

Abstract
Histone methyltransferases specific for the histone H3-lysine 9 residue, including Setdb1 (Set domain, bifurcated 1)/Eset/Kmt1e are associated with repressive chromatin remodeling and expressed in adult brain, but potential effects on neuronal function and behavior remain unexplored. Here, we report that transgenic mice with increased Setdb1 expression in adult forebrain neurons show antidepressant-like phenotypes in behavioral paradigms for anhedonia, despair, and learned helplessness. Chromatin immunoprecipitation in conjunction with DNA tiling arrays (ChIP-chip) revealed that genomic occupancies of neuronal Setdb1 are limited to <1% of annotated genes, which include the NMDA receptor subunit NR2B/Grin2B and other ionotropic glutamate receptor genes. Chromatin conformation capture and Setdb1-ChIP revealed a loop formation tethering the NR2B/Grin2b promoter to the Setdb1 target site positioned 30 kb downstream of the transcription start site. In hippocampus and ventral striatum, two key structures in the neuronal circuitry regulating mood-related behaviors, Setdb1-mediated repressive histone methylation at NR2B/Grin2b was associated with decreased NR2B expression and EPSP insensitivity to pharmacological blockade of NR2B, and accelerated NMDA receptor desensitization consistent with a shift in NR2A/B subunit ratios. In wild-type mice, systemic treatment with the NR2B antagonist, Ro25-6981 [R-(R,S)-α-(4-hydroxyphenyl)-β-methyl-4-(phenylmethyl)-1-piperidine propranol], and hippocampal small interfering RNA-mediated NR2B/Grin2b knockdown resulted in behavioral changes similar to those elicited by the Setdb1 transgene. Together, these findings point to a role for neuronal Setdb1 in the regulation of affective and motivational behaviors through repressive chromatin remodeling at a select set of target genes, resulting in altered NMDA receptor subunit composition and other molecular adaptations.
Introduction
Posttranslational histone modifications are involved in epigenetic regulation of gene expression and genome organization (Berger, 2007; Roth and Sweatt, 2009). Numerous studies highlighted the role of histone acetylation involved in various types of neuronal plasticity (Huang et al., 2002; Shahbazian et al., 2002; Brami-Cherrier et al., 2005; Thatcher and LaSalle, 2006; Weaver et al., 2006; Fischer et al., 2007; Lubin and Sweatt, 2007; Oliveira et al., 2007; Vecsey et al., 2007; Borrelli et al., 2008; Jiang et al., 2008a; Pandey et al., 2008; Crepaldi and Riccio, 2009; Guidotti et al., 2009). Furthermore, alterations in levels and activity of histone deacetylase enzymes profoundly affect depression-related behaviors in some preclinical models, which could point to novel targets for antidepressant drug development (Tsankova et al., 2006; Duman and Newton, 2007; Schroeder et al., 2007; Covington et al., 2009; Grayson et al., 2009; Gundersen and Blendy, 2009; Hobara et al., 2009). However, much less is known about the role of chromatin modifiers regulating histone modifications other than acetylation in the brain, including methylation (Gupta et al., 2010). Of note, various chromatin remodeling complexes associated with transcriptional repression involve histone methyltransferases (HMTs) for histone H3-lysine 9 (H3K9) and other lysine residues (Cedar and Bergman, 2009). A subset of H3K9 HMTs, including G9a and GLP/Eu-HMTAse1, assembled as heteromers (Tachibana et al., 2008), play a role for large scale chromatin organization during lineage restriction and cellular differentiation (Wen et al., 2009) and are essential for orderly brain development (Kleefstra et al., 2006). Additional members of the H3K9 HMT family, including Setdb1 (Set domain, bifurcated 1, also known as Eset/Kg1t/Kmt1e) and Suv39h1, show widespread expression in developing and adult brains (Lein et al., 2007). Although regulation of Suv39h1 expression in the nucleus accumbens (a core component in the brain's reward circuitry) is involved in stimulant addiction (Renthal et al., 2008), the role of Setdb1 for neuronal gene expression and behavior remains largely unexplored. Furthermore, although single genes, including p53BP2 and RASSF1A, were well described as Setdb1 targets in cancer cell lines (Li et al., 2006), the binding profile of Setdb1 on a genome-wide scale remains unexplored in the brain. To this end, we generated transgenic mouse lines with increased Setdb1 expression and activity, complemented by Setdb1 gene delivery studies in human cell lines derived from neural tissue. We use multiple lines of evidence to show that Setdb1 elicits changes in affective and motivational behaviors through a mechanism that involves partial repression of the NMDA receptor subunit NR2B/Grin2b. Notably, GRIN2B is strongly associated with genetic risk for bipolar affective disorder and schizophrenia in selected populations (Fallin et al., 2005; Avramopoulos et al., 2007; Allen et al., 2008), and furthermore, the nonselective NMDA antagonist ketamine and the NR2B-selective antagonist (1S, 2S)-1-(4-hydroxyphenyl)-2-(4-hydroxy-4-phenylpiperidino)-1-propanol (CP-101,606) were recently identified as fast-acting antidepressants in subjects with treatment-resistant depression (Berman et al., 2000; Zarate et al., 2006; Preskorn et al., 2008). Therefore, the findings presented here identify epigenetic fine-tuning of NMDA receptor gene expression as a new layer of regulation for the brain's affective and motivational states.
Materials and Methods
Animals
All experimental procedures were approved by the Institutional Animal Care and Use Committee of the University of Massachusetts Medical School. Animals were housed in groups of two to four per cage with food and water ad libitum under a 12 h light/dark cycle with lights on 7 A.M.
Generation of CaMKII-Setdb1 (CK-Setdb1) transgenic mice
A myc-tagged 4.6 kb mouse ESET/Setdb1 full-length cDNA fused to simian virus (SV40) late polyadenylation signal (SV40 pA) was isolated from plasmid pCS2-MT-ESET (a gift from Dr. Liu Yang, University of Arkansas for Medical Sciences, Little Rock, AR) (Yang et al., 2003) and ligated to the 8 kb CaMKII α promoter (Choi et al., 1991; Mayford et al., 1996) after insertion of a Kozak sequence, 5′-AGCCACCATGG-3′, to replace 5′-TTAAAGCTATGG-3′ in pCS2-MT-ESET. Linearized vector was injected into C57BL/6 × SJL F2 hybrid mouse embryos; two out of four different founder lines showed widespread expression of the transgene in forebrain. These two colonies were kept in different housing rooms. For each experiment, CK-Setdb1 +/0 mice were compared to gender-matched littermate controls housed in the same cage.
Immunohistochemistry
Coronal sections (18 μm thick) from perfusion-fixed (by phosphate-buffered 4% paraformaldehyde) adult brain were processed free-floating for anti-myc (A-14) (Santa Cruz Biotechnology; catalog #sc-789) immunoreactivity, which was detected with diaminobenzidine using an ABC kit (Vector Laboratories). This antibody recognizes epitopes of human c-myc, but cross-reactivity with the murine homolog was not detectable. For anti-myc immunofluorescence, fresh frozen brain sections were fixed with 100% methanol at −20°C for 10 min. Nuclei isolated from brain tissue homogenates in hypotonic solution were fixed, spread on slides, and air dried, and then stained for anti-myc and anti-NeuN (Millipore Bioscience Research Reagents; MAB377).
Immunoblotting
Samples were homogenized directly in 1× Laemmli buffer with 1× Complete Proteinase Inhibitor (Roche Applied Science; catalog #11697498001), incubated at 37°C for 10 min, and centrifuged at 13,500 × g at 4°C for 5 min. The supernatant was denatured at 95°C for 5 min electrophoresed on a 4–20% linear gradient Tris-HCl gel, and then transferred to nitrocellulose membrane. Immunostainings were performed with anti-myc (Santa Cruz Biotechnology; catalog #sc-789), two anti-Setdb1s (Ab-1, Millipore, catalog #07-378; Ab-2, Santa Cruz Biotechnology, catalog #sc-66884), anti-Grin2b (Millipore Bioscience Research Reagents; catalog #AB1557p), anti-Grin2a (Millipore Bioscience Research Reagents; catalog #AB1555p), and for loading controls, anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Abcam; catalog #ab9485) and anti-modification-independent histone H3 (Millipore; catalog #07-690). Immunoreactivity was detected with peroxidase-conjugated secondary antibody in conjunction with chemiluminesence-based film autoradiography. For quantification, Quantity-One (Bio-Rad) software was used.
Real-time reverse transcriptase PCR
Total RNA was extracted from cerebral cortex, striatum, or hippocampus using the RNeasy Lipid Tissue Mini kit (Qiagen; catalog #74804) and then subjected to SYBR green based one-step reverse transcriptase (RT)-PCR using Taqman One-Step RT-PCR Mix Reagents (Roche; catalog #4309169). Primers are listed in supplemental Table 1 (available at www.jneurosci.org as supplemental material). Ribosomal RNA (18S) was used as an internal control (Schroeder et al., 2007). Quantification was as described previously (Huang et al., 2006).
Chromatin immunoprecipitation
Samples were fixed in 1% formalin for 5 min at room temperature, sonicated at power level 6 (Branson Sonifier 250) on ice water (six runs of 1 min pulsing with 1 min resting interval), precleaned, and then subjected to anti-trimethyl H3K9 (H3K9me3; Millipore; catalog #07-442), anti-H3K9me2 (Millipore; catalog #07-441), anti-myc (Santa Cruz Biotechnology, catalog #sc-789), anti-Setdb1 (Santa Cruz Biotechnology; catalogs #c-66884), or anti-Krueppel-associated box domain (KRAB)-associated protein 1 (KAP-1; Abcam; catalog #ab22553) immunoprecipitation. Control samples were processed with normal rabbit IgG (Millipore; catalog #12-370) in parallel to samples with specific antibodies. For H3K9me3 chromatin immunoprecipitation (ChIP), input DNA and immunoprecipitated DNA were subjected to slot-blot hybridization using a 32P-labeled oligonucleotide probe, 5′ GGACC TGGAA TATGGC GAGAA A 3′, targeting mouse major satellite DNA. For PCR-based quantifications of ChIP assays, input DNA and immunoprecipitated DNA were subjected to SYBR-green-based real-time PCR using ChIP primers listed in supplemental Table 1 (available at www.jneurosci.org as supplemental material).
Chromatin immunoprecipitation followed by massively parallel sequencing
For two wild-type adult forebrains (mouse), anti-histone H3 trimethyl-lysine 4 (H3K4me3) ChIP was performed on micrococcal nuclease-prepared digests of nuclei as described previously (Huang et al., 2007). Subsequently, immunoprecipitated DNA was processed for deep sequencing by ligating the Genomic Adaptor Oligo Mix (Illumina) to fragments. After PCR amplification, ligated fragments around 250 bp were gel purified, and H3K4me3 ChIP libraries were deep sequenced by an Illumina Genome Analyzer (GA II). Genomic regions containing a significantly large number of reads, called peaks, were detected with MACS software (Zhang et al., 2008). Mappable tags were uploaded to the University of California Santa Cruz (UCSC) genome browser and peaks at the murine Grin2b locus (chromosome 6, 64.5 centimorgans) were extracted.
Chromosome conformation capture
To map chromosomal loop formations within the proximal 40 kb of the mouse Grin2b promoter, chromosome conformation capture (3C) was performed as described previously, with minor modifications (Miele and Dekker, 2009). In brief, forebrains of adult wild-type mice were homogenized in douncing buffer (Jiang et al., 2008b) with 2% formaldehyde to cross-link higher-order chromatin in its current conformation. Nuclei were extracted by ultracentrifugation under a sucrose gradient (Jiang et al., 2008b), and then 250 μl of 1× NEB2 (New England Biolabs) restriction enzyme buffer was added to the nuclei pellet. After resuspension, five 50 μl aliquots were prepared, and to each aliquot, 312 μl of 1× NEB2 and 38 μl of 1% SDS were added, and samples incubated at 65°C for 10 min to separate non-cross-linked proteins from chromatin. Then, 44 μl of 10% Triton X-100 was added to quench the SDS, gently mixed by pipetting, and then digested with 400 units of HindIII (New England Biolabs) at 37°C overnight under gentle horizontal shaking. Next, 86 μl of 10% SDS was added, and samples were incubated at 65°C for 30 min to inactivate HindIII restriction enzyme. Then, each sample was added to 7.61 ml of ligation mixture. The mixture consisted of 745 μl of 10% Triton X-100, 745 μl of 10× ligation buffer (1 m Tris HCl, pH 7.5, 1 m MgCl2, 1 m DTT), 80 μl of 10 mg/ml bovine serum albumin, 80 μl of 100 mm ATP, and 5960 μl of H2O. To each sample, 50 μl of T4 DNA ligase (1 U/μl) (Invitrogen) was added and incubated at 16°C for 4 h followed by reverse cross-linking at 65°C overnight with simultaneous proteinase K digestion (50 μl of 10 mg/ml) to remove chromatin-associated proteins. Then, for additional digestion, another 50 μl of proteinase K was added and incubated at 65°C for two additional hours. The ligated DNA fragments from pooled aliquots were purified by standard phenol/chloroform extraction and ethanol precipitation. To map for potential chromosomal interactions in cis around 40 kb of Grin2b, PCR was conducted with both “forward” and “reverse” primers positioned 5′ to 3′ on the sense strand (see Fig. 4 B, supplemental Table 1, available at www.jneurosci.org as supplemental material). Each primer was positioned ∼200 bp or less from the 3′ end of each HindIII restriction site, thereby representing a specific HindIII restriction fragment. PCR products were resolved in 2% Invitrogen ultrapure gels. In total, three series of 3C interaction maps were constructed by using a different restriction fragment anchor (primer) for each series (see Fig. 4 B). Two anchors were positioned around the Grin2b transcription start site (TSS), and the third anchor was ∼2 kb from the Setdb1 target sequence, separated by one HindIII restriction site. As a negative control, 3C libraries were generated from forebrain nuclei as described above, but without adding T4 ligase.
Figure 4.
