

Abstract
Signal transducer and activator of transcription 3 (STAT3) is activated in a variety of human cancers, including ovarian cancer. The molecular mechanism by which the STAT3 is activated in cancer cells is poorly understood. We observed that human ovarian xenograft tumors (A2780) in mice were severely hypoxic (pO2 ∼ 2 mmHg). We further observed that hypoxic exposure significantly increased the phosphorylation of STAT3 (pSTAT3) at the Tyr705 residue in A2780 cell line. The pSTAT3 (Tyr705) level was highly dependent on cellular oxygenation levels, with a significant increase at <2% O2, and without any change in the pSTAT3 (Ser727) or total STAT3 levels. The pSTAT3 (Tyr705) elevation following hypoxic exposure could be reversed within 12 hr after returning the cells to normoxia. The increased level of pSTAT3 was partly mediated by increased levels of reactive oxygen species generation in the hypoxic cancer cells. Conventional chemotherapeutic drugs cisplatin and taxol were far less effective in eliminating the hypoxic ovarian cancer cells suggesting a role for pSTAT3 in cellular resistance to chemotherapy. Inhibition of STAT3 by AG490 followed by treatment with cisplatin or taxol resulted in a significant increase in apoptosis suggesting that hypoxia-induced STAT3 activation is responsible for chemoresistance. The results have important clinical implications for the treatment of hypoxic ovarian tumors using STAT3-specific inhibitors.
Keywords: ovarian cancer, hypoxia, STAT3, ROS, cisplatin, taxol, AG490
Hypoxia, defined as subnormal levels of tissue oxygenation, has long been implicated in the development of resistance to radio- or chemotherapeutic treatment in several types of tumors.1 Clinical investigations have shown that most of the tumors are hypoxic and that the degree of hypoxia increases with tumor size. 1–3 Hypoxic tumor cells can be locally and systemically aggressive, with a decreased sensitivity to apoptotic and other cell-death signals, and increased signaling to promote angiogenesis, proliferation and systemic metastasis capacity.4 Experimental and spontaneous metastatic capacity is increased when tumor cells are subjected to hypoxia, and this can be secondary to genetic instability.3 Furthermore, hypoxia results in increased generation of reactive oxygen species (ROS) and several studies have suggested that ROS can act as secondary messengers and control various signaling cascades.5 Moreover, oxidative stress caused by ROS was reported to be a potent activator of nuclear factor κB (NF-κB), which is involved in the signaling pathways of many growth factors and cytokines.6
Several constitutively activated signal transducers and activators of transcription 3 (STAT3) proteins have been observed in a wide number of human cancer cell lines and primary tumors, including blood malignancies and solid ovarian cancers. 7–10 STAT3 is activated by phosphorylation at the Tyr705 residue, which induces dimerization, nuclear translocation and DNA binding.11 The resulting signal-transduction pathways permit them to play different roles in normal physiological cell processes, such as differentiation, proliferation, apoptosis and angiogenesis.9 However, aberrant activation of STAT3 signaling gives rise to different pathological events. Many studies suggest that STAT3 proteins could participate in tumorigenesis through upregulation of genes encoding apoptosis inhibitors (Mcl-1, Bcl-xL), cell-cycle regulators (cyclin D1/D2, c-Myc), and inducers of vascular endothelial growth factor (VEGF).2,12–15 It is reported using a human renal carcinoma cell line that hypoxia-mediated STAT3 activation was responsible for transcriptional activation of the VEGF promoter.16 Furthermore, active STAT3 interacted with HIF-1α, and increased HIF-1α accumulation in hypoxic cells.16,17 However, how the STAT3 is activated in hypoxic cancer cells is an intriguing question, and has not been studied.
Because activation of STAT3 directly regulates both cell-proliferation and survival genes that provide growth advantages to tumor cells by blocking proapoptotic genes, it is possible that the STAT3 could be activated by the tumor microenvironment, including growth under hypoxic conditions. Hence, the goal of this study was to determine the effect of hypoxia on the induction of STAT3 activation in human ovarian cancer cells. We observed that STAT3 activation was significantly increased by hypoxia, which was accompanied by increased ROS generation. The hypoxia-mediated STAT3 activation might be contributing to cellular resistance to anticancer drug-induced apoptosis, which would present a further and considerable clinical obstacle. Our results provide a rationale for new clinical perspectives on the development of future therapies for hypoxic ovarian cancers.
Material and methods
Materials
Dimethyl sulfoxide, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), dicarboxyfluorescein diacetate (DCF-DA), N-acetyl cysteine (NAC) and AG490 were obtained from Sigma (St. Louis, MO). Cell-culture media (RPMI 1640, DMEM), fetal bovine serum (FBS), antibiotics, sodium pyruvate, trypsin and phosphate-buffered saline (PBS) were purchased from Gibco BRL (Grand Island, NY). Polyvinylidene fluoride (PVDF) membrane and molecular weight markers were obtained from Bio-Rad. Antibodies directed against human pSTAT3 Tyr705 and Ser727, pSTAT1, pSTAT5 and pSTAT6 were purchased from Cell Signaling Technology (Beverly, MA). Antibodies specific for human VEGF, HIF-1α and STAT3, as well as STAT3 siRNA were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Lipofectamine kits were purchased from Invitrogen (Carlsbad, CA). RNAse was purchased from Promega Corporation (Madison, WI). Enhanced chemiluminescence (ECL) reagents were obtained from Amersham Pharmacia Biotech (Buckinghamshire, UK). All other reagents and compounds were of analytical grade.
Cell lines and culture
The human ovarian cancer cell line, A2780, that is, sensitive to treatment by the chemotherapy drug cisplatin was used.18 Cells were grown in RPMI 1640 medium supplemented with 10% FBS, 2% sodium pyruvate and 1% penicillin and streptomycin (PS). Cells were grown in a 100-mm dish to 70% confluence at 37°C in an atmosphere of 5% CO2 and 95% air. Cells were routinely trypsinized (0.05% trypsin/EDTA) and counted using a Nucleo-Counter (New Brunswick Scientific, Edison, NJ). Hypoxic exposure was achieved by incubating cells in a humidified atmosphere of 1% O2 and 5% CO2 in a specialized environmental chamber (C-Chamber and ProOx Model C21, BioSpherix, USA) contained within a standard cell culture incubator.
