

Abstract
Best vitelliform macular dystrophy (BVMD, also called Best disease) is a dominantly-inherited, juvenile-onset form of macular degeneration, which is characterized by abnormal accumulation of yellow pigment in the outer retina and a depressed electro-oculogram light peak (LP). Over 100 disease-causing mutations in human Bestrophin-1 (hBest1) are closely linked to BVMD and several other retinopathies. However, the physiological role of hBest1 and the mechanisms of retinal pathology remain obscure partly because hBest1 has been described as a protein with multiple functions including a Ca2+-activated Cl− channel, a Ca2+ channel regulator, a volume regulated Cl− channel, and a HCO3− channel. This review focuses on how dysfunction of hBest1 is related to accumulation of yellow pigment and a decreased LP. Dysfunction of hBest1 as a HCO3− channel or a volume regulated Cl− channel may be associated with defective regulation of the subretinal fluid or phagocytosis of photoreceptor outer segments by RPE cells, which may lead to fluid and pigment accumulation.
Keywords: bestrophin, Best vitelliform macular dystrophy, chloride channel, retinopathies
Introduction
Best vitelliform macular dystrophy (BVMD), also termed Best’s disease, is a dominantly inherited, juvenile-onset form of macular degeneration. Human bestrophin-1 (hBest1) has been identified as the gene responsible for BVMD [44,55]. So far, over 100 disease-causing mutations in hBest1 have been linked to BVMD and other retinopathies [31]. A key clinical feature of BVMD is a decreased electro-oculogram (EOG) light peak (LP) [2,14], which is believed to be caused by a defective Ca2+-activated Cl− channel in the basolateral membrane of RPE [22,23,33] where hBest1 is expressed [55,44,25,41]. Since hBest1 is clearly an anion channel [31,59,70,72,80,81,85,86] whose function is affected by disease-causing mutations, hBest1 is very likely to be the CaCC responsible for the LP. Thus, it is logical to conclude that loss of anion channel function caused by disease-causing mutations is responsible for the depressed LP in BVMD patients [70,31]. However, this idea has been seriously challenged by the finding that the LP and CaCCs are normal in mBest1 knockout mice [43]. This challenge is dependent on the presumption that human and mouse retina are the same and that hBest1 and mBest1 function similarly in vivo. However, human and mouse retina have different structures [11,71], and the identification and of the CaCCs responsible for the LP in human and mouse remain ambiguous.
BVMD is a type of retinal degeneration characterized by an abnormal accumulation of yellow pigment in the retinal pigment epithelium (RPE) [24,48,30,56,40]. It is mysterious how yellow pigment accumulation occurs in BVMD, although a similar yellow pigment accumulates in age-related macular degeneration (ARMD) [6,16]. A key component of the yellow pigment in ARMD is a pyridinium bis-retinoid called A2E, which originates from photoreceptor visual pigment. Marmorstein has provided some evidence that A2E is also a component of the yellow pigment in BVMD [3]. The presumed photoreceptor origin of the pigment has led to the suggestion that there may be a problem with turnover of photoreceptor outer segments (POS) [31,30,17]. Under normal conditions, POS are phagocytosed by the RPE and degraded in the phagolysosome. However, since hBest1 is not located on the apical side of the cell where phagocytosis of POS occurs [41], nor in the phagolysosome [37], it is unlikely that hBest1 is directly involved in POS phagocytosis. It may be that a more general disruption of fluid transport in RPE is linked to the development of BVMD [31]. Recent studies show that hBest1 has a high permeability to HCO3− [62], suggesting a potential role of pH regulation by hBest1 in BVMD. Furthermore, the regulation of pH and Ca2+ homeostasis by hBest1 is hypothesized to be involved in regulation of phagocytosis and lysosomal function in RPE [40].
Mutations in Best1 have also been linked to several other forms of retinopathies including adult onset macular dystrophy [67], autosomal dominant vitreochoidopathy [84], autosomal recessive bestrophinopathy [9], and canine multifocal retinopathy [26]. In addition, recently, it has been suggested that Best1 mutations are also responsible for a subset of retinitis pigmentosa [15]. Thus, the phenotype resulting from Best1 mutations may depend on multiple genetic and environmental factors. In this article, we review the recent studies on hBest1 functioning as a Ca2+-activated Cl− channel, a Ca2+ channel regulator, a volume regulated Cl− channel, and a HCO3− channel (summarized in Fig.1).
Fig. 1.

Summary of hBest1 functions. hBest1 is regulated directly by Ca2+ , either through the Ca2+ channel or Gq protein-coupled receptors. hBest1 can be phosphorylated by PKC to regulate channel rundown and dephosphorylated by PP2A activated by ceramide in response to hypertonic stress. hBest1 also inhibits Ca2+ channel though an SH3 binding domain. The intracellular pH can be regulated by hBest1 through its high permeability of HCO3−.
Disease-causing mutations of hBest1 are associated with Ca2+-activated Cl− channel dysfunction and decreased Light Peak
hBest1 functions as a Ca2+-activated Cl− channel
hBest1 was first identified as a Ca2+-activated Cl− channel in 2002 by Sun et al. [70], who found that expression of hBest1 in HEK cells induced Cl− currents that were regulated by Ca2+. The Ca2+ sensitivity of hBest1 is in the physiological range with a Kd of about 200 nM [80,31]. A Ca2+-binding site in hBest1 have been identified at the C-terminus immediately after the last transmembrane domain [80,36]. A central piece of evidence that bestrophin is a Cl− channel is shown by the findings that the gating and conductance of bestrophins are altered by mutagenesis of critical amino acids [61,60,59]. For example, the rectification of mBest2 can be altered in opposite directions by substitution of phenylalanine at position 80 with a positively charged arginine or a negatively charged glutamate [59]. Furthermore, mutation in the corresponding phenylalanine in dBest1 alters cation/anion selectivity [13], supporting the idea that bestrophin is an integral part of the channel pore. Using mutagenesis and cysteine-accessibility analysis, Qu et al. have shown that the second transmembrane domain (AA69-99) forms the pore and determines ion selectivity [59].
Mutations disrupt hBest1 functional domains
Many of the disease-causing mutations in hBest1 cause Cl− channel dysfunction [31,70,80,85,86]. The mutations are clustered in several functional regions of the protein. The highest density of mutations is located in a highly acidic region in the C-terminus immediately after the last transmembrane domain. Disease-causing mutations in this region render hBest1 nonfunctional as a Cl− channel and many of the mutations are dominant negative [58,70,80,86]. This region is important for Ca2+-sensing [80]. In a structure-function analysis of hBest1, all but two (F298W, and T307S) of 35 mutations introduced into this region (293-308) caused the channel to be incapable of being activated by Ca2+ [80]. Since these mutations do not affect cell surface expression [80,58], they likely alter Ca2+-binding. Qu et al. [58] have shown that the G299E, D301N and D302N mutations disrupt interaction between the N- and C-termini, which may be important for hBest1 functioning as a multimer in the plasma membrane. This suggests that the acidic region may participate in multiple important channel functions (Ca2+ sensing and subunit interaction).
Another hot spot for disease-causing mutations resides in the second transmembrane domain (TM2), which has been identified as the bestrophin channel pore [61,59,72]. Mutations in TM2 likely disrupt the pore structure, which results in a loss of channel function. Another hot spot is in the N-terminus, where some mutations (R19C, R25C and K30C) have been suggested to alter the interaction between N- and C- termini [58]. The remainder of the disease-causing mutations are located between TM2 and TM5, but it is not clear how these mutations affect channel function, partly because the topology of bestrophin is controversial in this region. The effects of disease-causing mutations on Cl− channel function were reviewed in detail in Hartzell et al. [31] and Boon et al. [7].