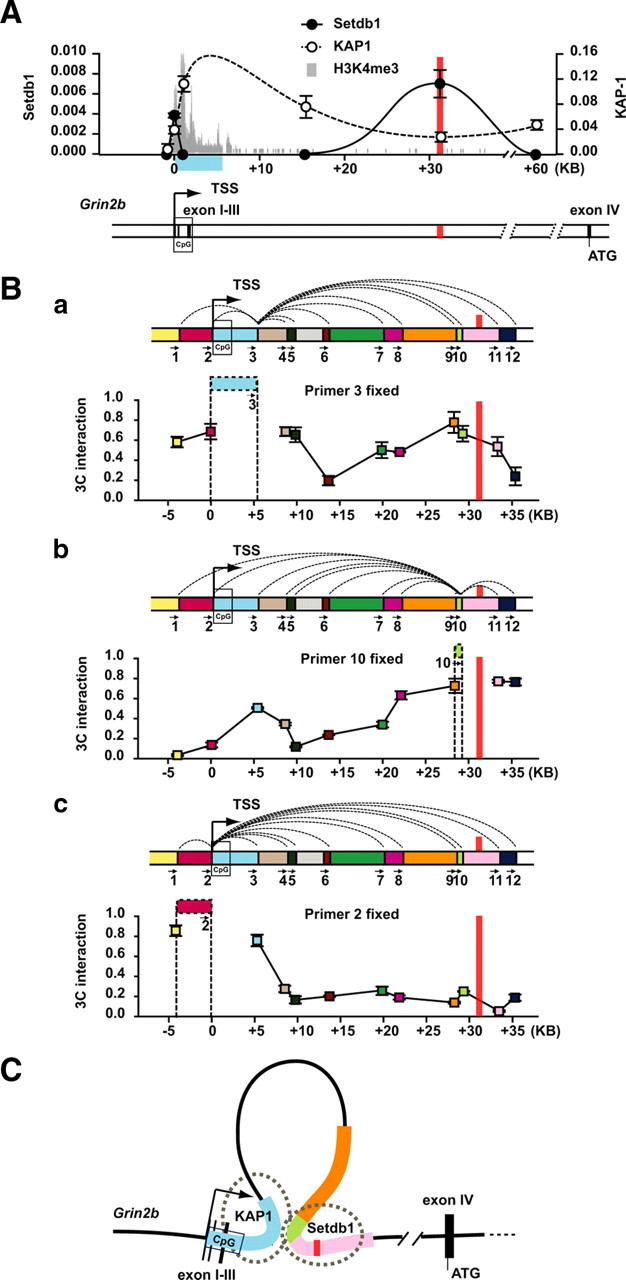
Chromatin architecture at the Grin2b locus in wild-type brain. A, Binding patterns of Setdb1 methyltransferase (black dots) and, for comparison, KAP-1 (white dots) in adult mouse forebrain (wild-type) within 60 kb from the Grin2b TSS as indicated. Data are expressed as the chip-to-input ratio (mean ± SEM); n = 3 chip experiments/antibody. Notice that Setdb1 occupancy is highest at the target site in intron III (marked by red line), corresponding to same target site shown in Figure 3 and Table 1. Notice further that KAP-1 occupancy is highest within first the 2–3 kb from the Grin2b TSS. Notice also that ChIP-seq (ChIP followed by massively parallel sequencing) H3K4me3 markings (shown in gray) at Grin2b are almost exclusively limited to this site. This portion of Grin2b is part of its most proximal HindIII fragment (blue box) and harbors a CpG island as indicated. B, 3C profiles from adult mouse forebrain (wild-type) for proximal 35 kb of Grin2b; colored bars each represent a specific HindIII restriction fragment. The position and orientation of 3C primers 1–12 is as indicated. Ba–Bc, Each 3C profile is anchored on specific restriction fragment/primer as indicated by colored horizontal bar in interaction map. The y-axes show the normalized pixel intensities (from agarose gel) for the specific PCR product from the anchoring primer and each of the remaining 11 primers as indicated by color code. Data are shown as the mean ± SEM from two 3C libraries processed in triplicate. Notice in Ba and Bb that there are comparatively high levels of interaction between 5 kb of the 5′ end of Grin2b (blue) and fragments 30 kb farther downstream from the TSS. The Setdb1 target site is shown as a red line. Notice also (Bc) that this interaction is not detected with the anchoring fragment immediately upstream of the TSS. C, Model for Grin2b chromatin loop, based on data shown in A and B. The loop tethers the 5′ end of the gene to intronic sequences >30 kb downstream from the TSS, including the Setdb1-target site. Notice that whereas the majority of Setdb1 is bound to the 3′ part of the loop, a smaller fraction is found at the 5′ part (see also A).
3C quantification
The level of interaction between any two regions along the genome is proportional to the extent of ligation between the two regions. This is measured semiquantitatively by the intensity of the PCR product generated using a specific primer pair representing the restriction fragments from the interacting regions. To correct for variations in specific primer pair efficiencies, each 3C-PCR product was normalized to the corresponding PCR product from a HindIII digested and then ligated bacterial artificial chromosome (BAC) representing ∼150 kb of genomic sequence surrounding the Grin2b TSS (BACPAC Resources Center). For 3C assays, the BAC template serves as a positive control and is expected to represent all possible fragment interactions in equal amounts, thereby controlling for primer efficiencies (Dekker, 2006; Miele et al., 2006; Miele and Dekker, 2009).
Mouse tiling array
A mouse tiling array, GeneChip Mouse Tiling Array 2.0R F (Affymetrix; catalog #900899), which is comprises ∼6.5 million 25 mer oligonucleotide probes covering chromosomes 6, 8, and 16, was used. After anti-myc ChIP, soluble DNA was amplified with a whole genome amplification system (Sigma; WGA2) according to a protocol from the Farnham Lab (http://www.epigenome-noe.net/researchtools/protocol.php?protid=30) with slight modifications. After library preparation, DNA samples were amplified by 14 cycles of unbiased PCR with universal primers, with 0.4 mm dUTP (Sigma; D0184). Amplified dUTP-incorporated DNA was then fragmented into around 69 bp by uracil DNA glycosylase digestion and end-labeled with biotin using the GeneChip WT Double Stranded DNA Terminal Labeling Kit (Affymetrix; catalog #900812). Approximately 7.5 μg of biotin-labeled DNA was hybridized to each tiling array. Hybridization, washing, staining, and scanning were conducted according to Affymetrix chromatin immunoprecipitation protocol (Protocol P/N 7002238, www.affymetrix.com/support).
Significant signals were calculated using the model-based analysis of tiling-arrays (MAT) algorithm as described previously (Johnson et al., 2006). Genomic windows of 600 bp were scored (MATscore), and p values were calculated based on the estimates of the nonenriched null distribution of the MAT scores. A false discovery rate for a region was calculated as the number of positive regions (above MATscore threshold) divided by the number of negative regions (below MATscore threshold). Genomic sites and genes with significantly higher signals in CK-Setdb1 than in wild-type samples are listed in Table 1 and supplemental Table S2 (available at www.jneurosci.org as supplemental material).
Table 1.
Gene hits from anti-myc ChIP-chip
| Gene symbol | Setdb1 occupancy |
Gene name | |
|---|---|---|---|
| a | b | ||
| Angpt2 | +10.9 | Int 1 | Angiopoietin 2 |
| Ano2 | +96.1 | Int 6 | Anoctamin 2 |
| Cadm2 | +462.4 | Int 1 | Cell adhesion molecule 2 |
| Cblb | +162.6 | Ex 17 | E3 ubiquitin-protein ligase Casitas B-lineage lymphoma B |
| Clec1a | +11.1 | Int 1 | C-type lectin domain, family 1, member A |
| Cntn4 | +563.3 | Int 5 | Contactin-4 |
| Cntn4 | +703.4 | Int 6 | Contactin-4 |
| Csmd1 | +1610.4 | Int 52 | CUB and sushi domain-containing protein 1 precursor |
| Eps8 | +27.0 | Int 1 | Epidermal growth factor receptor pathway substrate 8 |
| Gm156 | −5.6 | 5′ | Gene model 156 |
| Gpm6a | +97.7 | Int 4 | Neuronal membrane glycoprotein M6-a |
| Grid2 | +40.9 | Int 1 | Grid2 glutamate receptor, ionotropic, delta 2 |
| Grin2a | +289.3 | Int 5 | Glutamate receptor, ionotropic, NMDA2A |
| Grin2b | +31.4 | Int 3 | Glutamate receptor, ionotropic, NMDA2B |
| Il15 | +13.4 | Int 1 | Interleukin-15 |
| Kcnd2 | +143.3 | Int 1 | Potassium voltage-gated channel, Shal-related family, member 2 |
| Klra1 | −1.4 | 5′ | Killer cell lectin-like receptor, subfamily A, member 1 |
| Klra14 | −11.4 | 5′ | Killer cell lectin-like receptor, subfamily A, member 14 |
| Klra9 | +85.7 | Int 6 | Killer cell lectin-like receptor, subfamily A, member 9 |
| Klrk1 | +2.4 | Int 1 | Killer cell lectin-like receptor, subfamily K, member 1 |
| Lmo3 | +204.8 | Int 4 | Lin11, Isl-1, Mec-3 domain only 3 |
| Mcph1 | +134.9 | Int 12 | Microcephalin |
| Mtmr7 | +1.9 | Int 1 | Myotubularin-related protein 7 |
| Olfr203 | −1.6 | 5′ | Olfactory receptor 203 |
| Pik3c2g | +232.4 | Int 18 | Phosphatidylinositol 3-kinase, C2 domain containing, γ polypeptide |
| Ptprz1 | +154.4 | Int 17 | Protein tyrosine phosphatase, receptor type Z, polypeptide 1 |
| Robo2 | +239.9 | Int 2 | Roundabout homolog 2 precursor |
| Styk1 | +6.0 | Int 3 | Serine/threonine/tyrosine kinase 1 |
| Tmem39a | −1.5 | 5′ | Transmembrane protein 39A |
Int, Intron; Ex, exon; 5′, 5′ upstream of TSS. All p values are ≤0.00001; n = 3 CK-Setdb1 and, as negative control, 3 wild-type littermates.
a The length from gene hits to TSS (5′ to 3′; in kilobases).
b The length range of gene hits is from 624 to 1000 bp.
Setdb1 expression in U87-MG cells
mycSetdb1 full-length cDNA was cloned into an AAV vector backbone by ClaI and XbaI sites, and then transfected into the U87-MG human glioma cell line by using Lipofectamine 2000 (Invitrogen; catalog #11668) according to the manufacturer's protocols. In brief, Opti-MEM I Reduced Serum Medium was applied to cells with 80–90% confluence (0.2–0.25 × 106 cells per well of a six-well plate, 2 × 106 cells per 10 cm plate) 1 d before transfection. Four micrograms (six-well plate) or 24 μg (10 cm plate) of plasmid DNA was transfected. Cells that received Lipofectamine only were processed in parallel as negative controls. Medium was changed 4 h after transfection, and cells were kept in culture for another 20 h. Cells were then harvested and prepared for anti-myc XChIP, anti-Setdb1, and anti-Grin2b immunoblotting.
Electrophysiology: whole-cell recordings in hippocampal neurons
Hippocampus slices from age-matched postnatal day 18 (P18)–P21 CK-Setdb1 +/0 /CK-H2BeGFP +/0 and wild-type (CK-Setdb10/0/CK-H2BeGFP+/0) mice were prepared as reported previously (Martin and Siggins, 2002). In brief, animals were anesthetized and decapitated, and the brains rapidly transferred into an ice-cold (3–4°C) oxygenated, low-calcium HEPES-buffered salt solution containing the following (in mm): 234 sucrose, 2.5 KCl, 2 NaH2PO4, 11 glucose, 4 MgSO4, 2 CaCl2, and 1.5 HEPES. A tissue block containing the hippocampus was glued to a Teflon chuck and cut transversally with a vibroslicer (Vibratome 1500). The slices (350 μm thick) were incubated for up to 6 h at room temperature (20–22°C) in a gassed (95% O2 and 5% CO2) NaHCO3-buffered saline solution containing the following (in mm): 116.4 NaCl, 1.8 CaCl2, 0.4 MgSO4, 5.36 KCl, 0.89 NaH2PO4, 5.5 glucose, 24 NaHCO3, 100 glutathione, 1 nitro-arginine, and 1 kynurenic acid; pH was adjusted to 7.35 with NaOH, 300–305 mOsm/L. After 1 h of incubation, the region of the hippocampus was dissected out with the aid of a dissecting microscope, and the tissue was incubated for 15 min in an oxygenated (100% O2 with constant stirring) HEPES-buffered solution in the inner chamber of a Cell-Stirr flask (Wheaton) containing protease XIV (1 mg/ml) and the following (in mm): 136 NaCl, 0.44 KH2PO4, 2.2 KCl, 0.35 NaH2PO4, 5.5 glucose, 10 HEPES, 100 glutathione, 1 nitro-arginine, 1 kynurenic acid, and 1 pyruvic acid, pH 7.35 with NaOH, 300–305 mOsm. The temperature of this solution was kept constant (36°C) by a circulating water bath in the outer chamber of the flask.
After enzymatic digestion, the tissue was transferred into a centrifuge tube and rinsed twice with an Na+-isethionate solution. The tube was then filled with 5 ml of Na+-isethionate solution and after 10 min, the tissue was triturated using fire-polished Pasteur pipettes with successively smaller tip diameters. The supernatant was plated onto a 35 mm Petri dish placed on the stage of the inverted fluorescence microscope. The cells were allowed to attach to the bottom of the dish for at least 10 min before replacing the Na+-isethionate solution with normal Locke's solution composed of the following (in mm): 142 NaCl, 2 KCl, 1 CaCl2, 23 glucose, 15 HEPES, pH 7.35 with NaOH, osmolarity 300 mOsm/L.
Standard whole-cell recording methods were used as described previously (Mayford et al., 1996). Green fluorescent protein (GFP)-positive cells with pyramidal morphology were selected and applied for recording. Briefly, patch electrodes were pulled from borosilicate 1.5 mm capillary glass (Warner Instruments) on a Brown-Flaming P-97 puller (Sutter Instruments) to a final resistance of 2–4 MΩ. The electrodes were filled with a solution that consisted of the following (in mm): 120 KMeSO4, 11 EGTA, 10 HEPES, 0.5 CaCl2, 2Na2-ATP, 0.2 Na3-GTP, and 12 phosphocreatine, pH 7.35 with KOH, and osmolarity 270–275 mOsm. The capillaries were first filled through the tip and then backfilled with the recording solution. Recording was performed in voltage-clamp mode with an EPC-10 double amplifier from HEKA Elektronik, and filtered at 5 kHz and digitized at 1 kHz. Liquid junction potentials were not corrected but are estimated to be +4 mV.
Control and drug-containing solutions were applied by gravity at a rate of 1.5 ml/s using a rapid three-barrel capillary superfusion device (Warner Instruments) with the pipette tips placed about 200 μm from the recorded cell. Each capillary had a tip diameter of 500 μm, and the distance from center to center was 700 μm. The pipette assembly was attached to a motor, allowing fast lateral motions controlled by the acquisition software PatchMaster 2.32 (HEKA Elektronik). Dose–response curves were obtained as follows: after recording a stable current, pyramidal-like neurons were exposed for 4 s to increasing concentrations of NMDA (1 to 3000 μm) in the presence of a saturating (200 μm) concentration of glycine, a glycine receptor antagonist (picrotoxin, 1 μm), and 15 μm CNQX to block non-NMDA glutamate receptors, and peak current amplitude was measured. To avoid current inactivation, drugs were applied every 60 s. Glycine, glutamate, NMDA, and picrotoxin were purchased from Sigma. Dose–response curves were fitted with a Hill equation as follows: I = I max/[1 + (EC50/C)n], where I, I max, C, EC50, and n are the agonist-activated current, maximal agonist-activated current, agonist concentration, the concentration for 50% of the response, and the Hill coefficient, respectively. NMDA current desensitization was measured on NMDA currents evoked by coapplication (4 s) of 500 μm NMDA and 200 μm glycine at −60 mV holding potential. The current decay was fitted with a single exponential using an analysis software (FitMaster 2.35; HEKA Elektronik), and tau values were calculated to present the current desensitization rate.
EPSPs in medium spiny neurons
Brains (P18–P28), after removal from the skull, were submerged in ice-cold (∼1°C) artificial CSF (ACSF) containing the following (in mm): 125 sucrose, 53 NaCl, 2.5 KCl, 1 MgCl2, 1.25 NaH2PO4, 2 CaCl2, 26 NaHCO3, and 25 dextrose, osmolarity 310–315 mOsm, and 1 kynurenic acid, bubbled with 95% O2/5% CO2. Coronal slices (300 μm) cut with a VT1200 microtome (Leica) were moved to 35°C ACSF for 50 min, after which they were allowed to cool to room temperature in a regular ACSF for up to 8 h. All experiments were performed at room temperature (∼20°C). Patch pipettes were pulled with a final 4–6 MΩ resistance from 1.5 mm glass (World Precision Instruments) on a P-1000 Puller (Sutter Instruments) using 1.5 mm glass (WPI) and filled with the following (in mm): 130 KMeSO4, 7 KCl, 10 HEPES, 0.05 EGTA, 2 MgATP, 1 NaGTP, and 12 Na-phosphocreatine, adjusted with KOH to pH 7.3–7.4. Bipolar stimulating electrodes (FHC) were placed in close (<1 mm) proximity of the anterior commissure of the nucleus accumbens region. For each neuron, stimulation strength was adjusted until synaptic responses were of the desired amplitude (+10–20 mV). Acquisition of EPSPs started only after recording satisfactorily stable EPSPs. We performed all recordings in whole-cell current clamp in infrared–differential interference contrast mode using a Scientifica microscope equipped with Olympus optics, including a two-station objective nose (×10 and ×60). Neurons were visualized with a CCD video camera (Orca-03; Hamamatsu) connected to a focusing tower (50 mm motorized travel in both axis) equipped with a cube controller. Whole-cell current-clamp recordings were performed using an EPC-10 double amplifier (HEKA) controlled by PatchMaster 2.43, an acquisition software (HEKA), running on an Apple MacPro. Current and voltage traces were acquired at a 20 kHz sampling rate and filtered at 2 kHz. Series resistance (20–35 MΩ) and capacitance were compensated for and EPSPs amplitude was measured with FitMaster 2.43 (HEKA).