Cell proliferation assay
A2780 cells were seeded at a density of 1 × 104/ml (200 μl/well) into 96-well microplates with complete RPMI-1640 medium and cultured for 24 hr. The cells were incubated under normoxic (20% O2) or hypoxic (1% O2) conditions. After 24 hr of incubation, cell proliferation was assessed by BrdU assay methods. The method is based on incorporation of the pyrimidine analogue BrdU in place of thymidine into the DNA of proliferating cells. Experiments were performed in triplicate, and data were expressed as the mean of the triplicate determinations of a representative experiment in percentage of absorbance of samples compared with untreated cells. The absorbance at 350 nm was evaluated using a microplate reader.
Measurement of ROS
The intracellular detection of ROS was performed using DCF-DA, as previously described.19 A2780 cells were grown to 80% confluence on 6-mm glass cover-slips, and incubated under normoxic or hypoxic conditions. Following DCF-DA loading, the cells were incubated in the dark for 20 min, washed with protein-free medium and fluorescence images were immediately captured with a Nikon Eclipse TE2000-U camera system using excitation/emission at 495/520 nm. The captured images, 5–10 per slide, were then analyzed using image analysis software (MetaMorph).
Immunoblot analysis
A2780 cells were grown in RPMI-1640 medium under normoxic (20% O2) or hypoxic (1% O2) conditions for 24, 48 or 72 hr. Following hypoxic exposure, cell lysates were prepared in nondenaturing lysis buffer [10 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, 1 mM EGTA, 0.3 mM phenylmethylsulfonyl fluoride, 0.2 mM sodium orthovanadate, 0.5% NP40, aprotinin (1 μg/ml) and leupetin]. The cell lysates were centrifuged at 10,000g for 20 min at 4°C, and the supernatant was separated. The protein concentration in the lysates was determined using a Pierce detergent-compatible protein assay kit. For Western blotting, 25–50 μg of protein lysate per sample was denatured in 2× SDS-PAGE sample buffer and subjected to SDS-PAGE on a 10% or 12% tris-glycine gel. The separated proteins were transferred to a PVDF membrane and the membrane was blocked with 5% nonfat milk powder (w/v) in TBST (10 mM Tris, 100 mM NaCl, 0.1% Tween 20) for 1 hr at room temperature, or overnight at 4°C. The membranes were incubated with the primary antibodies mentioned previously. The bound antibodies were detected with horseradish peroxidase (HRP)-labeled sheep anti-mouse IgG or HRP-labeled donkey anti-rabbit IgG using an ECL detection system (ECL Advanced kit). Protein expression was quantified using Image Gauge version 3.45 software.
STAT3 siRNAs study
A2780 cells were transfected with STAT3 siRNA and negative control siRNA using Lipofectamine 2000 (Invitrogen) according to the manufacturer's recommended protocol. The negative control siRNA, which uses sequences that do not target any gene product, is essential for evaluating transfection efficiency and determining the effects of siRNA delivery on cells. One day after transfection, the complexes were removed and replaced with fresh culture medium. The cells were incubated for 24 hr in the hypoxic environment (1% O2), after which cell lysates were prepared, and subjected to immunoblot analysis for pSTAT3 and STAT3. The transfected cells were also subjected to cell proliferation assessment using BrdU incorporation, as mentioned previously in the cell proliferation assay. Cell viability was determined using the conversion of MTT to formazan via mitochondrial oxidation. One day after transfection, the transfection complexes were removed and replaced with culture medium, followed by incubation with cisplatin (100 μM) or taxol (50 μM) for 24 hr.
pO2 measurements in tumors
Human ovarian cancer xenograft tumors were grown in the hind limb of BALB/c nude mice by injecting cisplatin-sensitive (A2780) or cisplatin-resistant (A2780 cDDP) human ovarian cancer cells (5 × 106 cells in 60 μl of PBS) subcutaneously (s.c.), as we have reported.20 Similarly, radiation-induced fibrosarcoma (RIF-1) tumors were grown in C3H mice, as we have reported.2,21 When the tumors reached about 10–12 mm size in the largest dimension, tumor tissue oxygenation (pO2) was measured by electron paramagnetic resonance (EPR) oximetry using lithium octa-n-butoxy-naphthalocyanine (LiNc-BuO) microcrystals (10–30 μm size) as probe.22 Mice were anesthetized with 1.5% isoflurane-air mixture and LiNc-BuO crystals were implanted into the tumor or in the gastrocnemius muscle tissue of control (nontumor) C3H mice at a depth of about 3 mm. In vivo EPR measurements were performed 48 hr after implantation of the probe by using an L-band (1.2 GHz) EPR spectrometer (Magnettech, Germany) with a bridged loop-gap resonator.21 The peak-to-peak width of the EPR spectrum of the probe in the tissue was used to determine pO2 values using a precalibrated standard curve.22 The pO2 values are expressed as a mean ± standard error (SE) of data obtained from 5 mice per group.
Statistical analysis
All data were expressed as mean ± SE. Comparisons among groups were performed by a Student's t-test. The significance level was set at p < 0.05.
Results
Ovarian tumors are severely hypoxic
Hypoxic microenvironments are frequently found in many solid tumors, including ovarian tumors.1 However, the precise value of oxygenation in ovarian tumors is not known. We used EPR oximetry to measure oxygenation (pO2) in ovarian tumors. A2780 cells were transplanted and grown as a solid tumor in mice. When the tumor size reached about 12 mm in diameter, tumor oxygen levels were measured using EPR oximetry. The in vivo data (Fig. 1) showed that the murine ovarian tumor xenografts were severely hypoxic (A2780: 2.0 ± 0.7 mmHg; A2780 cDDP: 2.2 ± 1.1 mmHg) when compared RIF-1 tumor (7.8 ± 1.4 mmHg) or gastrocnemius muscle tissue (15.1 ± 1.6 mmHg).
Figure 1.