Association of hBest1 with LP generation
The characteristic feature of BVMD is the diminished LP of the EOG [2,14]. The LP is believed to be produced by activation of a Ca2+-activated Cl− channel in the basolateral membrane of the RPE by a “light peak substance” (LPS), which is released from photoreceptors stimulated by light [21,39] (Fig. 1). hBest1 is located in the basolateral membrane of the RPE [41,25,44,55], suggesting that hBest1 may be the CaCC responsible for the LP. The diminished LP in BVMD patients is consistent with a defective Cl− channel function caused by these disease-causing mutations [70,80,85,86].
If hBest1 is the CaCC responsible for the LP, how does the LPS activate hBest1? The identity of the LPS is not known, but ATP is a favorite candidate because extracellular ATP, when applied to the RPE cells, induces an increase in Cl− conductance across the RPE basal membrane, which is similar to effect of LPS, and LP and ATP-evoked response are blocked by a Cl− channel blocker DIDS [31,54,69]. It is hypothesized that ATP activates the Gq protein-coupled P2Y receptor, which activates PLC, subsequently producing two second messengers: IP3 which increases intracellular Ca2+, and DAG which activates PKC. Evidence supporting this hypothesis includes the observations that ATP induces an increase in intracellular Ca2+, and ATP-induced response is blocked by P2Y receptor blocker suramin, by ER Ca2+-ATPase inhibitor cyclopiazonic acid, and by Ca2+ buffer BAPTA [54].
Although Best1 has been a leading candidate for mediating the LP, there are several other Cl− channels in the basolateral membrane that should be considered. An increase in intracellular cAMP induces an activation of Cl− conductance, resulting in a decrease in basolateral membrane resistance, and an increase in transepithelial potentials [75,29,47]. CFTP is expressed in RPE cells [75,29,47], and can be activated by cAMP dependent activation of PKA. The LP is reduced in both CFTR knockout mice and mice with ΔF508 mutations, suggesting CFTR contributes to the generation of LP [78]. Since other RPE-related components of ERG are also reduced, CFTR mutations likely cause a general disruption of RPE cells [78]. However, there are no reports in human CF patients with abnormal LPs. The functions of CFTR in retinopathies have not been well studied yet.
CLC-2 has also been reported to be expressed in RPE cells [29,75,76]. Knockout of CLC2 leads to retinal degeneration, which might result from abnormal transepithelial ion and fluid transport, and subsequent photoreceptor impairment [8]. Furthermore, CLC-2 like currents have been recorded from RPE cells [29], and RPE short-circuit currents are reduced in CLC-2 knockout mice [8]. However, the precise role of CLC-2 in RPE ion and fluid transports largely remains unknown.
The newly discovered anoctamin family provides another candidate channel that might be responsible for the LP [32,10,66,83]. There are 10 anoctamins and Ano1 and 2 have been shown to be Ca2+-activated Cl− channels. The CaCC currents that have been recorded in RPE cells resemble Ano1 and Ano2 currents expressed in HEK cells [89], but the expression of Anos in RPE are unexplored and their role as CaCCs responsible for LP is only speculative. Therefore, the identification and function of CaCCs in RPE need to be further explored.
The conclusion that the LP is generated by the hBest1 Cl− channel is disfavored by the finding that patients with certain disease-causing mutations have normal or near-normal LPs. These mutations include T216T, A243V, ΔI295, D312N and L567F [86]. The two mutations T216I and L567F exhibit wild-type like Cl− currents, and may have been incorrectly identified as disease-causing mutations because no family history of BVMD was shown [86]. The A243V, ΔI295, and D312N mutations clearly show a defect in hBest1 Cl− channel function [85,86], and most patients, except a few, have depressed LPs. There are several possible explanations why some patients with these mutations have normal LPs. Although it is often said that these mutations exhibit reduced penetrance, the possibility exists that these mutations are actually null mutations that sometimes are disease-causing in the heterozygous state. This is supported by the finding that the D312N mutation in a compound homogygous condition with another recessive mutation (M325T) produces retinopathy associated with a severely diminished LP [9]. Alternatively, the patients with hBest1 mutations who have normal LPs could be explained if these individuals have some type of suppressor mutation or compensatory response.
The most challenging argument against hBest1 as the CaCC responsible for LP generation comes from mBest1 knockout mice, which have normal CaCCs in the RPE [43]. The LP in these animals is altered, but in a manner opposite to that predicted if mBest1 was generating the LP; namely, the relationship between the amplitude of the LP and light intensity is shifted to the left. Furthermore, mBest1 knockout mice do not exhibit ocular disease [43], further suggesting that mBest1 is not the CaCC responsible for the LP in the mouse RPE. However, recently it has been shown that a mouse harboring the W93C mutation does have eye disease that resembles BVMD [89]. The LP in W93C animals is enhanced at low light intensities and reduced in the middle of the intensity range, but the CaCC currents in the W93C RPE cells are normal. This suggests that although mBest1 is not responsible for the RPE CaCC, it somehow regulates the channel that is responsible for the LP. The difference in phenotype between the Best1 knockout and the W93C mutant mouse suggests that BVMD is caused by gain-of-function mutations in Best1. However, this conclusion is hard to reconcile with the clear loss of Cl− channel function associated with these mutations [70,86] and the loss of the LP in humans with autosomal recessive bestrophinopathy (ARB) which is thought to be caused by null mutations in hBest1. It is possible that hBest1 and mBest1 have different functions, but this explanation is disfavored because mBest1 functions as a CaCC when expressed heterologously [51] (but see below), and shares high homology with hBest1 including the Ca2+-binding region and the pore. However, it should be pointed out that disease-causing mutations have been evaluated only in hBest1 and not in mBest1.
It has been suggested that Best1 plays a role in regulation of intracellular Ca2+, which is proposed to be important in pathogenesis of BVMD [40,88]. This hypothesis is supported by the finding that hBest1 can inhibit voltage gated Ca2+ channels [64,87]. However, for reasons discussed below, it seems unlikely that mutations in hBest1 result in changes in intracellular Ca2+ solely through regulation of CaV1.3. Furthermore, unlike hBest1, mBest1 does not inhibit CaV1.3 [87], further suggesting that mBest1 may not function like hBest1. The finding that hBest1 is highly permeant to HCO3− [62] has raised the possibility that some of the effects of Best1 mutations may be related to changes in pH, but this has not yet been experimentally tested.
It should be noted that the mouse LP is considerably smaller than the cat, which has served as a model for the LP (Fig. 2). The LPs in human and cat are much larger than the C-wave [39], whereas the LP in mouse is usually smaller than the C-wave [43]. In rats, the LP is very small, if not completely absent [40]. This suggests that the ionic mechanism underlying the LP in human and rodents may be different. Furthermore, in our hands, transient expression of mBest1 in HEK cells does not induce Ca2+-activated Cl− currents (Fig. 3). This result is surprising, given the fact that two other laboratories have reported that mBest1 does induce currents [51,53]. Although we consistently observe robust expression of currents with hBest1 and mBest2, we have never seen currents with mBest1. The differences between our results and those of other labs may come from the different cell lines which may express different levels of mBest1 regulatory molecules, such as kinases and regulatory subunits. These molecules may affect mBest1 functional expression and regulation. However, this is only speculative, and has not been explored.
Fig. 2.

The difference in LP in mice and cats. The dc-ERG was recorded in (A) mice to a 7-min light stimulus [43] or (B) in cats to a 10-min light stimulus [39]. The LP is smaller than the C-wave in mice, while the LP in cats is much larger than the C-wave.
Fig. 3.