Striatal Setdb1-small interfering RNA injections
A set of three mouse Setdb1 Stealth small interfering RNA (siRNA), SETDB1MSS234817, SETDB1MSS234815, and SETDB1MSS294563 (Invitrogen), were tested in NIH mouse 3T3 cells. The most robust decrease (up to 70%) in Setdb1 expression at the mRNA level was detected 60 h after transfection of cells exposed to the Setdb1-siRNA SETDB1MSS294563 (D03 CCAACCUGUUUGUCCAGAAUGUGUU; D04 AACACAUUCUGGACAAACAGGUUGG), which was used for the following in vivo knockdown experiments. To deliver the Setdb1-siRNA into ventral striatum, P16–P20 CK-Setdb1 animals were anesthetized with a ketamine (100 mg/kg)/xylazine (10 mg/kg) mixture administered intraperitoneally, and thereafter placed on a stereotactic frame (Stoelting) equipped with a mouse adapter. Bilateral injection coordinates according to the mouse brain atlas (Franklin and Paxinos, 2001) were (from bregma) +1 mm anteroposterior (AP), ±1 mm mediolateral (ML), and 3.5 mm dorsoventral (DV). A 10 μl Hamilton syringe was used for injection. The syringe was vertically attached to a probe holder connected to a micropump (Stoelting) and filled with Setdb1-siRNA (SETDB1MSS294563) or vehicle control solution and inserted 3.5 mm deep into the mouse brain. Setdb1-siRNA (2 μg/μl) dissolved in 5% sterile glucose, mixed with six equivalent of linear 22 kDa polyethylenimine (ExGen 500; #RO521; Fermentas) as transfection reagent, was delivered into each side over a duration of 5 min. After each injection, the syringe was left in place for another 4 min to prevent backflow. Sixty hours after surgery, brain tissue adjacent to injection needle tracks was sliced for medium spiny neuron EPSPs recordings, anti-myc immunohistochemistry, and anti-Setdb1 immunoblotting as described above.
Hippocampal NR2B/Grin2b siRNA delivery
To deliver NR2B/Grin2b siRNA into the hippocampus of adult C57BL6/J mice, 22 gauge guidance cannulae (Plastics One) were mounted onto the skull with dental cement and closed with 28 gauge dummy cannulae to prevent tissue ingrowth. Coordinates were (from bregma) −1 mm AP, ±1 mm ML, and 2.0 mm DV. After surgery, mice were allowed to recover for 7 d. For siRNA injection, the dummy cannulae was replaced by the actual internal cannulae (28 gauge), which was attached via a 50-cm-long polyethylene tube to a Hamilton (10 μl) syringe fitted into a micropump (Soelting). Two microliters of siRNA mixture, comprising of equimolar ratios for three siRNAs (GRIN2BMSS204919, GRIN2BMSS204917, GRIN2BMSS204918; Invitrogen), or 2 μl of vehicle as a control was delivered bilaterally, over a duration of 2 min for each side. To avoid stress, during siRNA delivery, mice were unrestrained and freely moving in their home cage. After each injection, the internal cannulae was left in place for another 4 min to prevent backflow before the guidance cannulae was closed again with the guidance cannulae. The siRNA mixture was injected once daily for 3 d. Tail suspension was performed 24 h after the last injection, and then hippocampal tissue cubes (∼2 × 2 × 2 mm) surrounding the cannula track were harvested for anti-NR2B immunoblotting.
Behavioral assays
For each of the behavioral tests, CK-Setdb1 +/0 mice were housed in the same cage as their wild-type littermates (except for the sucrose preference test). Only adult male mice were used. All tests were scored blindly.
Sucrose preference test.
Hedonia/anhedonia was determined by a sucrose preference test in animals not deprived of food or water as described previously (Aonurm-Helm et al., 2008). Two days before the test, mice were single housed in regular Makrolon mouse cages (20 × 30.5 cm, 16.5 cm high). The test was performed over 10 d, during which mice were given the choice to drink either tap water or a 1% sucrose solution (sucrose dissolved in tap water). To avoid loss of fluid, the solutions were administered in bottles equipped with a no-leak, double-ball-point drinking mechanism. Every 24 h, all bottles were weighed to estimate the amount of liquid consumed by the mice. Solutions were freshly prepared every second day. Sucrose consumption was calculated as a percentage of the amount of total liquid consumed. Data were analyzed with a two-way ANOVA, with genotype (wild-type vs transgenic) as the between groups factor and day as the within groups factor, followed by Newman–Keuls post hoc test.
Tail suspension test.
Animals were suspended with a paper clip wrapped with tape around the tail, 0.75 cm from the tip, and then hung from a mounted hook 50 cm above the tabletop. Animals showing tail climbing behaviors were removed from this test. The whole test was videotaped, and the time spent immobile during a 6 min testing period was then scored by a trained observer.
Forced swim test.
Animals were placed into a 4 L Pyrex beaker (13 cm diameter, 24 cm height) filled with 22°C water, 17 cm deep. The time spent immobile during a 6 min testing period was measured. Immobility, evaluated by a blind observer, was defined according to the criteria as described previously (Schramm et al., 2001).
Learned helplessness.
A learned helplessness paradigm was conducted as described previously (Caldarone et al., 2004). Learned helplessness was induced in mice by administering 120 inescapable foot shocks (IESs) for 4 s each at 0.3 mA once every 26 s over a 1 h session in a closed compartment (Gemini Avoidance System; San Diego Instruments). In total, two sessions of IESs spaced ∼24 h apart were given. Twenty-four hours and 7 d after the second session, animals were tested in a shuttle escape task to determine their learned helplessness behavior. Animals were given 30 escape trials with 30 s intervals between each trial. The initial five trials featured a 0.3 mA shock with an open door to an adjacent “safe” compartment for escape. Shock stimulus was terminated at escape or after 26 s, whichever was shorter. The remaining trials proceeded similarly; however, the escape route (door opening) was presented 2 s after shock onset. Shock stimulus was terminated at escape or after 24 s, whichever was shorter. The latency to each escape was recorded, and the averaged latency from 30 trials was used to measure both learned helplessness as well as the subsequent recovery from helplessness.
Shock reactivity.
Animals were placed into an inescapable testing chamber and subjected to subsequent foot shocks with 2 s duration at 0.05, 0.1, 0.3, 0.5, and 0.7 mA with 28 s intervals. Animal behaviors induced by foot shock were monitored and scored as follows: flinching, 1; vocalizing, 2; running, 3; jumping, 4. Shock reactivity was presented as the total score reached after each trial of shock.
Passive avoidance.
The test apparatus consisted of two chambers divided by an automatic guillotine door: one chamber was illuminated by an overhead light and the other kept in dim light. On habituation day, animals were first introduced into the lighted chamber with the connecting door closed. After 2 s, the door was opened and the animals were allowed to freely explore for 5 min. On the training day (24 h after habituation day, preshock), the animals were introduced into the lighted chamber with the connecting door open. The latency to enter the dim chamber was recorded. Once the animal entered into the dim chamber, the door was closed, and an inescapable foot shock (2 s duration, 0.5 mA) was delivered in the dim chamber. Animals were left in the dim chamber for 10 additional seconds after shock and then returned to their home cages. Twenty-four hours or 7 d after training (after shock), animals were introduced into the light chamber with the connecting door open, and the latency to enter the dim chamber was recorded. A maximum testing time of 5 min was given.
Contextual and tone-induced fear conditioning.
A fear conditioning protocol was used as described previously (Reijmers et al., 2006), with minor modifications. On day 1, animals were subjected to a training trial by placing them in a chamber (Gemini Avoidance System; San Diego Instruments) with context A: methanol scented, grid floor, cue light on. After 3 min of habituation, four tone–shock pairings were presented 1 min apart. Each pairing consisted of a 20 s tone simultaneously ending with a 2 s, 0.5 mA inescapable foot shock. Animals remained in context A for 1 min after the last tone–shock pairing. On day 2, the day 1 training trial was repeated with the first 3 min used to measure context freezing (24 h after first context A exposure). On day 3, animals were tested for tone freezing (24 h after last tone–shock pairing) in a chamber with context B: ethanol scented, cue light off, left chamber wall covered with orange craft foam and back chamber wall with white/blue-striped contact paper. After a 3 min baseline, the tone was presented for 20 s, during which time freezing was measured. On day 7, animals were placed in context A for 3 min to measure context freezing (7 d after first context A exposure); on day 8, animals were placed in context B to measure tone freezing (7 d after last tone–shock pairing). In sham control groups, animals received tone but no shock stimulus in day 1 and day 2 trials. Behavior was video recorded and the total duration of freezing (bouts of >1 s of immobility) was scored during the indicated time interval.
Novelty-induced locomotion (ambulatory beam breaks).
Locomotion was monitored in a rectangular test chamber (19.1 × 29.2 × 12.7 cm) with clean bedding, using a photocell-beam-based computer detecting system (San Diego Instruments). Mice were placed into the test chamber for 90 min under standard room lighting conditions. The beam breaks were counted every 5 min to evaluate the spontaneous locomotor activity.
Rotarod.
Rotarod (Ugo Basile; 47600) was used to measure motor coordination in mice. The rotarod was set up with a slow acceleration mode, in which the rotation speed is accelerated from 2 to 40 rpm in 5 min. Mice were subjected to 10 consecutive 5 min trials with a 5 min intertrial interval. Twenty-four hours later, mice were tested by receiving three consecutive trials (at 5 min intervals), and the mean latency to fall off or, alternatively, to make a passive rotation was used to present the performance.
Light/dark box.
The box (15 × 15 × 23 cm) consisted of one brightly lit, open chamber connected by a small hole (4.5 cm) to a darkened, enclosed second chamber. Animals were introduced into the lit compartment, and the time spent in each compartment was manually recorded for 5 min after the first transition, with a 10 min maximum time.
Elevated plus maze.
The elevated plus maze (Med Associates) consisted of two open arms and two closed arms situated opposite each other and separated by a 6 cm square center platform. Each runway was 6 cm wide by 35 cm long. In addition, the open arms had lips 0.5 cm high to prevent the mice from slipping from the arm, and the closed arms were surrounded on three sides by 20 cm walls. The floors and walls were made from black polypropylene. For each test, the animal was introduced in the center square and then allowed to explore freely for 5 min. The time spent on open arms, in closed arms, or in the center square was recorded separately.
Results
Setdb1 occupancy in neuronal chromatin is limited to a small subset of genes
Setdb1 mRNA expression is widespread throughout the murine CNS, including a large majority of neurons (Lein et al., 2007). In adult mouse cerebral cortex, immunoreactivity for full-length, 180 kDa Setdb1 is readily detectable (Fig. 1 A). To explore the genomic targets of Setdb1 in neurons, we generated transgenic CK-Setdb1 mice expressing a functional, myc-tagged full-length Setdb1 cDNA under control of the CaMKII α (CK) promoter (Fig. 1 B). Two transgenic lines, from different founders, showed the expected, neuron-specific expression pattern, with nuclear localization, in cortical layers II–VI, hippocampus, striatum, and other telencephalic (and diencephalic) structures (Fig. 1 C,D).
Figure 1.
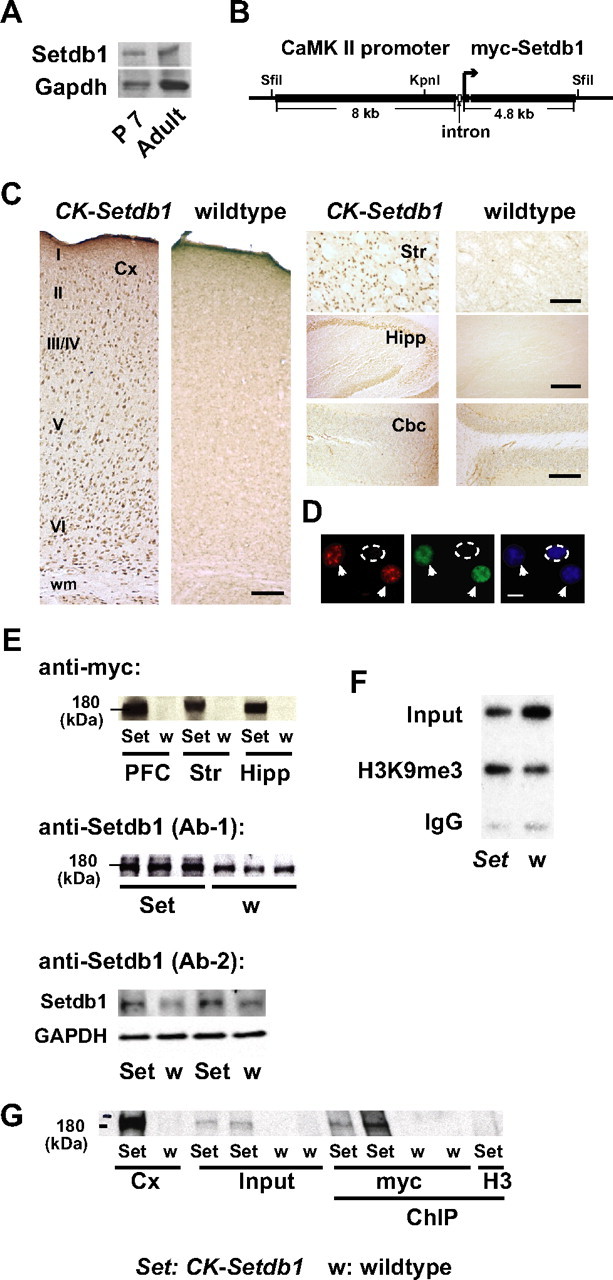
Expression and activity of Setdb1 in CK-Setdb1 and wild-type mice. A, Setdb1 expression in P7 and adult wild-type cerebral cortex. B, CK-Setdb1 transgene construct, including Sfil and KpnI restriction enzyme sites. C, Anti-myc immunoreactivity in brain sections of 8-week-old CK-Setdb1 and wild-type mice in cerebral cortex (Cx), striatum (Str), hippocampus (Hipp), and cerebellar cortex (Cbc). Scale bars: Cx, Str, 100 μm; Hipp, 200 μm; Cbc, 150 μm. D, Anti-myc (red) immunofluorescence in neuronal nuclei (anti-NeuN, green) (arrows), but not in nonneuronal nucleus (dotted line); DAPI (4′,6′-diamidino-2-phenylindole) is shown in blue. Scale bar, 10 μm. E, Immunoblots show 180 kDa anti-myc signal in prefrontal cortex (PFC), Str, and Hipp of CK-Setdb1 (Set) but not wild-type (w) mice (top), and increased 180 kDa anti-Setdb1 signal with two different antibodies, Ab-1 and Ab-2 (see Materials and Methods) in transgenic cerebral cortex (Ab-1) and hippocampus (Ab-2) (bottom), each lane loaded with 20 μg protein. F, Representative slot blot shows anti-H3K9me3 and IgG control ChIP hybridized with major satellite DNA probe. G, Representative blot shows mycSetdb1 immunoactivity in “Input” and anti-myc (myc) ChIP from CK-Setdb1 but not wild-type forebrains. Protein extracted from Cx and anti-histone H3 (H3) ChIP material were processed as positive and negative controls, respectively.