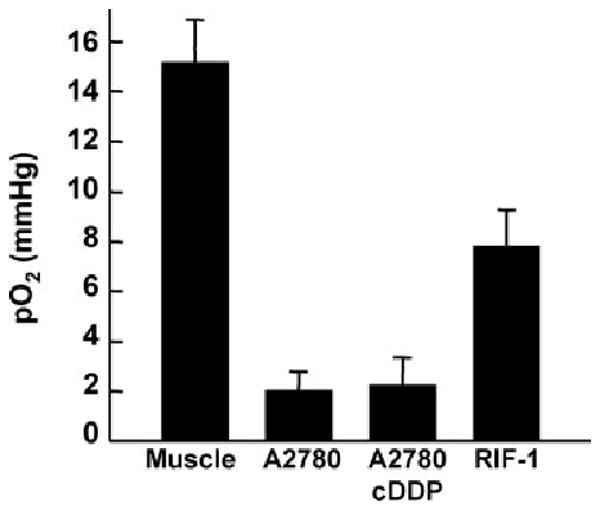
Partial pressure of oxygen (pO2) in A2780 xenograft tumors in mice. Shown are pO2 values obtained from A2780 (human ovarian), A2780 cDDP (cisplatin-resistant variant of A2780) and RIF-1 (radiation-induced fibrosarcoma) tumors grown by subcutaneous implantation of respective cells in the hind limb of mice. The measurements were performed by in vivo EPR oximetry when the tumor size was about 12 mm in diameter. For comparison, the pO2 value measured from the gastrocnemius muscle tissue of control (nontumor) mice is also shown. Data represent mean ± SE from 5 mice per group. The results show that the ovarian xenograft tumors are severely hypoxic when compared with RIF-1 tumor or muscle tissue.
STAT3 regulates ovarian cancer cell proliferation under hypoxic conditions
To understand the effect of hypoxia on the signaling proteins involved in the tumor progression and treatment, we performed in vitro experiments using ovarian cancer cell lines in culture. A2780 cells were grown under hypoxic (1% O2) and normoxic (20% O2) conditions. Western-blot assays revealed higher levels of HIF-1α, VEGF and pSTAT3 (Tyr705) in cells cultured under hypoxic conditions, when compared with cells grown under normoxic conditions (Fig. 2a). HIF-1β, total STAT3, and pSTAT3 (Ser727) levels were unchanged. The increase in pSTAT3 (Tyr705) level was 3-fold higher than increases in the expression of HIF-1α and VEGF (Fig. 2b). Ovarian cancer cell proliferation was not significantly affected by hypoxia treatment (Fig. 2c). Next, we determined whether inhibition of STAT3 had any effect on cell proliferation under hypoxic conditions. Suppression of STAT3 level by using STAT3 siRNA significantly affected cell proliferation when grown under hypoxic conditions when compared with cells grown under normoxic conditions (Fig. 2d). These results suggested that STAT3 may play a key role in the regulation of ovarian cancer cell proliferation under hypoxic conditions.
Figure 2.
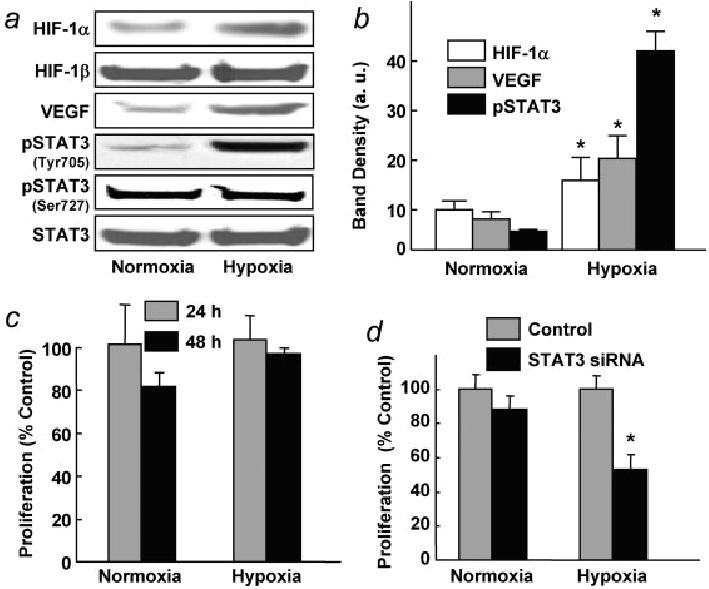
Effect of hypoxia on STAT3 activation in human ovarian cancer cells. Cells were cultured under normoxic (20% O2) or hypoxic (1% O2) conditions. (a) Representative Western blots of HIF-1α, VEGF, pSTAT3 (Tyr705), pSTAT3 (Ser727) and STAT3 are shown. (b) Quantification of HIF-1α, VEGF and pSTAT3 blots from triplicate experiments. All 3 protein levels were significantly upregulated (*p < 0.05) in the hypoxic cells. (c) Cell-proliferation data obtained using anti-BrdU assay at 24- and 48-hr exposure to hypoxia. (d) Effect of STAT3 siRNA transfection on the proliferation of cells cultured under normoxic and hypoxic conditions. STAT3 siRNA significantly (*p < 0.05) reduced cell proliferation in the hypoxic cells.
STAT3 activation in ovarian cancer cells is both time- and O2-dependent
We determined the effect of oxygen concentration and exposure time on the activation of STAT3 (Tyr705) in ovarian cancer cells. Figure 3a shows the increased levels of pSTAT3 in cells exposed to 1% O2 for 12, 24 and 48 hr. We also investigated the effect of hypoxic exposure on other pSTAT proteins (pSTAT1, pSTAT5 and pSTAT6), and did not observe any significant changes in the expression of these proteins (Fig. 3b). Furthermore, we checked pSTAT3 level in ovarian cancer cells cultured under increasingly hypoxic conditions (5, 2, 1 and 0.5% O2), and observed that the level was significantly increased in cells grown at 2% O2 or lower (Fig. 3c). In addition, we observed that the increase in pSTAT3 level could be reversed within 12 hr on returning the cells to a 20% O2 environment following hypoxic exposure (Fig. 3d). The results suggested that the hypoxia-induced activation was specific to STAT3 protein and modulated by the oxygen concentration in a time-dependent manner.
Figure 3.