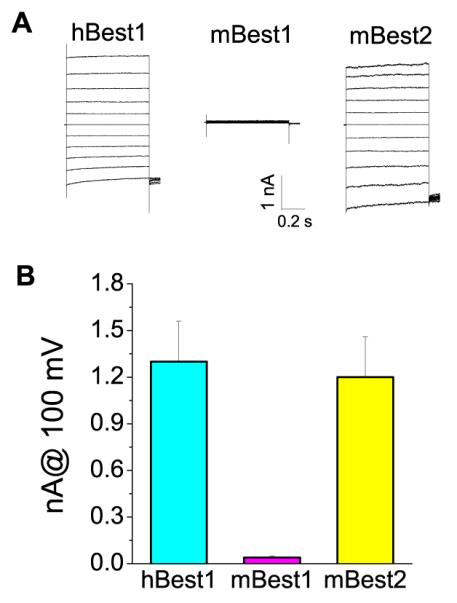
Expression of hBest1, mBest2 and mBest1 in HEK cells. A. Representative whole-cell current traces recorded from cells transfected with hBest1, mBest1 and mBest2 in presence of 10 μM Ca2+. B. Current amplitudes at 100 mV for hBest1 (n=13), mBest1 (n=10) and mBest2 (n=9).
Although there is evidence arguing against bestrophin as the CaCC responsible for LP generation, hBest1 remains a candidate since a majority of BVMD-causing mutations cause defects in Cl− channel function [31,70,80,85,86]. Furthermore, like BVMD patients, patients of autosomal recessive bestrophinophathy (ARB) also show depressed LPs, which is consistent with ARB-associated mutations causing defective Cl− channel function [9]. Unlike BVMD mutations, ARB mutations are not dominant negative, and people with heterozygous mutations in hBest1 have normal LPs [9]. This suggests that the LP is correlated with Cl− channel function of hBest1. Mutations in Best1 cause another recessive retinopathy in dog called canine mutifocal retinopathy (CMR) [26]. However, there is no report about LPs in CMR dogs. Mutations in hBest1 cause both dominant and recessive diseases, which are similar to myotonia-causing mutations in CLC-1. In recessive myotonia, some truncation mutations are unable to produce dominant-negative effects due to their inability to form a dimer with wild type subunits [34]. Similarly, a C73X stop mutation in CMR [26] and a R200X stop mutation in ABR [9] could also result from their inability to associate with wild type Best1. Other recessive mutations may also disrupt formation of multimers with wild type subunits. The D312N, V317M and M325T mutations that cause ABR are located in the EF hand-like structure responsible for Ca2+-binding. Other mutations in this region have been shown to disrupt interaction between N- and C-termini and suggest that this region may also be important in channel multimerization.
In summary, the fact that mBest1 knockout mice have normal LPs clearly show that mBest1 is not the CaCC responsible for mouse LPs [43]. However, this finding does not exclude the possibility that hBest1 is the CaCC responsible for the human LP since mBest1 does not mimic the function of hBestl.
hBest1 regulation by phosphorylation
The hBest1 C-terminus contains an EF-hand like structure that binds Ca2+ [80] and a PKC phosphorylation site (S358) that is believed to regulate channel function [81]. Thus, increases in intracellular Ca2+ probably activate hBest1 by binding to the EF hand-like structure. Following Ca2+ activation, hBest1 undergoes a time-dependent decrease in current amplitude, a process called rundown. The rundown can be inhibited by phosphorylation of the PKC phosphorylation site (S358) [81] (Fig.1). Therefore, hBest1 currents are activated by Ca2+, but PKC-dependent phosphorylation may be necessary to maintain the current. Although rundown has so far been studied only with hBest1 heterologously-expressed in HEK cells, it is possible that the mechanism responsible for rundown also operates in native RPE cells. We hypothesize that the processes underlying rundown modulates the timing of current turn-on and turn-off.
The PKC phosphorylation site (S358) is dephosphorylated by PP2A, which increases hBest1 channel rundown. This suggests that PP2A could switch off the LP signal, especially in the dark when less LPS is released, and thus less PKC is activated. Therefore, regulation of hBest1 phosphorylation by PKC and PP2A may underlie the time course of the LP. Though this hypothesis is based on the observations from heterologous expression of hBest1 in HEK cells, it may be physiologically relevant since hBest1 can be coimmunoprecipitated with PP2A from human RPE cells [42].
The PKC phosphorylation site (S358) appears not to be a PKA phosporylation site since both cAMP and forskolin show no effects on hBest1 [81]. However, S358 is likely a substrate for p21-activated kinase PAK2 since S358A mutation eliminates the phosphorylation in vitro by PAK2 [5]. Barro Soria et al [5] suggest that hBest1 facilitates Ca2+ release from ER by acting as a counterion pathway. PAK2-phosphorylation of hBest1 further promotes Ca2+ release from ER, and subsequently enhances Ca2+ activated Cl− and K+ channels. Since PAK2 shares the same phosphorylation site (S358) in hBest1 as PKC, PAK2 increases hBest1 function likely through inhibition of hBest1channel rundown.
Since hBest1 functions can be regulated by kinases such as PKC and PAK2, it is likely hBest1 function can be coupled to many types of receptors. However, until now, most studies shows that bestrophins are activated by Ca2+ in the patch pipettes in whole cell recordings, and not by GPCRs. Further experiments should be done to test whether bestrophins can be activated by GPCR through agonists in either overexpressing cells or native RPE cells. Furthermore, hBest1 can be dephosphorylated by PP2A through ceramide, which is coupled to several stress stimuli such as hyperosmotic stress [81].
Regulator of voltage-gated Ca2+ channels
In addition to functioning as a Cl− channel, Best1 has been shown to influence intracellular Ca2+ signaling by several different mechanisms, but results are often contradictory and do not simply explain the pathogenesis of BVMD. hBest1 can regulate voltage-gated Ca2+ channels [64,87,9] (Fig.1). Both Rosenthal et al [64] and Burgess et al [9] reported that hBest1 accelerated the activation of CaV1.3 currents, but Yu et al. [87] did not observe significant changes in current kinetics. Rather, Yu et al [87] reported that hBest1 expression significantly reduced the amplitude of CaV1.3 currents. This effect is explained by an SH3 binding domain (330-350) that binds CaV β subunits to regulate CaV1.3. The hBest1 residues P330 and P334 are critical for the regulation, since mutation of P330 and P334 abolishes this effect [87]. This SH3 binding domain lies between the Ca2+-binding site and a regulatory domain critical for channel rundown [80]. Some disease-causing mutations, G299R, G222E, and A146K, partly eliminate the inhibition of CaV1.3 by hBest1 [87]. However, other disease-causing mutants, D312N and R92S, have the same effect on CaV1.3 as wild type [87]. Effects of hBest1 disease-causing mutants on the activation and inactivation of CaV currents also seem inconsistent, or at least difficult to reconcile with a scheme involving regulation of CaV channels as a primary mechanism of Best1 action. Although wild type hBest1 accelerates CaV activation, the W93C mutation slows CaV1.3 activation and inactivation, the R213C mutation accelerates inactivation [64], and the R141H mutation eliminates the accelerating effect of hBest1 on CaV1.3 activation [9]. It is not obvious how these changes in regulation of CaV channels caused by mutations in hBest1 may be related to the disease phenotype. The inconsistent effects of different mutations on CaV1.3 suggest that BVMD is unlikely to be caused by changes in CaV1.3 function.
If hBest1 regulates CaV function through interaction of CaV β subunits, the question arises whether hBest1 function is affected by CaV β binding. This possibility is supported by the finding that the nonfunctional hBest1 350X mutant, which has the C-terminus deleted beyond amino acid 350 but contains the Ca2+ -binding site and the SH3 binding domain, can be activated by Ca2+ when coexpressed with Ca2+ channels in HEK cells (Fig. 4). This suggests that the ligation of the SH3 domain could alter the conformation of hBest1.
Fig. 4.
