

Abstract
Purpose
The epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKI) gefitinib and erlotinib benefit some non–small cell lung cancer (NSCLC) patients, but most do not respond (primary resistance) and those who initially respond eventually progress (acquired resistance). EGFR TKI resistance is not completely understood and has been associated with certain EGFR and K-RAS mutations and MET amplification.
Experimental Design
We hypothesized that dual inhibition of the vascular endothelial growth factor (VEGF) and EGFR pathways may overcome primary and acquired resistance. We investigated the VEGF receptor/EGFR TKI vandetanib, and the combination of bevacizumab and erlotinib in vivo using xenograft models of EGFR TKI sensitivity, primary resistance, and three models of acquired resistance, including models with mutated K-RAS and secondary EGFR T790M mutation.
Results
Vandetanib, gefitinib, and erlotinib had similar profiles of in vitro activity and caused sustained tumor regressions in vivo in the sensitive HCC827 model. In all four resistant models, vandetanib and bevacizumab/erlotinib were significantly more effective than erlotinib or gefitinib alone. Erlotinib resistance was associated with a rise in both host and tumor-derived VEGF but not EGFR secondary mutations in the KRAS mutant-bearing A549 xenografts. Dual inhibition reduced tumor endothelial proliferation compared with VEGF or EGFR blockade alone, suggesting that the enhanced activity of dual inhibition is due at least in part to antiendothelial effects.
Conclusion
These studies suggest that erlotinib resistance may be associated with a rise in both tumor cell and host stromal VEGF and that combined blockade of the VEGFR and EGFR pathways can abrogate primary or acquired resistance to EGFR TKIs. This approach merits further evaluation in NSCLC patients.
Non–small cell lung cancer (NSCLC) is the leading cause of cancer deaths in the United States (1) and worldwide, with a 5-year survival rate of only 15% for all stages combined (2). Because conventional chemotherapy regimens have had limited efficacy, targeted therapies such as those that inhibit epidermal growth factor receptor (EGFR) or vascular endothelial growth factor (VEGF) signaling pathways are being extensively evaluated (3). In a phase III study, the EGFR tyrosine kinase inhibitor (TKI) erlotinib significantly improved overall survival relative to supportive care for refractory stage IIIB/IV NSCLC (4). However, objective tumor responses were observed only in 8.9% of treated patients and even patients who initially responded ultimately developed progressive disease.
Translational Relevance.
Epidermal growth factor receptor (EGFR) inhibitors have shown clinical benefit for only a subset of non–small cell lung cancer (NSCLC) patients, and even patients who do initially experience a major response eventually develop therapeutic resistance. For this reason, considerable effort has been focused on understanding the mechanisms regulating primary and acquired resistance to EGFR inhibitors. In the present report, we investigated the efficacy of dual targeting of the vascular endothelial growth factor receptor (VEGFR) and EGFR pathways in vivo using xenograft models of EGFR TKI sensitivity, primary resistance, and three models of acquired resistance. Our results indicate that combined VEGFR/EGFR pathway blockade can abrogate primary or acquired resistance to EGFR inhibitors in all four models. Furthermore, in models lacking EGFR secondary mutations, EGFR TKI resistance is associated with increased tumor- and host-derived VEGF. These findings suggest that dual VEGFR/EGFR blockade is an approach that merits further investigation for treating primary or acquired resistance to EGFR TKIs.
Efforts to understand the mechanism of sensitivity and resistance to EGFR inhibitors have led to the discovery of important biological differences among NSCLC tumor subgroups. Sensitivity to EGFR TKIs is associated with somatic mutations in EGFR, most commonly the exon 19 deletion or the L858R point mutation (5–7) or amplification of the EGFR gene (8). Several mechanisms associated with resistance to EGFR inhibitors have been identified. In tumors without somatic mutations of EGFR, primary resistance to erlotinib has been associated with K-RAS mutations (9) or EGFR-independent activation of the PI3K/Akt pathway (10). Acquired resistance to EGFR TKIs has also been associated with a secondary mutation in the EGFR TK domain, T790M (11–13), as well as amplification of the MET proto-oncogene (14).
VEGF is a key regulator of angiogenesis and a validated target for NSCLC (15, 16). The VEGF and EGFR pathways are known to be interrelated (3). For example, VEGF is down-regulated by EGFR inhibition, likely through both hypoxia-induciable factor-α–dependent and independent mechanisms (17–22), and EGFR, like VEGF receptor (VEGFR)-2, may be expressed on tumor-associated endothelium (23–25). Furthermore, in xenograft models, acquired resistance to cetuximab, a monoclonal antibody targeting EGFR, was associated with increased VEGF levels and increased tumor angiogenesis in vivo (26). These studies suggest that dual blockade of the VEGF and EGFR pathways would be more effective than either approach alone and may also have activity in tumors with acquired resistance to EGFR inhibitors. However whether this finding extends to resistance to EGFR TKIs is unknown.
Clinically, inhibition of both VEGF and EGFR has been investigated using the combination of bevacizumab and erlotinib or a single multitargeted TKIs such as vandetanib. Both approaches have shown promising results in phase II testing in NSCLC patients (27–30). Vandetanib prolonged progression-free survival (PFS) compared with gefitinib. Furthermore, in two separate studies vandetanib combined with chemotherapy prolonged PFS compared with chemotherapy alone (28, 30), although in one of these studies vandetanib monotherapy was inferior to doublet chemotherapy (30). Vandetanib is currently undergoing phase III testing in NSCLC patients.
The aim of this study was to explore the potential utility of combined VEGFR/EGFR pathway inhibition in four different clinically relevant xenograft models of EGFR TKI resistance – one of primary resistance and three of acquired resistance – in order to investigate whether there are specific situations in which dual blockade may provide a significant advantage over monotherapy.
Materials and Methods
Cell lines
All NSCLC cell lines were obtained from the American Type Culture Collection and were maintained at 37°C with 5% CO2, in media supplemented with 10% fetal bovine serum and 1% L-glutamine-penicillin-streptomycin. A549 cells (wild-type EGFR) were cultured with F12 media. Calu-6 cells (wild-type EGFR) were maintained in MEM also supplemented with 1% nonessential amino acids. The H3255 cell line (L858R mutation EGFR) was grown in ACL4 media (Life Technologies, Inc.), whereas H1975 cells (L858R and T790M mutation EGFR) were grown in RPMI 1640 media. Both H1650 and HCC827 cells (exon 19 deletion EGFR) were cultured with RPMI 1640 media, and DFCI-011 cells (exon 19 deletion EGFR) were grown in DMEM. NIH3T3 cells were cultured with DMEM and stably transfected to express EGFR harboring an exon 19 deletion with or without L858R mutation. Lewis lung cancer cells were obtained from Dr. Isaiah Fidler (University of Texas, M. D. Anderson Cancer Center).
Materials
Vandetanib (ZD6474) was kindly provided by AstraZeneca. Bevacizumab, erlotinib, and gefitinib were obtained from the pharmacies at Dana-Farber Cancer Institute or M. D. Anderson Cancer Center.
Cell growth assays
The following NSCLC cell lines were seeded (2,000 or 5,000 cells per well, depending on cell type) onto 96-well plates: A549, Calu-6, H1650, H1975, H3255, and HCC827. After a 24-h incubation in serum-containing media, cells were treated with vandetanib, erlotinib, gefitinib (0-10 μmol/L), or bevacizumab (0-10 ng/mL) for 72 h in serum-containing media. MTT solution (Sigma) was added to each well to a final concentration of 0.1 mg/mL, and plates were incubated for 2 h at 37°C. After carefully aspirating the media and MTT from each well, the formazan crystals were dissolved with 100 μL of DMSO and absorbance was read at 570 nm.
