

Summary
Cholesteryl ester accumulation by macrophages is a critical early event in atherogenesis. To test the hypothesis that sterol loading promotes foam cell formation and vascular disease by perturbing a network of interacting proteins, we used a global approach to identify proteins that are differentially expressed when macrophages are loaded with cholesterol in vivo. Our analysis revealed a sterol-responsive network that is highly enriched in proteins with known physical interactions, established roles in vesicular transport, and demonstrated atherosclerotic phenotypes in mice. Pharmacologic intervention with a statin or rosiglitazone and use of mice deficient in LDL receptor or apolipoprotein E implicated the network in atherosclerosis. Biochemical fractionation revealed that most of the sterol-responsive proteins resided in microvesicles, providing a physical basis for the network's functional and biochemical properties. These observations identify a highly integrated network of proteins whose expression is influenced by environmental, genetic, and pharmacological factors implicated in atherogenesis.
Introduction
Macrophages are essential effectors of innate and adaptive immunity (Gordon and Taylor, 2005). Via their scavenger receptors and other mechanisms, they clear bacterial pathogens and apoptotic cells (Suzuki et al., 1997) and release a wide range of cytokines and chemokines that orchestrate inflammation. Recent studies have also implicated macrophages in tumor progression, adipose tissue expansion, and insulin resistance (de Luca and Olefsky, 2008; Wellen and Hotamisligil, 2005). Moreover, these cells promote atherosclerosis by mediating the uptake of modified lipoproteins into the artery wall (Li and Glass, 2002).
When macrophages take up and degrade more lipoprotein-derived cholesterol than they excrete, they convert free cholesterol to cholesteryl ester, which accumulates as cytosolic lipid droplets that appear foamy under the light microscope (Brown and Goldstein, 1983). Genetic, biochemical, and clinical studies provide compelling evidence that macrophage foam cells are of central importance in atherogenesis (Li and Glass, 2002).
One key source of this excess sterol in humans is low density lipoprotein (LDL). Moreover, mice with targeted disruption of the LDL-receptor gene (Ldlr) have markedly elevated LDL levels when fed a Western-type diet rich in cholesterol and saturated fat (though mice normally have low levels of LDL and are resistant to atherogenesis). Consequently, Ldlr-/- mice rapidly develop atherosclerosis (Ishibashi et al., 1994).
Because macrophages are key players in atherosclerosis and many other conditions, it is likely that broad networks of interacting genes and proteins (Ghazalpour et al., 2004; Schadt and Lum, 2006; Lusis et al., 2008)—rather than simple linear pathways that affect only sterol balance—promote foam cell formation and atherogenesis. We therefore combined proteomics with bioinformatics to obtain a global view of macrophages' roles in vascular disease. This approach uncovered a network of proteins that is regulated when macrophages become foam cells. Dietary studies, pharmacological interventions, and work with genetically engineered mice linked this highly interconnected sterol-responsive network to atherosclerosis.
Results
Identification of macrophage sterol-responsive proteins
Previous studies have demonstrated that peritoneal macrophages of Ldlr-/- mice can be loaded with cholesteryl ester in vivo and that this model is relevant for studying the roles of foam cells in atherosclerosis (Li et al., 2004). We therefore placed Ldlr-/- mice on a chow (low-fat) or Western-type (high-fat) diet for 14 weeks (Fig. 1a), and then harvested macrophages from the peritoneal cavity. Compared with control cells, macrophages isolated from the mice on the Western diet had markedly higher levels of cellular cholesteryl ester mass (Fig. 1b), the biochemical hallmark of foam cells (Brown and Goldstein, 1986). Oil-red O staining and light microscopy confirmed that these macrophages were loaded with neutral lipid (Fig. 1c,d).
Figure 1. Proteomics analysis of conditioned medium harvested from control and sterol-loaded macrophages.
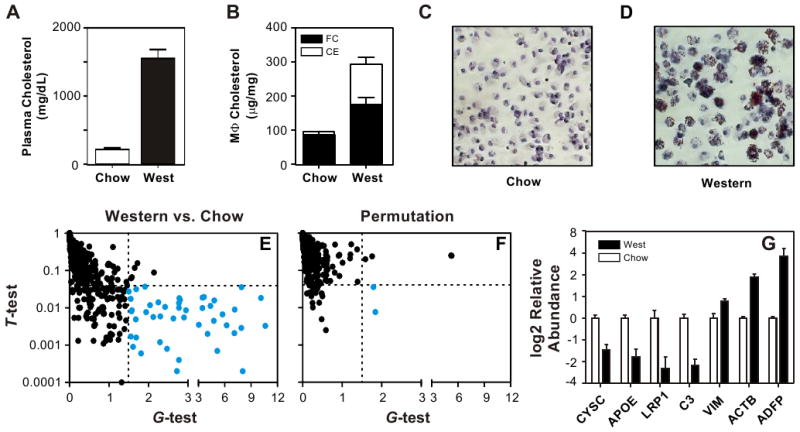
Macrophages were isolated from the peritoneum of male Ldlr-/- mice fed a chow (low fat) or Western (high fat) diet for 14 weeks. Panel A: Plasma cholesterol levels. Panel B: Macrophage (Mϕ) cholesterol levels. FC, free cholesterol; CE, cholesteryl ester. Panels C,D: Oil-red O staining of macrophages. Panel E: Medium conditioned by macrophages isolated from mice fed a chow or Western diet was digested with trypsin and analyzed by LC-ESI-MS/MS. Proteins were quantified by the total number of unique peptides (spectral counts) detected by MS/MS. Confidence intervals (dotted lines; G>1.5 and α=0.04) and the FDR were established by permutation analysis. Proteins differing in relative abundance between control and foam cells (blue ●) were identified as those that exhibited a significant difference in the total number of peptides, as assessed by both the t-test (p-value) and G-test (G-test). Panel F: Representative example of a randomly selected permutation analysis (false positive result, blue ●). Panel G: Biochemical validation of differential protein expression by macrophage foam cells. Equal amounts of protein from medium conditioned by control cells or foam cells were subjected to SDS-PAGE, using 4%–12% gradient gels. Proteins were transferred to PVDF membranes, and probed with antibodies raised against murine CYSC, APOE, C3, VIM, ACTB, LRP1, or ADFP. Immunoblots were quantified by densitometry and statistical significance was assessed using a two-tailed, Student's t-test. Results are means and standard deviations. See also Table S1.
We used LC-ESI-MS/MS analysis to identify 777 proteins with high confidence in the medium of control and/or sterol-loaded macrophages (Table S1). To determine which of those proteins changed their relative abundance when macrophages were sterol-loaded in vivo, we used the G-test and t-test to find significant differences in spectral counts (Fu et al., 2008; Liu et al., 2004; Old et al., 2005), a measure of relative protein concentration (Fig. 1e). We estimated the false discovery rate (FDR) by using the same statistical tests with all possible permutations of the data. Permutation analysis revealed that G>1.5 (G-test) and p<0.04 (t-test) yielded the most true-positive protein identifications, with an estimated FDR of 6.3% (data not shown). Using these stringent statistical criteria (Fig. 1e), we identified 46 proteins that were expressed at a different level in medium of macrophage foam cells than in medium of control cells isolated from Ldlr-/- mice fed a chow diet (Fig. 2, Table S1). In contrast, only 2 proteins resided in this region of the graph in a representative random permutation analysis (Fig. 1f), which is consistent with our estimated FDR.