The CK-Setdb1 mice showed a several-fold increase in Setdb1 mRNA levels in multiple areas of the forebrain, compared to wild-type littermates (fold change in CK-Setdb1, mean ± SEM: cortex, 2.7 ± 0.1; striatum, 1.9 ± 0.4; hippocampus, 5.7 ± 1.4, n = 4–5/group; p < 0.05–0.01; Mann–Whitney test). Expression of the myc-tagged transgenic protein was associated with a robust increase in full-length, ∼180 kDa Setdb1 immunoreactivity, as determined with two different anti-Setdb1 antibodies (Fig. 1 E). This reflected an approximately twofold increase in Setdb1 protein, when normalized to GAPDH “housekeeping” protein (mean ± SEM: CK-Setdb1, 0.49 ± 0.01; wild-type, 0.19 ± 0.14, n = 3/group; p = 0.05; Mann–Whitney test). To test whether increased Setdb1 protein results in upregulated HMT activity, including H3K9 methylation, chromatin extracts from anti-H3K9me3 immunoprecipitates were probed—without previous PCR amplification—with a major satellite sequence to label pericentric repeats. These are defined by A/T rich major satellite repeats composed of ∼105 copies of a 234 bp unit (Waterston et al., 2002) and high levels of H3K9 methylation (Martens et al., 2005), which in part is regulated by Setdb1 (Lee et al., 2008; Loyola et al., 2009; Fritsch et al., 2010). Indeed, pericentric H3K9me3 in CK-Setdb1 forebrain was increased by >50% from wild-type (Fig. 1 F) (chip to input, mean ± SEM: CK-Setdb1, 0.41 ± 0.10; wild-type, 0.26 ± 0.06; n = 12/group; p < 0.05; Wilcoxon signed rank), thereby suggesting that H3K9 HMT activity is upregulated in pericentric heterochromatin in the transgenic animals. This H3K9 hypermethylation of the major satellite repeats in CK-Setdb1 forebrain was specific, because H3K9me3 immunoreactivity in tissue extracts, which include the entire fraction of chromatin-bound and free histones, were not significantly different between transgenic and wild-type hippocampus, or prefrontal cortex or striatum (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). The observation that total H3K9 methylation is not increased in CK-Setdb1 forebrain is not too surprising, however, given that the murine genome contains at least six or seven H3K9-specific methyltransferase enzymes (Kouzarides, 2007), many of which are expressed in adult mouse brain (Lein et al., 2007).
The pull-down of cross-linked chromatin with an anti-myc antibody confirmed the presence of 180 kDa mycSetdb1 immunoreactivity in CK-Setdb1 forebrain, but not in wild-type mice (Fig. 1 G). Thus, the approach to immunoprecipitate myc-tagged chromatin proteins, although originally described in yeast (Ren et al., 2000), is also suitable for mouse brain. We then profiled genomic occupancies of neuronal Setdb1 in adult forebrain by chromatin immunoprecipitation using anti-myc antibody in conjunction with an Affymetrix (genomic DNA) tiling array (chip-chip) covering murine chromosomes 6, 8, and 16 excluding repeats. The initial rationale in choosing this particular chromosomal array was to include mouse chromosome 16 sequences syntenic to a portion of human 22q11.2 conferring high risk for affective disorder and psychosis as a microdeletion (DiGeorge/Velo-cardio-facial/22qDS) syndrome (Mukai et al., 2008). Significant peaks, calculated by MATscores with a 600 bp sliding window, were limited to altogether 70 sites across chromosomes 6, 8, and 16, but no Setdb1 signal was observed within the 22qDS homolog (supplemental Table S2, available at www.jneurosci.org as supplemental material). Instead, among these 70 sites were at least 29 annotated gene hits from a total of ∼4000 genes of chromosomes 6, 8, and 16 represented on the array. These included 13 of 29 sites located approximately 30 kb or less from the nearest TSS (Table 1). This finding is consistent with the notion that Setdb1 is involved in transcriptional regulation (Schultz et al., 2002; Seum et al., 2007). Strikingly, among the 29 genes targeted by Setdb1 (out of ∼4000 genes represented on the array) were (i) Grin2a and Grin2b, the sole two NMDA receptor subunits on the array, and (ii) Grid2, encoding the glutamate delta-2 receptor (Table 1). Because three of the altogether four ionotropic glutamate receptor genes represented on different portions of chromosomes 6, 8, and 16 (Grid2, Grik1, Grin2a, Grin2b) were targeted by Setdb1, there was a highly significant, >200-fold enrichment for multiple Gene Ontology (GO) categories involving glutamate receptors and excitatory neurotransmission, including GO:0060079 (regulation of excitatory postsynaptic membrane potential) and GO:051899 (membrane depolarization). We conclude that chromatin surrounding glutamate receptor genes is targeted by Setdb1, which otherwise is bound to <1% of annotated genes in nuclei of mature neurons. In addition, Setdb1 located to several distinct sequences within a gene cluster on chromosome 6 encoding natural killer cell immunoglobulin-like receptors (Table 1, supplemental Table S2, available at www.jneurosci.org as supplemental material).
Setdb1 regulates the expression of NMDA receptor subunit Grin2b
Next, we tested whether the Setdb1-mediated increased H3K9 HMT activity in transgenic forebrain (Fig. 1 G) would result in repressive chromatin remodeling and downregulated expression of NMDA receptor subunits that are targeted by Setdb1 (Table 1). To this end, we measured in adult CK-Setdb1 mice and their wild-type littermates the mRNA levels of the constitutive subunit Grin1 (NR1) and the two Setdb1 target genes, Grin2a and Grin2b, with the latter gene being of particular interest given that Setdb1 is positioned only 30 kb from its TSS (Table 1). Notably, adult CK-Setdb1 mice, compared to wild-type littermates, showed a highly significant, 20–50% reduction in Grin2b mRNA and protein levels in hippocampus, and a similar tendency in prefrontal cortex (Fig. 2 A,B). In contrast, levels of Grin2a (which, unlike Grin2b, is targeted by Setdb1 at the 3′ end, >270 kb downstream of the TSS) (Table 1) were not significantly altered (Fig. 2 A,B). Likewise, expression of the constitutive subunit, Grin1 (NR1), remained unaltered in CK-Setdb1 animals (Fig. 2 A). The lower level of Grin2b in CK-Setdb1 brain could be attributable to transcriptional repression, because H3K9 methylation at Grin2b's Setdb1 target site was significantly increased in the transgenic animals (CK-Setdb1, 1.20 ± 0.18; wild-type, 0.78 ± 0.07; n = 11/group; p < 0.05; Mann–Whitney test).
Figure 2.
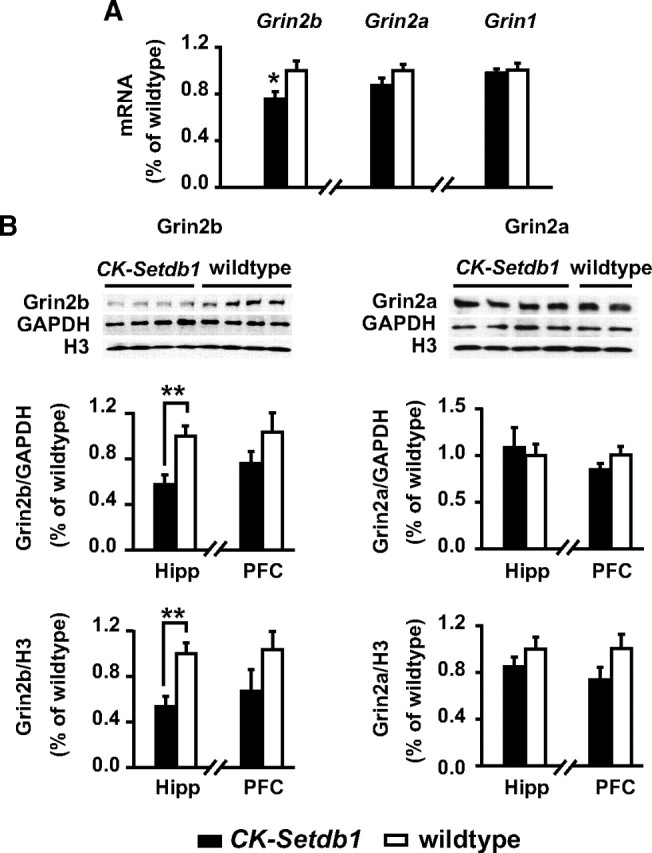
Decreased Grin2b expression in CK-Setdb1 mouse brain. A, Real-time RT-PCR quantification of Grin2b, Grin2a, and Grin1 mRNAs in CK-Setdb1 and wild-type hippocampus, as indicated. n = 6–8/group. *p < 0.05 (Mann–Whitney test). B, Western blot analysis of Grin2b and Grin2a proteins from hippocampus (Hipp) and prefrontal cortex (PFC) of adult CK-Setdb1 and wild-type littermates. Relative protein levels were normalized to GAPDH and histone H3, respectively. n = 7–15/group. **p < 0.01 (Mann–Whitney test). All data are shown as mean ± SEM.
To test whether Setdb1 elicits an inhibitory effect on Grin2b transcription in other species, including human, we transfected U87MG glioma cells, which express low to moderate levels of GRIN2B and other glutamate receptor genes (Stepulak et al., 2009), with mycSetdb1 cDNA. Indeed, three of three mycSetdb1 transfected cultures, compared to controls, showed a more than twofold increase (mean ± SEM, 2.3 ± 0.1) in Setdb1 occupancy at proximal intronic GRIN2B sequences, which are homolog to the Setdb1 target site within the murine Grin2b (Fig. 3 A–C). This was associated with a significant, ∼20% reduction in GRIN2B expression, compared to controls with histone H3 as a loading control (mean ± SEM: Setdb1 transfected cultures, 0.78 ± 0.04; control cultures, 1.00 ± 0.02; n = 3/group; p < 0.05; Mann–Whitney test) (Fig. 3 D). Together, these findings suggest that expression of GRIN2B is sensitive to changes in SETDB1 levels and activity.
Figure 3.

Setdb1 negatively regulates GRIN2B expression in human U87-MG glioma cells. A, B, Setdb1 target sites at Grin2b (mouse) and GRIN2B (human). A, Top, Distribution of scores calculated with MAT software in the 1.3 Mb region of mouse chromosome 6 enclosing the Grin2b gene. Scores are calculated over a 600 bp interval and assigned to the tiling probe at the center of the interval. Scores are plotted as a custom track in the UCSC genome browser (http://genome.ucsc.edu/). The significance threshold is 3.66 for p < 1e−5. Bottom, Sequence alignment between mouse (Ms) and human (Hu) GRIN2B gene loci at Setdb1 target sites. Conserved sequences are indicated with asterisks. B, Schematic overview shows Setdb1 target sites in 5′-untranslated regions from mouse (solid arrow) and human (dashed arrow) Grin2b/GRIN2B loci. C, Representative blot shows mycSetdb1 immunoreactivity in human glioma (U87-MG) culture transfected with mycSetdb1 cDNA (Set+), but not control (C). Histone H3 was used as loading control. D, Representative immunoblots show Setdb1 and GRIN2B immunoreactivity in Set+ and control U87-MG cultures.
Chromatin architectures at the Grin2b locus
The studies above demonstrate that transgenic Setdb1 preferentially targets, both in human and mouse, GRIN2B/Grin2b intronic sequences that are ∼30 kb distal from the TSS. To rule out that this genomic occupancy is an off-target effect of the transgene, we checked Setdb1 occupancies at Grin2b in forebrain of wild-type mice with anti-Setdb1 ChIP. Remarkably, highest levels of Setdb1 were measured at the same sequence (in intron III) that had emerged as the primary target of transgenic Setdb1 (Fig. 4 A), essentially ruling out that this is an off-target. Furthermore, a second, smaller wild-type Setdb1 ChIP signal was detected at the gene's TSS (Fig. 4 A). This distinct binding profile of Setdb1 was highly specific, when compared to the genomic occupancies of the KAP-1 transcriptional corepressor (also known as TRIM28/TIF1b/KRIP1), which is a putative binding partner of Setdb1 in repressive chromatin remodeling complexes (Ayyanathan et al., 2003). KAP-1, in striking contrast to Setdb1, showed peak levels within the first 2–3 kb from Grin2b's TSS followed by a broad but weak distribution spread at least 60 kb into the Grin2b gene (Fig. 4 A). Of note, the KAP-1 peak matched the location of the sole CpG island found at the 5′ end of Grin2b. Furthermore, when the genome-wide distribution of H3K4me3, an epigenetic mark that is sharply regulated around TSSs and associated with positive or negative regulation of gene expression (Shilatifard, 2008), was mapped in wild-type forebrain, the signal at the Grin2b locus was almost exclusively confined to the first few kilobases downstream of its TSS (Fig. 4 A). Therefore, the distribution pattern both of the KAP-1 repressor and of the H3K4me3 mark, and the position of Grin2b's sole CpG island at its 5′ end, together highlight the potential importance of the first 2–3 kbs following this gene's TSS for transcriptional regulation.
However, the studies above also demonstrate that Setdb1 is primarily found at Grin2b intronic sequences that are removed (30 kb) from the gene's TSS. To understand how this binding pattern could be associated with Setdb1's negative regulation of Grin2b expression (Figs. 2, 3), we used chromosome conformation capture technology (Dekker, 2008) to map physical interactions within 40 kb of Grin2b TSS. We prepared 3C libraries from DNA ligase-treated HindIII digests of formalin-cross-linked nuclei isolated from adult mouse forebrain (wild-type), as described in Materials and Methods. Then, 3C interaction maps were generated with specific HindIII fragment as anchors (Fig. 4 Ba–Bc). In 3C maps, the interaction frequency between chromatin fragments is measured as the relative intensity of the corresponding PCR products, compared to Grin2b BAC reference templates, which are thought to represent all possible fragment interactions in equal amounts (Dekker, 2006; Miele et al., 2006) (see Materials and Methods).
In 3C experiments, the degree of interaction typically is very high between adjacent 3C fragments, but then rapidly declines to very low or undetectable levels when probed with fragments/primers further removed from the anchor (Dekker, 2006, 2008). This is also what we observed, as shown in the 3C map anchored on the HindIII fragment positioned 5′ from the Grin2b TSS (Fig. 4 Bc, primer 2). This would suggest that chromatin upstream from Grin2b's TSS shows very little interaction with chromatin structures other than those in its immediate proximity. In striking contrast, the 3C map anchored on the HindIII fragment composed of the first 5 kb following Grin2b's TSS (Fig. 4 Ba, primer 3) revealed two peaks. There was, in addition to the expected interaction with neighboring primers, a second 3C peak from PCR products with primers positioned 30 kb downstream from the TSS (Fig. 4 Ba), which included the aforementioned Setdb1 target sequence in intron III (Fig. 4 A,Ba–Bc, red line). The specificity of the above peak was further confirmed in “reciprocal” 3C maps now anchored on the +30 kb portion of intron III (Fig. 4 Bb, primer 10), which again showed comparatively high levels of interaction with the HindIII fragment/primer representing the TSS and +5 kb of Grin2b (Fig. 4, blue), whereas the interaction with the surrounding fragments was much lower (Fig. 4 Bb). No products were obtained from PCRs of 3C libraries that were not treated with DNA ligase (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). These findings together suggest that there is a chromatin loop caused by a physical interaction between sequences 30 kb downstream from TSS, which are targeted by Setdb1 (Fig. 4 C), and the 5′ end of Grin2b, which represents part of the promoter including the TSS and the sole CpG island in that region as well as high levels of a Setdb1 binding partner, the KAP-1 repressor. These findings, which were obtained from wild-type brain, further highlight the potential role of Setdb1 for Grin2b expression.