Western blotting
Cell lines were seeded into 6-well plates at a concentration of 2 × 105 cells per well. After 24 h of growth in serum-containing media, cells were incubated in serum-free media for 24 h. Vandetanib or gefitinib (0-10 μmol/L) was then added for 3 h, at which time cells were stimulated for 15 min with 10 ng/mL EGF (Sigma). Western blots were done on whole-cell extracts using a phospho-specific EGFR antibody (pY1068, 1:1,000 dilution; Cell Signaling Technology). The membranes were stripped and reblotted with antibody against EGFR (1:2,000 dilution; Santa Cruz Biotechnology) to measure total protein expression.
Tumor sections were snap-frozen in liquid nitrogen for protein isolation, and EGFR signal transduction was evaluated by Western blot as previously described (10). Primary antibodies included the following: EGFR (NeoMarkers), Y1068 p-EGFR, Akt, S473 p-Akt, p-42/44 MAPK, Y1289 p-HER3 (Cell Signaling Technology); and HER3 (Santa Cruz Biotechnology Inc.).
Multiplex bead assay
A549, HCC827, and H1975 cells were plated in serum-containing media. After a 24-h incubation period they were serum-starved overnight. Cells were pretreated with 1 μmol/L erlotinib or vandetanib in serum-free media for 12 h and then media containing EGF (60 ng/mL) alone (control) or EGF in the presence of 1 μmol/L erlotinib or vandetanib was added. After 15 min, cells were washed in ice-cold PBS and whole-cell protein lysates were collected. Phosphorylated EGFR was quantified using a bead-based Bio-Plex phosphorylation assay (Bio-Rad) according to the manufacturer's instructions.
Detection of plasma VEGF
VEGF was assayed in EDTA plasma using the Luminex Multiplex assay with Bio-Plex Cytokine assay reagents (Bio-Rad Laboratories) on a Bio-Plex Luminex xMAP (Bio-Rad Laboratories) as described by the manufacturer.
VEGF ELISA assay
3 × 105 cells were plated in 6-well dishes. Cells were serum-starved for 24 h and media were collected for ELISA after 8 h. VEGF ELISA was purchased from R&D and analysis was conducted according to the manufacturer's instructions.
Real-ime PCR
Total RNA was prepared from cell culture and frozen xenografts tumor samples using RNeasy Mini kit (QIAGEN) according to the manufacturer's instructions. Human GAPDH was used as an endogenous control for human VEGF and mouse tubulin was used as an endogenous control for murine VEGF. Lewis lung carcinoma cells grown under normoxic or hypoxic conditions were used as a negative and positive control for mouse VEGF, respectively. A549 cells cultured in normoxic or hypoxic conditions were used as a negative and positive control for human VEGF, respectively. We used SuperScriptTM III RNase H-Reverse Transcriptase (Invitrogen) to convert RNA into cDNA. Real-time quantitative PCR was done using TaqMan one-step RT-PCR master mix kit and gene specific taqman gene expression assay kits (Applied Biosystems, Inc.; Hs99999905_mL Gapdh, Mm00495804_1Tubb5, Hs00900054_m1VEGF, and Mm00437306_m1VEGFa) in a 7300 real-time PCR system (Applied Biosystems). Samples were prepared in duplicate, and 200 ng mRNA were added to each PCR tube containing the reaction mixture. Real-time cylcer conditions were programmed according to the recommended protocols as follows: 48 degrees for 30 min, 95 degrees for 10 min followed by 45 cycles of 95 degrees for 15 sec and 60 degree for 1 min.
Xenograft models
All mice used in these experiments were cared for in accordance with the standards of the Institutional Animal Care and Use Committee under a protocol approved by the Animal Care and Use Committee of the Children's Hospital Boston. Nude mice (6-8 weeks old; Charles River Laboratories) were inoculated s.c. in the lower rear flank with human NSCLC cells (HCC827, HCC827-T790M, A549, or H1975 cell lines) suspended in 0.2 mL of PBS (5 × 106 cells per mouse inoculation). All mice were monitored for tumor growth at the site of inoculation by palpation. Once tumors became palpable (∼50 mm3), tumor size was measured twice weekly using calipers, and the tumor volume was calculated using the formula (length × width2 × 0.52). When the group mean tumor volume reached the indicated starting volume, the mice were randomized to different treatment arms (at least 6 mice per treatment arm). For A549 tumors, an initial mean volume of ∼200 mm3 was used; in the A549 erlotinib-progression experiment, mice in the initial erlotinib-treated group with progression (i.e. mean volume doubled to ∼400 mm3) were then crossed over to different treatment arms. Drugs were administered at doses/schedules (equal to or below the maximum tolerated doses) previously reported to be effective in tumor growth inhibition (31). The mice were treated using the following drug doses and treatment schedules: vandetanib 50 mg/kg oral daily; erlotinib 100 mg/kg oral daily; gefitinib 150 mg/kg oral daily; bevacizumab 5 mg/kg i.p. twice weekly. Doses were reduced if mice lost ≥10% body weight. Control mice were treated with vehicle alone (1% Tween 80 in PBS). Tumors were excised and fixed in formalin or frozen in optimal cutting temperature for further histologic analysis.
Histology
Immunohistochemical double-staining was done on formalin-fixed and paraffin-embedded material. Five-micrometer sections were deparaffinized and subjected to antigen retrieval at 125°C for 5 min in 1 mmol/L EDTA buffer, pH 8.0, in a pressure cooker. Sections were incubated at 4°C overnight with 1:25 rabbit monoclonal antibody ab40815 (Abcam) specific for the EGFR phosphorylated at tyrosine 1068. Specific staining was detected by RMR622 rabbit on rodent polymer (Biocare) and visualized by 3-amino-9-ethylcarbazole (Dako). Sequentially, after a brief denaturing step with denaturing solution DNS001 (Biocare), sections were incubated with rat anti-CD34 ab8158 (Abcam) 1:50 for 2 h at room temperature. This stain was detected by alkaline phosphatase–conjugated goat antirat secondary antibody sc3824 (SantaCruz) and visualized by Ferangi blue (Biocare). For the Ki67 and von Willebrand factor double-staining a simultaneous method was applied. After heat-induced antigen retrieval in Target Retrieval Solution pH 6 (Dako), sections were incubated with mouse specific rat anti–Ki67 M7249 (Dako) 1:200 and rabbit anti–von Willebrand factor A0082 (Dako) 1:200. Specific staining was detected with a mixture of goat antirabbit horseradish peroxidase polymer Envision K4008 (Dako) and alkaline phosphatase–conjugated goat antirat secondary antibody sc3824 (SantaCruz).
A semiquantitative staining index was used to evaluate the expression of phosphorylated EGFR in tumor cells and tumor-associated endothelial cells separately, as previously published (32). Briefly, the staining index was calculated as a product of staining intensity (score 0–3) and the proportion of immunopositive tumor or endothelial cells (≤10%=1, 10-50%=2, >50%=3). The effects of drug treatments on tumor endothelium proliferation were quantified using a Ki67 (mouse specific) and von Willebrand factor dual immunohistochemical staining, as previously reported (33, 34). Quantifications were done using proliferating microvessel density, which represents the number of Ki67-positive microvessels per field of view. Quantification was assessed at microvessel density hotspots using at least 5 to 10 fields of view for each tumor at 400× magnification. In general, proliferating microvessel density counts are order of magnitude lower than traditional microvessel density counts because a single microvessel can be either Ki67 positive or negative as unit.
To evaluate VEGF by immunohistochemistry, paraffin-embedded sections were deparaffinized and incubated in pepsin at 37° for 20 min. Endogenous peroxidases were blocked using a solution of 3% H2O2 in methanol. Sections were incubated in rabbit anti-VEGF antibodies (1:200; Santa Cruz) overnight. Slides were washed in PBS, incubated in horseradish peroxidase–conjugated secondary antibody and incubated in stable diaminovenzidine (Research Genetics) to visualize a positive reaction.
