an aggregation number, Na, of approximately 100 Fos-10 molecules per micelle.
Rh,0 coincides closely with the Rm value calculated for 102 mM Fos-10 concentration (Fig. 6), which is in the range where we obtained optimal NMR spectra (Fig. 5A). Since the OmpX concentration in the NMR sample was 1.0 mM, we predict an aggregation number of approximately 90, which is based on the Fos-10 CMC of 10.8 mM and the assumption that the detergent molecules are either present as monomers in solution or incorporated into the mixed micelles. This prediction is in good agreement with the experimentally determined value of Na = 100. These findings imply that for optimal sample conditions, the detergent molecules should either be bound to mixed micelles or monomeric in solution, so that the optimal Fos-10 concentration, , can be estimated by
| (13) |
For lower Fos-10 concentrations the amount of detergent is too small to reconstitute all the OmpX molecules into mixed micelles so that some OmpX is not NMR-observable. This interpretation of our data at low detergent concentration (Figs. 4A and 5A) is in line with results from Opella and coworkers.5 The deterioration of the NMR spectra at high detergent content can be explained by an increase of the viscosity due to the excess detergent in the form of empty micelles, which slows down the diffusion of the protein–detergent mixed-micelles, and thus causes line-broadening and reduced signal intensity of the OmpX NMR spectra.
Conclusions
The important conclusion from the present work for future studies with membrane proteins is the finding that high quality NMR spectra can be obtained only over a very limited range of detergent concentrations. Using our microcoil-NMR platform, this key result could be rationalized by investigations of the hydrodynamic properties of the IMP/detergent mixed micelles at discrete detergent concentrations in the NMR samples. The size of the OmpX/Fos-10 mixed micelles was thus found to be maintained at approximately 50 kDa over a large range of detergent concentrations. The deterioration of the NMR spectra at high detergent concentrations can be rationalized by a crowding effect due to the formation of empty Fos-10 micelles, which slows down the molecular tumbling of the mixed micelles, resulting in faster spin relaxation.
References
- 1.(a) Faham S, Watanabe A, Besserer GM, Cascio D, Specht A, Hirayama BA, Wright EM, Abramson J. Science. 2008;321:810–814. doi: 10.1126/science.1160406. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hilf RJC, Dutzler R. Nature. 2008;452:375–379. doi: 10.1038/nature06717. [DOI] [PubMed] [Google Scholar]; (c) Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EYT, Lane JR, Izerman AP, Stevens RC. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Kadaba NS, Kaiser JT, Johnson E, Lee A, Rees DC. Science. 2008;321:250–3. doi: 10.1126/science.1157987. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Murakami M, Kouyama T. Nature. 2008;453:363–367. doi: 10.1038/nature06925. [DOI] [PubMed] [Google Scholar]; (f) Park JH, Scheerer P, Hofmann KP, Choe HW, Ernst OP. Nature. 2008;454:183–187. doi: 10.1038/nature07063. [DOI] [PubMed] [Google Scholar]; (g) Remaut H, Tang C, Henderson NS, Pinkner JS, Wang T, Hultgren SJ, Thanassi DG, Waksman G, Li H. Cell. 2008;133:640–652. doi: 10.1016/j.cell.2008.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Scheerer P, Park JH, Hildebrand PW, Kim YJ, Krausz N, Choe HW, Hofmann KP, Ernst OP. Nature. 2008:455. doi: 10.1038/nature07330. [DOI] [PubMed] [Google Scholar]; (i) Stouffer AL, Acharya R, Salom D, Levine AS, Di Costanzo L, Soto CS, Tereshko V, Nanda V, Stayrook S, DeGrado WF. Nature. 2008;451:596–599. doi: 10.1038/nature06528. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Tsukazaki T, Mori H, Fukai S, Ishitani R, Mori T, Dohmae N, Perederina A, Sugita Y, Vassylyev DG, Ito K, Nureki O. Nature. 