Functional alterations of the NMDA receptor complex in CK-Setdb1 hippocampus
The NMDA receptor subunit, NR2B/Grin2b, is assembled into synaptic and extrasynaptic NMDA receptor complexes and is involved in a wide range of functions including various types of neuronal plasticity (Thomas et al., 2006). To explore whether Setdb1, by decreasing Grin2b expression, alters NMDA receptor function in CK-Setdb1 mice, we recorded NMDA currents in whole-cell patch clamp on somata of acutely dissociated hippocampal neurons from P18–P21 animals. To visualize mycSetdb1-expressing neurons in the dish, we crossed CK-Setdb1 mice with a transgenic line expressing GFP-conjugated histone H2B under control of the CK promoter (Jiang et al., 2008b). Coexpression of the two transgenes was apparent in hippocampal sections (Fig. 5 A) and confirmed by counting of nuclei extracted from hippocampus (including the CA sectors), with 99.4% (723 of 727 counted) GFP-positive neuronal nuclei expressing mycSetdb1. Recordings were performed on GFP-positive cells with pyramidal neuron-like morphology (Fig. 5 B). When NMDA currents were evoked with NMDA concentrations ranging from 1 μm to 3 mm, in the presence of 15 μm CNQX to block non-NMDA glutamate receptors, current amplitudes were maximum with 1 mm NMDA and rapidly declined with lower concentrations (Fig. 5 C). Dose–response curves, including the EC50, were nearly equivalent between wild-type (324 μm) and CK-Setdb1 mice (310 μm) (Fig. 5 C). Because the EC50 for NMDA is higher for receptors containing Grin2a/NR2A and Grin2b/NR2B, as compared to the remaining NR2 subunits, Grin2c/NR2C and Grin2d/NR2D (which are expressed at low levels in P18 and older hippocampus) (Laurie and Seeburg, 1994), we conclude that CK-Setdb1 animals do not show a generalized loss of NMDA receptor function. However, heteromeric receptors comprised of Grin2b subunits (together with the constitutive subunit Grin1) show slower current decay rates compared to Grin2a (Vicini et al., 1998; Tovar et al., 2000). Therefore, we compared Ca2+-independent desensitization kinetics in CK-Setdb1 and wild-type neurons. NMDA currents were evoked around −60 mV holding potential by coapplication of 500 μm NMDA with saturating concentrations of glycine (200 μm), and fast and slow current decay were best fit by a single exponential (Fig. 5 D). Indeed, the averaged τ for GFP-positive neurons from CK-Setdb1 mice (0.76 ± 0.13 s; n = 16) was significantly decreased compared to those from the control group (genotype CK-Setdb10/0/CK-H2BeGFP +/0; 1.21 ± 0.22 s; n = 8) (Fig. 5 D). Together, these findings suggest that Setdb1 represses Grin2b expression in neurons, thereby affecting NMDA receptor subunit composition and desensitization kinetics.
Figure 5.
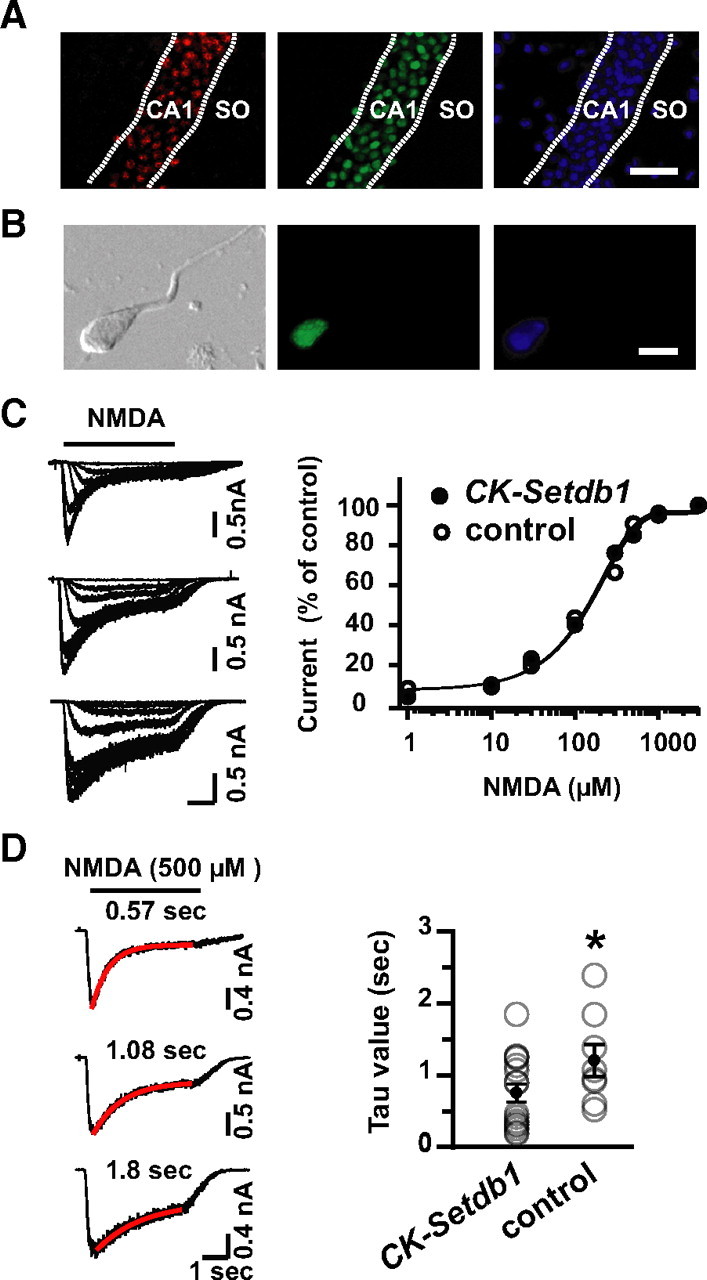
Accelerated NMDA current desensitization in CK-Setdb1 hippocampal neurons. A, Representative images show coexpression of myc (red) immunoreactivity and GFP (green) in pyramidal neuron layer of the hippocampal CA1 field from CK-Setdb1/CK-H2BeGFP mice. Neither transgene is expressed in stratum oriens (SO) that harbors primarily GABAergic neurons; blue shows DAPI (4′,6′-diamidino-2-phenylindole) counterstain. B, Example of an acutely dissociated GFP-positive neuron from hippocampus of CK-Setdb1/CK-H2BeGFP mice at postnatal day 18. C, Left, NMDA currents evoked by NMDA from 1 μm to 3 mm for different hippocampal neurons. Horizontal bar represents timing of drug exposure. Right, Dose–response plot of peak NMDA currents from CK-Setdb1 (CK-Setdb1 +/0 /CK-H2BeGFP +/0; filled circle) and controls (CK-Setdb10/0/CK-H2BeGFP +/0; open circle); n = 8–10/genotype. Notice that dose–response curves are indistinguishable between CK-Setdb1 and control mice. D, Left, Representative individual currents of three hippocampal neurons in response to 500 μm NMDA. Red lines represent single exponential fits of current desensitization; values (seconds) are indicated above the traces. Right, Scatter plot shows individual NMDA current desensitization rate (open circle) and averaged rate (black dots) for GFP-positive neurons from CK-Setdb1 and control mice. Notice the significantly faster desensitization in GFP-positive CK-Setdb1 neurons in comparison to control neurons. n = 8–19/group. *p < 0.05 (one-tailed Mann–Whitney test). Scale bars: A, 40 μm; B, 10 μm. Data are mean ± SEM.
Medium spiny neuron EPSPs of CK-Setdb1 mice are not sensitive to NR2B antagonist
The findings above suggest that general NMDA receptor signaling is preserved in CK-Setdb1 forebrain, although the partial decrease in NR2B/Grin2b expression could, in addition to changing biophysical properties of the receptor (see above), render the transgenic brain less sensitive to the effect of specific NR2B antagonist drugs. To explore this, we assessed the acute effects of the NR2B-selective antagonist drug ifenprodil on NMDA EPSPs of striatal medium spiny neurons (MSNs) located in the nucleus accumbens/ventral striatum, which, like the hippocampus, is part of the neuronal circuitry regulating affective and motivational behaviors.
The current–voltage relationship and other biophysical properties that define MSNs (Martin et al., 1997) were indistinguishable between CK-Setdb1 and control mice (Fig. 6 A). Thus, the rectification in the hyperpolarized range of potential was clearly visible, as was the ramp in response to depolarizing current steps. Furthermore, MSNs action potential firing patterns in both wild-type and CK-Setdb1 mice showed little adaptation and displayed a fast afterhyperpolarization whose amplitude similarly declined as a function of the number of action potentials. Then, we evoked NMDA EPSPs every 20 s for up to 10 min before (baseline) (Fig. 6 C, y-axis) and during 100 μm ifenprodil exposure in the presence of 10 μm CNQX. Representative traces from wild-type mice show that ifenprodil strongly inhibits NMDA-EPSP amplitudes (Fig. 6 B). When monitored 5 min after ifenprodil exposure, the drug had reduced the NMDA-EPSP amplitudes by nearly 50% in five of five wild-type neurons (Fig. 6 B,C). In contrast, four of five neurons from transgenic striatum were essentially insensitive to ifenprodil's effect under these experimental conditions (Fig. 6 B,C). Only one of five MSNs from CK-Setdb1 mice was sensitive to ifenprodil's inhibitory effect on NMDA-EPSP amplitudes; the neuron-to-neuron variability in ifenprodil sensitivities could be attributable to differences in Setdb1 transgene expression. These data further confirm that NMDA receptor subunit composition and function is altered because of a downregulation of Grin2b in CK-Setdb1 animals.
Figure 6.
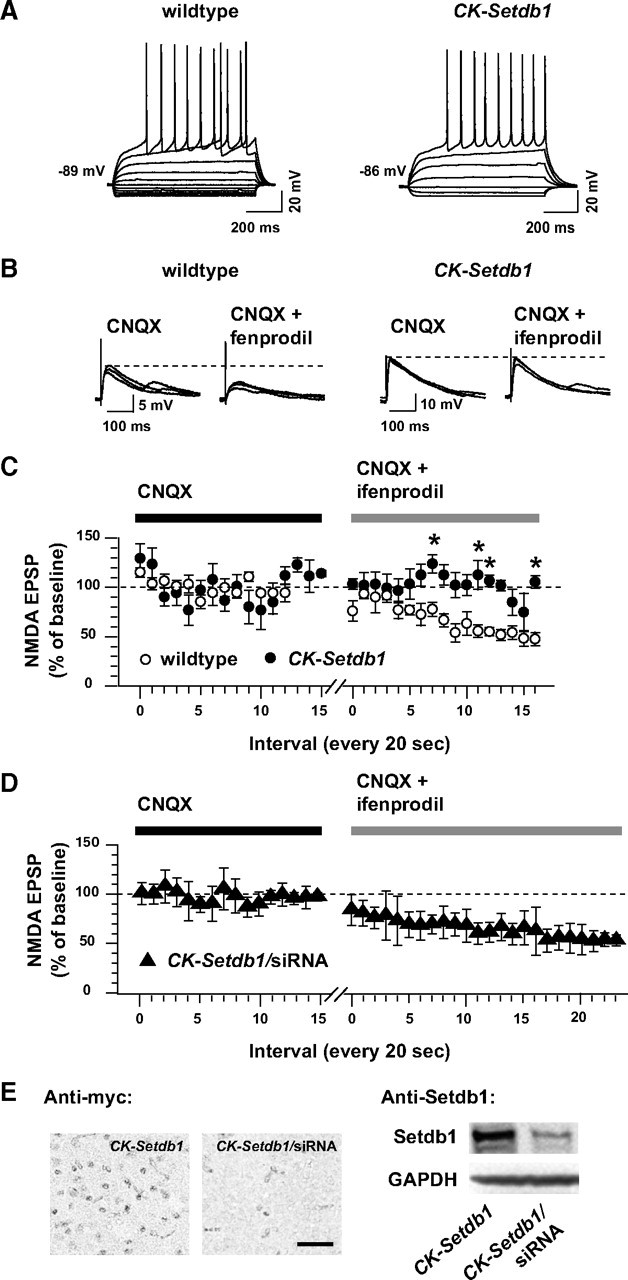
Ifenprodil-mediated inhibition of NMDA EPSPs in CK-Setdb1 and wild-type MSNs. A, Current–voltage (I–V) relationship of representative nucleus accumbens/ventral striatum neurons from wild-type and CK-Setdb1 mice recorded in whole-cell patch clamp in a slice of tissue. Notice the strong rectification in the hyperpolarizing range, the large action potential overshoot, presence of a ramp, and hyperpolarized resting potential (approximately −87 mV) that identify these neurons as MSNs. B, Representative NMDA EPSPs from wild-type and transgenic neurons, recorded at resting membrane potential before (10 μm CNQX) and during ifenprodil perfusion (10 μm CNQX + 100 μm ifenprodil). Notice that the NMDA-EPSP amplitude remained unchanged in CK-Setdb1 neuron after ifenprodil exposure. C, Mean amplitudes of NMDA EPSPs evoked every 20 s before (CNQX) and during ifenprodil exposure (CNQX + ifenprodil) in slices from wild-type (open circles) and CK-Setdb1 mice (filled circles). EPSPs were evoked in an Mg2+-free ACSF, and their amplitudes were expressed as a percentage of baseline (CNQX). Data (mean ± SEM) are shown for four transgenic and five wild-type neurons (animals). A fifth CK-Setdb1 neuron was removed as an outlier (Grubbs' test; two-way ANOVA, genotype, F (1,6) = 105.2; p < 0.001). *p < 0.05 (Newman–Keuls post hoc test). D, Same recording as in C but from CK-Setdb1 mice treated with Setdb1-siRNA (filled triangles). n = 4 neurons. Notice that in C, wild-type but not CK-Setdb1 neurons show reduced NMDA EPSPs from baseline (dotted line) in the presence of ifenprodil. Notice also that in D, siRNA treatment restores NMDA-EPSP sensitivity to ifenprodil in CK-Setdb1 neurons. E, Representative examples of anti-myc immunohistochemistry (left) and anti-Setdb1 immunoblots (right) from the ventral striatum of untreated and Setdb1-siRNA-treated CK-Setdb1 mice. Scale bar, 100 μm. Notice the robust decrease of myc and Setdb1 immunoreactivity after siRNA treatment. Data in D and E are from brains harvested 60 h after siRNA delivery into striatum.
Next, we wanted to explore whether the Setdb1-mediated downregulation of NR2B/Grin2b in striatum is reversible. We therefore repeated the neuronal recordings in the striatal slice preparation in CK-Setdb1 animals that were exposed to a single injection of Setdb1-siRNA 60 h before tissue harvest. Pilot studies in 3T3 cells identified a specific siRNA that mediated a strong downregulation of Setdb1 transcript (see Materials and Methods), and this siRNA mediated a robust decrease in striatal Setdb1 protein at 60 h after injection (Fig. 6 E). Indeed, the siRNA-mediated knockdown of Setdb1 in transgenic striatum was sufficient to restore the neuronal response to ifenprodil by reducing NMDA EPSPs ∼50% from baseline (Fig. 6 D), which is of similar magnitude when compared to the drug's reducing effects of NMDA EPSPs in wild-type striatum (Fig. 6 C). Based on this, we conclude that Setdb1's inhibitory effect on Grin2b-mediated neurotransmission is reversible.