EGFR and K-RAS mutation analysis
Genomic DNA was isolated from 4-μm sections using the PicoPure DNA extraction kit (Molecular Devices) according to the manufacturer's instructions. Exons 18 through 21 of EGFR and exon 1 of KRAS were PCR-amplified using intron-based primers. All PCR products were directly sequenced using the Applied Biosystems PRISM dye terminator cycle sequencing method. All sequence variants were confirmed by independent PCR amplifications from at least two independent microdissections and sequenced in both directions, as previously reported (35).
Statistics
In vivo data are expressed as mean ± SE. In vitro data are expressed as mean ± SD. Statistical significance was assessed using Student's t-test. P < 0.05 was considered statistically significant. All statistical tests were two-sided. Mann-Whitney rank sum test was used when data failed normality.
Results
Sensitivity of EGFR to inhibition by vandetanib in vitro
We examined the ability of vandetanib to inhibit wild-type and mutated EGFR phosphorylation in vitro, and compared it with the tyrosine kinase inhibitors gefinitib and erlotinib. NIH3T3 cells, which bear no detectable endogenous EGF receptor, were transfected with EGFR bearing exon 19 deletions (Fig. 1A) or L858R mutations (Fig. 1B). These two mutations are known to promote constitutive activation of the receptor, and to be sensitive to small molecule EGFR TKIs gefitinib and erlotinib. EGFR phosphorylation was detected by Western analysis. Vandetanib inhibited mutated EGFR in a pattern similar to that of gefitinib, although gefitinib inhibited EGFR activation at lower concentrations in the NIH3T3 cell lines harboring the exon 19 deletion or the L858R mutation.
Fig. 1.
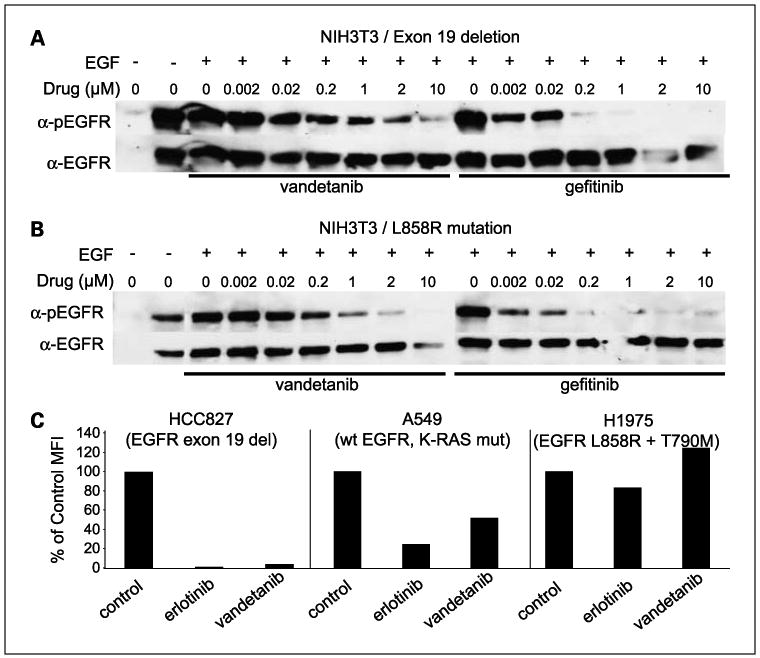
Vandetanib inhibits EGFR in a manner similar to gefitinib and erlotinib. NIH3T3 cells were transfected to express EGFR bearing exon 19 deletions (A) or L858R mutations (B) and treated with EGF and increasing concentrations of vandetanib or gefitinib. Expression of activated EGFR was evaluated by Western analysis. C, inhibition of EGFR activation in HCC827, A549, and H1975 NSCLC cell lines by erlotinib or vandetanib was evaluated by multiplex bead assay.
Multiplex bead assay was used to measure the ability of vandetanib and erlotinib to inhibit EGFR phosphorylation in NSCLC cell lines including A549 cells that express wild-type EGFR, HCC827 cells that harbor EGFR activating mutations, and H1975 cells that express mutated EGFR and the T790M resistance mutation. Treatment of HCC827 cells (EGFR exon 19 del.) with 1 μmol/L erlotinib resulted in complete inhibition of EGFR activation (Fig. 1C). Similarly, vandetanib almost completely diminished EGFR phosphorylation in HCC827 cells. In wild-type EGFR-expressing A549 cells, erlotinib (4.4-fold decrease) and vandetanib (2-fold decrease) caused only a partial reduction in EGFR activation. Neither erlotinib nor vandetanib treatment resulted in significant decreases in EGFR phosphorylation in H1975 cells harboring L858R and T790M mutations. Consistent with these findings, neither vandetanib nor gefitinib inhibited EGFR bearing L858R and T790M mutations after transient transfection into Cos-7 cells (data not shown).
Vandetanib inhibits the in vitro growth of cell lines bearing mutated EGFR at concentrations comparable with gefitinib and erlotinib
The in vitro effect of gefitinib, erlotinib, vandetanib, and bevacizumab on the viability of various NSCLC cell lines bearing wild-type or mutated EGFR was evaluated using a standard MTT assay (Fig. 2A to D). Consistent with earlier studies of EGFR TKIs, cell lines bearing mutated EGFR (HCC827, H3255) were more sensitive to vandetanib than those bearing wild-type EGFR (A549, Calu-6), which required concentrations >1 μmol/L of drug for 50% inhibition (IC50). H1975 cells, which have both L858R and T790M mutations and H1650 cells were relatively resistant to vandetanib treatment. Overall, the pattern of in vitro sensitivity correlated with that observed for gefitinib and erlotinib, although higher concentrations of vandetanib were required for inhibition of the sensitive HCC827 and H3255 cell lines (Fig. 2A to C). As expected, none of the tested NSCLC cell lines were sensitive to inhibition of the VEGF pathway alone (Fig. 2D).
Fig. 2.

Effect of EGFR/VEGFR inhibitors on tumor cell viability in vitro. MTT assay was done to evaluate the effect of gefitinib (A), erlotinib (B), vandetanib (C), and bevacizumab (D) on the growth of NSCLC cell lines. These four compounds were tested on EGFR wild-type cell lines (A549 and Calu-6), cells with EGFR-activating mutations (HCC827, H3255), and H1975 cells that express both the EGFR-activating mutation and the T790M resistance mutation. Cells were treated with gefitinib, erlotinib, vandetanib (0.001-10 nmol/L), or bevacizumab (0.001-10 μg/mL) for 72 h. The percentage of viable cells is shown relative to that of untreated control. Results are shown as mean values with SD based on results from at least three replicate wells.
Effects of EGFR and VEGFR/EGFR inhibition in EGFR TKI-sensitive HCC827 xenograft model
We assessed the effects of EGFR inhibition using gefitinib, erlotinib, and VEGFR/EGFR inhibition using vandetanib in the HCC827 xenograft model. HCC827 tumor cells have been previously shown to be sensitive to EGFR TKIs in vitro (36, 37) and to cetuximab when grown as xenografts (38). Established s.c. tumors with a mean tumor volume of approximately 400 mm3 (n = 6-7 mice per group) were treated with gefitinib, erlotinib, vandetanib, or vehicle control. Gefitinib, erlotinib, and vandetanib treatments resulted in significant regressions of established tumors within 1 week of treatment, to a mean tumor volume of <50 mm3 for each group (Fig. 3A). On day 38 of treatment, all mean tumor volumes were statistically different when compared with the control group (all comparisons P ≤ 0.005). After more than 60 days of treatment there was no evidence of tumor regrowth.
Fig. 3.