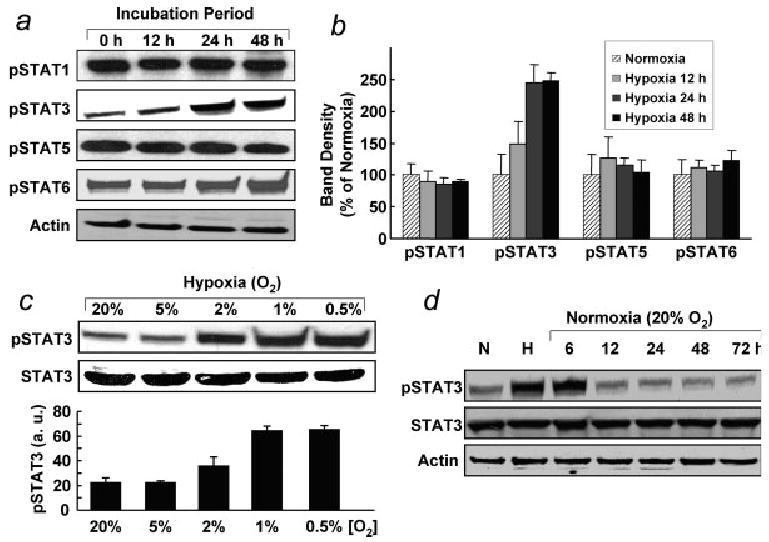
Activation of STAT proteins in ovarian cancer cells cultured under hypoxic conditions. (a) Phosphorylated STATs after exposure to hypoxia for 0 (normoxia), 12, 24 and 48 hr. There was no change in the levels of pSTAT1, pSTAT5 and pSTAT6 when compared with cells cultured under normoxic conditions, whereas pSTAT3 level was enhanced in a time-dependent manner. (b) Quantification of band density of pSTATs from triplicate experiments. (c) Magnitude of pSTAT3 as a function of hypoxic exposure level. The pSTAT3 level was significantly enhanced in cells cultured at 2, 1 and 0.5% O2. (d) After 48 hr of incubation under hypoxia (H, 1% O2), cells were returned to normoxic culture (20% O2) for 72 hr. The level of pSTAT3 was reversed after 12 hr or more on return to normoxic culture.
Hypoxic exposure increases ROS production in A2780 cells
ROS regulate angiogenesis and tumor growth through VEGF and STAT3 expression/upregulation.5 The increase in pSTAT3 level in cells cultured under hypoxic conditions prompted us to examine the cellular ROS levels using DCF-DA staining. We observed significantly increased fluorescence intensity by intracellular ROS in cells exposed to hypoxic conditions for 12 hr, when compared with cells grown under normoxic conditions (Fig. 4a). The ROS levels were significantly higher in the hypoxic cells than the normoxic cells, as determined by quantification of fluorescence intensity (Fig. 4b). We determined whether the inhibition of ROS by NAC would affect pSTAT3 levels in hypoxic ovarian cancer cells. Figures 4c–4d shows that cells cultured under hypoxic (1% O2) conditions and treated with 100 μM NAC had significantly lower pSTAT3 levels than untreated controls. The results implicated the involvement of hypoxia-mediated ROS generation in the activation of STAT3.
Figure 4.
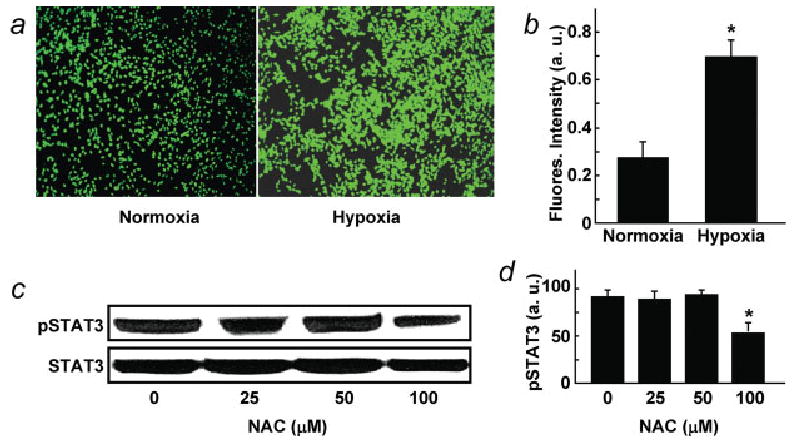
ROS levels in ovarian cancer cells cultured under normoxic (20% O2) and hypoxic (1% O2) conditions for 24 hr. (a) Dicarboxy-fluorescein diacetate (DCF-DA) fluorescence images of normoxic and hypoxic-cultured cells. (b) DCF-DA fluorescence intensity showing the ROS level in hypoxic cells was significantly (*p < 0.05) greater than normoxic cells. (c) Western blots of pSTAT3 in cells cultured under hypoxic conditions in presence of N-acetyl cysteine (NAC), a known ROS inhibitor. (d) Quantification of band density of C from triplicate experiments. Inhibition pSTAT3 upregulation was significant at 100 μM NAC concentration (*p < 0.05).
Hypoxic exposure increases drug resistance in A2780 cells
Activated STAT3 has been known to be associated with drug resistance in ovarian cancer cells.7 To determine the effect of hypoxia-induced activation of STAT3 on ovarian cancer therapy, we treated A2780 cells grown under hypoxic conditions using cisplatin (100 μM) or taxol (50 μM) for 48 hr. Flow cytometry determination of the subG1 population (Fig. 5) showed that cells grown under hypoxic (1% O2) conditions were more resistant to treatment than cells grown under normoxic culture conditions. The results suggested that hypoxic exposure of A2780 cells increased drug resistance to conventional chemotherapeutics.
Figure 5.
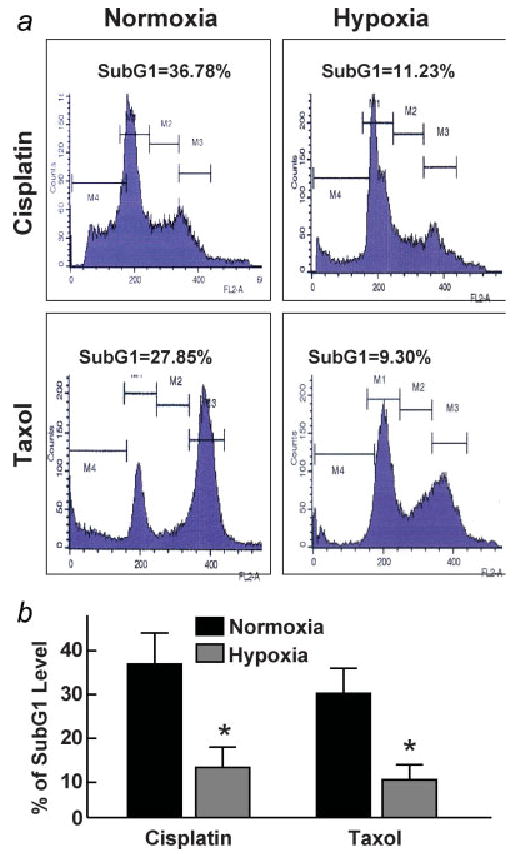
Effect of cisplatin or taxol exposure on the subG1 population of ovarian cancer cells cultured under normoxic or hypoxic conditions for 24 hr. Cells were treated with cisplatin (100 μM) or taxol (50 μM) for 48 hr. The cells were then collected and analyzed for subG1 levels using flow cytometry. (a) Representative flow-cytogram of cells. (b) Quantification of subG1 level as a percentage of total cells in triplicate experiments. The data show a significant difference in the chemotherapeutic efficacy between the hypoxic and normoxic cells. *p < 0.05 versus corresponding normoxic group.