Figure 2. Proteins differentially expressed by macrophage foam cells.

Proteins differing in relative abundance in the conditioned medium of control and foam cells were identified as described in the legend to Fig. 1. A positive or negative value for the G-test indicates an increased or decreased level of protein expression relative to the control. See also Table S1.
We assessed the effectiveness of our analytical strategy by using commercially available antibodies to quantify the relative abundance of 7 of the 46 macrophage proteins that met our statistical criteria: apolipoprotein E (APOE), cystatin C (CYSC), complement factor C3 (C3), vimentin (VIM), β-actin (ACTB), LDL receptor-related protein-1 (LRP1), and adipophilin (ADFP). Immunoblot analysis of conditioned media of control and cholesteryl ester-loaded macrophages (Fig. 1g) verified significant differences in the levels of all 7 proteins (two-tailed, Student's t-test: APOE, p=0.001; CYSC, p=0.002; C3, p=0.001; VIM, p=0.008; ACTB, p=0.0006; LRP1, p=0.03; ADFP, p=0.02).
Differentially expressed proteins form a network
We call the 46 proteins that are differentially expressed by foam cells “sterol-responsive,” because their relative abundance changes significantly when Ldlr-/- macrophages become loaded with cholesteryl ester in vivo. We assessed the potential functional significance of these proteins in three ways. First, we searched for known physical interactions among them, using Ingenuity systems (Calvano et al., 2005) and the BIND, DIP, MIPS, IntAct, BioGRID, and MINT databases. These analyses revealed a protein-protein interaction network of 26 proteins (nodes) and 30 interactions (edges) (Fig. 3), predicting multiple interactions among many of the 46 sterol-responsive proteins.
Figure 3. The macrophage sterol-responsive network (MSRN).

Panel A: A protein–protein interaction network was constructed, using the 46 proteins that were differentially expressed (upregulated, red; downregulated, green) by macrophage foam cells isolated from Ldlr-/- mice. GO analysis of the network revealed modules enriched in proteins implicated in lipid binding, cytoskeletal regulation, and vesicle-mediated transport (p=0.01, 0.001 and 0.003, respectively; Fisher's exact test with Benjamini-Hochberg correction). Proteins that associate with an atherosclerotic phenotype (circled in blue) in genetically engineered mice or with myeloid specific expression/deletion as assessed by bone marrow transplantation (BMT, **) were identified with PubMatrix. Note that 10 of 16 sterol-responsive proteins that were not previously shown to interact physically or functionally with other MSRN proteins, termed previously unassigned, reside in the microvesicle fraction. Panel B: Comparison of MSRN protein expression in media and isolated microvesicles. A positive or negative value for the G-test indicates an increased or decreased level of protein expression relative to the control. See also Table S2.
Second, we used Gene Ontology annotations to organize the 46 proteins into functional modules (Fig. 3). This approach identified significant enrichment (relative to the entire mouse genome) in 3 modules: cytoskeletal regulation (p=0.001), vesicle-mediated transport (p=0.003), and lipid binding (p=0.01). While alterations in lipid metabolism are known to regulate foam cells, the potential roles of the cytoskeleton and vesicular transport in foam cell formation have received little attention, though vesicular transport is thought to play a key role in the intracellular trafficking of lipids.
Third, we used an algorithm to systematically search PubMed for documented atherosclerotic phenotypes in mice with genetic manipulations (e.g., targeted mutagenesis, transgenic overexpression) of proteins we detected in macrophage-conditioned media. Remarkably, mice that overexpress or lack any of 10 of the 46 sterol-responsive proteins have atherosclerotic phenotypes (Fig. 3; indicated by a blue border). In contrast, only 21 of the other 731 proteins detected in macrophage-conditioned media have demonstrated atherogenic phenotypes in mice (Table S2). This greater than 8-fold enrichment of proteins associated with atherosclerosis in the sterol-responsive proteins was highly significant (p=3×10-6, Fisher's exact test). Moreover, previous studies have shown that the atherosclerotic phenotypes of 6 of the 10 sterol-responsive proteins were recapitulated in mice by transplanting bone marrow from genetically engineered mice into wild-type mice. Thus, macrophage-specific expression of these proteins may contribute to atherogenesis.
Collectively, these observations identify a set of 46 proteins that respond to sterol-loading of macrophages in vivo in a coordinated fashion. We term this set the “macrophage sterol-responsive network” (MSRN; Fig. 3a), because these proteins are: i) coordinately regulated when Ldlr-/- macrophages become foam cells in vivo, ii) integrated through predicted protein–protein interactions, iii) functionally overrepresented in proteins involved in lipid binding, cytoskeletal regulation, and vesicle-mediated transport, and iv) highly enriched in proteins with causative roles in atherogenesis.
Most MSRN proteins localize to vesicles
Shed microvesicles are enriched in cholesterol and proteins that are involved in vesicular transport and cytoskeletal regulation (Simpson et al., 2008), and these two functional categories are significantly enriched in the MSRN. To test the hypothesis that a subset of MSRN proteins resides in microvesicles, we centrifuged macrophage-conditioned medium at 100,000×g and analyzed the pelleted material by LC-ESI-MS/MS. The microvesicle fraction contained 32 of the 46 MSRN proteins (Table S3). Moreover, 8 of 12 proteins that lacked previously known binding or functional associations with other MSRN proteins (Fig. 3a) were detected in the pelleted material, suggesting that they were associated with other members of the MSRN in microvesicles. Remarkably, the relative abundance of 26 of 32 MSRN proteins in the microvesicle fraction changed significantly in a manner that mirrored that of macrophage-conditioned medium (Fig. 2). These observations strongly suggest that most sterol-responsive proteins in macrophage-conditioned medium are physically co-assembled into one or more populations of vesicles. This hypothesis offers a structural explanation for the MSRN's enrichment in known physical interactions and functional annotations.
Anti-atherosclerotic interventions target the MSRN
If the MSRN orchestrates a molecular network involved in atherogenesis, interventions aimed at treating atherosclerosis should specifically affect that network. To test this notion, we harvested macrophages from Ldlr-/- mice that had received 100 mg/kg/day simvastatin or 10 mg/kg/day rosiglitazone for the last 2 weeks of the 14-week Western diet. Other investigators have demonstrated that these interventions retard atherosclerosis in Ldlr-/- mice without altering levels of circulating lipoproteins (Chen et al., 2002; Li et al., 2000).