Antidepressant-like behavioral changes in CK-Setdb1 mice
Next, we hypothesized that NR2B/Grin2b-regulated behaviors are altered in CK-Setdb1 mice. A partial decrease in Grin2b expression is thought to underlie some of the behavioral plasticity after repeated amphetamine exposure (Mao et al., 2009), and moreover, there is evidence that the antidepressant-like effects of nonspecific NMDA receptor antagonists in human and rodent are mimicked by drugs that selectively target the Grin2b subunit (Maeng et al., 2008; Preskorn et al., 2008). Importantly, in these models for (partial) Grin2b inhibition, general cognition and memory remained intact. In contrast, mice with a complete genetic ablation of Grin2b in hippocampus and other corticolimbic circuitry results show deficits in synaptic plasticity and learning and memory (von Engelhardt et al., 2008). Therefore, we predicted that Setdb1-mediated partial repression of Grin2b could alter the animal's motivational and affective states without necessarily compromising memory or general neurological functions. Indeed, CK-Setdb1 mice exhibited, compared to wild-type littermates, antidepressive phenotypes in behavioral paradigms for anhedonia (increased sucrose consumption over the course of 10 d without changes in total fluid intake in CK-Setdb1 line 1) (Fig. 7 A) and despair, as evidenced by decreased immobility in tail suspension and Porsolt swim tests (mean ± SEM, tail suspension: CK-Setdb1 (line 1), 134 ± 8 s; wild-type, 172 ± 16 s; forced swim: CK-Setdb1 (line 2), 145 ± 9 s; wild-type, 211 ± 12 s; n = 8–11/group; p < 0.05–0.01, Mann–Whitney test). Importantly, similar behavioral changes could be elicited in wild-type (C57BL/6J) mice treated with the NR2B selective antagonist “R-(R,S)-α-(4-hydroxyphenyl)-β-methyl-4-(phenylmethyl)-1-piperidine propranol (Ro25-6981; 5 mg/kg, i.p.) These include the aforementioned sucrose consumption paradigm after subchronic treatment (Fig. 8 A) and behavioral despair when measured 30 min after acute treatment (mean ± SEM, tail suspension: Ro25-6981, 76 ± 9.2 s; saline, 191 ± 9.4 s, n = 10/treatment; p < 0.01; Mann–Whitney test). A similar antidepressant-like phenotype was observed in C57BL6/J mice treated with bilateral daily injections, for 3 d, of Grin2b-siRNA into the hippocampus, which resulted in a robust, ∼50% decrease in NR2B/Grin2 protein (Fig. 8 B) (mean ± SEM, tail suspension: Grin2b-siRNA, 150 ± 20 s; vehicle, 188 ± 11 s; n = 6–7/group; p < 0.05 by Student's t test; p < 0.07 by Mann–Whitney test). To further confirm the antidepressive phenotype of our CK-Setdb1 animals, we applied the learned helplessness paradigm, in which the state of ”helplessness“ was measured by the escape latencies (in testing sessions when flight becomes possible) 1 or 7 d after 2 d of exposure to mild but inescapable repeated foot shocks (training sessions). One day after the end of training, both transgenic and littermate control mice showed escape latencies approaching, on average, 20 s (Fig. 7 B). However, 7 d after training, CK-Setdb1 but not their littermate controls showed a significant, >50% reduction in escape latencies as compared to day 1, indicating accelerated recovery from learned helplessness in the transgenic animals. The difference between genotypes in escape latencies on day 7 was significant (Fig. 7 B).
Figure 7.
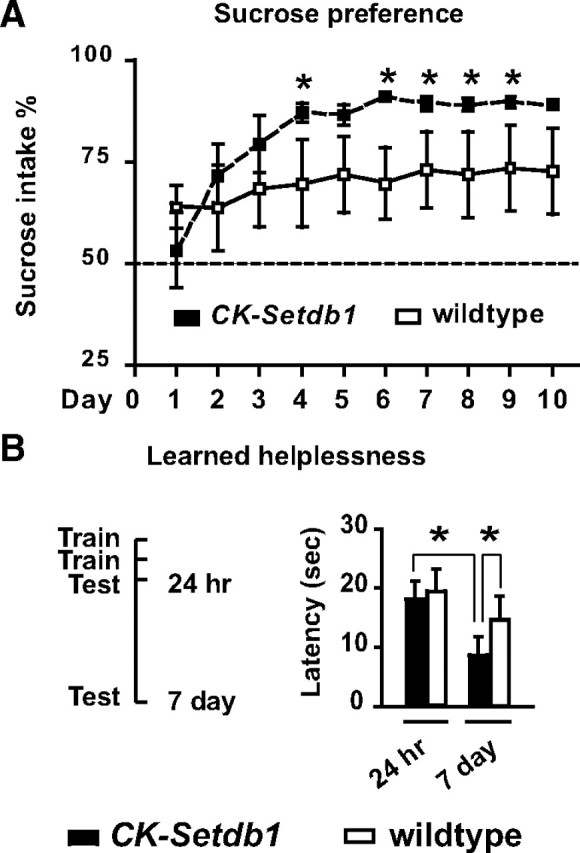
Antidepressive phenotypes in CK-Setdb1 mice. A, Sucrose consumption (transgenic line 1), expressed as a percentage of total fluid intake, in CK-Setdb1 and wild-type mice over the course of 10 d (n = 10–11/group; two-way ANOVA, day by genotype: F (9,162) = 2.16, p < 0.05; *p < 0.05, Newman–Keuls post hoc test). B, Accelerated extinction of learned helplessness (transgenic lines 1 and 2) in 2- to10-month-old male CK-Setdb1 mice compared to wild-type littermates. Left, Time line shows shock training and escape behavior testing. Right, Bar graph shows averaged escape latencies 24 h and 7 d after the end of training. Notice the significant reduction in escape latency in CK-Setdb1 mice on day 7; n = 8–9 per group. *p ≤ 0.05; Newman–Keuls post hoc after two-way ANOVA.
Figure 8.
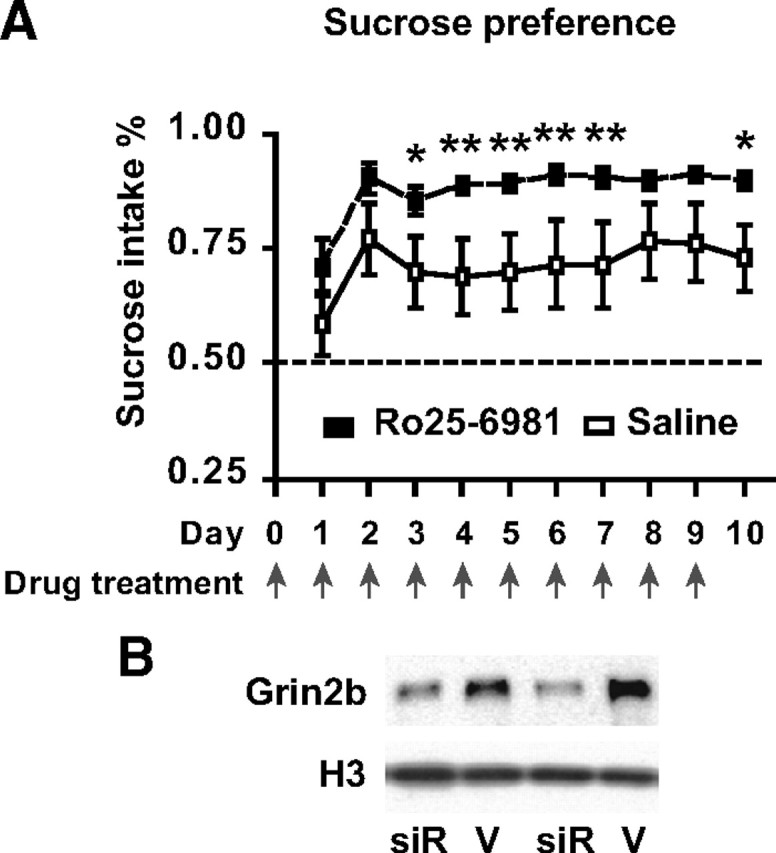
Antidepressant-like behaviors in wild-type (C57BL/6J) mice treated with NR2B selective antagonist Ro25-6981 or hippocampal knockdown of NR2B/Grin2b. A, Sucrose consumption in mice treated with Ro25-6981 or saline once daily for 10 d. First dose was given at day 0 (n = 10/treatment; two-way ANOVA: day, F (9,153) = 6.38, p < 0.01; treatment, F (1,17) = 4.28, p < 0.05; *p < 0.05, **p < 0.01, Newman–Keuls post hoc test). B, Representative hippocampal immunoblots showing NR2B/Grin2b and histone H3 (pan) immunoreactivity in animals exposed to Grin2b-siRNA (siR) or vehicle (V).
Of note, the decrease in despair-related immobility and increased sucrose consumption in the Setdb1 transgenic mice is comparable to similar behavioral changes observed in wild-type mice exposed to acute or chronic treatment with conventional antidepressants (Berton and Nestler, 2006), suggesting that upregulation of Setdb1 in neurons mediates an antidepressant-like behavioral effect. This phenotype, which was observed in two independent transgenic lines, was not associated with a more generalized alteration in the retention of foot shock-associated memories (as measured in passive avoidance and fear conditioning paradigms) (Fig. 9 B,C), or a deficit in shock sensitivity or perception (Fig. 9 A). Furthermore, the maintenance of body weight, novelty-induced locomotion activity, and anxiety-related behaviors remained unaltered in the transgenic animals, and only a subtle decrease (<7% from controls) was observed in rotarod performance (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). These findings indicate that the observed antidepressant-like phenotype in the CK-Setdb1 mice is not explained by a generalized alteration in neurological functions.
Figure 9.
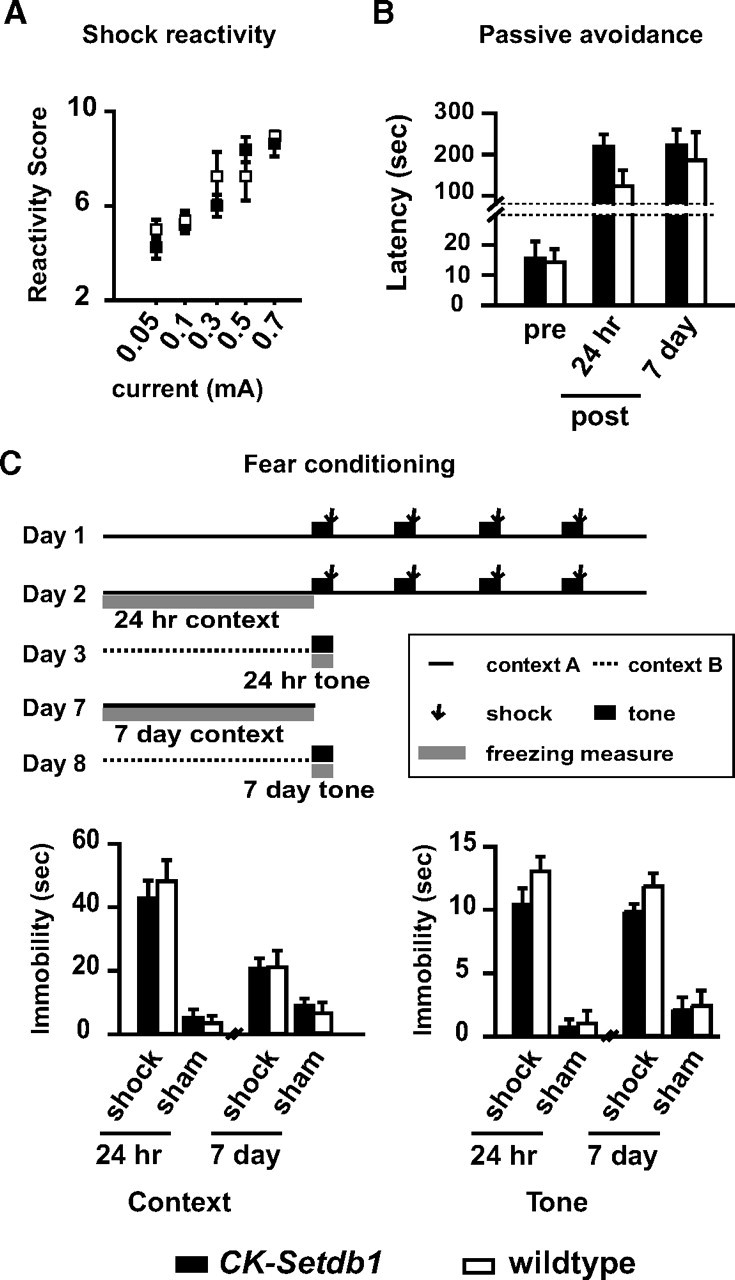
Shock-associated reactivity and memory are preserved in CK-Setdb1 mice. A, Scatter plot shows shock reactivity scores (flinching, vocalization, running, and jumping) in CK-Setdb1 and littermate control mice exposed to an escalating current (0.05–0.7 mA, 2 s each) as indicated. B, Passive avoidance. The bar graph shows the latency (seconds) to enter dim chamber before (pre) and 24 h or 7 d after (post) single foot shock. In A and B, n = 8–11/genotype. C, Fear conditioning. Top, Experimental design, including time lines for context and tone. Bottom, Bar graphs show context- (left) and tone-induced (right) total immobility/freezing in CK-Setdb1 and wild-type littermates over the course of 180 and 20 s for context and tone, respectively, 24 h and 7 d after shock or sham exposure, as indicated. Notice there are no significant differences between transgenic and control mice. All data are mean ± SEM.
Discussion
The present study demonstrates that increased expression and activity of Setdb1 histone methyltransferase in forebrain neurons is associated with an antidepressant-like phenotype in behavioral paradigms related to anhedonia, despair, and helplessness. These behavioral effects occurred in the context of a limited genomic occupancy of Setdb1 in neuronal chromatin, which was confined to a very small set (<1%) of genes and, unexpectedly, included the two NMDA receptor subunits, NR2A/Grin2a and NR2B/Grin2b. Although expression of Grin2a and overall NMDA receptor function was preserved in CK-Setdb1 mice, these animals were affected by a partial, 20–50% decrease in Grin2b levels, which resulted in altered NMDA desensitization kinetics and insensitivity to the effect of NR2B-selective antagonist drugs. Therefore, these studies provide the first evidence that a member of the H3K9 HMT family of molecules regulates specific motivational and affective behaviors, and the expression of a neurotransmitter receptor system that is of crucial importance for neuronal signaling and plasticity.
Presently, the molecular mechanisms mediating the selectivity of Setdb1 target sites in neuronal chromatin, including the unexpected preference for multiple ionotropic glutamate receptor subunits, remain unknown. However, these observations from mouse forebrain resonate with recent reports in the fruit fly. Of note, the Drosophila ortholog of the mammalian Setdb1, Egg/dSetdb1, preferentially binds to the fourth chromosome, a small structure comprised of 4.2 Mb that harbors only 80 genes, including the fly's sole metabotropic glutamate receptor gene (Seum et al., 2007; Tzeng et al., 2007; Riddle et al., 2009). This affinity of Egg/dSetdb1 could be explained by the high density of repeats on the fly's fourth chromosome, which is five times higher than on the other autosomes (Riddle et al., 2009). The affinity of Setdb1 for repeat sequences was also apparent in the present study, as evidenced by increased H3K9 methylation in pericentric heterochromatin of transgenic animals (Fig. 1 F), and it is noteworthy that some of the Setdb1-binding sites on mouse chromosomes 6, 8, and 16, including the Grin2b target, are positioned within large introns and flanked by repeat sequences (Fig. 3 A). It is unlikely, however, that repeat density is the sole determinant governing Setdb1's unusual occupancy pattern in neuronal chromatin, and a mechanistic understanding of this apparently highly regulated process awaits further investigations. Based on our 3C and Setdb1-ChIP experiments at the Grin2b locus, Setdb1 is found in an intronic chromatin segment involved in a loop formation via interaction with the Grin2b promoter (Fig. 4). Whether or not these higher-order chromatin structures at Grin2b are representative for other Setdb1 target sites in the genome remains to be clarified. It is likely that chromatin loopings at Grin2b and many other genomic loci play a critical role for neuronal function. Although the findings reported here represent some of the first studies on three-dimensional chromatin architecture for a glutamate receptor gene, the overall importance of higher order chromatin is best illustrated by the fact that simple nucleosome/core histone templates compact DNA about sevenfold, which is more than two orders of magnitude below the level of genome compaction observed in a vertebrate cell nucleus (Ho and Crabtree, 2010).