Inhibition of STAT3 increases chemosensitivity in hypoxic A2780 cells
To determine if the induction of chemoresistance in hypoxic cells was due to the activated STAT3, we studied the effect of AG490, an indirect inhibitor of STAT3, on chemosensitivity. A2780 cells were grown under hypoxic (1% O2) conditions to 80% confluence, and incubated for 12 hr in the presence of 50 μM AG490. After this period, we added cisplatin (100 μM) or taxol (50 μM) and incubated the cells for an additional 24 hr. The cells were then collected and analyzed by flow cytometry to determine the subG1 (apoptotic) levels. The suppression of STAT3 by AG490, followed by treatment with cisplatin or taxol, resulted in a significantly higher level of apoptosis when compared with cells that were not treated with AG490 (Figs. 6a and 6b). Further, we used siRNA transfection studies to see whether STAT3 suppression would increase the chemosensitivity in hypoxic cells. A2780 cells transfected with STAT3 siRNA showed 80–90% suppression of STAT3 levels (data not shown). Silencing of STAT3 by STAT3 siRNA followed by treatment with cisplatin or taxol under hypoxic conditions showed a significantly decreased (>70%) cell viability when compared with control cells treated with cisplatin or taxol alone (Fig. 6c). These results confirmed that induction of chemoresistance in hypoxic cells was due to STAT3 activation in the hypoxic cells.
Figure 6.
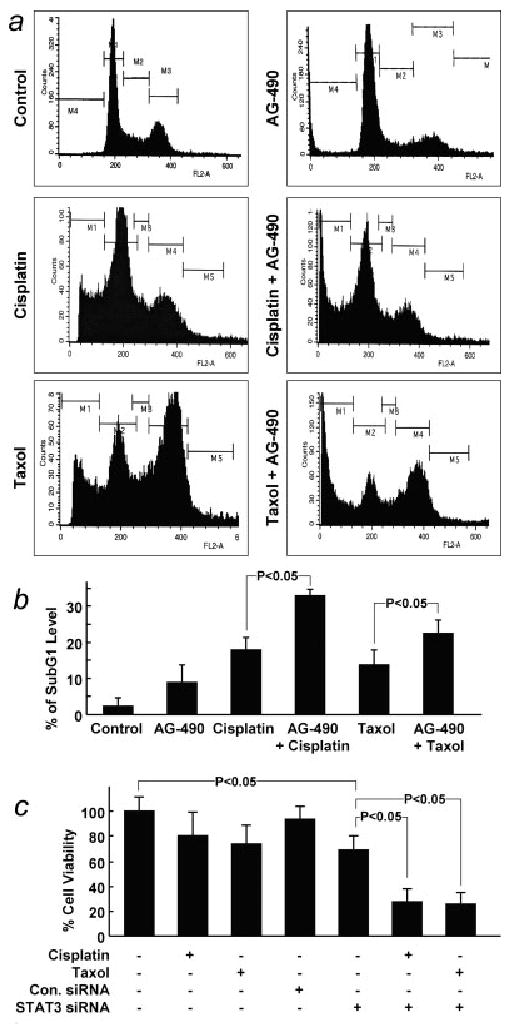
Effect of STAT3 inhibition on the subG1 population and viability of ovarian cancer cells cultured under hypoxic conditions for 24 hr. AG490 (50 μM), an indirect inhibitor of STAT3, was used to inhibit STAT3. Cells were treated with AG490 alone, cisplatin (100 μM), taxol (50 μM), cisplatin + AG490 or taxol + AG490 for 24 hr in hypoxic culture (1% O2). (a) Representative flow cytogram of cells. (b) Quantification of subG1 level as a percentage of total cells in triplicate experiments. (c) The effect of STAT3 suppression, achieved by transfection with STAT3 siRNA, on the viability (by MTT assay) of cells treated with cisplatin (100 μM) or taxol (50 μM) under hypoxic conditions.
Discussion
Tumor hypoxia is not only a major problem for radiation anticancer therapy, it has also been implicated in the development of resistance to many conventional chemotherapeutic agents.4,23 Because hypoxia is an inherent characteristic of many solid tumors, hypoxic stress may provide a common signal that induces a prolonged increase in angiogenic gene expression during tumorigenesis through the activation of oncogenes, including STAT3.12,24 We observed that solid tumor xenografts of ovarian cancer were severely hypoxic and that ovarian cancer cells grown under hypoxic conditions exhibited significantly activated STAT3 (Tyr705) and higher levels of ROS generation when compared with ovarian cancer cells grown in normoxic culture. We further observed that activation of STAT3 is partially regulated by ROS induction and inhibition of STAT3 leads to reduced cellular proliferation and increased induction of apoptosis following treatment with anticancer compounds.
An intriguing observation of the presence study was the oxygen level of the ovarian xenograft tumors, which were severely hypoxic (∼2 mmHg) when compared with RIF-1 tumor (∼8 mmHg), a representative case of hypoxic tumors. The pO2 data were obtained by EPR oximetry, which used a paramagnetic oxygen-sensing probe (LiNc-BuO) that was surgically implanted in the tumor 48 hr before the measurements were made. The waiting time was necessary for the tissue to recover from any trauma associated with the surgical procedure.25 The probe is biocompatible and stable in tissues, thus enabling subsequent measurements to be made noninvasively and repeatedly over long periods of time, if desired.22 Over the years, EPR oximetry has been used to measure pO2 in the heart, brain, and tumor of a variety of animal models and diseases.26 However, this is the first report of pO2 measurement in an ovarian xenograft tumor. Oxygenation of RIF-1 tumors has been extensively studied by several methods, including EPR oximetry, and the results were found to be fairly consistent.21,25,27–29 Our present data on RIF-1 tumor is in good agreement with a recent report by Hou et al.,30 who used EPR oximetry with a similar probe. However, it should be noted that the same method and probe may give a range (distribution) of pO2 values up on EPR imaging of oxygen, as we have reported.2 Although heterogeneity in the oxygen levels is known for solid tumors, this study used only a single-point measurement and hence may not rule out the possibility of any heterogeneous distribution of oxygen concentration in the ovarian tumor.