Neither statin nor rosiglitazone therapy affected plasma cholesterol levels or plasma lipoprotein profiles (Fig. S1), but they reduced macrophage cholesterol accumulation by ∼40% (Fig. 4a). In striking contrast, both interventions markedly altered the expression pattern of MRSN proteins in macrophages isolated from Ldlr-/- mice. Indeed, only 2 (statin) and 4 (rosiglitazone) of the 46 MSRN proteins were still differentially expressed by macrophages isolated from Ldlr-/- mice that had received the high-fat diet and one of the two interventions (Fig. 4b,c). Neither intervention appreciably altered levels of non-MSRN proteins in macrophage- conditioned medium (Fig. 4d), indicating that the effect on the MSRN was specific. These data demonstrate that two different pharmacological interventions, each of which inhibits atherosclerosis without affecting plasma cholesterol levels, specifically target the MSRN. Thus, dysregulation of this network might promote the development of atherogenesis in vivo, and pharmacologic interventions that retard atherosclerosis might act in part by normalizing expression of the MSRN.
Figure 4. Impact of anti-atherosclerotic interventions on the MSRN.

Ldlr-/- mice were fed a Western diet for 14 weeks with or without simvastatin (Statin) or rosiglitazone (Rosi) for the final 2 weeks. Panel A: Macrophage (Mϕ) cholesterol levels; FC, free cholesterol; CE, cholesteryl ester. Results are means and standard deviations. Panels B,C: Differentially expressed proteins of macrophages isolated from Ldlr-/- mice fed a chow diet vs. simvastatin-treated (Panel B) or rosiglitazone-treated (Panel C) Ldlr-/- mice fed a Western diet. Proteins identified as differentially expressed by macrophages isolated from chow and Western diet-fed mice (Fig. 1e) are shown in blue. Panel D: Relative abundance of proteins detected in conditioned media of macrophages was assessed by the G-test. Results represent chow vs. Western diet (center axis), Western diet plus statin vs. Western diet (left axis), or Western diet plus rosiglitazone vs. Western diet (right axis). A positive or negative value for the G-test indicates an increased or decreased level of protein expression in the first sample relative to the second. Proteins in all panels are sorted in descending order based on G-test values of the Western vs. chow diets (center axis). Bars at the same vertical level correspond to the same protein. Red and green areas highlight proteins that reside in the MSRN. See also Table S1.
Apolipoprotein E is a key protein in the MSRN
Because simvastatin or rosiglitazone therapy markedly alters the expression pattern of MSRN proteins in macrophages isolated from Ldlr-/- mice while only modestly affecting macrophage sterol levels (Fig. 4a), cholesterol accumulation alone is unlikely to explain MSRN regulation. Instead, a protein that interacts with many other proteins might exert widespread effects on its parent network if its expression changed (Chen et al., 2008). APOE, which is underexpressed in the MSRN and is highly anti-atherogenic in mice (Bellosta et al., 1995; Linton et al., 1995), might be one such protein. To test the proposal that APOE regulates the MSRN, we harvested macrophages from Apoe-/- mice fed a low-fat or Western diet, incubated them for 6 h, and analyzed the conditioned media with proteomic methods.
Diet-induced changes in plasma cholesterol and lipoproteins (Fig. S1) were similar in the Apoe-/- and Ldlr-/- mice. Moreover, >96% of the proteins detected in media conditioned by macrophages from the two mouse strains were the same. Thus, the overall pattern of protein expression was similar in the two genotypes. In contrast, Apoe-/- macrophages isolated from mice on the Western diet displayed significantly higher cholesteryl ester levels than Ldlr-/- macrophages (Fig. 5a). This observation is consistent with APOE's well-established role in promoting cholesterol efflux from macrophages (Langer et al., 2000).
Figure 5. Role of APOE in regulation of the MSRN.
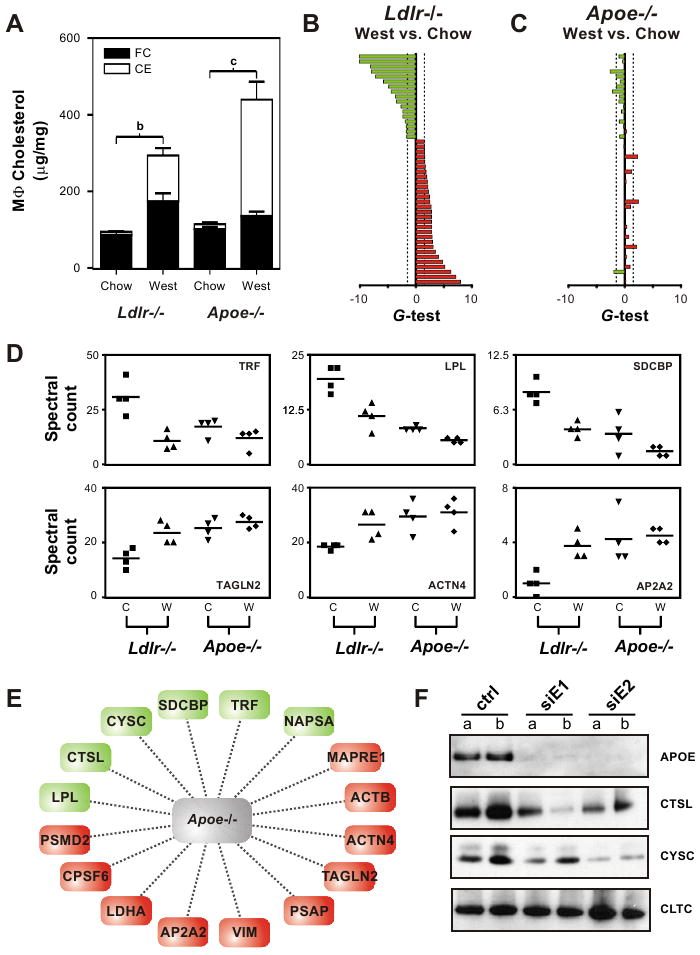
Macrophages were isolated from the peritoneum of Ldlr-/- or Apoe-/- mice fed a chow or Western diet for 14 weeks. Panel A: Macrophage cholesterol (MϕC) levels. FC; free cholesterol; CE, cholesteryl ester. Panels B,C: Differential expression of MSRN proteins. Red, increased protein level; green, decreased protein level. Proteins are in the same order, top to bottom, as in Fig. 2. Panel B: Ldlr-/- macrophages, chow vs. Western diet. Panel C: Apoe-/- macrophages, chow vs. Western diet. Panel D: Quantification of MSRN proteins by LC-ESI-MS/MS and spectral counting of macrophages harvested from Ldlr-/- or Apoe-/- mice fed a chow (C) or Western (W) diet for 14 weeks. Panel E: Subset of MSRN proteins that are dysregulated in macrophages harvested from Apoe-/- mice fed a low-fat diet. Panel F: Immunoblot analysis of conditioned medium of macrophages harvested from C57BL/6J mice and treated with siRNA to APOE. See also Figure S3.
Despite this difference in cholesteryl ester content, only 6 proteins responded to sterol accumulation in Apoe-/- macrophages (Table S4; estimated FDR=6.4%). Moreover, the extent of MSRN regulation imposed by cholesterol loading was substantially attenuated in Apoe-/- mice (compare Figs. 5b and 5c).