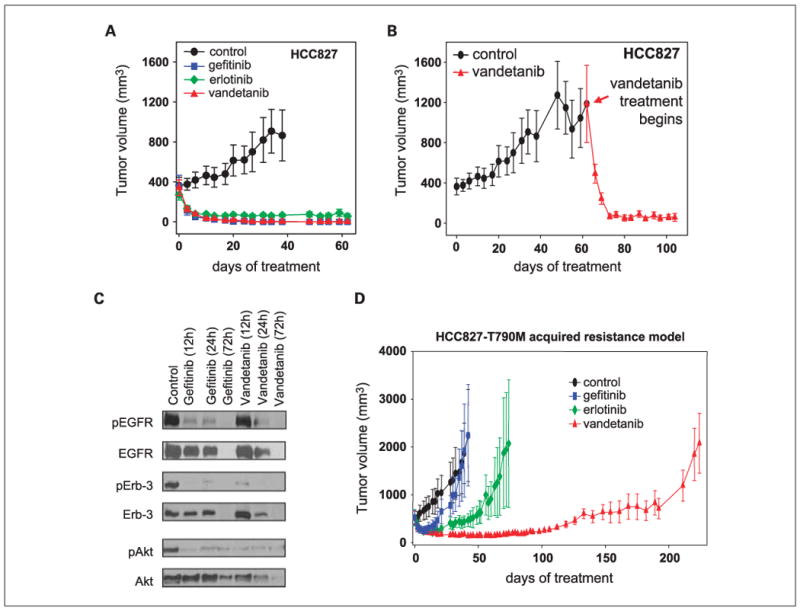
EGFR TKI sensitive xenograft model of NSCLC. A, HCC827 xenografts (wild-type K-RAS, EGFR exon 19 del.; n = 6-7 mice per group) were treated with either gefitinib (blue squares), erlotinib (green diamonds), or vandetanib (red triangles). All mice bearing HCC827 tumors (∼ 400 mm3) responded to gefitinib, erlotinib, and vandetanib treatment within 5 d, and tumors remained at a microscopic size for >60 d of continuous treatment. In contrast, tumor growth progressed in all vehicle-treated mice. B, Vandetanib effectively reduced even large (∼1,200 mm3) HCC827 tumors to a microscopic size within ∼10 d. C, HCC827 tumor xenografts from mice treated with vehicle, gefitinib, or vandetanib for 12, 24, or 72 h were isolated and protein lysates were evaluated for EGFR phosphorylation and downstream signaling by Western blot. D, HCC827 cells were transfected to express copies of mutated EGFR bearing the T790M resistance mutation, injected into nude mice and treated with either vehicle (black circles), erlotinib (blue squares), gefitinib (green diamonds), or vandetanib (red triangles). Vandetanib treatment effectively inhibited tumor growth by day 100, followed by gradual resistance to treatment by day 230. Results are shown as mean tumor volumes and SE at each time point.
In a separate experiment, vandetanib caused complete and sustained regressions of large tumors with a mean volume greater than 1,000 mm3 (Fig. 3B) at the time of treatment initiation. Within 4 days of vandetanib treatment mean tumor volume was reduced by half (to 492 ± 94 mm3) and by 11 days of treatment all tumors were barely palpable (mean tumor volume 66 ± 23 mm3). Together these results indicate that both EGFR TKIs erlotinib and gefitinib, and the dual VEGFR/EGFR inhibitor vandetanib, are able to induce significant regression or total tumor eradication of established xenografts bearing EGFR exon 19 deletions. To assess the dynamic effects of these agents on EGFR signaling in tumors in vivo, we isolated protein from HCC827 tumors treated with gefitinib or vandetanib for 12 h, 24 h, and 72 h, and evaluated EGFR activation and downstream signal transduction by Western blot. Both vandetanib and gefitinib completely inhibited phosphorylation of EGFR as well as Erb-3 and AKT by 72 h although these changes occurred more rapidly with gefitinib (Fig. 3C).
Establishment of an EGFR TKI-resistant xenograft model by transfection of EGFR L858R/T790M into HCC827 cells
Acquired resistance to EGFR TKIs has been associated with the emergence of tumor cells bearing the mutated EGFR with a secondary T790M mutation. To determine whether expression of T790M mutation would induce EGFR TKI resistance in vivo, HCC827 cells were retrovirally transduced with EGFR/del19 and T790M mutation (HCC827-T790M) or a control vector expressing green fluorescent protein (HCC827-GFP). HCC827-GFP xenografts remained highly sensitive to EGFR TKIs comparable with the parental HCC827 xenografts (data not shown). In contrast, all HCC827-T790M xenografts developed acquired resistance to gefitinib within 25 days of treatment (n = 5 mice) and erlotinib within 50 days (n = 5 mice) despite an initial sensitivity to these treatments (Fig. 3D). Although resistance to gefitinib and erlotinib (EGFR TKIs) emerged within the first 25 days, dual inhibition of the EGFR and VEGFR pathways with vandetanib delayed the appearance of resistant tumors past 120 days (n = 5 mice).
Treatment of A549 xenografts as a model of primary resistance
The effect of EGFR TKI inhibition and dual VEGFR/EGFR inhibition in A549 (K-RAS G12S mutation, wt EGFR) xenografts was evaluated as a model of primary resistance to EGFR TKIs. Despite our finding that A549 cells were resistant to EGFR inhibitors in tissue culture, A549 tumors growing in nude mice were moderately sensitive to gefitinib (54% tumor inhibition on day 38 of treatment, data not shown) and erlotinib (50% tumor inhibition; Fig. 4). However, blockade of both the VEGF and EGFR pathways either by vandetanib (78% tumor inhibition) or combination treatment of erlotinib and bevacizumab (85% tumor inhibition) was more effective than targeting either pathway alone.
Fig. 4.
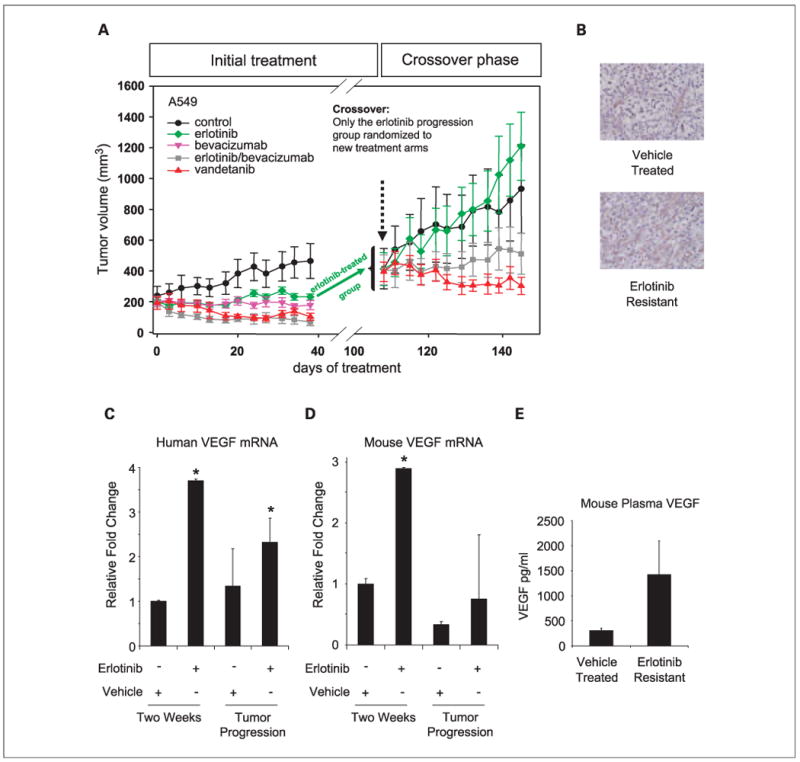
Xenograft model of NSCLC primary resistance. A, animals bearing A549 (K-RAS mutant, EGFR wild-type) xenografts were treated with erlotinib (green diamonds), bevacizumab (pink upside-down triangles), erlotinib and bevacizumab combination (gray squares), and vandetanib (red triangles). A549 tumors responded moderately to EGFR TKI (erlotinib) and VEGF inhibition (bevacizumab) single-agent treatments. However, combined targeting of EGFR and VEGFR pathways using erlotinib and bevacizumab combination treatment or vandetanib inhibited tumor growth better than erlotinib or bevacizumab monotherapies. Erlotinib-treated mice were allowed to progress (i.e. to become erlotinib-resistant) to a mean tumor volume of ∼400 mm3 and were randomized into four new treatment groups: control (black circles), erlotinib (green diamonds), erlotinib/bevacizumab combination group (gray squares), and vandetanib (red triangles). Erlotinib-treated and control tumors continued growth with similar kinetics after treatment crossover. However, dual inhibition of EGFR and VEGFR (erlotinib/bevacizumab combination and vandetanib treatments) resulted in stable disease where tumors remained at ∼400 mm3 for approximately 40 d. Results are shown as mean tumor volumes and SE at each time-point. B, representative immunohistochemical images of VEGF staining in A549 vehicle-treated and erlotinib-resistant tumors. C and D, expression of human (C) and mouse (D) VEGF RNA in A549 tumors treated with vehicle or with erlotinib for 2 wk or after tumors became resistant to therapy. Data are graphed as the mean ± SD. * P < 0.05 (E) VEGF is elevated in the plasma of mice with tumors resistant to erlotinib. Data are graphed as the mean ± SE.