One attractive model (Fig. 4 C) would be that, via the loop, Setdb1-enriched chromatin associates with the Grin2b promoter through an interaction with its binding partner, the KAP-1 (or TRIM28/TIF1b/KRIP1) transcriptional corepressor (Ayyanathan et al., 2003). Interestingly, mice with forebrain-specific ablation of KAP-1 exhibit elevated levels of anxiety and stress-related impairments in memory tasks (Jakobsson et al., 2008). Therefore, the broadly opposing patterns of behavioral changes after KAP-1 loss of function (Jakobsson et al., 2008) and Setdb1 gain of function (present study), which were not associated with generalized neurological defects in these different mouse models, further highlight the importance of the KAP-1-Setdb1 repressor complex for the regulation of affective behaviors.
Among the Setdb1-regulated genes likely to mediate the mood-related effect of this HMT, the NMDA receptor subunit NR2B/Grin2b could play a prominent role. First, GRIN2B carries strong genetic risk for bipolar disorder in the Ashkenazi (Fallin et al., 2005; Avramopoulos et al., 2007). Second, in the present study, behavioral changes in wild-type mice treated with an NR2B antagonist, or with siRNA-mediated NR2B knockdown in hippocampus, were similar to those observed in (drug-naive) transgenic Setdb1 animals. The findings presented here are also in good agreement with the recent reports on antidepressant-like effects of NR2B antagonists in rodent and human (Maeng et al., 2008; Preskorn et al., 2008). Although these and other NMDA-related drug studies (Berman et al., 2000; Zarate et al., 2006; Salvadore et al., 2009) are extremely promising, the side-effect profile of NMDA receptor antagonists, including confusion and impaired cognition and memory, prohibits wider clinical applications. Based on the findings presented here, upregulation of Setdb1 methyltransferase activity may provide an alternative antidepressant strategy, because Setdb1-mediated partial downregulation of NR2B/Grin2b expression is much less likely to be detrimental for neuronal functions compared to NMDA antagonist drug treatment (Darrah et al., 2008) or the complete genetic ablation of the Grin2b gene, which results in significant memory deficits (von Engelhardt et al., 2008). Our finding that Grin2b expression in human glioma cells decreased after upregulation of Setdb1 occupancy within this gene, in conjunction with a previous study reporting that histone methylation levels at NMDA receptor and many other glutamate receptor genes are subject to developmental and region-specific regulation in the human brain (Stadler et al., 2005), further speaks to the therapeutic potential of Setdb1 and other HMT-mediated chromatin remodeling mechanisms.
The mechanism(s) through which NR2B/Grin2b downregulation results in antidepressant action are still unclear. It has been proposed that enhancing non-NMDA, specifically AMPA, relative to NMDA ionotropic glutamate receptor signaling could underly the therapeutic benefits of NMDA antagonists (Maeng et al., 2008). In the CK-Setdb1 mice of the present study, overall NMDA receptor density and signaling was maintained at normal levels (Fig. 5 C). Instead, a shift in NMDA receptor subunit composition caused by a selective decrease in NR2B/Grin2b expression (Fig. 5 D) could explain the change in depression related behaviors. In support of this hypothesis, alterations in NR2A/NR2B ratios strongly affect synaptic plasticity and circuitry formation in the developing cerebral cortex (Philpot et al., 2007; Zhao and Constantine-Paton, 2007; Cho et al., 2009).
Furthermore, it is very likely that the Setdb1-mediated antidepressant-like phenotype reported here involves additional genes that are not directly related to the NMDA receptor system. For example, Gpm6a is (like Grin2b and Grin2a) among the list of 29 Setdb1 gene targets on chromosomes 6, 8, and 16 (Table 1). Gpm6a encodes a glycoprotein on neuronal membranes, and genetic polymorphisms within GPM6A confer a significant risk for depression in subjects with psychosis (Boks et al., 2008). A more comprehensive assessment of Setdb1 target genes will require chromatin profiling across all murine chromosomes.
It is remarkable that upregulation of histone acetylation and of H3K9 methylation, two types of histone modifications enriched in different portions of the genome that either define open (acetylation) or, in case of the trimethylated H3K9, repressed and silenced chromatin (Berger, 2007; Wang et al., 2008; Roth and Sweatt, 2009), both result in antidepressant-like phenotypes. Of note, >40% of affected individuals show an incomplete response to conventional antidepressants (Berton and Nestler, 2006), and therefore drugs acting as specific Setdb1 activators and other epigenetic regulators of gene expression, including class I/II histone deacetylase inhibitors (Tsankova et al., 2006; Duman and Newton, 2007; Schroeder et al., 2007; Covington et al., 2009; Grayson et al., 2009; Gundersen and Blendy, 2009; Hobara et al., 2009) could potentially benefit a substantial portion of these hitherto treatment-resistant cases. Of note, chronic restraint stress (a preclinical model for depression) induces the downregulation of the H3K9me3 mark in the dentate gyrus of the hippocampus, and this can be reversed by treatment with a prototype antidepressant and serotonin reuptake inhibitor, fluoxetine (Hunter et al., 2009). Finally, it will be interesting to explore whether interference with the orderly regulation of other types of histone modifications, including arginine methylation, serine phosporylation, and lysine ubiquitinylation and SUMOylation (Berger, 2007; Roth and Sweatt, 2009), induces changes in the brain's affective and motivational states. It is very likely that the rich cache of epigenetic regulators expressed in the brain will include chromatin-modifying proteins that could provide promising targets for novel antidepressant treatment strategies.
Footnotes
This work was supported by grants from the National Institutes of Health. We thank Yin Guo, Anouch Matevossian, and Catheryne Whittle for excellent technical assistance, Dr. Steve Jones and Staff from the University of Massachusetts Medical School (UMMS) Transgenic Core Facility, Dr. Ellen Kittler and Maria Zapp from the UMMS Deep Sequencing Core, Dr. Liu Yang for providing Setdb1 cDNA, Dr. Paul Gardner for providing U87-MG cells, Dr. Andrew Tapper for advice with regard to behavioral studies, and Dr. Job Dekker and Dr. Nynke Van Berkum for expert input on chromosome conformation capture studies.
References
- Allen NC, Bagade S, McQueen MB, Ioannidis JP, Kavvoura FK, Khoury MJ, Tanzi RE, Bertram L. Systematic meta-analyses and field synopsis of genetic association studies in schizophrenia: the SzGene database. Nat Genet. 2008;40:827–834. doi: 10.1038/ng.171. [DOI] [PubMed] [Google Scholar]
- Aonurm-Helm A, Jurgenson M, Zharkovsky T, Sonn K, Berezin V, Bock E, Zharkovsky A. Depression-like behaviour in neural cell adhesion molecule (NCAM)-deficient mice and its reversal by an NCAM-derived peptide, FGL. Eur J Neurosci. 2008;28:1618–1628. doi: 10.1111/j.1460-9568.2008.06471.x. [DOI] [PubMed] [Google Scholar]
- Avramopoulos D, Lasseter VK, Fallin MD, Wolyniec PS, McGrath JA, Nestadt G, Valle D, Pulver AE. Stage II follow-up on a linkage scan for bipolar disorder in the Ashkenazim provides suggestive evidence for chromosome 12p and the GRIN2B gene. Genet Med. 2007;9:745–751. doi: 10.1097/gim.0b013e318159a37c. [DOI] [PubMed] [Google Scholar]
- Ayyanathan K, Lechner MS, Bell P, Maul GG, Schultz DC, Yamada Y, Tanaka K, Torigoe K, Rauscher FJ., 3rd Regulated recruitment of HP1 to a euchromatic gene induces mitotically heritable, epigenetic gene silencing: a mammalian cell culture model of gene variegation. Genes Dev. 2003;17:1855–1869. doi: 10.1101/gad.1102803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–412. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, Krystal JH. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47:351–354. doi: 10.1016/s0006-3223(99)00230-9. [DOI] [PubMed] [Google Scholar]
- Berton O, Nestler EJ. New approaches to antidepressant drug discovery: beyond monoamines. Nat Rev Neurosci. 2006;7:137–151. doi: 10.1038/nrn1846. [DOI] [PubMed] [Google Scholar]
- Boks MP, Hoogendoorn M, Jungerius BJ, Bakker SC, Sommer IE, Sinke RJ, Ophoff RA, Kahn RS. Do mood symptoms subdivide the schizophrenia phenotype? Association of the GMP6A gene with a depression subgroup. Am J Med Genet B Neuropsychiatr Genet. 2008;147B:707–711. doi: 10.1002/ajmg.b.30667. [DOI] [PubMed] [Google Scholar]
- Borrelli E, Nestler EJ, Allis CD, Sassone-Corsi P. Decoding the epigenetic language of neuronal plasticity. Neuron. 2008;60:961–974. doi: 10.1016/j.neuron.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brami-Cherrier K, Valjent E, Herve D, Darragh J, Corvol JC, Pages C, Arthur SJ, Girault JA, Caboche J. Parsing molecular and behavioral effects of cocaine in mitogen- and stress-activated protein kinase-1-deficient mice. J Neurosci. 2005;25:11444–11454. doi: 10.1523/JNEUROSCI.1711-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldarone BJ, Harrist A, Cleary MA, Beech RD, King SL, Picciotto MR. High-affinity nicotinic acetylcholine receptors are required for antidepressant effects of amitriptyline on behavior and hippocampal cell proliferation. Biol Psychiatry. 2004;56:657–664. doi: 10.1016/j.biopsych.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet. 2009;10:295–304. doi: 10.1038/nrg2540. [DOI] [PubMed] [Google Scholar]
- Cho KK, Khibnik L, Philpot BD, Bear MF. The ratio of NR2A/B NMDA receptor subunits determines the qualities of ocular dominance plasticity in visual cortex. Proc Natl Acad Sci U S A. 2009;106:5377–5382. doi: 10.1073/pnas.0808104106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi T, Huang M, Gorman C, Jaenisch R. A generic intron increases gene expression in transgenic mice. Mol Cell Biol. 1991;11:3070–3074. doi: 10.1128/mcb.11.6.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covington HE, 3rd, Maze I, LaPlant QC, Vialou VF, Ohnishi YN, Berton O, Fass DM, Renthal W, Rush AJ, 3rd, Wu EY, Ghose S, Krishnan V, Russo SJ, Tamminga C, Haggarty SJ, Nestler EJ. Antidepressant actions of histone deacetylase inhibitors. J Neurosci. 2009;29:11451–11460. doi: 10.1523/JNEUROSCI.1758-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crepaldi L, Riccio A. Chromatin learns to behave. Epigenetics. 2009;4:23–26. doi: 10.4161/epi.4.1.7604. [DOI] [PubMed] [Google Scholar]
- Darrah JM, Stefani MR, Moghaddam B. Interaction of N-methyl-d-aspartate and group 5 metabotropic glutamate receptors on behavioral flexibility using a novel operant set-shift paradigm. Behav Pharmacol. 2008;19:225–234. doi: 10.1097/FBP.0b013e3282feb0ac. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker J. The three ‘C’s of chromosome conformation capture: controls, controls, controls. Nat Methods. 2006;3:17–21. doi: 10.1038/nmeth823. [DOI] [PubMed] [Google Scholar]
- Dekker J. Gene regulation in the third dimension. Science. 2008;319:1793–1794. doi: 10.1126/science.1152850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duman RS, Newton SS. Epigenetic marking and neuronal plasticity. Biol Psychiatry. 2007;62:1–3. doi: 10.1016/j.biopsych.2007.04.037. [DOI] [PubMed] [Google Scholar]
- Fallin MD, Lasseter VK, Avramopoulos D, Nicodemus KK, Wolyniec PS, McGrath JA, Steel G, Nestadt G, Liang KY, Huganir RL, Valle D, Pulver AE. Bipolar I disorder and schizophrenia: a 440-single-nucleotide polymorphism screen of 64 candidate genes among Ashkenazi Jewish case-parent trios. Am J Hum Genet. 2005;77:918–936. doi: 10.1086/497703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A, Sananbenesi F, Wang X, Dobbin M, Tsai LH. Recovery of learning and memory is associated with chromatin remodelling. Nature. 2007;447:178–182. doi: 10.1038/nature05772. [DOI] [PubMed] [Google Scholar]
- Franklin KB, Paxinos G. Sydney: Academic; 2001. The mouse brain in stereotaxic coordinates. [Google Scholar]
- Fritsch L, Robin P, Mathieu JR, Souidi M, Hinaux H, Rougeulle C, Harel-Bellan A, Ameyar-Zazoua M, Ait-Si-Ali S. A subset of the histone H3 lysine 9 methyltransferases Suv39h1, G9a, GLP, and SETDB1 participate in a multimeric complex. Mol Cell. 2010;37:46–56. doi: 10.1016/j.molcel.2009.12.017. [DOI] [PubMed] [Google Scholar]
- Grayson DR, Kundakovic M, Sharma RP. Is there a future for HDAC inhibitors in the pharmacotherapy of psychiatric disorders? Mol Pharmacol. 2009;77:126–135. doi: 10.1124/mol.109.061333. [DOI] [PubMed] [Google Scholar]
- Guidotti A, Dong E, Kundakovic M, Satta R, Grayson DR, Costa E. Characterization of the action of antipsychotic subtypes on valproate-induced chromatin remodeling. Trends Pharmacol Sci. 2009;30:55–60. doi: 10.1016/j.tips.2008.10.010. [DOI] [PubMed] [Google Scholar]
- Gundersen BB, Blendy JA. Effects of the histone deacetylase inhibitor sodium butyrate in models of depression and anxiety. Neuropharmacology. 2009;57:67–74. doi: 10.1016/j.neuropharm.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Kim SY, Artis S, Molfese DL, Schumacher A, Sweatt JD, Paylor RE, Lubin FD. Histone methylation regulates memory formation. J Neurosci. 2010;30:3589–3599. doi: 10.1523/JNEUROSCI.3732-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho L, Crabtree GR. Chromatin remodelling during development. Nature. 2010;463:474–484. doi: 10.1038/nature08911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobara T, Uchida S, Otsuki K, Matsubara T, Funato H, Matsuo K, Suetsugi M, Watanabe Y. Altered gene expression of histone deacetylases in mood disorder patients. J Psychiatr Res. 2009;44:263–270. doi: 10.1016/j.jpsychires.2009.08.015. [DOI] [PubMed] [Google Scholar]
- Huang HS, Matevossian A, Jiang Y, Akbarian S. Chromatin immunoprecipitation in postmortem brain. J Neurosci Methods. 2006;156:284–292. doi: 10.1016/j.jneumeth.2006.02.018. [DOI] [PubMed] [Google Scholar]