We have reported, for the first time, that constitutive activation of STAT3 (Tyr705) was detected in ovarian cancer cells grown under hypoxic conditions, and that pSTAT3 (Ser727) levels remained unchanged. Tyrosine phosphorylation has been considered to be more important than serine phosphorylation in the activation of STAT3.9,24 Recent evidence has shown that STAT3, through hypoxia-induced tyrosine phosphorylation, directly binds to HIF-1α and upregulates HIF-1α stability by delaying protein degradation and accelerating protein synthesis in human renal carcinoma cells.16 The activation of STAT3 also promotes angiogenesis through the induction of VEGF.31 In this study, we observed that the increase in STAT3 activation was 3 times higher than increases in the more traditional hypoxia markers HIF-1α and VEGF in ovarian cancer cells grown under hypoxic conditions. In addition, this work demonstrated that STAT3 is required for ovarian cancer cell proliferation and that the blocking of STAT3 expression by siRNA significantly reduces cell proliferation capacity under hypoxic conditions. The exact mechanism by which the expression and regulation of STAT3 play a role in ovarian cancer cell proliferation and migration under hypoxic conditions remains unclear.
The induction of ROS production has long been associated with tumor progression. It has been suggested that the elimination of excessive ROS by chemicals or antioxidants may decrease the metastatic potential of various types of cancer, and this information has opened up new areas of research for cell biologists.32 In this study, we observed increased ROS production in ovarian cancer cells cultured under hypoxic conditions. High levels of ROS production have also been observed in other human cancer cells.5,33 However, the biological role of ROS in cancer cells has yet to be determined. We have shown that hypoxia-induced ROS activates STAT3, which may lead to downstream proliferative responses. The multiple effects mediated by ROS could account for tumorigenesis through STAT3 activation, as well as DNA mutation.34 We have also shown that inhibition of ROS by NAC was unable to completely inhibit STAT3 activation, implying that additional signaling mechanisms may also be at work. Another possible mechanism for STAT3 activation in hypoxic ovarian cancer cells may be through activation of MAPK pathways.35
Because inhibition of STAT3 activation causes tumor cell apoptosis, STAT3 is considered to be a promising target for anticancer therapy.36 Pharmacologic inhibitors of STAT3, such as AG490, have been shown to significantly suppress cellular pSTAT3 levels and induce apoptosis in a variety of human cancer cells, including breast, prostate, colon and ovarian cancers.37–39 Traditional chemotherapeutic agents commonly used for ovarian cancer treatment, such as cisplatin or taxol, often become ineffective due to chemoresistance, particularly under hypoxic conditions.40,41 The exact mechanism for this hypoxia-induced chemoresistance has not yet been well established. Given the results of our study, one possible mechanism may be through activation of the STAT3-signaling pathway. In in vitro experiments, we have demonstrated that an indirect STAT3 inhibitor, AG490, partially overcame this hypoxia-induced chemoresistance to cisplatin or taxol treatment.
Recent evidence indicates that STAT3 inhibitors are potentially powerful therapeutic agents for various cancers, including ovarian cancer.15,36,42,43 Our findings suggest a new, novel function of activated STAT3 as a key promoter of hypoxia-induced resistance to common anticancer drugs in human ovarian cancer cells. Therefore, the inhibition of STAT3 may be of particular use in clinical therapeutics for patients with solid tumors, particularly those that have become resistant to chemotherapeutic intervention. Further, through the use of antioxidant compounds, it may be possible to decrease the levels of oxidative stress incurred by cells and tissues, further reducing STAT3 activation in hypoxic tumor cells. Our study clearly supports the hypothesis that by targeting and reducing STAT3 activation in cancer cells, it may be possible to regulate angiogenic protein expression, reduce or overcome chemotherapeutic resistance in hypoxic tumors, and to reduce tumor growth, not only in ovarian cancer, but in other cancers as well.
Acknowledgments
NIH; Grant number: CA-102264; Grant sponsor: Kaleidoscope of Hope Foundation, NJ.