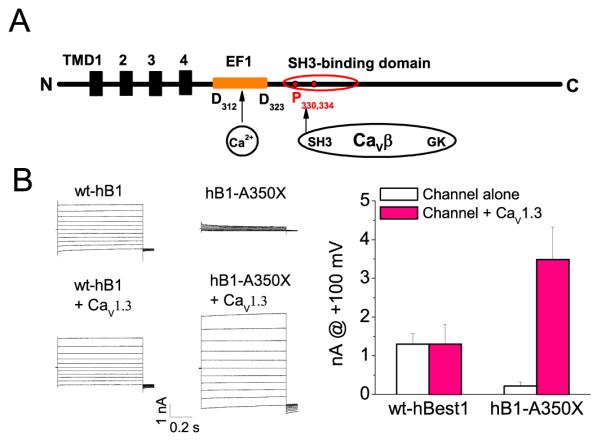
Regulation of hBest1 by Ca2+ channel through an SH3 binding domain. A. Model of functional domains in hBest1. Orange EF1 is an EF hand like structure (D312-D323) critical for Ca2+ binding. The red circle represents an SH3 binding domain, which binds to β subunits of CaV1.3, and inhibits CaV1.3. Two prolines (P330 and P334 in red) are critical for this regulation. Representative current traces (B) and current amplitudes (C) recorded from cells transfected with wild type hBest1 or the 350X mutant with and without CaV1.3. The 350X mutation was made by introducing a stop codon at position 350, thus deleting C-terminus beyond 350, but containing Ca2+ -binding site and the SH3 binding domain. The 350X can not be activated by Ca2+, but this nonfunctional channel was rescued by CaV1.3.
Regardless of the role of Best1, voltage-gated Ca2+ channels clearly play a role in mouse LP generation [43]. The LP is reduced by the Ca2+ channel blocker nimodipine [43], and deletion of the genes for CaV1.3 [43] or β4 eliminate the light peak [79]. Whether Ca2+ influx through the CaV channel is involved in CaCC activation and generation of the LP is unknown. Alternatively, interaction of CaV1.3 with Best1 may alter Best1 function. However, unlike hBest1, mBest1 does not inhibit CaV1.3 [87], suggesting mBest1 does not directly regulate the Ca2+ channel to modulate intracellular Ca2+ concentration.
In addition, hBest1 also regulates Ca2+ release from the ER. RPE cells from mBest1 knockout mice show a greater increase in intracellular Ca2+ concentration in response to ATP than wild type mice [40,89], suggesting mBest1 has an inhibitory effect on release of Ca2+ from internal stores. However, Barro-Soria et al. [5] suggest that hBest1 is an ER-resident protein that facilitates Ca2+ release from ER by acting as a counterion pathway. A possible explanation for this discrepancy might be differences between mBest1 and hBest1 or the use of knockout mice vs. transfected cells.
It is unclear how mouse models can be extrapolated to the function of human RPE, since mice, unlike human, have no obvious macula, and BVMD is clearly a macular disease. Mouse retina contains predominantly rod photoreceptors with only 2-3% of cone photorecetoprs [11,71]. Thus, mouse retina is more similar to human peripheral retina than macula, where cone photoreceptors are concentrated. It is unknown why BVMD primarily affects human macula since hBest1 is expressed more in peripheral retina than in macula [49]. In addition, the high density of cone photoreceptors in the macula may require high phagocytic function of RPE in response to light.
Lipofuscin accumulation in BVMD
Clinical and histopathological analysis of BVMD patients’ eyes shows accumulation of yellow pigment, often referred to as lipofuscin, in the RPE [24,31,40,48,56]. The major fluorescent component of lipofuscin is A2E (N-retinylidene-N-retinylethanolamine), which is the metabolite of retinal, a visual pigment from photoreceptors. Accumulation of A2E has been found to promote apoptosis of RPE [68], and to disrupt lysosomal function in the RPE [18]. The photoreceptor origin of A2E suggests that accumulation of lipofuscin may be related to a problem with the phagocytosis of photoreceptor and/or phagolysosome function [17,30,31,40]. However, it is unclear how dysfunction of hBest1 is related to accumulation of lipofuscin.
Although a large fraction of hBest1 is expressed intracellularly [70,80,85,86], hBest1 is concentrated in the ER [5] and is not coexpressed with lysosomal markers [37]. Furthermore, hBest1 is expressed on the basolateral side of the RPE [41,44], not the apical side where phagocytosis of POS occurs. Thus, it is unlikely that hBest1 directly participates in regulation of phagocyotsis or phagolysosome function, but may have an indirect effect. A common feature of diseases caused by Best1 mutations is edema and accumulation of fluid in the outer retina [46,56,73,57]. Disruption of fluid transport in RPE due to the dysfunction of hBest1is hypothesized to be the mechanism of BVMD [31]. Here we review evidence that hBest1 functions as a volume regulated Cl− channel and a HCO3− channel.
Volume regulated anion channel
Bestrophins including hBest1, mBest2 and dBest1 are regulated by cell volume [13,81,12,19]. dBest1 has been demonstrated to be a native volume regulated Cl− channel in Drosophila S2 cells [13,12]. The dBest1 Cl− currents are activated by hyposmolarity and inhibited by hyperosmolarity. The osmotically sensitive currents are knocked down by dBest1 RNAi, and can be rescued by transfection of both wild type dBest1 and a mutant dBest1 with altered ion permeability [13,12]. However, bestrophin is not the classic volume regulated anion channel (VRAC) in mammals because the VRAC is unaffected in macrophages from mBest1 and mBest2 knockout mice [13]. Furthermore, the classic VRAC currents show outward rectification and inactivation at positive potentials in a time-dependent manner, while bestrophins exhibit little outward rectification and no inactivation at positive potentials. Thus, bestrophins may be one of the several Cl− channels that play a role in regulation of cell volume.
Bestrophins can be regulated by both Ca2+ and cell volume. The two pathways can be independent of each other, but can interact with each other [12]. Ca2+ potentiates dBest1 current amplitudes activated by increases in cell volume [12]. hBest1 currents activated by Ca2+ can be inhibited by decreases in cell volume [19,81] (Fig.1). Furthermore, increases in cell volume often result in increases in intracellular Ca2+ [45]. Therefore, hBest1 could be activated by hyposmotic swelling through Ca2+, since hBest1 currents are activated by intracellular Ca2+ with an EC50 of 140 nM [80], which can be reached during cell swelling in response to hyposmotic treatment. The direct studies of hBest1 activation by hyposmolarity are thwarted by endogenous VRACs, which are ubiquitously expressed in cell lines such as HEK.
The mechanism underlying the regulation of hBest1 by cell volume has been studied in a heterologous expression system [19,81]. hBest1 currents expressed in HEK, HeLa, and ARPE-19 cell lines are strongly inhibited by small increases in extracellular osmolarity [19,81]. The inhibition of hBest1 by hypertonic solution is mediated through dephosphorylation of a PKC phosphorylation site (S358) [81] (Fig.1). Furthermore, ceramide, a PP2A activator, mimics the effects of hypertonic solution, and a neutral sphingomyelinase inhibitor blocks the effects of hypertonic stress, suggesting that hypertonic stress activates neutral sphingomyelinase to release ceramide, which dephosphorylates S358 to accelerate channel rundown [81].
It is unclear how regulation of hBest1 by hypertonic solution in a heterologous expression system could be extrapolated to function of hBest1 in vivo. However, it is interesting that like hBest1 currents, the LP is inhibited by hypertonic condition [50]. In addition, hBest1 has been identified to be coimmunoprecipitated with PP2A in human RPE cells [42], further suggesting that dephosphorylation of hBest1 by PP2A may play an important role in hypertonic inhibition of hBest1 in vivo.
One very speculative idea is presented here. RPE cells play an important role in removing the daily shed photoreceptor outer segments (POS) through phagocytosis, which is crucial for photoreceptor survival. Just before POS are shed, a large amount of osmolytes such taurine, glutamate, aspartate and glycine are leaked from POS, accumulate in the extracellular space, and subsequently are transported into RPE. RPE will undergo swelling as water follows the uptake of osmolytes into the cells [31]. Cell swelling could activate hBest1 or classical VRACs either directly or through increase of intracellular Ca2+. Opening of Cl− channels will cause a Cl− efflux, which could cause the cells to undergo regulatory volume decrease (RVD) and shrink. The mechanical force following cell swelling and shrinkage could play a role in RPE phagocytosis. Shrinkage will subsequently decrease Cl− efflux by inhibiting hBest1, possibly through production of ceramide (see below) [81]. Then, RPE cells will undergo a process of regulatory volume increase (RVI) to resume their original cell volumes [38]. Thus, hBest1 is hypothesized to control POS phagocytic process through regulating cell volume (summarized in Fig. 5).