To further investigate the effectiveness of EGFR versus VEGFR/EGFR targeted treatments after the development of EGFR TKI resistance, a group of mice bearing A549 tumors which had progressed during prolonged (∼110 days) treatment with erlotinib was utilized. A large cohort of mice (n = 32 mice) bearing A549 tumors was treated with erlotinib as part of the experiment shown in Fig. 4 (initial treatment panel). When the mean tumor size approximately doubled during erlotinib treatment, from mean size of 200 mm3 to approximately 400 mm3 (erlotinib progression model), mice were randomized to four groups receiving treatment with either vehicle alone (control), erlotinib, vandetanib, or erlotinib combined with bevacizumab (n = 5-6 mice per group; Fig. 4; “Crossover phase” panel). In this model, erlotinib no longer caused significant inhibition of tumor growth as compared with vehicle-treated controls (P = 0.468, t-test), confirming that these tumors had indeed acquired erlotinib resistance. The addition of bevacizumab to erlotinib significantly inhibited tumor growth by 58% (P = 0.027). Vandetanib was the most effective agent in this model, significantly inhibiting tumor growth by 75%, compared with controls and erlotinib groups (P = 0.008, t-test, vandetanib versus erlotinib groups). Bevacizumab alone could not be tested after the crossover because there were not sufficient tumors that had progressed at the time of the crossover randomization.
Because EGFR TKI resistance is associated with a secondary mutation in the EGFR TK domain T790M (11–13), we next evaluated whether EGFR mutational changes occurred in this model of acquired resistance. We collected DNA from A549 parental and erlotinib-resistant tumors and tested for alterations in the EGFR and K-RAS genes. In the A549 model, all resistant tumors had the expected K-RAS mutation and none had EGFR mutations in exons 18 to 21 suggesting that the resistance was mediated by an alternative mechanism. We also investigated EGFR phosphorylation by immunohistochemistry in the A549 model and observed that pEGFR immunoreactivity was low in the controls and was further reduced in tumors from erlotinib-treated mice after 2 weeks of treatment and in the resistant tumors (data not shown).
Given our finding that A549 tumors with acquired resistance to erlotinib were sensitive to dual EGFR/VEGFR inhibition and that vandetanib was more effective than the bevacizumab/erlotinib combination we next evaluated changes in VEGF in erlotinib-sensitive and -resistant tumors. By immunohistochemistry, we observed an increase in VEGF expression in erlotinib-resistant A549 tumors compared with vehicle-treated controls (Fig. 4B). This antibody did not distinguish between mouse and human VEGF. Next, to quantitatively assess whether the increased VEGF was human (tumor cell)- or mouse (host)-derived, we did real-time PCR using mouse- and human-specific primers. The specificity of the primers was confirmed using human A549 and mouse Lewis lung carcinoma tumor cells (data not shown). We observed a significant increase in tumor cell-derived (human) VEGF RNA after two weeks of erlotinib treatment (P = 0.0002) and in erlotinib-resistant tumors (P = 0.0406; Fig. 4C). In addition, we detected a significant rise in mouse VEGF RNA following two-week erlotinib treatment (Fig. 4D; P = 0.001). Furthermore, we evaluated changes in circulating VEGF in the plasma of animals bearing A549 tumors treated with vehicle or treated with erlotinib until resistance occurred, and observed >3-fold increase in circulating VEGF associated with erlotinib resistance (P = 0.184; Fig. 4E).
NSCLC animal model of acquired resistance to EGFR TKI: H1975 cells
The efficacy of EGFR and VEGFR inhibitors in H1975 xenografts (bearing EGFR L858R and T790M mutations) was assessed. Ten days following treatment, H1975 tumors were not significantly inhibited by erlotinib (P = 0.792 compared with control) or gefitinib (P = 0.159 compared with control; Fig. 5). However, treatment with bevacizumab as a single agent, or in combination with erlotinib, inhibited tumor growth by 68% and 75%, respectively. Vandetanib inhibited tumor growth by 80%, which was significantly greater than erlotinib inhibition (P ≤ 0.001). There was no statistical difference in tumor growth inhibition among the vandetanib, bevacizumab, and bevacizumab/erlotinib groups (P = 0.544).
Fig. 5.

Xenograft model of NSCLC secondary resistance. H1975 NSCLC cells bearing an EGFR-activating mutation as well as the T790M mutation were injected into nude mice. Established tumors were treated with gefitinib (blue squares), erlotinib (green diamonds), vandetanib (red triangles), or bevacizumab as a single agent (pink upside-down triangles) or in combination with erlotinib (gray squares). Results are shown as mean tumor volumes and SE at each time point.
Differential expression of EGFR and VEGF in A549 and H1975 xenografts
Although both A549 and H1975 cell lines were similarly resistant to EGFR TKIs in tissue culture, A549 cells displayed moderate in vivo sensitivity to EGFR TKIs when grown as xenografts, whereas H1975 cells did not. In an effort to understand the mechanism behind this disparity, we evaluated the expression of phosphorylated EGFR (p1068) within A549 and H1975 tumor cells and tumor-associated endothelial cells by immunohistochemistry (Fig. 6A, B, and E). As shown in Fig. 6E, levels of pEGFR were similar in tumor cells in both A549 and H1975 xenografts. In contrast, almost twice the relative level pEGFR was detectable in the endothelial cells within A549 tumors as compared with the endothelium from H1975 tumors (Fig. 6E). These findings suggested that the observed in vivo differences in sensitivity may be due at least in part due to effects of EGFR inhibition on tumor endothelium. We next evaluated the effect of these drugs on A549 tumor endothelium by dual immunohistochemistry using antibodies against Ki67 and von Willebrand factor (Fig. 6C, D, and F). Inhibition of EGFR or VEGFR pathways alone resulted in only a slight decrease (2-fold decrease, t-test control versus gefitininb, P = 0.032; control versus bevacizumab, P = 0.003) in Ki67-positive endothelial cells whereas blockade of both pathways caused a significant decrease (5- to 13-fold decrease; t- test, control versus erlotinib/bevacizumab or vandetanib, P < 0.001) in vascular proliferation (proliferating microvessel density) as assessed using Ki67 and von Willebrand factor dual immunohistochemical staining (Fig. 6F). The erlotinib group was not included in this analysis because these mice went on to the erlotinib progression model. These findings suggested that tumor endothelium within the A549 xenografts may be dependent on both the VEGFR and EGFR pathways, whereas the H1975 tumor endothelium seems to be more dependent on the VEGF pathway alone. To further investigate this, we collected tumor cell conditioned media and evaluated the in vitro VEGF production by A549 and H1975 cell lines by ELISA assay. Although both A549 and H1975 cells secreted detectable levels of VEGF, H1975 cells secreted over 2-fold more VEGF compared with A549 cells (Fig. 6G). Collectively, these data suggest that angiogenesis within A549 tumors is dependent on both the VEGF and EGFR pathways, whereas angiogenesis within H1975 tumors is to a greater extent VEGF-dependent.