2008;455:988–991. doi: 10.1038/nature07421. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Wang W, Black SS, Edwards MD, Miller S, Morrison EL, Bartlett W, Dong C, Naismith JH, Booth IR. Science. 2008;321:1179–1183. doi: 10.1126/science.1159262. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Warne T, Serrano-Vega MJ, Baker JG, Moukhametzianov R, Edwards PC, Henderson R, Leslie AGW, Tate CG, Schertler GFX. Nature. 2008;454:486–491. doi: 10.1038/nature07101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (m) Weyand S, et al. Science. 2008;322:709–713. doi: 10.1126/science.1164440. [DOI] [PMC free article] [PubMed] [Google Scholar]; (n) Zimmer J, Nam Y, Rapoport TA. Nature. 2008;455:936–943. doi: 10.1038/nature07335. [DOI] [PMC free article] [PubMed] [Google Scholar]; (o) Gerber S, Comellas-Bigler M, Goetz BA, Locher KP. Science. 2008;321:246–50. doi: 10.1126/science.1156213. [DOI] [PubMed] [Google Scholar]; (p) Singh SK, Piscitelli CL, Yamashita A, Gouaux E. Science. 2008;322:1655–61. doi: 10.1126/science.1166777. [DOI] [PMC free article] [PubMed] [Google Scholar]; (q) Shinoda T, Ogawa H, Cornelius F, Toyoshima C. Nature. 2009;459:446–50. doi: 10.1038/nature07939. [DOI] [PubMed] [Google Scholar]; (r) Aller SG, Yu J, Ward A, Weng Y, Chittaboina S, Zhuo R, Harrell PM, Trinh YT, Zhang Q, Urbatsch IL, Chang G. Science. 2009;323:1718–22. doi: 10.1126/science.1168750. [DOI] [PMC free article] [PubMed] [Google Scholar]; (s) Shaffer PL, Goehring A, Shankaranarayanan A, Gouaux E. Science. 2009;325:1010–14. doi: 10.1126/science.1176088. [DOI] [PMC free article] [PubMed] [Google Scholar]; (t) Gao X, Lu F, Zhou L, Dang S, Sun L, Li X, Wang J, Shi Y. Science. 2009;324:1565–8. doi: 10.1126/science.1173654. [DOI] [PubMed] [Google Scholar]; (u) Fang Y, Jayaram H, Shane T, Kolmakova-Partensky L, Wu F, Williams C, Xiong Y, Miller C. Nature. 2009;460:1040–43. doi: 10.1038/nature08201. [DOI] [PMC free article] [PubMed] [Google Scholar]; (v) Bocquet N, Nury H, Baaden M, Le Poupon C, Changeux JP, Delarue M, Corringer PJ. Nature. 2009;457:111–114. doi: 10.1038/nature07462. [DOI] [PubMed] [Google Scholar]; (w) Gonzales EB, Kawate T, Gouaux E. Nature. 2009;460:599–604. doi: 10.1038/nature08218. [DOI] [PMC free article] [PubMed] [Google Scholar]; (x) Hearn EM, Patel DR, Lepore BW, Indic M, van den Berg B. Nature. 2009;458:367–370. doi: 10.1038/nature07678. [DOI] [PMC free article] [PubMed] [Google Scholar]; (y) Hilf RJC, Dutzler R. Nature. 2009;457:115–118. doi: 10.1038/nature07461. [DOI] [PubMed] [Google Scholar]; (z) Kawate T, Michel JC, Birdsong WT, Gouaux E. Nature. 2009;460:592–598. doi: 10.1038/nature08198. [DOI] [PMC free article] [PubMed] [Google Scholar]; (aa) Maeda S, Nakagawa S, Suga M, Yamashita E, Oshima A, Fujiyoshi Y, Tsukihara T. Nature. 2009;458:597–602. doi: 10.1038/nature07869. [DOI] [PubMed] [Google Scholar]; (ab) Mueller M, Grauschopf U, Maier T, Glockshuber R, Ban N. Nature. 2009;459:726–730. doi: 10.1038/nature08026. [DOI] [PubMed] [Google Scholar]; (ac) Ressl S, Terwisscha van Scheltinga AC, Vonrhein C, Ott V, Ziegler C. Nature. 2009;458:47–52. doi: 10.1038/nature07819. [DOI] [PubMed] [Google Scholar]; (ad) Stein A, Weber G, Wahl MC, Jahn R. Nature. 2009;460:525–528. doi: 10.1038/nature08156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Hiller S, Garces RG, Malia TJ, Orekhov VY, Colombini M, Wagner G. Science. 2008;321:1206–1210. doi: 10.1126/science.1161302. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Van Horn WD, Kim HJ, Ellis CD, Hadziselimovic A, Sulistijo ES, Karra MD, Tian C, Sonnichsen FD, Sanders CR. Science. 2009;324:1726–1729. doi: 10.1126/science.1171716. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Schnell JR, Chou JJ. Nature. 2008;451:591–595. doi: 10.1038/nature06531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235.(b) 203 membrane protein structures were deposited in the PDB up to 9/9/2009.