These observations suggest that macrophages need to express APOE to modulate MSRN expression in vivo, raising the possibility that APOE is an important node in the network. To test this proposal, we compared protein expression levels in conditioned media of macrophages isolated from Apoe-/- and Ldlr-/- mice. The expression of many MSRN proteins in the Apoe-/- macrophages from mice on the chow diet strongly resembled that in Ldlr-/- macrophages loaded with cholesteryl ester (Fig. 5d). Thus, levels of ∼40% of the MSRN proteins in Ldlr-/- foam cells and Apoe-/- control cells were statistically indistinguishable (p>0.1, two-tailed, Student's t-test; Fig. 5e, Table S5). This suggests that APOE deficiency regulates the expression—either stimulation or repression—of a subset of MSRN proteins even in the absence of cholesterol loading.
To further test the hypothesis that APOE regulates the MSRN in the absence of cholesterol loading, we isolated macrophages from wild-type C57BL/6J mice. We then acutely lowered the cells' APOE level by treating them with siRNA duplexes, and monitored changes in APOE, CTSL, CYSC, and CLTC levels in the medium. As predicted from the proteomic analysis of Apoe-/- macrophages, immunoblot analysis confirmed that CTSL and CYSC levels fell when APOE release into medium was suppressed, whereas the level of CLTC, a protein that was not regulated by APOE (Table S5), remained unchanged (Fig. 5f). We obtained similar results when we isolated macrophages from Ldlr-/- mice, suggesting that LDLR is not an important regulator of the MSRN (Fig. S2).
These observations provide evidence for coordinate regulation of MSRN proteins. As a further test, we treated peritoneal macrophages isolated from Ldlr-/- mice with control siRNA duplexes or siRNA duplexes specific for CTSL, CYSC, or C3. Then we used immunoblotting to monitor the expression of ACTB, APOE, CLTC, CTSL, CYSC, and C3 protein in macrophage-conditioned media (Fig. S3). siRNA-mediated knockdown of CYSC significantly reduced ACTB (p=0.02), CLTC (p=0.006), and C3 (p=0.04) levels without altering APOE or CTSL levels. On the other hand, downregulation of CTSL lowered C3 expression (p=0.04) without affecting ACTB, APOE, CLTC, or CYSC levels. Finally, reducing C3 expression elevated APOE levels (p=0.05) and lowered CTSL expression (p=0.003), but had no effect on ACTB, C3, CLTC, and CYSC levels. Taken together, these studies provide strong evidence for coordinate regulation of MSRN proteins.
Statins normalize the MSRN in Ldlr-/- but not Apoe-/- macrophages
Our observations indicate that APOE is an important regulator of the MSRN. Moreover, previous studies have demonstrated that simvastatin retards atherosclerosis in Ldlr-/- mice but not in Apoe-/- mice (Wang et al., 2002). Thus, anti-atherosclerotic interventions might normalize the network by restoring APOE levels.
To test this idea, we compared the effects of simvastatin therapy (100 mg/kg/day for 2 weeks) on the MSRN in macrophages isolated from Ldlr-/- and Apoe-/- mice fed a Western diet. Statin therapy reversed the expression pattern of MSRN proteins in macrophages isolated from Ldlr-/- mice but not in those of Apoe-/- mice (Fig. 6a,b). This difference could not be explained by differential cholesterol accumulation, because simvastatin therapy reduced macrophage cholesterol levels to a similar extent (∼40%) in both genetic backgrounds (Fig. S4). These findings are consistent with the proposal that APOE regulates the MSRN to inhibit atherogenesis.
Figure 6. APOE expression in atherosclerotic lesions of Ldlr-/- mice.
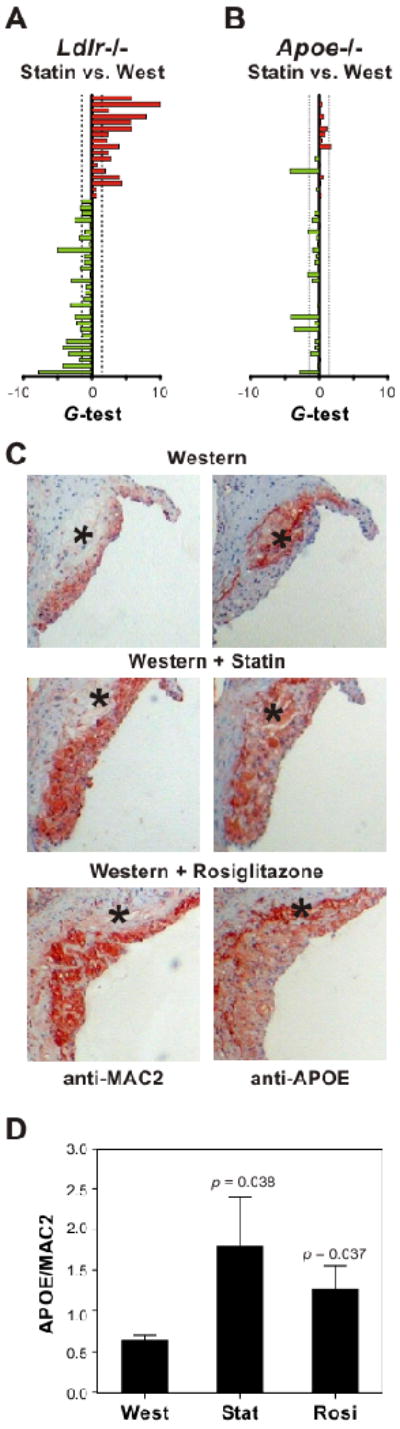
Ldlr-/- or Apoe-/- mice were fed a Western diet for 14 weeks or a Western diet plus simvastatin (Statin) or rosiglitazone (Rosi) for the final 2 weeks of the regimen. Panels A,B: Differential expression of MSRN proteins by macrophages isolated from statin-treated Ldlr-/- or Apoe-/- mice fed a Western diet. Red, increased protein level; green, decreased protein level. Proteins are in the same order, top to bottom, as in Fig. 2. Panel C: Immunohistochemical staining of aortic sinus sections isolated from Ldlr-/- mice on the different regimens. Adjacent sections were immunostained with antibodies to APOE or MAC2 (a macrophage marker). *, necrotic core. Images, 10×. Panel D: Quantification of immunohistochemical staining for APOE and MAC2. Results (N=5 per group) are means and standard deviations. See also Figure S5.
Anti-atherosclerotic interventions restore lesion APOE expression
Taken together, our data suggest that restoring levels of macrophage APOE normalizes the MSRN expression pattern and that this normalization is a critical component of simvastatin's action. We also found that APOE was one of the most strongly downregulated MSRN proteins in cells harvested from Ldlr-/- mice fed a Western diet. To explore the possibility that simvastatin and rosiglitazone inhibit atherosclerosis partly by increasing APOE levels in artery wall macrophages, we used immunohistochemistry to investigate expression of APOE and the macrophage marker MAC2 in aortic lesions of Ldlr-/- mice fed a Western diet for 14 weeks.