Fig. 6.
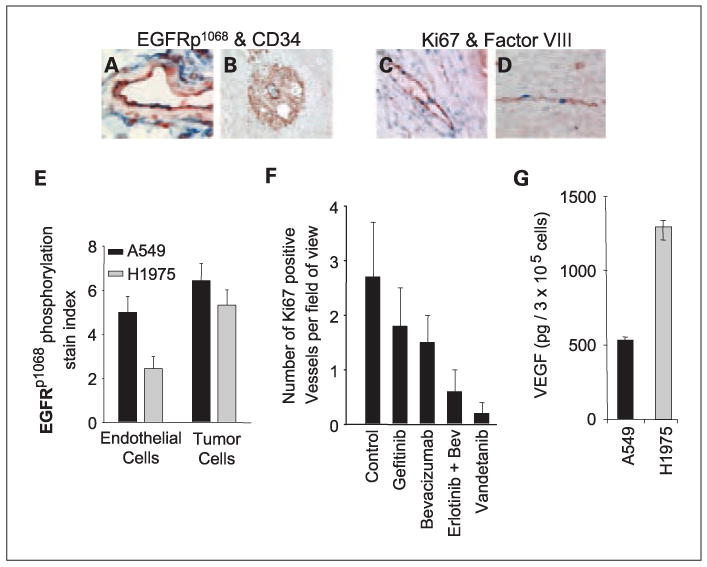
Differential expression of EGFR and VEGF in A549 and H1975 xenografts. Expression of phosphorylated EGFR on A549 and H1975 tumor cells and tumor-associated endothelial cells was evaluated by double-staining. In A549 tumors colocalization of pEGFR and CD34 was frequently observed (A). However, in H1975 tumors endothelium was weak or negative for pEGFR staining (B). Panel B also shows a representative vital pEGFR-positive zone comprising tumor cells (red) supported by CD34 positive (blue) vessels. To evaluate endothelial cell proliferation, A549 tumors were immunostained with antibodies directed against Ki67 (blue, mouse specific) and von Willebrand factor (red). Peritumoral vessels (C) and intratumoral vessels (D) showed positive endothelial cell nuclei, indicating active angiogenesis. E, nearly twice as much pEGFR was detected on endothelial cells within A549 tumors as compared with H1975 tumors. pEGFR staining in tumor cells did not differ significantly between the two cell types. Results are shown as mean ± SD. F, Ki67 and von Willebrand factor staining was quantified to evaluate endothelial cell proliferation within A549 tumors. Dual inhibition of EGFR/VEGFR pathways dramatically reduced the number of proliferating endothelial cells. Data are graphed as mean ± SD. G, in vitro VEGF secretion by A549 and H1975 cells was evaluated by ELISA assay and graphed as mean ± SD.
Discussion
EGFR inhibitors have shown clinical benefit for NSCLC and other tumor types, but <10% of previously treated NSCLC patients have an objective tumor response (4, 39) and even patients who do initially experience a major response eventually develop therapeutic resistance. For this reason, considerable effort has been focused on understanding the mechanisms regulating primary and acquired resistance to EGFR inhibitors. Secondary EGFR mutations (T790M mutation), amplification of the MET proto-oncogene, K-RAS mutations, and increased angiogenesis are associated with resistance to EGFR TKIs (9–14, 26). Elevated VEGF levels have been associated with resistance to the EGFR-targeting monoclonal antibody cetuximab (26). However, whether this extends to EGFR TKI resistance was unknown. Given the coregulation of the EGFR and VEGFR pathways and the association of elevated VEGF levels with acquired resistance, we hypothesized that dual inhibition of VEGF and EGFR pathways may be one strategy to overcome resistance to EGFR TKIs. Here we investigated this hypothesis using four xenograft models that represent different potential types of EGFR inhibitor resistance. We found that combining VEGFR and EGFR inhibition either through single-agent or combined therapy is effective in overcoming both primary and secondary resistance to EGFR inhibitors in all models tested.
We showed that vandetanib has a similar spectrum of in vitro activity against wild-type and mutated EGFR as gefitinib and erlotinib, albeit with slightly lower potency. Similar to gefitinib and erlotinib, vandetanib did not inhibit phosphorylation of EGFR bearing the T790M secondary mutation in cell lines (Fig. 1). Vandetanib was also similar to the EGFR TKIs in its ability to inhibit cell proliferation (Fig. 2), and EGFR signaling in vivo (Fig. 3C). This indicates that the greater in vivo potency of dual VEGF/EGFR inhibition using vandetanib (or the combination of bevacizumab and erlotinib) compared with erlotinib or gefitinib alone does not stem from a greater ability of these agents to inhibit EGFR activity in the tumor cells and is more likely due to the additional VEGFR component of treatment.
Through tumor xenograft experiments, we examined the effects of EGFR therapy alone versus the combination of VEGFR and EGFR therapy. In a NSCLC model using HCC827 cells highly sensitive to EGFR inhibition, all of the inhibitors were able to cause tumor regression, and even large tumors (>1,000 mm3) could be completely regressed by vandetanib. When mutated EGFR bearing a secondary T790M mutation was retrovirally transduced into HCC827 cells, the resulting xenografts became resistant to EGFR TKIs, providing direct evidence that T790M is able to mediate resistance to EGFR TKIs. Interestingly, in this model, vandetanib more than doubled the time required for resistance to emerge although all tumors did eventually become resistant.
In the three other models of primary or secondary EGFR TKI resistance, dual inhibition was more effective than EGFR TKIs alone although significant differences in response to these two approaches were observed. In the H1975 tumor model, EGFR TKIs gefitinib and erlotinib did not significantly inhibit growth in vitro or in vivo, whereas targeting VEGF alone using the VEGF monoclonal antibody bevacizumab yielded tumor regressions that were not significantly different from the combination of bevacizumab and erlotinib or vandetanib. This suggested that blockade of the VEGF pathway was primarily responsible for the tumor growth inhibition in this model. In the A549 model, by contrast, cells were resistant to EGFR inhibitors in vitro but were moderately sensitive in vivo, and combined inhibition was more effective than either VEGF or EGFR blockade alone.
One possible explanation for these results is that the EGFR TKIs not only block receptor activation on tumor cells but also on stromal cells, including the tumor vasculature. Consistent with this possibility, studies by Fidler and colleagues showed that tumor cell–derived transforming growth factor-α activates EGFR on endothelial cells, and treatment with an EGFR inhibitor results in apoptosis of tumor-associated endothelial cells and thus the surrounding tumor cells (40, 41). Additional studies have revealed that although tumor-associated endothelial cells express EGFR and respond to ligand stimulation in some tumors, normal endothelial cells lack EGFR, suggesting that EGFR kinase inhibitors may be effective at targeting the tumor vasculature but not normal endothelial cells (25). In agreement with these findings, we observed that EGFR was indeed activated on endothelial cells within A549 tumors to a greater extent than in H1975 tumors. Furthermore, EGFR or VEGF blockade alone caused a decrease in proliferating tumor vasculature in the A549 model, whereas the combination showed a greater decrease, suggesting an additive or synergistic antiendothelial effect in this model.