Fig. 5.

Model of hBest1 as a volume regulated Cl− channel in regulation of phagocytosis of POS. A. In the dark, RPE and photoreceptors are not closely attached. B. After the lights turn on, a large amount of osmolytes such glutamate, aspartate and glycine are leaked from POS, accumulate in the extracellular space, and subsequently are transported into RPE. Water is passively absorbed into RPE. C. RPE swells as water enters into the cells. Cell swelling could activate hBest1. D. Opening of hBest1 Cl− channels will cause a Cl− efflux, which could cause the cells to undergo regulatory volume decrease (RVD) and shrink. Photoreceptors are attached to RPE cells following cell swelling and shrinkage. E. Phagocytosis of POS into RPE.
Ceramide may play an important role in hBest1 regulation and in the response of RPE to osmotic changes. Ceramide mimics the effect of hypertonic stress, and a neutral sphingomyelinase inhibitor blocks hypertonic inhibition of hBest1 current, suggesting that hyperosmolarity-induced cell shrinkage could activate sphingomyelinase to release cermide, which mediates the hypertonic inhibition of hBest1[81]. Inhibition of hBest1 Cl− current will result in accumulation of Cl− and water, which causes cells to undergo RVI to recover their initial volume. Therefore, hBest1 plays a key role in cell volume regulation through ceramide. Ceramide, a lipid signaling molecule mediating several cellular responses to stress stimuli such as cytokines and oxidative stress [65,52,27,28,35], promote RPE apoptosis [4], and is implicated in photoreceptor apoptosis during several kinds of retinopathies including macular degeneration, diabetic retinopathy, and retinal detachment [1,20,63,82].
HCO3− channel function of bestrophins
Bestrophins including hBest1-4 and mBest2 are highly permeable to HCO3− [62]. The relative permeability of HCO3− to Cl− is larger than most of other anion channels such CFTR, CLC and ligand-gated anion channels. It is likely that bestrophins conduct both Cl− and HCO3− through the same pore since mutation of a pore residue (V78C) changes the relative HCO3− premability [62]. Furthermore, the disease-causing mutations (Y85H, R92C and W93C) abolish both Cl− and HCO3− currents equally, and Ca2+ is required for activation of both Cl− and HCO3− currents. Thus, it is possible that physiologically bestrophins function as both Cl− channels and HCO3− channels (Fig.1).
Since hBest1 disease-causing mutations also abolish the conductance of HCO3−, it raises the question whether abnormal HCO3− transport in the RPE may contribute to Best’s disease. Metabolically active photoreceptors produce large amounts of CO2 which could lower pH to inhibit photoreceptor function [74,77]. Thus, removal of HCO3− from the subretinal space is important for photoreceptor function. Furthermore, altered intracellular pH in RPE cells is hypothesized to be important in engulfment of POS, maturation and acidification of lysosome, and delivery of lysosomal enzyme [40]. Altered phagocytosis and phagolysomal function is proposed to increase formation of A2E and other lipofuscin substances [40]. Therefore, dysfunction of hBest1 as HCO3− channels may result in abnormal accumulation of lipofuscin in BVMD.
Conclusions and significance
Bestrophins have clearly been shown to be CaCCs in overexpressing cells, but evidence in native RPE cells is inconclusive. The CaCC in mouse RPE cells is not changed in Best1 knockout mice and W93C Best1 knock-in mice [43,89]. Further studies to identify how hBest1 is regulated by these signaling molecules in RPE cell as well as in animal model would be helpful in understanding of the bestrophin-related retinopathies.
Bestrophin has a large HCO3− conductance, suggesting it may have a physiological role as a HCO3− channel. mBest2 has been identified as a HCO3− channel in the distal colon [88]. Whether bestrophin functions as HCO3− channel in RPE cells is to be determined. Direct measurement of HCO3− currents as well as pH in RPE cells from wild type and knockout mice could help to elucidate physiological function of bestrophins in RPE.
It is still unclear how dysfunction of hBest1 causes the pathology of BVMD such as lipofuscin accumulation. Recently, the BVMD-causing mutation W93C was introduced in mice [89]. These mice show a phenotype resembling BVMD and have a suppressed response to ATP-induced increase in intracellular Ca2+ concentration. It is unclear why knock-in of this mutation causes lipofuscin accumulation and how knockin of this mutation alters Ca2+ signaling. Identification of the signaling pathway involving bestrophin is critical for unraveling the pathology of BVMD. In addition, mutations in Best1 have also been linked to several other forms of retinopathies including adult onset macular dystrophy [67], autosomal dominant vitreochoidopathy [85], autosomal recessive bestrophinopathy [9], and canine multifocal retinopathy [26]. This suggests that dysfunction of hBest1 in different retinopathies can cause different disease phenotypes, and it is likely that multiple genetic and environmental factors may be involved in these retinopathies. Discovery of the genes and/or proteins which interact with bestrophins would be a potential target for therapy of these bestrophin-related retinopathies.
Acknowledgments
This work is supported by NIH grants GM60448, EY014852, and a Core Grant for Vision Research P30-EY006360. Q.Xiao is supported by an American Heart Association postdoctoral fellowship.
References
- 1.Acharya JK, Dasgupta U, Rawat SS, Yuan C, Sanxaridis PD, Yonamine I, Karim P, Nagashima K, Brodsky MH, Tsunoda S, Acharya U. Cell-nonautonomous function of ceramidase in photoreceptor homeostasis. Neuron. 2008;57:69–79. doi: 10.1016/j.neuron.2007.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arden GB. Alterations in the standing potential of the eye associated with retinal disease. Trans Ophthalmol Soc U K. 1962;82:63–72. [PubMed] [Google Scholar]
- 3.Bakall B, Radu RA, Stanton JB, Burke JM, McKay BS, Wadelius C, Mullins RF, Stone EM, Travis GH, Marmorstein AD. Enhanced accumulation of A2E in individuals homozygous or heterozygous for mutations in BEST1 (VMD2) Exp Eye Res. 2007;85:34–43. doi: 10.1016/j.exer.2007.02.018. [DOI] [PubMed] [Google Scholar]
- 4.Barak A, Morse LS, Goldkorn T. Ceramide: a potential mediator of apoptosis in human retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 2001;42:247–254. [PubMed] [Google Scholar]
- 5.Barro-Soria R, Aldehni F, Almaca J, Witzgall R, Schreiber R, Kunzelmann K. ER-localized bestrophin 1 activates Ca(2+)-dependent ion channels TMEM16A and SK4 possibly by acting as a counterion channel. Pflugers Arch. 2010;459:485–497. doi: 10.1007/s00424-009-0745-0. [DOI] [PubMed] [Google Scholar]