- Huang HS, Matevossian A, Whittle C, Kim SY, Schumacher A, Baker SP, Akbarian S. Prefrontal dysfunction in schizophrenia involves mixed-lineage leukemia 1-regulated histone methylation at GABAergic gene promoters. J Neurosci. 2007;27:11254–11262. doi: 10.1523/JNEUROSCI.3272-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Doherty JJ, Dingledine R. Altered histone acetylation at glutamate receptor 2 and brain-derived neurotrophic factor genes is an early event triggered by status epilepticus. J Neurosci. 2002;22:8422–8428. doi: 10.1523/JNEUROSCI.22-19-08422.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter RG, McCarthy KJ, Milne TA, Pfaff DW, McEwen BS. Regulation of hippocampal H3 histone methylation by acute and chronic stress. Proc Natl Acad Sci U S A. 2009;49:20912–20917. doi: 10.1073/pnas.0911143106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsson J, Cordero MI, Bisaz R, Groner AC, Busskamp V, Bensadoun JC, Cammas F, Losson R, Mansuy IM, Sandi C, Trono D. KAP1-mediated epigenetic repression in the forebrain modulates behavioral vulnerability to stress. Neuron. 2008;60:818–831. doi: 10.1016/j.neuron.2008.09.036. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Langley B, Lubin FD, Renthal W, Wood MA, Yasui DH, Kumar A, Nestler EJ, Akbarian S, Beckel-Mitchener AC. Epigenetics in the nervous system. J Neurosci. 2008a;28:11753–11759. doi: 10.1523/JNEUROSCI.3797-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Matevossian A, Huang HS, Straubhaar J, Akbarian S. Isolation of neuronal chromatin from brain tissue. BMC Neurosci. 2008b;9:42. doi: 10.1186/1471-2202-9-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson WE, Li W, Meyer CA, Gottardo R, Carroll JS, Brown M, Liu XS. Model-based analysis of tiling-arrays for ChIP-chip. Proc Natl Acad Sci U S A. 2006;103:12457–12462. doi: 10.1073/pnas.0601180103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleefstra T, Brunner HG, Amiel J, Oudakker AR, Nillesen WM, Magee A, Genevieve D, Cormier-Daire V, van Esch H, Fryns JP, Hamel BC, Sistermans EA, de Vries BB, van Bokhoven H. Loss-of-function mutations in euchromatin histone methyl transferase 1 (EHMT1) cause the 9q34 subtelomeric deletion syndrome. Am J Hum Genet. 2006;79:370–377. doi: 10.1086/505693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Laurie DJ, Seeburg PH. Ligand affinities at recombinant N-methyl-d-aspartate receptors depend on subunit composition. Eur J Pharmacol. 1994;268:335–345. doi: 10.1016/0922-4106(94)90058-2. [DOI] [PubMed] [Google Scholar]
- Lee J, Hagerty S, Cormier KA, Kim J, Kung AL, Ferrante RJ, Ryu H. Monoallele deletion of CBP leads to pericentromeric heterochromatin condensation through ESET expression and histone H3 (K9) methylation. Hum Mol Genet. 2008;17:1774–1782. doi: 10.1093/hmg/ddn067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, Chen L, Chen L, Chen TM, Chin MC, Chong J, Crook BE, Czaplinska A, Dang CN, Datta S, Dee NR, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- Li H, Rauch T, Chen ZX, Szabo PE, Riggs AD, Pfeifer GP. The histone methyltransferase SETDB1 and the DNA methyltransferase DNMT3A interact directly and localize to promoters silenced in cancer cells. J Biol Chem. 2006;281:19489–19500. doi: 10.1074/jbc.M513249200. [DOI] [PubMed] [Google Scholar]
- Loyola A, Tagami H, Bonaldi T, Roche D, Quivy JP, Imhof A, Nakatani Y, Dent SY, Almouzni G. The HP1alpha-CAF1-SetDB1-containing complex provides H3K9me1 for Suv39-mediated K9me3 in pericentric heterochromatin. EMBO Rep. 2009;10:769–775. doi: 10.1038/embor.2009.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubin FD, Sweatt JD. The IkappaB kinase regulates chromatin structure during reconsolidation of conditioned fear memories. Neuron. 2007;55:942–957. doi: 10.1016/j.neuron.2007.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeng S, Zarate CA, Jr, Du J, Schloesser RJ, McCammon J, Chen G, Manji HK. Cellular mechanisms underlying the antidepressant effects of ketamine: role of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. Biol Psychiatry. 2008;63:349–352. doi: 10.1016/j.biopsych.2007.05.028. [DOI] [PubMed] [Google Scholar]
- Mao LM, Wang W, Chu XP, Zhang GC, Liu XY, Yang YJ, Haines M, Papasian CJ, Fibuch EE, Buch S, Chen JG, Wang JQ. Stability of surface NMDA receptors controls synaptic and behavioral adaptations to amphetamine. Nat Neurosci. 2009;12:602–610. doi: 10.1038/nn.2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens JH, O'Sullivan RJ, Braunschweig U, Opravil S, Radolf M, Steinlein P, Jenuwein T. The profile of repeat-associated histone lysine methylation states in the mouse epigenome. EMBO J. 2005;24:800–812. doi: 10.1038/sj.emboj.7600545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin G, Siggins GR. Electrophysiological evidence for expression of glycine receptors in freshly isolated neurons from nucleus accumbens. J Pharmacol Exp Ther. 2002;302:1135–1145. doi: 10.1124/jpet.102.033399. [DOI] [PubMed] [Google Scholar]
- Martin G, Nie Z, Siggins GR. mu-Opioid receptors modulate NMDA receptor-mediated responses in nucleus accumbens neurons. J Neurosci. 1997;17:11–22. doi: 10.1523/JNEUROSCI.17-01-00011.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayford M, Bach ME, Huang YY, Wang L, Hawkins RD, Kandel ER. Control of memory formation through regulated expression of a CaMKII transgene. Science. 1996;274:1678–1683. doi: 10.1126/science.274.5293.1678. [DOI] [PubMed] [Google Scholar]
- Miele A, Dekker J. Mapping cis- and trans- chromatin interaction networks using chromosome conformation capture (3C) Methods Mol Biol. 2009;464:105–121. doi: 10.1007/978-1-60327-461-6_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miele A, Gheldof N, Tabuchi TM, Dostie J, Dekker J. Mapping chromatin interactions by chromosome conformation capture. Curr Protoc Mol Biol. 2006 doi: 10.1002/0471142727.mb2111s74. Chapter 21, Unit 21.11. [DOI] [PubMed] [Google Scholar]
- Mukai J, Dhilla A, Drew LJ, Stark KL, Cao L, MacDermott AB, Karayiorgou M, Gogos JA. Palmitoylation-dependent neurodevelopmental deficits in a mouse model of 22q11 microdeletion. Nat Neurosci. 2008;11:1302–1310. doi: 10.1038/nn.2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira AM, Wood MA, McDonough CB, Abel T. Transgenic mice expressing an inhibitory truncated form of p300 exhibit long-term memory deficits. Learn Mem. 2007;14:564–572. doi: 10.1101/lm.656907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey SC, Ugale R, Zhang H, Tang L, Prakash A. Brain chromatin remodeling: a novel mechanism of alcoholism. J Neurosci. 2008;28:3729–3737. doi: 10.1523/JNEUROSCI.5731-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philpot BD, Cho KK, Bear MF. Obligatory role of NR2A for metaplasticity in visual cortex. Neuron. 2007;53:495–502. doi: 10.1016/j.neuron.2007.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preskorn SH, Baker B, Kolluri S, Menniti FS, Krams M, Landen JW. An innovative design to establish proof of concept of the antidepressant effects of the NR2B subunit selective N-methyl-d-aspartate antagonist, CP-101,606, in patients with treatment-refractory major depressive disorder. J Clin Psychopharmacol. 2008;28:631–637. doi: 10.1097/JCP.0b013e31818a6cea. [DOI] [PubMed] [Google Scholar]
- Reijmers LG, Coats JK, Pletcher MT, Wiltshire T, Tarantino LM, Mayford M. A mutant mouse with a highly specific contextual fear-conditioning deficit found in an N-ethyl-N-nitrosourea (ENU) mutagenesis screen. Learn Mem. 2006;13:143–149. doi: 10.1101/lm.98606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren B, Robert F, Wyrick JJ, Aparicio O, Jennings EG, Simon I, Zeitlinger J, Schreiber J, Hannett N, Kanin E, Volkert TL, Wilson CJ, Bell SP, Young RA. Genome-wide location and function of DNA binding proteins. Science. 2000;290:2306–2309. doi: 10.1126/science.290.5500.2306. [DOI] [PubMed] [Google Scholar]
- Renthal W, Carle TL, Maze I, Covington HE, 3rd, Truong HT, Alibhai I, Kumar A, Montgomery RL, Olson EN, Nestler EJ. Delta FosB mediates epigenetic desensitization of the c-fos gene after chronic amphetamine exposure. J Neurosci. 2008;28:7344–7349. doi: 10.1523/JNEUROSCI.1043-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddle NC, Shaffer CD, Elgin SC. A lot about a little dot—lessons learned from Drosophila melanogaster chromosome 4. Biochem Cell Biol. 2009;87:229–241. doi: 10.1139/o08-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth TL, Sweatt JD. Regulation of chromatin structure in memory formation. Curr Opin Neurobiol. 2009;19:336–342. doi: 10.1016/j.conb.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvadore G, Cornwell BR, Colon-Rosario V, Coppola R, Grillon C, Zarate CA, Jr, Manji HK. Increased anterior cingulate cortical activity in response to fearful faces: a neurophysiological biomarker that predicts rapid antidepressant response to ketamine. Biol Psychiatry. 2009;65:289–295. doi: 10.1016/j.biopsych.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramm NL, McDonald MP, Limbird LE. The alpha(2a)-adrenergic receptor plays a protective role in mouse behavioral models of depression and anxiety. J Neurosci. 2001;21:4875–4882. doi: 10.1523/JNEUROSCI.21-13-04875.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder FA, Lin CL, Crusio WE, Akbarian S. Antidepressant-like effects of the histone deacetylase inhibitor, sodium butyrate, in the mouse. Biol Psychiatry. 2007;62:55–64. doi: 10.1016/j.biopsych.2006.06.036. [DOI] [PubMed] [Google Scholar]
- Schultz DC, Ayyanathan K, Negorev D, Maul GG, Rauscher FJ., 3rd SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 2002;16:919–932. doi: 10.1101/gad.973302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seum C, Reo E, Peng H, Rauscher FJ, 3rd, Spierer P, Bontron S. Drosophila SETDB1 is required for chromosome 4 silencing. PLoS Genet. 2007;3:e76. doi: 10.1371/journal.pgen.0030076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahbazian M, Young J, Yuva-Paylor L, Spencer C, Antalffy B, Noebels J, Armstrong D, Paylor R, Zoghbi H. Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron. 2002;35:243–254. doi: 10.1016/s0896-6273(02)00768-7. [DOI] [PubMed] [Google Scholar]
- Shilatifard A. Molecular implementation and physiological roles for histone H3 lysine 4 (H3K4) methylation. Curr Opin Cell Biol. 2008;20:341–348. doi: 10.1016/j.ceb.2008.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler F, Kolb G, Rubusch L, Baker SP, Jones EG, Akbarian S. Histone methylation at gene promoters is associated with developmental regulation and region-specific expression of ionotropic and metabotropic glutamate receptors in human brain. J Neurochem. 2005;94:324–336. doi: 10.1111/j.1471-4159.2005.03190.x. [DOI] [PubMed] [Google Scholar]
- Stepulak A, Luksch H, Gebhardt C, Uckermann O, Marzahn J, Sifringer M, Rzeski W, Staufner C, Brocke KS, Turski L, Ikonomidou C. Expression of glutamate receptor subunits in human cancers. Histochem Cell Biol. 2009 doi: 10.1007/s00418-009-0613-1. [DOI] [PubMed] [Google Scholar]
- Tachibana M, Matsumura Y, Fukuda M, Kimura H, Shinkai Y. G9a/GLP complexes independently mediate H3K9 and DNA methylation to silence transcription. EMBO J. 2008;27:2681–2690. doi: 10.1038/emboj.2008.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thatcher KN, LaSalle JM. Dynamic changes in Histone H3 lysine 9 acetylation localization patterns during neuronal maturation require MeCP2. Epigenetics. 2006;1:24–31. doi: 10.4161/epi.1.1.2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas CG, Miller AJ, Westbrook GL. Synaptic and extrasynaptic NMDA receptor NR2 subunits in cultured hippocampal neurons. J Neurophysiol. 2006;95:1727–1734. doi: 10.1152/jn.00771.2005. [DOI] [PubMed] [Google Scholar]
- Tovar KR, Sprouffske K, Westbrook GL. Fast NMDA receptor-mediated synaptic currents in neurons from mice lacking the epsilon2 (NR2B) subunit. J Neurophysiol. 2000;83:616–620. doi: 10.1152/jn.2000.83.1.616. [DOI] [PubMed] [Google Scholar]
- Tsankova NM, Berton O, Renthal W, Kumar A, Neve RL, Nestler EJ. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci. 2006;9:519–525. doi: 10.1038/nn1659. [DOI] [PubMed] [Google Scholar]
- Tzeng TY, Lee CH, Chan LW, Shen CK. Epigenetic regulation of the Drosophila chromosome 4 by the histone H3K9 methyltransferase dSETDB1. Proc Natl Acad Sci U S A. 2007;104:12691–12696. doi: 10.1073/pnas.0705534104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vecsey CG, Hawk JD, Lattal KM, Stein JM, Fabian SA, Attner MA, Cabrera SM, McDonough CB, Brindle PK, Abel T, Wood MA. Histone deacetylase inhibitors enhance memory and synaptic plasticity via CREB:CBP-dependent transcriptional activation. J Neurosci. 2007;27:6128–6140. doi: 10.1523/JNEUROSCI.0296-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicini S, Wang JF, Li JH, Zhu WJ, Wang YH, Luo JH, Wolfe BB, Grayson DR. Functional and pharmacological differences between recombinant N-methyl-d-aspartate receptors. J Neurophysiol. 1998;79:555–566. doi: 10.1152/jn.1998.79.2.555. [DOI] [PubMed] [Google Scholar]
- von Engelhardt J, Doganci B, Jensen V, Hvalby O, Gongrich C, Taylor A, Barkus C, Sanderson DJ, Rawlins JN, Seeburg PH, Bannerman DM, Monyer H. Contribution of hippocampal and extra-hippocampal NR2B-containing NMDA receptors to performance on spatial learning tasks. Neuron. 2008;60:846–860. doi: 10.1016/j.neuron.2008.09.039. [DOI] [PubMed] [Google Scholar]
- Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, Cui K, Roh TY, Peng W, Zhang MQ, Zhao K. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet. 2008;40:897–903. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, Antonarakis SE, Attwood J, Baertsch R, Bailey J, Barlow K, Beck S, Berry E, Birren B, Bloom T, Bork P, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- Weaver IC, Meaney MJ, Szyf M. Maternal care effects on the hippocampal transcriptome and anxiety-mediated behaviors in the offspring that are reversible in adulthood. Proc Natl Acad Sci U S A. 2006;103:3480–3485. doi: 10.1073/pnas.0507526103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen B, Wu H, Shinkai Y, Irizarry RA, Feinberg AP. Large histone H3 lysine 9 dimethylated chromatin blocks distinguish differentiated from embryonic stem cells. Nat Genet. 2009;41:246–250. doi: 10.1038/ng.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Mei Q, Zielinska-Kwiatkowska A, Matsui Y, Blackburn ML, Benedetti D, Krumm AA, Taborsky GJ, Jr, Chansky HA. An ERG (ets-related gene)-associated histone methyltransferase interacts with histone deacetylases 1/2 and transcription co-repressors mSin3A/B. Biochem J. 2003;369:651–657. doi: 10.1042/BJ20020854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarate CA, Jr, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, Charney DS, Manji HK. A randomized trial of an N-methyl-d-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. 2006;63:856–864. doi: 10.1001/archpsyc.63.8.856. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nussbaum C, Myers RM, Brown M, Li W, Liu XS. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao JP, Constantine-Paton M. NR2A−/− mice lack long-term potentiation but retain NMDA receptor and L-type Ca2+ channel-dependent long-term depression in the juvenile superior colliculus. J Neurosci. 2007;27:13649–13654. doi: 10.1523/JNEUROSCI.3153-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]