A549 NSCLC xenografts that initially responded to EGFR inhibition but eventually progressed were found to be resistant to EGFR inhibition, but sensitive to blockade of the VEGFR pathway. Secondary EGFR mutations were not observed in these tumors, and they were found to have the expected KRAS mutation in both vehicle-treated and erlotinib-treated tumors. The mechanism(s) underlying this acquired resistance are not completely understood. One possible explanation is that prolonged treatment with an EGFR inhibitor shifts the tumor cell population towards a less EGFR-dependent phenotype and more VEGF-dependent angiogenesis. Supporting this concept, our studies show that both tumor-derived and host-derived VEGF are elevated in association with erlotinib resistance. In the initial phase of this model, vandetanib treatment was equivalent to the erlotinib/bevacizumab combination. However, after tumors became erlotinib-resistant, vandetanib was superior to erlotinib/bevacizumab. Vandetanib inhibits VEGFR on both human and mouse cells, whereas bevacizumab binds human, but not mouse VEGF, supporting a role for stromal-derived VEGF in EGFR TKI resistance. These findings illustrate a possible stromal mechanism of drug resistance. Consistent with our findings, others have reported that in xenograft models of colon cancer tumors with acquired resistance to EGFR TKIs, VEGF is elevated, and tumors are sensitive to ZD6474 (42). Additionally, resistance of A431 xenografts to EGFR monoclonal antibodies is associated with elevated VEGF production (26), and in cell culture studies, tumor cells resistant to EGFR inhibitors exhibited elevated VEGFR-1 expression and increased VEGF secretion (43). Furthermore, we have recently observed that treatment with an EGFR inhibitor is able to shift tumor endothelium from dependency on EGFR to VEGFR in a melanoma xenograft model (44). These findings may have important implications regarding the treatment of patients who develop resistance to EGFR inhibitors, as they may retain or even have enhanced sensitivity to VEGF pathway blockade.
Clinical testing of dual VEGFR/EGFR inhibition has yielded promising results to date. In a randomized phase II study, the combination of erlotinib and bevacizumab was compared with chemotherapy or chemotherapy with bevacizumab in 120 previously treated nonsquamous NSCLC patients (27). Patients treated with erlotinib and bevacizumab showed a trend towards prolonged PFS compared with those receiving chemotherapy. In a phase II trial of 127 previously treated NSCLC patients evaluating two doses of vandetanib (100 mg or 300 mg) in combination with docetaxel for previously treated NSCLC patients, the combination of vandetanib (100 mg)/docetaxel showed prolonged PFS (18.7 versus 12 weeks) and a higher objective response rate (26% versus 12%; ref. 28) compared with docetaxel alone. Additionally, vandetanib was compared with gefitinib in 168 patients with platinum-refractory NSCLC (29). There was a statistically significant improvement in median PFS with vandetanib compared with gefitinib (11 versus 8 weeks). Finally, vandetanib with paclitaxel and carboplatin (VPC) was compared with paclitaxel and carboplatin (PC) in previously untreated patients with NSCLC. Compared with the PC arm, patients receiving VPC had a slightly longer PFS (hazard ratio, 0.76; 24 versus 23 weeks in control PC arm) although vandetanib by itself was inferior to PC (30). When considered along with the clinical data showing efficacy of combining VEGFR and EGFR treatment for NSCLC, our data suggest that the use of either single- or multi-agent therapy to target both the VEGF and EGFR pathways may be more effective than either VEGF or EGFR inhibition alone in some (but not all) cases and that dual inhibition may be a useful strategy for treating, or delaying the emergence of, at least some forms of primary and acquired resistance to EGFR inhibitors.
Acknowledgments
We thank Adi Gazdar (University of Texas Southwestern Dallas, Texas) for providing the HCC827 cells and Anderson Ryan (AstraZeneca Phamaceuticals, Macclesfield, UK) for providing vandetanib and for helpful discussions. The authors would also like to thank Dr. Folkman for his inspiration and involvement with this project.
Grant support: This work was supported by DFCI Lung SPORE NIH grant P20 CA090578, the University of Texas Southwestern Medical Center and M. D. Anderson Cancer Center SPORE NIH grant P50 CA070907, and HHMI-SPORE Pilot Research Project grant. J.V. Heymach is a Damon Runyon-Lilly Clinical Investigator supported in part by the Damon Runyon Cancer Research Foundation (CI 24-04), and the Physician Scientist Program at M. D. Anderson.
Footnotes
Disclosure of Potential Conflicts of Interest: J. V. Heymach, commercial reserch grant, AstraZeneca; consultant, AstraZeneca, Genentech.
References
- 1.Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics, 2007. CA Cancer J Clin. 2007;57:43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 2.Cancer Facts and Figures. American Cancer Society. 2006 [Google Scholar]
- 3.Byers LA, Heymach JV. Dual targeting of the vascular endothelial growth factor and epidermal growth factor receptor pathways: rationale and clinical applications for non-small-cell lung cancer. Clin Lung Cancer. 2007;8 2:S79–85. doi: 10.3816/clc.2007.s.006. [DOI] [PubMed] [Google Scholar]
- 4.Shepherd FA, Rodrigues Pereira J, Ciuleanu T, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353:123–32. doi: 10.1056/NEJMoa050753. [DOI] [PubMed] [Google Scholar]
- 5.Paez JG, Janne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science New York (NY) 2004;304:1497–500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 6.Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–39. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 7.Pao W, Miller V, Zakowski M, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101:13306–11. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hirsch FR, Varella-Garcia M, McCoy J, et al. Increased epidermal growth factor receptor gene copy number detected by fluorescence in situ hybridization associates with increased sensitivity to gefitinib in patients with bronchioloalveolar carcinoma subtypes: a Southwest Oncology Group Study. J Clin Oncol. 2005;23:6838–45. doi: 10.1200/JCO.2005.01.2823. [DOI] [PubMed] [Google Scholar]
- 9.Pao W, Wang TY, Riely GJ, et al. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005;2:e17. doi: 10.1371/journal.pmed.0020017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Engelman JA, Janne PA, Mermel C, et al. ErbB-3 mediates phosphoinositide 3-kinase activity in gefitinib-sensitive non-small cell lung cancer cell lines. Proc Natl Acad Sci U S A. 2005;102:3788–93. doi: 10.1073/pnas.0409773102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kobayashi S, Boggon TJ, Dayaram T, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–92. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 12.Kobayashi S, Ji H, Yuza Y, et al. An alternative inhibitor overcomes resistance caused by a mutation of the epidermal growth factor receptor. Cancer Res. 2005;65:7096–101. doi: 10.1158/0008-5472.CAN-05-1346. [DOI] [PubMed] [Google Scholar]
- 13.Pao W, Miller VA, Politi KA, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–43. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 15.Sandler A, Gray R, Perry MC, et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med. 2006;355:2542–50. doi: 10.1056/NEJMoa061884. [DOI] [PubMed] [Google Scholar]
- 16.Nilsson M, Heymach JV. Vascular endothelial growth factor (VEGF) pathway. J Thorac Oncol. 2006;1:768–70. [PubMed] [Google Scholar]
- 17.Swinson DE, Jones JL, Cox G, Richardson D, Harris AL, O'Byrne KJ. Hypoxia-inducible factor-1 α in non small cell lung cancer: relation to growth factor, protease and apoptosis pathways. Int J Cancer. 2004;111:43–50. doi: 10.1002/ijc.20052. [DOI] [PubMed] [Google Scholar]