- 6.Bok D. New insights and new approaches toward the study of age-related macular degeneration. Proc Natl Acad Sci U S A. 2002;99:14619–14621. doi: 10.1073/pnas.242607899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boon CJ, Klevering BJ, Leroy BP, Hoyng CB, Keunen JE, den Hollander AI. The spectrum of ocular phenotypes caused by mutations in the BEST1 gene. Prog Retin Eye Res. 2009;28:187–205. doi: 10.1016/j.preteyeres.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 8.Bosl MR, Stein V, Hubner C, Zdebik AA, Jordt SE, Mukhopadhyay AK, Davidoff MS, Holstein AF, Jentsch TJ. Male germ cells and photoreceptors, both dependent on close cell-cell interactions, degenerate upon ClC-2 Cl(−) channel disruption. EMBO J. 2001;20:1289–1299. doi: 10.1093/emboj/20.6.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burgess R, Millar ID, Leroy BP, Urquhart JE, Fearon IM, De BE, Brown PD, Robson AG, Wright GA, Kestelyn P, Holder GE, Webster AR, Manson FD, Black GC. Biallelic mutation of BEST1 causes a distinct retinopathy in humans. Am J Hum Genet. 2008;82:19–31. doi: 10.1016/j.ajhg.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Caputo A, Caci E, Ferrera L, Pedemonte N, Barsanti C, Sondo E, Pfeffer U, Ravazzolo R, Zegarra-Moran O, Galietta LJ. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science. 2008;322:590–594. doi: 10.1126/science.1163518. [DOI] [PubMed] [Google Scholar]
- 11.Carter-Dawson LD, LaVail MM. Rods and cones in the mouse retina. I. Structural analysis using light and electron microscopy. J Comp Neurol. 1979;188:245–262. doi: 10.1002/cne.901880204. [DOI] [PubMed] [Google Scholar]
- 12.Chien LT, Hartzell HC. Drosophila Bestrophin-1 Chloride Current Is Dually Regulated by Calcium and Cell Volume. J Gen Physiol. 2007;130:513–534. doi: 10.1085/jgp.200709795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chien LT, Hartzell HC. Rescue of volume-regulated anion current by bestrophin mutants with altered charge selectivity. J Gen Physiol. 2008;132:537–546. doi: 10.1085/jgp.200810065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cross HE, Bard L. Electro-oculography in Best’s macular dystrophy. Am J Ophthalmol. 1974;77:46–50. doi: 10.1016/0002-9394(74)90603-5. [DOI] [PubMed] [Google Scholar]
- 15.Davidson AE, Millar ID, Urquhart JE, Burgess-Mullan R, Shweikh Y, Parry N, O’Sullivan J, Maher GJ, McKibbin M, Downes SM, Lotery AJ, Jacobson SG, Brown PD, Black GC, Manson FD. Missense mutations in a retinal pigment epithelium protein, bestrophin-1, cause retinitis pigmentosa. Am J Hum Genet. 2009;85:581–592. doi: 10.1016/j.ajhg.2009.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Jong PT. Age-related macular degeneration. N Engl J Med. 2006;355:1474–1485. doi: 10.1056/NEJMra062326. [DOI] [PubMed] [Google Scholar]
- 17.Eldred GE. Lipofuscin fluorophore inhibits lysosomal protein degradation and may cause early stages of macular degeneration. Gerontology. 1995;41:15–28. doi: 10.1159/000213722. [DOI] [PubMed] [Google Scholar]
- 18.Finnemann SC, Leung LW, Rodriguez-Boulan E. The lipofuscin component A2E selectively inhibits phagolysosomal degradation of photoreceptor phospholipid by the retinal pigment epithelium. Proc Natl Acad Sci USA. 2002;99:3842–3847. doi: 10.1073/pnas.052025899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fischmeister R, Hartzell C. Volume-Sensitivity of the Bestrophin Family of Chloride Channels. J Physiol. 2005;552.2:477–491. doi: 10.1113/jphysiol.2004.075622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fox TE, Han X, Kelly S, Merrill AH, Martin RE, Anderson RE, Gardner TW, Kester M. Diabetes alters sphingolipid metabolism in the retina: a potential mechanism of cell death in diabetic retinopathy. Diabetes. 2006;55:3573–3580. doi: 10.2337/db06-0539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gallemore RP, Griff ER, Steinberg RH. Evidence in support of a photoreceptoral origin for the “light-peak substance”. Invest Ophthalmol Vis Sci. 1988;29:566–571. [PubMed] [Google Scholar]
- 22.Gallemore RP, Hughes BA, Miller SS. Retinal pigment epithelial transport mechanisms and their contributions to the electroretinogram. Prog Retinal Eye Res. 1997;16:509–566. [Google Scholar]
- 23.Gallemore RP, Hughes BA, Miller SS. Light-induced responses of the retinal pigment epithelium. In: Marmor MF, Wolfensberger TJ, editors. The Retinal Pigment Epithelium. Oxford Univ. Press; Oxford, UK: 1998. pp. 175–198. [Google Scholar]
- 24.Gass JDM. Stereoscopic Atlas of Macular Diseases: diagnosis and treatment. Mosby; St. Louis, MO: 1987. [Google Scholar]
- 25.Gouras P, Braun K, Ivert L, Neuringer M, Mattison JA. Bestrophin detected in the basal membrane of the retinal epithelium and drusen of monkeys with drusenoid maculopathy. Graefes Arch Clin Exp Ophthalmol. 2009;247:1051–1056. doi: 10.1007/s00417-009-1091-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guziewicz KE, Zangerl B, Lindauer SJ, Mullins RF, Sandmeyer LS, Grahn BH, Stone EM, Acland GM, Aguirre GD. Bestrophin Gene Mutations Cause Canine Multifocal Retinopathy: A Novel Animal Model for Best Disease. Invest Ophthalmol Vis Sci. 2007;48:1959–1967. doi: 10.1167/iovs.06-1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hannun YA. Functions of ceramide in coordinating cellular responses to stress. Science. 1996;274:1855–1859. doi: 10.1126/science.274.5294.1855. [DOI] [PubMed] [Google Scholar]
- 28.Hannun YA, Luberto C. Ceramide in the eukaryotic stress response. Trends Cell Biol. 2000;10:73–80. doi: 10.1016/s0962-8924(99)01694-3. [DOI] [PubMed] [Google Scholar]
- 29.Hartzell HC, Qu Z. Chloride currents in acutely isolated Xenopus retinal pigment epithelial cells. J Physiol. 2003;549:453–469. doi: 10.1113/jphysiol.2003.040428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hartzell HC, Qu Z, Putzier I, Artinian L, Chien L-T, Cui Y. Looking chloride channels straight in the eye: bestrophins, lipofuscinosis, and retinal degeneration. Physiol. 2005;20:292–302. doi: 10.1152/physiol.00021.2005. [DOI] [PubMed] [Google Scholar]
- 31.Hartzell HC, Qu Z, Yu K, Xiao Q, Chien LT. Molecular physiology of bestrophins: multifunctional membrane proteins linked to best disease and other retinopathies. Physiol Rev. 2008;88:639–672. doi: 10.1152/physrev.00022.2007. [DOI] [PubMed] [Google Scholar]
- 32.Hartzell HC, Yu K, Xiao Q, Chien LT, Qu Z. Anoctamin/TMEM16 family members are Ca2+-activated Cl- channels. J Physiol. 2009;587:2127–2139. doi: 10.1113/jphysiol.2008.163709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hughes BA, Gallemore RP, Miller SS. Transport Mechanisms in the Retinal Pigment Epithelium. In: Marmor MF, Wolfensberger TJ, editors. The Retinal Pigment Epithelium. Oxford Univ. Press; Oxford, UK: 1998. pp. 103–134. [Google Scholar]
- 34.Jentsch TJ, Poet M, Fuhrmann JC, Zdebik AA. Physiological functions of CLC Cl-channels gleaned from human genetic disease and mouse models. Annu Rev Physiol. 2005;67:779–807. doi: 10.1146/annurev.physiol.67.032003.153245. [DOI] [PubMed] [Google Scholar]
- 35.Kolesnick RN, Kronke M. Regulation of ceramide production and apoptosis. Annu Rev Physiol. 1998;60:643–665. doi: 10.1146/annurev.physiol.60.1.643. [DOI] [PubMed] [Google Scholar]