- 4.Wallin E, von Heijne G. Protein Sci. 1998;7:1029–38. doi: 10.1002/pro.5560070420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McDonnell PA, Opella SJ. J Magn Reson. 1993;B102:120–125. [Google Scholar]
- 6.Krueger-Koplin RD, Sorgen PL, Krueger-Koplin ST, Rivera-Torres IO, Cahill SM, Grinius L, Krulwicz TA, Grivin ME. J Biomol NMR. 2004;17:43–57. doi: 10.1023/B:JNMR.0000012875.80898.8f. [DOI] [PubMed] [Google Scholar]
- 7.(a) Marassi FM, Opella SJ. Curr Op Struct Biol. 1998;8:640–648. doi: 10.1016/s0959-440x(98)80157-7. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sanders RS, Oxenoid K. Biochim Biophys Acta. 2000;1508:129–145. doi: 10.1016/s0005-2736(00)00308-4. [DOI] [PubMed] [Google Scholar]; (c) Sanders RS, Sonnichsen F. Magn Reson Chem. 2006:S24–S40. doi: 10.1002/mrc.1816. [DOI] [PubMed] [Google Scholar]
- 8.(a) Arora A, Abildgaard F, Bushweller JH, Tamm LK. Nature. 2001;8:334–338. doi: 10.1038/86214. [DOI] [PubMed] [Google Scholar]; (b) Hwang PM, Choy W, Lo EI, Chen L, Forman-Kay JD, Raetz CRH, Prive GG, Bishop RE, Kay LE. Proc Natl Acad Sci USA. 2002;99:13560–13565. doi: 10.1073/pnas.212344499. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Fernandez C, Hilty C, Wider G, Güntert P, Wüthrich K. J Mol Biol. 2004;336:1211–1221. doi: 10.1016/j.jmb.2003.09.014. [DOI] [PubMed] [Google Scholar]; (d) Oxenoid K, Chou JJ. Proc Natl Acad Sci USA. 2005;102:10870–10875. doi: 10.1073/pnas.0504920102. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Teriete P, Franzin CM, Choi J, Marassi FM. Biochemistry. 2007;46:6774–6783. doi: 10.1021/bi700391b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Liang B, Tamm LK. Proc Natl Acad Sci USA. 2007;104:16140–16145. doi: 10.1073/pnas.0705466104. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Zhou Y, Cierpicki T, Jimenez RHF, Lukasik SM, Ellena JF, Cafiso DS, Kadokura H, Beckwith J, Bushweller JH. Mol Cell. 2008;31:896–908. doi: 10.1016/j.molcel.2008.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Q, Horst R, Geralt M, Ma X, Hong W, Finn MG, Stevens RC, Wüthrich K. J Am Chem Soc. 2008;130:7357–7363. doi: 10.1021/ja077863d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pervushin K, Riek R, Wider G, Wüthrich K. Proc Nat Acad Sci USA. 1997;94:12366–12377. doi: 10.1073/pnas.94.23.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bartels C, Xia T, Billeter M, Güntert P, Wüthrich K. J Biomol NMR. 1995;6:1–10. doi: 10.1007/BF00417486. [DOI] [PubMed] [Google Scholar]
- 12.Lee D, Hilty C, Wider G, Wüthrich K. J Magn Reson. 2006;178:72–76. doi: 10.1016/j.jmr.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 13.Horst, R.; Wüthrich, K. to be submitted.
- 14.Mills R. J Phys Chem. 1973;77:685–688. [Google Scholar]
- 15.(a) Fernandez C, Adeishvili K, Wüthrich K. Proc Nat Acad Sci USA. 2001;98:2358–2363. doi: 10.1073/pnas.051629298. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Fernandez C, Hilty C, Wider G, Wüthrich K. Proc Nat Acad Sci USA. 2002;99:13533–13537. doi: 10.1073/pnas.212515099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lipfert J, Columbus L, Chu VB, Lesley SA, Doniach S. J Phys Chem B. 2007;111:12427–12438. doi: 10.1021/jp073016l. [DOI] [PubMed] [Google Scholar]
- 17.Cantor CR, Schimmel PR. Biophysical Chemistry. Part 2. Freeman; San Francisco : 1980. [Google Scholar]
- 18.(a) Strop P, Brunger AT. Protein Sci. 2005;14:2207–2211. doi: 10.1110/ps.051543805. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Maslennikov I, Kefala G, Johnson C, Riek R, Choe S, Kwiatkowski W. BMC Structural Biology. 2007;7:74. doi: 10.1186/1472-6807-7-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.(a) Ericsson CA, Soderman O, Garamus VM, Bergstrom M, Ulvenlund S. Langmuir. 2004;20:1401–1408. doi: 10.1021/la035613e. [DOI] [PubMed] [Google Scholar]; (b) Zulauf M. In: Crystallization of Membrane Proteins. Michel H, editor. CRC Press; Boca Raton, FL, USA: 1991. [Google Scholar]
- 20.(a) Piazza R, Degiorgio V, Corti M, Stavans J. Physical Review B. 1990;42:4885–4888. doi: 10.1103/physrevb.42.4885. [DOI] [PubMed] [Google Scholar]; (b) Cichocki B, Ekiel-Jezewska ML, Wajnryb E. J Chem Phys. 1999;111:3265–3273. [Google Scholar]; (c) Bernardo P, Garcia de la Torre J, Pons M. J Mol Recognit. 2004;17:397–497. doi: 10.1002/jmr.694. [DOI] [PubMed] [Google Scholar]
- 21.De Marco A, Wüthrich K. J Magn Reson. 1976;24:201–204. [Google Scholar]