The lesions contained high levels of APOE. However, the bulk of immunoreactive material localized to their necrotic cores (Fig. 6c, Fig. S5). Treating the animals for 2 weeks with simvastatin or rosiglitazone markedly increased macrophage APOE protein levels (simvastatin, p=0.04; rosiglitazone, p=0.03) without appreciably altering the amount of APOE detected in the necrotic core (Fig. 6c-d, Fig. S5). Collectively, these observations provide strong evidence that macrophage foam cells in the artery wall, like foam cells isolated from the peritoneal cavity of hypercholesterolemic Ldlr-/- mice, have abnormally low APOE levels that are normalized by anti-atherosclerotic interventions.
Discussion
In contrast to traditional biochemical studies that focus on individual proteins, our approach seeks to identify protein networks that coherently respond to environmental, pharmacological, and genetic interventions that promote cardiovascular disease. We therefore used tandem mass spectrometry in concert with bioinformatics to obtain a comprehensive view of the shed and secreted proteome of macrophage foam cells generated in vivo. By determining the known physical interactions and biological functions of differentially expressed proteins, we identified the macrophage sterol-responsive network (MSRN), a highly integrated network of proteins that are coordinately regulated by sterol loading.
Networks of highly interconnected proteins can develop new, disease-causing properties when strongly perturbed (Lusis et al., 2008; Chen et al., 2008), and two lines of evidence suggest that modulating the expression of key MSRN proteins can influence atherosclerosis. First, relative to the overall shed and secreted macrophage proteome, the MSRN was greatly enriched in proteins linked to established atherosclerotic phenotypes in transgenic and knock-out mice. Moreover, levels of APOE, C3, CYSC, LRP1, LYZ, and MFGE8 were lower when macrophages were loaded with sterol in vivo, and atherosclerosis is enhanced in mice that lack any of those proteins (Bellosta et al., 1995; Linton et al., 1995; Buono et al., 2002; Bengsston et al., 2005; Overton et al., 2007; Liu et al., 2006; Ait-Ouefella et al., 2007). In contrast, ADFP levels were enhanced in cholesterol-loaded macrophages, and transgenic expression of ADFP promotes atherosclerosis in mice (Paul et al., 2008). These observations further support our proposal that MSRN proteins are coordinately regulated and that network dysregulation is important for foam cell formation and atherogenesis.
Second, treating Ldlr-/- mice with rosiglitazone, which retards atherosclerosis without affecting plasma cholesterol levels in this genetic background (Li et al., 2000), reversed the protein expression pattern that is seen when macrophages are loaded with sterol in vivo. We observed a similar reversal in macrophages isolated from statin-treated Ldlr-/- mice but not in macrophages isolated from statin-treated Apoe-/- mice. Importantly, statins are known to inhibit atherosclerosis in Ldlr-/- but not Apoe-/- mice (Wang et al., 2002). Thus, our results with two different pharmacological interventions in two different genetic backgrounds support a model in which sterol-responsive proteins in macrophages form a highly interconnected network, the MSRN, that promotes atherosclerosis when dysregulated.
APOE, which plays a key role in macrophage cholesterol homeostasis, is a particularly striking example of an MSRN protein. When this protein is missing from macrophages, mice suffer from accelerated atherosclerosis (Bellosta et al., 1995; Linton et al., 1995). Classical studies of cultured macrophages have demonstrated that APOE secretion is upregulated by sterol loading with acetyl-LDL (Brown and Goldstein, 1983). In contrast, we found that APOE was one of the most downregulated proteins in the MSRN of macrophage foam cells harvested from the peritoneum. However, APOE expression increased markedly when we incubated cultured peritoneal macrophages of Ldlr-/- mice with acetyl-LDL (Fig. S6). Other investigators have also noted significant differences in the expression patterns of cultured macrophages and foam cells generated in vivo. For example, Li et al. found that PPARα, PPARβ, and PPARγ agonists induced cultured macrophages to express ABCA1 and LXRα (Li et al., 2000). In striking contrast, all three agonists failed to influence the expression of those genes in atherosclerotic lesions or in foam cells derived from hypercholesterolemic mice. These observations support the proposal that macrophages of Ldr-/- mice loaded with cholesteryl ester in vivo provide a relevant model for studying foam cells' roles in atherosclerosis.
Because APOE is one of the most downregulated MSRN proteins, statins and thiazolidinediones might inhibit atherogenesis in part by increasing APOE expression by artery wall macrophages. Indeed, in immunohistochemical studies of Ldlr-/- mice fed a Western diet, APOE was readily visualized in the acellular necrotic core of atherosclerotic lesions but was essentially undetectable in macrophages. In contrast, it was readily detectable in both locations in statin- or rosiglitazone-treated animals. These results imply that the APOE levels are likely to be diminished in artery wall foam cells of hypercholesterolemic Ldlr-/- mice and that this effect is partly reversed by statin or rosiglitazone treatment. Thus, our observations support the proposal that pharmacological interventions target the MSRN in Ldlr-/- mice and that the ability to regulate the network is one atheroprotective effect of statin and rosiglitazone in this model of hypercholesterolemia.
Unexpectedly, we found that expression of one-third of the MSRN proteins was dysregulated in macrophages isolated from Apoe-/- mice on the low-fat diet. These cells showed little evidence of sterol accumulation, implying that APOE modulates the expression of a subset of MSRN proteins and that the network is dysfunctional in macrophages lacking APOE. Genetic ablation of other highly interconnected proteins within the MSRN might similarly potentiate network dysregulation and atherogenesis. This hypothesis is strengthened by the demonstration that mice deficient in ADFP (Paul et al., 2008), APOE (Bellosta et al., 2008), LPL (Babaev et al., 2008), LRP1 (Overton et al., 2008), LYZ (Liu et al., 2008), or MFGE8 (Ait-Ouefella et al., 2008), all of which map to the network, have macrophage-specific phenotypes. Moreover, we found that attenuating levels of individual proteins in the MSRN with siRNA affected the expression of other network proteins.
Proteins involved in lipid binding, cytoskeletal regulation, and vesicle-mediated transport were overrepresented in the MSRN, and mice lacking proteins implicated in those functions have atherosclerotic phenotypes (Buono et al., 2002; Overton et al., 2007; Ait-Oufella et al., 2007; Feil et al., 2004). The network was also greatly enriched in proteins with known physical interactions, raising the possibility that macromolecular assemblies might partly account for the link between sterol-responsive proteins and functional categories. Remarkably, when macrophage-conditioned medium was centrifuged at 100,000×g, more than two-thirds of the MSRN proteins localized to the pellet, indicating that they likely resided in the microvesicle fraction. Colocalization in microvesicles, which transfer proteins and RNA between neighboring cells and has been implicated in thrombosis, cytokine release, and inflammation (Cocucci et al., 2009), would readily account for the enrichment of physical and functional interactions we observed in MSRN proteins.