- 18.Swinson DE, O'Byrne KJ. Interactions between hypoxia and epidermal growth factor receptor in non-small-cell lung cancer. Clin Lung Cancer. 2006;7:250–6. doi: 10.3816/CLC.2006.n.002. [DOI] [PubMed] [Google Scholar]
- 19.Luwor RB, Lu Y, Li X, Mendelsohn J, Fan Z. The antiepidermal growth factor receptor monoclonal antibody cetuximab/C225 reduces hypoxia-inducible factor-1 á, leading to transcriptional inhibition of vascular endothelial growth factor expression. Oncogene. 2005;24:4433–41. doi: 10.1038/sj.onc.1208625. [DOI] [PubMed] [Google Scholar]
- 20.Ciardiello F, Bianco R, Damiano V, et al. Anti-angiogenic and antitumor activity of anti-epidermal growth factor receptor C225 monoclonal antibody in combination with vascular endothelial growth factor antisense oligonucleotide in human GEO colon cancer cells. Clin Cancer Res. 2000;6:3739–47. [PubMed] [Google Scholar]
- 21.Ciardiello F, Caputo R, Bianco R, et al. Inhibition of growth factor production and angiogenesis in human cancer cells by ZD1839 (Iressa), a selective epidermal growth factor receptor tyrosine kinase inhibitor. Clin Cancer Res. 2001;7:1459–65. [PubMed] [Google Scholar]
- 22.Pore N, Jiang Z, Gupta A, Cerniglia G, Kao GD, Maity A. EGFR tyrosine kinase inhibitors decrease VEGF expression by both hypoxia-inducible factor (HIF)-1-independent and HIF-1-dependent mechanisms. Cancer Res. 2006;66:3197–204. doi: 10.1158/0008-5472.CAN-05-3090. [DOI] [PubMed] [Google Scholar]
- 23.Bruns CJ, Solorzano CC, Harbison MT, et al. Blockade of the epidermal growth factor receptor signaling by a novel tyrosine kinase inhibitor leads to apoptosis of endothelial cells and therapy of human pancreatic carcinoma. Cancer Res. 2000;60:2926–35. [PubMed] [Google Scholar]
- 24.Solorzano CC, Baker CH, Tsan R, et al. Optimization for the blockade of epidermal growth factor receptor signaling for therapy of human pancreatic carcinoma. Clin Cancer Res. 2001;7:2563–72. [PubMed] [Google Scholar]
- 25.Amin DN, Hida K, Bielenberg DR, Klagsbrun M. Tumor endothelial cells express epidermal growth factor receptor (EGFR) but not ErbB3 and are responsive to EGF, to EGFR. kinase inhibitors Cancer Res. 2006;66:2173–80. doi: 10.1158/0008-5472.CAN-05-3387. [DOI] [PubMed] [Google Scholar]
- 26.Viloria-Petit A, Crombet T, Jothy S, et al. Acquired resistance to the antitumor effect of epidermal growth factor receptor-blocking antibodies in vivo: a role for altered tumor angiogenesis. Cancer Res. 2001;61:5090–101. [PubMed] [Google Scholar]
- 27.Herbst RS, O'Neill VJ, Fehrenbacher L, et al. Phase II study of efficacy and safety of bevacizumab in combination with chemotherapy or erlotinib compared with chemotherapy alone for treatment of recurrent or refractory non small-cell lung cancer. J Clin Oncol. 2007;25:4743–50. doi: 10.1200/JCO.2007.12.3026. [DOI] [PubMed] [Google Scholar]
- 28.Heymach JV, Johnson BE, Prager D, et al. Randomized, placebo-controlled phase II study of vandetanib plus docetaxel in previously treated non small-cell lung cancer. J Clin Oncol. 2007;25:4270–7. doi: 10.1200/JCO.2006.10.5122. [DOI] [PubMed] [Google Scholar]
- 29.Natale R, Bodkin D, Govindan R, et al. ZD6474 versus gefitinib in patients with advanced NSCLC: Final results from a two-part, double-blind, randomized phase II trial. J Clin Oncol, 2006 ASCO Annual Meeting Proceedings. 2006;24:7000. [Google Scholar]
- 30.Heymach JV, Paz-Ares L, De Braud F, et al. Randomized phase II study of vandetanib alone or with paclitaxel and carboplatin as first-line treatment for advanced non-small-cell lung cancer. J Clin Oncol. 2008;26:5407–15. doi: 10.1200/JCO.2008.17.3138. [DOI] [PubMed] [Google Scholar]
- 31.Wedge SR, Ogilvie DJ, Dukes M, et al. ZD6474 inhibits vascular endothelial growth factor signaling, angiogenesis, and tumor growth following oral administration. Cancer Res. 2002;62:4645–55. [PubMed] [Google Scholar]
- 32.Straume O, Akslen LA. Expresson of vascular endothelial growth factor, its receptors (FLT-1, KDR) and TSP-1 related to microvessel density and patient outcome in vertical growth phase melanomas. Am J Pathol. 2001;159:223–35. doi: 10.1016/S0002-9440(10)61688-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weidner N, Semple JP, Welch WR, Folkman J. Tumor angiogenesis and metastasis–correlation in invasive breast carcinoma. N Engl J Med. 1991;324:1–8. doi: 10.1056/NEJM199101033240101. [DOI] [PubMed] [Google Scholar]
- 34.Stefansson IM, Salvesen HB, Akslen LA. Vascular proliferation is important for clinical progress of endometrial cancer. Cancer Res. 2006;66:3303–9. doi: 10.1158/0008-5472.CAN-05-1163. [DOI] [PubMed] [Google Scholar]
- 35.Tang X, Shigematsu H, Bekele BN, et al. EGFR tyrosine kinase domain mutations are detected in histologically normal respiratory epithelium in lung cancer patients. Cancer Res. 2005;65:7568–72. doi: 10.1158/0008-5472.CAN-05-1705. [DOI] [PubMed] [Google Scholar]
- 36.Mukohara T, Engelman JA, Hanna NH, et al. Differential effects of gefitinib and cetuximab on non-small-cell lung cancers bearing epidermal growth factor receptor mutations. J Natl Cancer Inst. 2005;97:1185–94. doi: 10.1093/jnci/dji238. [DOI] [PubMed] [Google Scholar]
- 37.Amann J, Kalyankrishna S, Massion PP, et al. Aberrant epidermal growth factor receptor signaling and enhanced sensitivity to EGFR inhibitors in lung cancer. Cancer Res. 2005;65:226–35. [PubMed] [Google Scholar]
- 38.Steiner P, Joynes C, Bassi R, et al. Tumor growth inhibition with cetuximab and chemotherapy in non-small cell lung cancer xenografts expressing wild-type and mutated epidermal growth factor receptor. Clin Cancer Res. 2007;13:1540–51. doi: 10.1158/1078-0432.CCR-06-1887. [DOI] [PubMed] [Google Scholar]
- 39.Thatcher N, Chang A, Parikh P, et al. Gefitinib plus best supportive care in previously treated patients with refractory advanced non-small-cell lung cancer: results from a randomised, placebo-controlled, multicentre study (Iressa Survival Evaluation in Lung Cancer) Lancet. 2005;366:1527–37. doi: 10.1016/S0140-6736(05)67625-8. [DOI] [PubMed] [Google Scholar]
- 40.Baker CH, Pino MS, Fidler IJ. Phosphorylated epidermal growth factor receptor on tumor-associated endothelial cells in human renal cell carcinoma is a primary target for therapy by tyrosine kinase inhibitors. Neoplasia. 2006;8:470–6. doi: 10.1593/neo.06172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Busby JE, Kim SJ, Yazici S, et al. Therapy of multidrug resistant human prostate tumors in the prostate of nude mice by simultaneous targeting of the epidermal growth factor receptor and vascular endothelial growth factor receptor on tumor-associated endothelial cells. Prostate. 2006;66:1788–98. doi: 10.1002/pros.20519. [DOI] [PubMed] [Google Scholar]
- 42.Ciardiello F, Bianco R, Caputo R, et al. Antitumor activity of ZD6474, a vascular endothelial growth factor receptor tyrosine kinase inhibitor, in human cancer cells with acquired resistance to antiepidermal growth factor receptor therapy. Clin Cancer Res. 2004;10:784–93. doi: 10.1158/1078-0432.ccr-1100-03. [DOI] [PubMed] [Google Scholar]
- 43.Bianco R, Rosa R, Damiano V, et al. Vascular endothelial growth factor receptor-1 contributes to resistance to anti-epidermal growth factor receptor drugs in human cancer cells. Clin Cancer Res. 2008;14:5069–80. doi: 10.1158/1078-0432.CCR-07-4905. [DOI] [PubMed] [Google Scholar]
- 44.Amin DN, Bielenberg DR, Lifshits E, Heymach JV, Klagsbrun M. Targeting EGFR activity in blood vessels is sufficient to inhibit tumor growth and is accompanied by an increase in VEGFR-2 dependence in tumor endothelial cells. Microvasc Res. 2008;76:15–22. doi: 10.1016/j.mvr.2008.01.002. [DOI] [PubMed] [Google Scholar]