- 36.Kranjc A, Grillo FW, Rievaj J, Boccaccio A, Pietrucci F, Menini A, Carloni P, Anselmi C. Regulation of bestrophins by Ca2+: a theoretical and experimental study. PLoS One. 2009;4:e4672. doi: 10.1371/journal.pone.0004672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kunzelmann K, Milenkovic VM, Spitzner M, Soria RB, Schreiber R. Calcium-dependent chloride conductance in epithelia: is there a contribution by Bestrophin? Pflugers Arch. 2007;454:879–889. doi: 10.1007/s00424-007-0245-z. [DOI] [PubMed] [Google Scholar]
- 38.Lang F, Busch GL, Ritter M, Volkl H, Waldegger S, Gulbins E, Haussinger D. Functional significance of cell volume regulatory mechanisms. Physiol Rev. 1998;78:247–306. doi: 10.1152/physrev.1998.78.1.247. [DOI] [PubMed] [Google Scholar]
- 39.Linsenmeier RA, Steinberg RH. Origin and sensitivity of the light peak in the intact cat eye. J Physiol. 1982;331:653–673. doi: 10.1113/jphysiol.1982.sp014396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marmorstein AD, Cross HE, Peachey NS. Functional roles of bestrophins in ocular epithelia. Prog Retin Eye Res. 2009;28:206–226. doi: 10.1016/j.preteyeres.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marmorstein AD, Marmorstein LY, Rayborn M, Wang X, Hollyfield JG, Petrukhin K. Bestrophin, the product of the Best vitelliform macular dystrophy gene (VMD2), localizes to the basolateral membrane of the retinal pigment epithelium. Proc Natl Acad Sci USA. 2000;97:12758–12763. doi: 10.1073/pnas.220402097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marmorstein LY, McLaughlin PJ, Stanton JB, Yan L, Crabb JW, Marmorstein AD. Bestrophin interacts physically and functionally with protein phosphatase 2A. J Biol Chem. 2002;277:30591–30597. doi: 10.1074/jbc.M204269200. [DOI] [PubMed] [Google Scholar]
- 43.Marmorstein LY, Wu J, McLaughlin P, Yocom J, Karl MO, Neussert R, Wimmers S, Stanton JB, Gregg RG, Strauss O, Peachey NS, Marmorstein AD. The light peak of the electroretinogram is dependent on voltage-gated calcium channels and antagonized by bestrophin (best-1) J Gen Physiol. 2006;127:577–589. doi: 10.1085/jgp.200509473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marquardt A, Stohr H, Passmore LA, Kramer F, Rivera A, Weber BH. Mutations in a novel gene, VMD2, encoding a protein of unknown properties cause juvenile-onset vitelliform macular dystrophy (Best’s disease) Hum Mol Genet. 1998;7:1517–1525. doi: 10.1093/hmg/7.9.1517. [DOI] [PubMed] [Google Scholar]
- 45.McCarty NA, O’Neil RG. Calcium signaling in cell volume regulation. Physiol Rev. 1992;72:1037–1061. doi: 10.1152/physrev.1992.72.4.1037. [DOI] [PubMed] [Google Scholar]
- 46.Men G, Batioglu F, Ozkan SS, Atilla H, Ozdamar Y, Aslan O. Best’s vitelliform macular dystrophy with pseudohypopyon: an optical coherence tomography study. Am J Ophthalmol. 2004;137:963–965. doi: 10.1016/j.ajo.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 47.Miller S, Farber D. Cyclic AMP modulation of ion transport across frog retinal pigment epithelium. Measurements in the short-circuit state. J Gen Physiol. 1984;83:853–874. doi: 10.1085/jgp.83.6.853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mohler CW, Fine SL. Long-term evaluation of patients with Best’s vitelliform dystrophy. Ophthalmol. 1981;88:688–692. doi: 10.1016/s0161-6420(81)34965-3. [DOI] [PubMed] [Google Scholar]
- 49.Mullins RF, Kuehn MH, Faidley EA, Syed NA, Stone EM. Differential macular and peripheral expression of bestrophin in human eyes and its implication for best disease. Invest Ophthalmol Vis Sci. 2007;48:3372–3380. doi: 10.1167/iovs.06-0868. [DOI] [PubMed] [Google Scholar]
- 50.Niemeyer G. Retinal research using the perfused mammalian eye. Prog Retinal Eye Res. 2001;20:289–318. doi: 10.1016/s1350-9462(00)00029-x. [DOI] [PubMed] [Google Scholar]
- 51.O’Driscoll KE, Leblanc N, Hatton WJ, Britton FC. Functional properties of murine bestrophin 1 channel. Biochem Biophys Res Commun. 2009;384:476–481. doi: 10.1016/j.bbrc.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Obeid LM, Linardic CM, Karolak LA, Hannun YA. Programmed cell death induced by ceramide. Science. 1993;259:1769–1771. doi: 10.1126/science.8456305. [DOI] [PubMed] [Google Scholar]
- 53.Park H, Oh SJ, Han KS, Woo DH, Park H, Mannaioni G, Traynelis SF, Lee CJ. Bestrophin-1 encodes for the Ca2+-activated anion channel in hippocampal astrocytes. J Neurosci. 2009;29:13063–13073. doi: 10.1523/JNEUROSCI.3193-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Peterson WM, Meggyesy CF, Yu KF, Miller SS. Extracellular ATP activates calcium signaling, ion, and fluid transport in retinal pigment epithelium. J Neurosci. 1997;17:2324–2337. doi: 10.1523/JNEUROSCI.17-07-02324.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Petrukhin K, Koisti MJ, Bakall B, Li W, Xie G, Marknell T, Sandgren O, Forsman K, Holmgren G, Andreasson S, Vujic M, Bergen AAB, McGarty-Dugan V, Figueroa D, Austin CP, Metzker ML, Caskey CT, Wadelius C. Identification of the gene responsible for Best macular dystrophy. Nat Genet. 1998;19:241–247. doi: 10.1038/915. [DOI] [PubMed] [Google Scholar]
- 56.Pianta MJ, Aleman TS, Cideciyan AV, Sunness JS, Li Y, Campochiaro BA, Campochiaro PA, Zack DJ, Stone EM, Jacobson SG. In vivo micropathology of Best macular dystrophy with optical coherence tomography. Exp Eye Res. 2003;76:203–211. doi: 10.1016/s0014-4835(02)00280-4. [DOI] [PubMed] [Google Scholar]
- 57.Pierro L, Tremolada G, Introini U, Calori G, Brancato R. Optical coherence tomography findings in adult-onset foveomacular vitelliform dystrophy. Am J Ophthalmol. 2002;134:675–680. doi: 10.1016/s0002-9394(02)01685-9. [DOI] [PubMed] [Google Scholar]
- 58.Qu Z, Cheng W, Cui Y, Cui Y, Zheng J. Human disease-causing mutations disrupt an N-C-terminal interaction and channel function of bestrophin 1. J Biol Chem. 2009;284:16473–16481. doi: 10.1074/jbc.M109.002246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Qu Z, Chien LT, Cui Y, Hartzell HC. The anion-selective pore of the bestrophins, a family of chloride channels associated with retinal degeneration. J Neurosci. 2006;26:5411–5419. doi: 10.1523/JNEUROSCI.5500-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Qu Z, Fischmeister R, Hartzell C. Mouse bestrophin-2 is a bona fide Cl(−) channel: identification of a residue important in anion binding and conduction. J Gen Physiol. 2004;123:327–340. doi: 10.1085/jgp.200409031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Qu Z, Hartzell C. Determinants of anion permeation in the second transmembrane domain of the mouse bestrophin-2 chloride channel. J Gen Physiol. 2004;124:371–382. doi: 10.1085/jgp.200409108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Qu Z, Hartzell HC. Bestrophin Cl− channels are highly permeable to HCO3−. Am J Physiol Cell Physiol. 2008;294:C1371–C1377. doi: 10.1152/ajpcell.00398.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ranty ML, Carpentier S, Cournot M, Rico-Lattes I, Malecaze F, Levade T, Delisle MB, Quintyn JC. Ceramide production associated with retinal apoptosis after retinal detachment. Graefes Arch Clin Exp Ophthalmol. 2009;247:215–224. doi: 10.1007/s00417-008-0957-6. [DOI] [PubMed] [Google Scholar]