We therefore propose that the atherogenic actions of cholesterol-loaded macrophages are an emergent property that results when the normal balance of MSRN proteins in microvesicles is perturbed. We further suggest that certain dietary factors or genetic variations can disturb this network, thereby promoting vascular disease. By integrating mouse and human data, we hope to better understand the MSRN's role in foam cell formation, with the long-term goal of identifying therapeutic interventions for targeting networks rather than individual proteins.
Experimental Procedures
Macrophage foam cell formation
Male Ldlr-/- and Apoe-/- mice on the C57BL/6J genetic background received a chow (4% fat (w/w); PicoLab, Rodent diet 20:50503) or Western diet (21% fat, 1.25% cholesterol; Harlan Teklad, #TD96121) for 14 weeks. For anti-atherosclerotic interventions, animals were fed the Western diet to which simvastatin (100 mg/kg/day; Merck & Co Inc.) or rosiglitazone (10 mg/kg/day; GlaxoSmithKline) were added during the last 2 weeks of the diet. Four independent biological replicates were obtained for each experimental condition. Peritoneal macrophages were harvested from the mice 5 days after thioglycolate was injected11. Cells were washed with phosphate-buffered saline (PBS), seeded into T-75 flasks (20×106/flask), incubated at 37°C for 2 h in serum-free Dulbecco's minimum essential medium (DMEM), and washed 3 times with PBS. Macrophages were then cultured for 6 h in DMEM.
Protein isolation
Macrophage-conditioned medium (∼10 μg protein/mL) was collected, clarified by centrifugation (5 min at 1,000×g), supplemented with 0.02% sodium deoxycholate and 20% trichloroacetic acid, and incubated overnight at 4°C. Proteins were harvested by centrifugation (15,000×g for 30 min at 4°C). The protein pellet was washed twice with ice-cold acetone, reconstituted in digestion buffer (0.1% Rapigest (Waters Corp.), 50 mM Tris-HCl, pH 8.8), and then reduced, alkylated, and digested overnight at 37°C with sequencing-grade trypsin (1:50, w/w, trypsin/protein; Promega). Tryptic digests were mixed with acetic acid (1:1, v/v) and subjected to solid-phase extraction on a C18 column (HLB, 1 mL; Waters Corp.) according to the manufacturer's protocol. Fractions containing peptides were dried under vacuum and resuspended in 0.3% acetic acid/5% acetonitrile (1 mg protein/mL) for analysis.
Liquid chromatography-electrospray ionization-tandem MS (LC-ESI-MS/MS)
Tryptic digests (2 μg protein) were injected in duplicate into a trap column (Paradigm Platinum Peptide Nanotrap, 0.15×50 mm; Michrom Bioresources, Inc.) and desalted for 5 min with 5% acetonitrile, 0.1% formic acid (50 μL/min). Peptides were then eluted onto an analytical reverse-phase column (0.150×150 mm, 5 μm beads; Magic C18AQ, Michrom Bioresources, Inc.) and separated at a flow rate of 1 μL/min over 180 min, using a linear gradient of 5% to 35% buffer B (90% acetonitrile, 0.1% formic acid) in buffer A (5% acetonitrile, 0.1% formic acid). Mass spectra were acquired in the positive ion mode, using electrospray ionization and a linear ion trap mass spectrometer (LTQ, Thermo Electron Corp.) with data-dependent acquisition (Vaisar et al., 2007). MS/MS scans were obtained on the 8 most abundant peaks in each survey MS scan.
Peptide and protein identification
MS/MS spectra were searched against the mouse International Protein Index (IPI) database (version 2006/04/18) (Kerset et al., 2004), using the SEQUEST search engine with the following search parameters: unrestricted enzyme specificity, 2.8 amu precursor ion mass tolerance, 1.0 amu fragment ion mass tolerance, fixed Cys alkylation, and variable Met oxidation. SEQUEST results were further validated with PeptideProphet (Keller et al., 2002) and ProteinProphet (Nesvizhskii et al., 2003), using an adjusted probability of ≥0.90 for peptides and ≥0.96 for proteins. Proteins considered for analysis had to be identified in every biological replicate of at least one biological condition. When MS/MS spectra could not differentiate between protein isoforms, all isoforms were included in the analysis.
Protein quantification
Proteins detected by LC-ESI-MS/MS were quantified by spectral counting (the total number of MS/MS spectra detected for a protein). Two replicate injections for each sample were averaged to obtain spectral counts.
Microvesicles
Microvesicles were isolated from macrophage-conditioned medium, using differential centrifugation (Hegmans et al., 2004). Conditioned medium was pre-cleared by centrifugation at 1000×g for 10 min, two spins at 4000×g for 10 min, and one spin at 10,000×g for 30 min. Microvesicles were isolated from the supernatant by ultracentrifugation (two 100,000×g spins for 1 h). The protein pellet was washed with PBS after each spin. Microvesicles were reconstituted in digestion buffer (0.1% Rapigest (Waters Corp.), 50 mM Tris-HCl, pH 8.8), digested with trypsin, and analyzed with MS/MS. Results are the average of duplicate analyses of 4 pooled microvesicle isolations for each condition. Differences in spectral counts between the conditions were assessed with the G-test (Sohkal and Rohlf, 1995).
Biochemical assays
Cholesterol levels were determined using cholesterol oxidase and a fluorescence-based peroxidase assay (Invitrogen). Protein concentration of macrophage-conditioned medium was determined by the Bradford assay, with albumin as the standard.
siRNA
Adhesion purified peritoneal macrophages from wild-type C57BL/6J mice were transfected with control siRNA (30 nM, Ambion) or two siRNAs specific for APOE (30 nM, Ambion) using the DeliverX Plus siRNA transfection kit (Panomics). Cells were washed 3 times with PBS and incubated in serum-free DMEM for 24 h. Macrophage-conditioned medium was then collected and levels of APOE, CTSL, CYSC, and CLTC quantified by immunoblot analysis.
Immunoblot analysis
Macrophage-conditioned medium was subjected to SDS-PAGE on 4%–12% gradient gels, transferred to PVDF membranes, and probed with antibodies (0.5 μg/mL) raised against murine ACTB (Abcam), ADFP (Abcam), APOE (Abcam), C3 (Abcam), CTSL (R&D Systems), CYSC (Abcam), and LRP1 (Abcam). Proteins were quantified by densitometry, using Quantity One software (Biorad).
Analysis of atherosclerotic lesions
For immunocytochemical staining, deparaffinized sections from lesions in the aortic sinus were rinsed in PBS, incubated for 10 min in Peroxo-Block (Zymed Laboratories Inc., San Francisco, California, USA), and boiled for 10 min in 0.01 M citrate buffer (pH 6.0). Adjacent sections were incubated overnight at 4°C with rat anti-MAC2 (Cedarlane, 1:2000) or rabbit anti-APOE (Abcam, 1:500) antibody in PBS containing 1% BSA. Antibodies were detected with a peroxidase-chromagen system (Dako NovaRed) in sections counterstained with hematoxylin and eosin. APOE and MAC2 immunoreactivity were quantified in lesions using Image Pro Plus (Media Cybernetics).