Abbreviations
- DCF-DA
dicarboxyfluorescein diacetate
- EPR
electron paramagnetic resonance
- HIF-1
hypoxia-inducible factor 1
- LiNc-BuO
lithium octa-n-butoxy-naphthalocyanine
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NAC
N-acetyl cysteine
- NF-κB
nuclear factor-κB
- pO2
partial pressure of oxygen
- RIF-1
radiation-induced fibrosarcoma-1
- ROS
reactive oxygen species
- STAT3
signal transducer and activator of transcription 3
- VEGF
vascular endothelial growth factor
References
- 1.Brown JM, Wilson WR. Exploiting tumour hypoxia in cancer treatment. Nat Rev Cancer. 2004;4:437–47. doi: 10.1038/nrc1367. [DOI] [PubMed] [Google Scholar]
- 2.Bratasz A, Pandian RP, Deng Y, Petryakov S, Grecula JC, Gupta N, Kuppusamy P. In vivo imaging of changes in tumor oxygenation during growth and after treatment. Magn Reson Med. 2007;57:950–9. doi: 10.1002/mrm.21212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Krohn KA, Link JM, Mason RP. Molecular imaging of hypoxia. J Nucl Med. 2008;49(Suppl 2):S129–48. doi: 10.2967/jnumed.107.045914. [DOI] [PubMed] [Google Scholar]
- 4.Brown JM. Exploiting the hypoxic cancer cell: mechanisms and therapeutic strategies. Mol Med Today. 2000;6:157–62. doi: 10.1016/s1357-4310(00)01677-4. [DOI] [PubMed] [Google Scholar]
- 5.Xia C, Meng Q, Liu LZ, Rojanasakul Y, Wang XR, Jiang BH. Reactive oxygen species regulate angiogenesis and tumor growth through vascular endothelial growth factor. Cancer Res. 2007;67:10823–30. doi: 10.1158/0008-5472.CAN-07-0783. [DOI] [PubMed] [Google Scholar]
- 6.Kratsovnik E, Bromberg Y, Sperling O, Zoref-Shani E. Oxidative stress activates transcription factor NF-kB-mediated protective signaling in primary rat neuronal cultures. J Mol Neurosci. 2005;26:27–32. doi: 10.1385/jmn:26:1:027. [DOI] [PubMed] [Google Scholar]
- 7.Rosen DG, Mercado-Uribe I, Yang G, Bast RC, Jr, Amin HM, Lai R, Liu J. The role of constitutively active signal transducer and activator of transcription 3 in ovarian tumorigenesis and prognosis. Cancer. 2006;107:2730–40. doi: 10.1002/cncr.22293. [DOI] [PubMed] [Google Scholar]
- 8.Bromberg J. Stat proteins and oncogenesis. J Clin Invest. 2002;109:1139–42. doi: 10.1172/JCI15617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, Darnell JE., Jr Stat3 as an oncogene. Cell. 1999;98:295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 10.Sommer VH, Clemmensen OJ, Nielsen O, Wasik M, Lovato P, Brender C, Eriksen KW, Woetmann A, Kaestel CG, Nissen MH, Ropke C, Skov S, et al. In vivo activation of STAT3 in cutaneous T-cell lymphoma. Evidence for an antiapoptotic function of STAT3. Leukemia. 2004;18:1288–95. doi: 10.1038/sj.leu.2403385. [DOI] [PubMed] [Google Scholar]
- 11.Darnell JE, Jr, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264:1415–21. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- 12.Yu H, Jove R. The STATs of cancer—new molecular targets come of age. Nat Rev Cancer. 2004;4:97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- 13.Bhardwaj A, Sethi G, Vadhan-Raj S, Bueso-Ramos C, Takada Y, Gaur U, Nair AS, Shishodia S, Aggarwal BB. Resveratrol inhibits proliferation, induces apoptosis, and overcomes chemoresistance through down-regulation of STAT3 and nuclear factor-kappaB-regulated antiapoptotic and cell survival gene products in human multiple myeloma cells. Blood. 2007;109:2293–302. doi: 10.1182/blood-2006-02-003988. [DOI] [PubMed] [Google Scholar]
- 14.Pathak AK, Bhutani M, Nair AS, Ahn KS, Chakraborty A, Kadara H, Guha S, Sethi G, Aggarwal BB. Ursolic acid inhibits STAT3 activation pathway leading to suppression of proliferation and chemosensitization of human multiple myeloma cells. Mol Cancer Res. 2007;5:943–55. doi: 10.1158/1541-7786.MCR-06-0348. [DOI] [PubMed] [Google Scholar]
- 15.Selvendiran K, Bratasz A, Tong L, Ignarro LJ, Kuppusamy P. NCX-4016, a nitro-derivative of aspirin, inhibits EGFR and STAT3 signaling and modulates Bcl-2 proteins in cisplatin-resistant human ovarian cancer cells and xenografts. Cell Cycle. 2008;7:81–8. doi: 10.4161/cc.7.1.5103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jung JE, Lee HG, Cho IH, Chung DH, Yoon SH, Yang YM, Lee JW, Choi S, Park JW, Ye SK, Chung MH. STAT3 is a potential modulator of HIF-1-mediated VEGF expression in human renal carcinoma cells. Faseb J. 2005;19:1296–8. doi: 10.1096/fj.04-3099fje. [DOI] [PubMed] [Google Scholar]
- 17.Xu Q, Briggs J, Park S, Niu G, Kortylewski M, Zhang S, Gritsko T, Turkson J, Kay H, Semenza GL, Cheng JQ, Jove R, et al. Targeting Stat3 blocks both HIF-1 and VEGF expression induced by multiple oncogenic growth signaling pathways. Oncogene. 2005;24:5552–60. doi: 10.1038/sj.onc.1208719. [DOI] [PubMed] [Google Scholar]
- 18.Bratasz A, Weir NM, Parinandi NL, Zweier JL, Sridhar R, Ignarro LJ, Kuppusamy P. Reversal to cisplatin sensitivity in recurrent human ovarian cancer cells by NCX-4016, a nitro derivative of aspirin. Proc Natl Acad Sci USA. 2006;103:3914–9. doi: 10.1073/pnas.0511250103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bhimani RS, Troll W, Grunberger D, Frenkel K. Inhibition of oxidative stress in HeLa cells by chemopreventive agents. Cancer Res. 1993;53:4528–33. [PubMed] [Google Scholar]
- 20.Bratasz A, Selvendiran K, Wasowicz T, Bobko A, Khramtsov VV, Ignarro LJ, Kuppusamy P. NCX-4040, a nitric oxide-releasing aspirin, sensitizes drug-resistant human ovarian xenograft tumors to cisplatin by depletion of cellular thiols. J Transl Med. 2008;6:9. doi: 10.1186/1479-5876-6-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ilangovan G, Li H, Zweier JL, Krishna MC, Mitchell JB, Kuppusamy P. In vivo measurement of regional oxygenation and imaging of redox status in RIF-1 murine tumor: effect of carbogen-breathing. Magn Reson Med. 2002;48:723–30. doi: 10.1002/mrm.10254. [DOI] [PubMed] [Google Scholar]