- 64.Rosenthal R, Bakall B, Kinnick T, Peachey N, Wimmers S, Wadelius C, Marmorstein A, Strauss O. Expression of bestrophin-1, the product of the VMD2 gene, modulates voltage-dependent Ca2+ channels in retinal pigment epithelial cells. FASEB J. 2006;20:178–180. doi: 10.1096/fj.05-4495fje. [DOI] [PubMed] [Google Scholar]
- 65.Ruvolo PP. Ceramide regulates cellular homeostasis via diverse stress signaling pathways. Leukemia. 2001;15:1153–1160. doi: 10.1038/sj.leu.2402197. [DOI] [PubMed] [Google Scholar]
- 66.Schroeder BC, Cheng T, Jan YN, Jan LY. Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell. 2008;134:1019–1029. doi: 10.1016/j.cell.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Seddon JM, Afshari MA, Sharma S, Bernstein PS, Chong S, Hutchinson A, Petrukhin K, Allikmets R. Assessment of mutations in the best macular dystrophy (VMD2) gene in patients with adult-onset foveomacular vitelliform dystrophy, age-related maculopathy, and bull’s-eye maculopathy. Ophthalmol. 2001;108:2060–2067. doi: 10.1016/s0161-6420(01)00777-1. [DOI] [PubMed] [Google Scholar]
- 68.Shaban H, Borras C, Vina J, Richter C. Phosphatidylglycerol potently protects human retinal pigment epithelial cells against apoptosis induced by A2E, a compound suspected to cause age-related macula degeneration. Exp Eye Res. 2002;75:99–108. doi: 10.1006/exer.2001.1192. [DOI] [PubMed] [Google Scholar]
- 69.Strauss O. The Retinal Pigment Epithelium in Visual Function. Physiol Rev. 2005;85:845–881. doi: 10.1152/physrev.00021.2004. [DOI] [PubMed] [Google Scholar]
- 70.Sun H, Tsunenari T, Yau K-W, Nathans J. The vitelliform macular dystrophy protein defines a new family of chloride channels. Proc Natl Acad Sci USA. 2002;99:4008–4013. doi: 10.1073/pnas.052692999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Szel A, Rohlich P, Caffe AR, van VT. Distribution of cone photoreceptors in the mammalian retina. Microsc Res Tech. 1996;35:445–462. doi: 10.1002/(SICI)1097-0029(19961215)35:6<445::AID-JEMT4>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 72.Tsunenari T, Sun H, Williams J, Cahill H, Smallwood P, Yau K-W, Nathans J. Structure-function analysis of the bestrophin family of anion channels. J Biol Chem. 2003;278:41114–41125. doi: 10.1074/jbc.M306150200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vedantham V, Ramasamy K. Optical coherence tomography in Best’s disease: an observational case report. Am J Ophthalmol. 2005;139:351–353. doi: 10.1016/j.ajo.2004.07.039. [DOI] [PubMed] [Google Scholar]
- 74.Wangsa-Wirawan ND, Linsenmeier RA. Retinal oxygen: fundamental and clinical aspects. Arch Ophthalmol. 2003;121:547–557. doi: 10.1001/archopht.121.4.547. [DOI] [PubMed] [Google Scholar]
- 75.Weng TX, Godley BF, Jin GF, Mangini NJ, Kennedy BG, Yu AS, Wills NK. Oxidant and antioxidant modulation of chloride channels expressed in human retinal pigment epithelium. Am J Physiol Cell Physiol. 2002;283:C839–C849. doi: 10.1152/ajpcell.00445.2001. [DOI] [PubMed] [Google Scholar]
- 76.Wills NK, Weng T, Mo L, Hellmich HL, Yu A, Wang T, Buchheit S, Godley BF. Chloride channel expression in cultured human fetal RPE cells: response to oxidative stress. Invest Ophthalmol Vis Sci. 2000;41:4247–4255. [PubMed] [Google Scholar]
- 77.Winkler BS. Buffer dependence of retinal glycolysis and ERG potentials. Exp Eye Res. 1986;42:585–593. doi: 10.1016/0014-4835(86)90048-5. [DOI] [PubMed] [Google Scholar]
- 78.Wu J, Marmorstein AD, Peachey NS. Functional abnormalities in the retinal pigment epithelium of CFTR mutant mice. Exp Eye Res. 2006;83:424–428. doi: 10.1016/j.exer.2006.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wu J, Marmorstein AD, Striessnig J, Peachey NS. Voltage-Dependent Calcium Channel CaV1.3 Subunits Regulate the Light Peak of the Electroretinogram. J Neurophysiol. 2007;97:3731–3735. doi: 10.1152/jn.00146.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Xiao Q, Prussia A, Yu K, Cui YY, Hartzell HC. Regulation of bestrophin Cl channels by calcium: role of the C terminus. J Gen Physiol. 2008;132:681–692. doi: 10.1085/jgp.200810056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Xiao Q, Yu K, Cui YY, Hartzell HC. Dysregulation of human bestrophin-1 by ceramide-induced dephosphorylation. J Physiol. 2009;587:4379–4391. doi: 10.1113/jphysiol.2009.176800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yamada Y, Tian J, Yang Y, Cutler RG, Wu T, Telljohann RS, Mattson MP, Handa JT. Oxidized low density lipoproteins induce a pathologic response by retinal pigmented epithelial cells. J Neurochem. 2008;105:1187–1197. doi: 10.1111/j.1471-4159.2008.05211.x. [DOI] [PubMed] [Google Scholar]
- 83.Yang YD, Cho H, Koo JY, Tak MH, Cho Y, Shim WS, Park SP, Lee J, Lee B, Kim BM, Raouf R, Shin YK, Oh U. TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature. 2008;455:1210–1215. doi: 10.1038/nature07313. [DOI] [PubMed] [Google Scholar]
- 84.Yardley J, Leroy BP, Hart-Holden N, Lafaut BA, Loeys B, Messiaen LM, Perveen R, Reddy MA, Bhattacharya SS, Traboulsi E, Baralle D, De Laey JJ, Puech B, Kestelyn P, Moore AT, Manson FD, Black GC. Mutations of VMD2 splicing regulators cause nanophthalmos and autosomal dominant vitreoretinochoroidopathy (ADVIRC) Invest Ophthalmol Vis Sci. 2004;45:3683–3689. doi: 10.1167/iovs.04-0550. [DOI] [PubMed] [Google Scholar]
- 85.Yu K, Cui Y, Hartzell HC. The bestrophin mutation A243V, linked to adult-onset vitelliform macular dystrophy, impairs its chloride channel function. Invest Ophthalmol Vis Sci. 2006;47:4956–4961. doi: 10.1167/iovs.06-0524. [DOI] [PubMed] [Google Scholar]
- 86.Yu K, Qu Z, Cui Y, Hartzell HC. Chloride channel activity of bestrophin mutants associated with mild or late-onset macular degeneration. Invest Ophthalmol Vis Sci. 2007;48:4694–4705. doi: 10.1167/iovs.07-0301. [DOI] [PubMed] [Google Scholar]
- 87.Yu K, Xiao Q, Cui G, Lee A, Hartzell HC. The best disease-linked Cl− channel hBest1 regulates Ca V 1 (L-type) Ca2+ channels via src-homology-binding domains. J Neurosci. 2008;28:5660–5670. doi: 10.1523/JNEUROSCI.0065-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yu K, Lujan R, Marmorstein A, Gabriel S, Hartzell HC. Bestrophine-2 mediates bicarbonate transport by goblet cells in mammalian colon. J Clin Invest. 2010;120(5) doi: 10.1172/JCI41129. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang Y, Stanton JB, Wu J, Yu K, Hartzell HC, Peachey NS, Marmorstein LY, Marmorstein AD. Suppression of Ca2+ Signaling in a Mouse Model of Best Disease. Hum Mol Genet. 2010;19:1108–1118. doi: 10.1093/hmg/ddp583. http://hmg.oxfordjournals.org/cgi/content/full/ddp583v2. [DOI] [PMC free article] [PubMed] [Google Scholar]