Functional annotation
Functional enrichments in gene ontology annotations in the MSRN, relative to the mouse genome, were identified using the Bingo 2.0 plugin in Cytoscape (V2.5.2) (Maere et al., 2005). Statistical significance was assessed using the Fisher's exact test with Benjamini-Hochberg false discovery rate correction (Benjamini and Hochberg, 1995). To identify macrophage proteins associated with an atherosclerotic phenotype in mice, we conducted keyword searches, using PubMatrix (Becker et al., 2003). The search term was “protein name”, and the modifier terms were “atherosclerosis and knockout”, “atherosclerosis and deficiency”, and “atherosclerosis and expression”. All positive results were verified by manual inspection.
Protein interaction networks
Protein interaction networks were established from a manually compiled database based on >200,000 full-text, peer-reviewed articles with Ingenuity Systems (Calvano et al., 2005). Interactions were validated using the BIND, DIP, MIPS, IntAct, BioGRID, and MINT databases.
Statistical analysis
Spectral count differences between foam cells and control cells were assessed using a two-tailed, Student's t-test and the G-test (Old et al., 2005). The G-test evaluates the null hypothesis that observed values are due to random sampling from a distribution with a given expected value, using an approximation of the χ2 distribution with one degree of freedom. Permutation analysis was used to empirically estimate the false positive rate. The false discovery rate, FDR, was estimated as the ratio of the false positive rate to that of the sum of the false and true positive rates (Benjamini and Hochberg, 1995). Significance cutoff values for the G-statistic and t-test were determined by minimizing the empirical FDR and maximizing the number of differentially expressed proteins.
Supplementary Material
Acknowledgments
We thank Alan Attie (University of Wisconsin) and Stan Fields and Bill Parks at the University of Washington for critical review of the manuscript. This research was supported by grants from the National Institutes of Health (HL030086, HL018645, HL074223, HL078527, HL086798). LB and TV were supported by a Canadian Institutes of Health Research Fellowship Award (LB) and a Pilot and Feasibility Award (TV) from the Diabetes and Endocrinology Research Center. Mass spectrometry experiments were supported by the Mass Spectrometry Resource (Department of Medicine), the Proteome Resource (School of Medicine), and the Mass Spectrometry Core (Diabetes and Endocrinology Research Center) at the University of Washington.
Abbreviations
- apo
apolipoprotein
- FDR
false-discovery rate
- LC-ESI-MS/MS
liquid chromatography-electrospray ionization tandem MS
- LDL
low density lipoprotein
- MS
mass spectrometry
- MSRN
macrophage sterol-responsive network
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ait-Oufella H, Kinugawa K, Zoll J, Simon T, Boddaert J, Heeneman S, Blanc-Brude O, Barateau V, Potteaux S, Merval R, et al. Lactadherin deficiency leads to apoptotic cell accumulation and accelerated atherosclerosis in mice. Circulation. 2007;115:2168–2177. doi: 10.1161/CIRCULATIONAHA.106.662080. [DOI] [PubMed] [Google Scholar]
- Babaev VR, Patel MB, Semenkovich CF, Fazio S, Linton MF. Macrophage lipoprotein lipase promotes foam cell formation and atherosclerosis in low density lipoprotein receptor-deficient mice. J Biol Chem. 2000;275:26293–26299. doi: 10.1074/jbc.M002423200. [DOI] [PubMed] [Google Scholar]
- Becker KG, Hosack DA, Dennis G, Jr, Lempicki RA, Bright TJ, Cheadle C, Engel J. PubMatrix: a tool for multiplex literature mining. BMC Bioinformatics. 2003;4:61. doi: 10.1186/1471-2105-4-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellosta S, Mahley RW, Sanan DA, Murata J, Newland DL, Taylor JM, Pitas RE. Macrophage-specific expression of human apolipoprotein E reduces atherosclerosis in hypercholesterolemic apolipoprotein E-null mice. J Clin Invest. 1995;96:2170–2179. doi: 10.1172/JCI118271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengtsson E, To F, Håkansson K, Grubb A, Brånén L, Nilsson J, Jovinge S. Lack of the cysteine protease inhibitor cystatin C promotes atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2005;25:2151–2156. doi: 10.1161/01.ATV.0000179600.34086.7d. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. J R Statist Soc B. 1995;57:289–300. [Google Scholar]
- Brown MS, Goldstein JL. Lipoprotein metabolism in the macrophage: implications for cholesterol deposition in atherosclerosis. Annu Rev Biochem. 1983;52:223–261. doi: 10.1146/annurev.bi.52.070183.001255. [DOI] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science. 1986;232:34–47. doi: 10.1126/science.3513311. [DOI] [PubMed] [Google Scholar]
- Buono C, Come CE, Witztum JL, Maguire GF, Connelly PW, Carroll M, Lichtman AH. Influence of C3 deficiency on atherosclerosis. Circulation. 2002;105:3025–3031. doi: 10.1161/01.cir.0000019584.04929.83. [DOI] [PubMed] [Google Scholar]
- Calvano SE, Xiao W, Richards DR, Felciano RM, Baker HV, Cho RJ, Chen RO, Brownstein BH, Cobb JP, Tschoeke SK, et al. A network-based analysis of systemic inflammation in humans. Nature. 2005;437:1032–1037. doi: 10.1038/nature03985. [DOI] [PubMed] [Google Scholar]
- Chen Y, Zhu J, Lum PY, Yang X, Pinto S, MacNeil DJ, Zhang C, Lamb J, Edwards S, Sieberts SK, et al. Variations in DNA elucidate molecular networks that cause disease. Nature. 2008;452:429–435. doi: 10.1038/nature06757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Fukutomi T, Zago AC, Ehlers R, Detmers PA, Wright SD, Rogers C, Simon DI. Simvastatin reduces neointimal thickening in low-density lipoprotein receptor-deficient mice after experimental angioplasty without changing plasma lipids. Circulation. 2002;106:20–23. doi: 10.1161/01.cir.0000022843.76104.01. [DOI] [PubMed] [Google Scholar]
- Cocucci E, Racchetti G, Meldolesi J. Shedding microvesicles: artefacts no more. Trends Cell Biol. 2009;19:43–51. doi: 10.1016/j.tcb.2008.11.003. [DOI] [PubMed] [Google Scholar]
- de Luca C, Olefsky JM. Inflammation and insulin resistance. FEBS Lett. 2008;582:97–105. doi: 10.1016/j.febslet.2007.11.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feil S, Hofmann F, Feil R. SM22alpha modulates vascular smooth muscle cell phenotype during atherogenesis. Circ Res. 2004;94:863–865. doi: 10.1161/01.RES.0000126417.38728.F6. [DOI] [PubMed] [Google Scholar]