- 22.Pandian RP, Parinandi NL, Ilangovan G, Zweier JL, Kuppusamy P. Novel particulate spin probe for targeted determination of oxygen in cells and tissues. Free Radic Biol Med. 2003;35:1138–48. doi: 10.1016/s0891-5849(03)00496-9. [DOI] [PubMed] [Google Scholar]
- 23.Kunz M, Ibrahim SM. Molecular responses to hypoxia in tumor cells. Mol Cancer. 2003;2:23. doi: 10.1186/1476-4598-2-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee MY, Joung YH, Lim EJ, Park JH, Ye SK, Park T, Zhang Z, Park DK, Lee KJ, Yang YM. Phosphorylation and activation of STAT proteins by hypoxia in breast cancer cells. Breast. 2006;15:187–95. doi: 10.1016/j.breast.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 25.Vikram DS, Bratasz A, Ahmad R, Kuppusamy P. A comparative evaluation of EPR and OxyLite oximetry using a random sampling of pO(2) in a murine tumor. Radiat Res. 2007;168:308–15. doi: 10.1667/RR0854.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Swartz HM. Using EPR to measure a critical but often unmeasured component of oxidative damage: oxygen. Antioxid Redox Signal. 2004;6:677–86. doi: 10.1089/152308604773934440. [DOI] [PubMed] [Google Scholar]
- 27.Ilangovan G, Bratasz A, Kuppusamy P. Non-invasive measurement of tumor oxygenation using embedded microparticulate EPR spin probe. Adv Exp Med Biol. 2005;566:67–73. doi: 10.1007/0-387-26206-7_10. [DOI] [PubMed] [Google Scholar]
- 28.Ilangovan G, Li H, Zweier JL, Kuppusamy P. Effect of carbogen-breathing on redox status of the RIF-1 tumor. Adv Exp Med Biol. 2003;510:13–7. doi: 10.1007/978-1-4615-0205-0_3. [DOI] [PubMed] [Google Scholar]
- 29.Kuppusamy P, Afeworki M, Shankar RA, Coffin D, Krishna MC, Hahn SM, Mitchell JB, Zweier JL. In vivo electron paramagnetic resonance imaging of tumor heterogeneity and oxygenation in a murine model. Cancer Res. 1998;58:1562–8. [PubMed] [Google Scholar]
- 30.Hou H, Lariviere JP, Demidenko E, Gladstone D, Swartz H, Khan N. Repeated tumor pO(2) measurements by multi-site EPR oximetry as a prognostic marker for enhanced therapeutic efficacy of fractionated radiotherapy. Radiother Oncol. 2009;91:126–31. doi: 10.1016/j.radonc.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen H, Ye D, Xie X, Chen B, Lu W. VEGF, VEGFRs expressions and activated. STATs in ovarian epithelial carcinoma. Gynecol Oncol. 2004;94:630–5. doi: 10.1016/j.ygyno.2004.05.056. [DOI] [PubMed] [Google Scholar]
- 32.Galanis A, Pappa A, Giannakakis A, Lanitis E, Dangaj D, Sandaltzopoulos R. Reactive oxygen species and HIF-1 signalling in cancer. Cancer Lett. 2008;266:12–20. doi: 10.1016/j.canlet.2008.02.028. [DOI] [PubMed] [Google Scholar]
- 33.Choi SO, Cho YS, Kim HL, Park JW. ROS mediate the hypoxic repression of the hepcidin gene by inhibiting C/EBPalpha and STAT3. Biochem Biophys Res Commun. 2007;356:312–7. doi: 10.1016/j.bbrc.2007.02.137. [DOI] [PubMed] [Google Scholar]
- 34.Simon AR, Rai U, Fanburg BL, Cochran BH. Activation of the JAK-STAT pathway by reactive oxygen species. Am J Physiol. 1998;275:C1640–52. doi: 10.1152/ajpcell.1998.275.6.C1640. [DOI] [PubMed] [Google Scholar]
- 35.Chatterjee M, Jain S, Stuhmer T, Andrulis M, Ungethum U, Kuban RJ, Lorentz H, Bommert K, Topp M, Kramer D, Muller-Hermelink HK, Einsele H, et al. STAT3 and MAPK signaling maintain overexpression of heat shock proteins 90alpha and beta in multiple myeloma cells, which critically contribute to tumor-cell survival. Blood. 2007;109:720–8. doi: 10.1182/blood-2006-05-024372. [DOI] [PubMed] [Google Scholar]
- 36.Klampfer L. Signal transducers and activators of transcription (STATs): novel targets of chemopreventive and chemotherapeutic drugs. Curr Cancer Drug Targets. 2006;6:107–21. doi: 10.2174/156800906776056491. [DOI] [PubMed] [Google Scholar]
- 37.Barton BE, Karras JG, Murphy TF, Barton A, Huang HF. Signal transducer and activator of transcription 3 (STAT3) activation in prostate cancer: direct STAT3 inhibition induces apoptosis in prostate cancer lines. Mol Cancer Ther. 2004;3:11–20. [PubMed] [Google Scholar]
- 38.Burdelya L, Catlett-Falcone R, Levitzki A, Cheng F, Mora LB, Sotomayor E, Coppola D, Sun J, Sebti S, Dalton WS, Jove R, Yu H. Combination therapy with AG-490 and interleukin 12 achieves greater antitumor effects than either agent alone. Mol Cancer Ther. 2002;1:893–9. [PubMed] [Google Scholar]
- 39.Burke WM, Jin X, Lin HJ, Huang M, Liu R, Reynolds RK, Lin J. Inhibition of constitutively active Stat3 suppresses growth of human ovarian and breast cancer cells. Oncogene. 2001;20:7925–34. doi: 10.1038/sj.onc.1204990. [DOI] [PubMed] [Google Scholar]
- 40.Wang J, Zhou JY, Wu GS. ERK-dependent MKP-1-mediated cisplatin resistance in human ovarian cancer cells. Cancer Res. 2007;67:11933–41. doi: 10.1158/0008-5472.CAN-07-5185. [DOI] [PubMed] [Google Scholar]
- 41.Zhong XS, Liu LZ, Skinner HD, Cao Z, Ding M, Jiang BH. Mechanism of vascular endothelial growth factor expression mediated by cisplatin in human ovarian cancer cells. Biochem Biophys Res Commun. 2007;358:92–8. doi: 10.1016/j.bbrc.2007.04.083. [DOI] [PubMed] [Google Scholar]
- 42.Selvendiran K, Koga H, Ueno T, Yoshida T, Maeyama M, Torimura T, Yano H, Kojiro M, Sata M. Luteolin promotes degradation in signal transducer and activator of transcription 3 in human hepatoma cells: an implication for the antitumor potential of flavonoids. Cancer Res. 2006;66:4826–34. doi: 10.1158/0008-5472.CAN-05-4062. [DOI] [PubMed] [Google Scholar]
- 43.Selvendiran K, Tong L, Vishwanath S, Bratasz A, Trigg NJ, Kutala VK, Hideg K, Kuppusamy P. EF24 induces G2/M arrest and apoptosis in cisplatin-resistant human ovarian cancer cells by increasing PTEN expression. J Biol Chem. 2007;282:28609–18. doi: 10.1074/jbc.M703796200. [DOI] [PMC free article] [PubMed] [Google Scholar]