- Fu X, Gharib SA, Green PS, Aitken ML, Frazer DA, Park DR, Vaisar T, Heinecke JW. Spectral index for assessment of differential protein expression in shotgun proteomics. J Proteome Res. 2008;7:845–854. doi: 10.1021/pr070271+. [DOI] [PubMed] [Google Scholar]
- Ghazalpour A, Doss S, Yang X, Aten J, Toomey EM, Van Nas A, Wang S, Drake TA, Lusis AJ. Thematic review series: The pathogenesis of atherosclerosis. Toward a biological network for atherosclerosis. J Lipid Res. 2004;45:1793–1805. doi: 10.1194/jlr.R400006-JLR200. [DOI] [PubMed] [Google Scholar]
- Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–964. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- Hegmans JP, Bard MP, Hemmes A, Luider TM, Kleijmeer MJ, Prins JB, Zitvogel L, Burgers SA, Hoogsteden HC, Lambrecht BN. Proteomic analysis of exosomes secreted by human mesothelioma cells. Am J Pathol. 2004;164:1807–1815. doi: 10.1016/S0002-9440(10)63739-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi S, Goldstein JL, Brown MS, Herz J, Burns DK. Massive xanthomatosis and atherosclerosis in cholesterol-fed low density lipoprotein receptor-negative mice. J Clin Invest. 1994;93:1885–1893. doi: 10.1172/JCI117179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem. 2002;74:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- Kersey PJ, Duarte J, Williams A, Karavidopoulou Y, Birney E, Apweiler R. The International Protein Index: an integrated database for proteomics experiments. Proteomics. 2004;4:1985–1988. doi: 10.1002/pmic.200300721. [DOI] [PubMed] [Google Scholar]
- Langer C, Huang Y, Cullen P, Wiesenhütter B, Mahley RW, Assmann G, von Eckardstein A. Endogenous apolipoprotein E modulates cholesterol efflux and cholesteryl ester hydrolysis mediated by high-density lipoprotein-3 and lipid-free apolipoproteins in mouse peritoneal macrophages. J Mol Med. 2000;78:217–227. doi: 10.1007/s001090000096. [DOI] [PubMed] [Google Scholar]
- Li AC, Binder CJ, Gutierrez A, Brown KK, Plotkin CR, Pattison JW, Valledor AF, Davis RA, Willson TM, Witztum JL, et al. Differential inhibition of macrophage foam-cell formation and atherosclerosis in mice by PPARalpha, beta/delta, and gamma. J Clin Invest. 2004;114:1564–1576. doi: 10.1172/JCI18730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li AC, Brown KK, Silvestre MJ, Willson TM, Palinski W, Glass CK. Peroxisome proliferator-activated receptor gamma ligands inhibit development of atherosclerosis in LDL receptor-deficient mice. J Clin Invest. 2000;106:523–531. doi: 10.1172/JCI10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li AC, Glass CK. The macrophage foam cell as a target for therapeutic intervention. Nat Med. 2002;8:1235–1242. doi: 10.1038/nm1102-1235. [DOI] [PubMed] [Google Scholar]
- Linton MF, Atkinson JB, Fazio S. Prevention of atherosclerosis in apolipoprotein E-deficient mice by bone marrow transplantation. Science. 1995;267:1034–1037. doi: 10.1126/science.7863332. [DOI] [PubMed] [Google Scholar]
- Liu H, Sadygov RG, Yates JR., 3rd A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal Chem. 2004;76:4193–4201. doi: 10.1021/ac0498563. [DOI] [PubMed] [Google Scholar]
- Liu H, Zheng F, Li Z, Uribarri J, Ren B, Hutter R, Tunstead JR, Badimon J, Striker GE, Vlassara H. Reduced acute vascular injury and atherosclerosis in hyperlipidemic mice transgenic for lysozyme. Am J Pathol. 2006;169:303–313. doi: 10.2353/ajpath.2006.050885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusis AJ, Attie AD, Reue K. Metabolic syndrome: from epidemiology to systems biology. Nat Rev Genet. 2008;9:819–830. doi: 10.1038/nrg2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maere S, Heymans K, Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics. 2005;21:3448–3449. doi: 10.1093/bioinformatics/bti551. [DOI] [PubMed] [Google Scholar]
- Nesvizhskii AI, Keller A, Kolker E, Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem. 2003;75:4646–4658. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- Old WM, Meyer-Arendt K, Aveline-Wolf L, Pierce KG, Mendoza A, Sevinsky JR, Resing KA, Ahn NG. Comparison of label-free methods for quantifying human proteins by shotgun proteomics. Mol Cell Proteomics. 2005;4:1487–1502. doi: 10.1074/mcp.M500084-MCP200. [DOI] [PubMed] [Google Scholar]
- Overton CD, Yancey PG, Major AS, Linton MF, Fazio S. Deletion of macrophage LDL receptor-related protein increases atherogenesis in the mouse. Circ Res. 2007;100:670–677. doi: 10.1161/01.RES.0000260204.40510.aa. [DOI] [PubMed] [Google Scholar]
- Paul A, Chang BH, Li L, Yechoor VK, Chan L. Deficiency of adipose differentiation-related protein impairs foam cell formation and protects against atherosclerosis. Circ Res. 2008;102:1492–1501. doi: 10.1161/CIRCRESAHA.107.168070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadt EE, Lum PY. Thematic review series: systems biology approaches to metabolic and cardiovascular disorders. Reverse engineering gene networks to identify key drivers of complex disease phenotypes. J Lipid Res. 2006;47:2601–2613. doi: 10.1194/jlr.R600026-JLR200. [DOI] [PubMed] [Google Scholar]
- Simpson RJ, Jensen SS, Lim JW. Proteomic profiling of exosomes: current perspectives. Proteomics. 2008;8:4083–4099. doi: 10.1002/pmic.200800109. [DOI] [PubMed] [Google Scholar]
- Sokal RR, Rohlf FJ. Biometry: The principles and practice of statistics in biological research. 3rd. W.H. Freeman; New York: 1995. [Google Scholar]
- Suzuki H, Kurihara Y, Takeya M, Kamada N, Kataoka M, Jishage K, Ueda O, Sakaguchi H, Higashi T, Suzuki T, et al. A role for macrophage scavenger receptors in atherosclerosis and susceptibility to infection. Nature. 1997;386:292–296. doi: 10.1038/386292a0. [DOI] [PubMed] [Google Scholar]
- Vaisar T, Pennathur S, Green PS, Gharib SA, Hoofnagle AN, Cheung MC, Byun J, Vuletic S, Kassim S, Singh P, et al. Shotgun proteomics implicates protease inhibition and complement activation in the antiinflammatory properties of HDL. J Clin Invest. 2007;117:746–56. doi: 10.1172/JCI26206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YX, Martin-McNulty B, Huw LY, da Cunha V, Post J, Hinchman J, Vergona R, Sullivan ME, Dole W, Kauser K. Anti-atherosclerotic effect of simvastatin depends on the presence of apolipoprotein E. Atherosclerosis. 2002;162:23–31. doi: 10.1016/s0021-9150(01)00678-5. [DOI] [PubMed] [Google Scholar]
- Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest. 2005;115:1111–1119. doi: 10.1172/JCI25102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.