

Abstract
We explored the crosstalk between cell survival (phosphatidylinositol 3-kinase (PI3K)/Akt) and mitogenic (Ras/Raf/MEK/extracellular signal-regulated kinase (ERK)) signaling pathways activated by an epidermal growth factor (EGF) and analyzed their sensitivity to small molecule inhibitors in the PI3K-mutant estrogen receptor (ER)-positive MCF7 and T47D breast cancer cells. In contrast to MCF7 cells, ERK phosphorylation in T47D cells displayed resistance to MEK inhibition by several structurally different compounds, such as U0126, PD 098059 and PD 198306, MEK suppression by small interfering RNA (siRNA) and was also less sensitive to PI3K inhibition by wortmannin. Similar effect was observed in PI3K-wild type ER-positive BT-474 cells, albeit to a much lesser extent.
MEK-independent ERK activation was induced only by ErbB receptor ligands and was resistant to inhibition of several kinases and phosphatases that are known to participate in the regulation of Ras/mitogen-activated protein kinase (MAPK) cascade. Although single agents against PDK1 or Akt did not affect EGF-induced ERK phosphorylation, a combination of PI3K/Akt and MEK inhibitors synergistically suppressed ERK activation and cellular growth. siRNA-mediated silencing of class I PI3K or Akt1/2 genes also significantly decreased U0126-resistant ERK phosphorylation.
Our data suggest that in T47D cells ErbB family ligands induce a dynamic, PI3K/Akt-sensitive and MEK-independent compensatory ERK activation circuit that is absent in MCF7 cells. We discuss candidate proteins that can be involved in this activation circuitry and suggest that PDZ-Binding Kinase/T-LAK Cell-Originated Protein Kinase (PBK/TOPK) may play a role in mediating MEK-independent ERK activation.
Keywords: EGF signaling, ERK activation, PI3K/Akt, crosstalk, breast cancer resistance, MEK inhibition
INTRODUCTION
Infiltrative ductal carcinomas often show uncontrolled anchorage-independent growth, increased invasiveness and survival, which can be attributed to the enhanced autocrine growth factor production, overexpression of their cognate receptors and dysregulation of intracellular signal transduction pathways. Intrinsic or acquired resistance of breast tumor cells to anticancer drugs, including anti-estrogens, can emerge from compensatory circuits and engagement of redundant signaling pathways [1].
The epidermal growth factor (EGF)1 receptor, a member of the ErbB family of receptor tyrosine kinases, regulates normal mammary gland growth and development [2] and is overexpressed in 15-20% of breast carcinomas [3]. EGF-evoked signals are generally transmitted to the Raf/MEK/ERK (mitogen-activated protein kinase (MAPK)) cascade through the small GTPase Ras and Src family tyrosine kinases. EGF also facilitates the activation of class I phosphoinositide 3-kinases (PI3K). PI3K phosphorylates phosphatidylinositol lipids to generate PI(3,4,5)P3 or other phosphoinositides, which are recognized by pleckstrin homology (PH) domains of various proteins, including the downstream effector serine/threonine kinase Akt. Membrane targeted Akt is subsequently phosphorylated and activated by phosphatidylinositol-dependent kinase 1 (PDK1). In addition, PDK1 activates some protein kinase C (PKC) isozymes and together with Akt regulates p70 ribosomal S6 kinase (p70S6K) activation and the phosphorylation of ribosomal S6 protein (S6RP), implicated in the control of the translational machinery [4]. The translocation of phosphorylated ERK, Akt and their substrates into the cell nucleus leads to the expression of specific sets of genes that determine relevant biological responses to extracellular cues: namely, cell division, proliferation, differentiation, adhesion or migration, cytoskeletal rearrangements, changes in metabolism, DNA repair, survival or death (apoptosis) [5].
Cell survival (PI3K/Akt) and mitogenic (Ras/MAPK) linear cascades rarely act as independent parallel pathways; rather they influence each other at different points and phases of signal propagation in both negative and positive manners, resulting in dynamic and complex crosstalk. This crosstalk is mediated by various kinases and phosphatases as shown in Fig. 1. The occurrence of multiple regulatory feedbacks depends on cell type, the stage of cell differentiation, ligand type and dose. Thus, the separate inhibition of one or another module may not always lead to the desired suppression of tumor growth.
Fig. 1.

Scheme of interactions between cell surivival (PI3K/Akt) and mitogenic (Ras/MAPK) signaling pathways (A) and modulation of ERK activity by phosphatases and their upstream effectors (B). Arrows show activatory- and blunt-end lines - inhibitory interactions
In previous studies we focused on the crosstalk of PI3K/Akt and Ras/MAPK in normal cells without mutations in these pathways resulting from concerted stimulation by EGF and insulin [6, 7]. Significant crosstalk between the PI3K/Akt and Ras/MAPK pathways in tumor cells makes pathway activation robust to perturbations, which relates to different drug sensitivity profiles [8-10]. In this study, we use small molecule agents and siRNA treatments to investigate whether the PI3K/Akt and Ras/MAPK signaling pathways display similar sensitivities to inhibition in tumorigenic T47D and MCF7 breast cancer cells upon EGF stimuli. Derived from metastatic pleural effusions of invasive ductal carcinoma, these cell lines express ER-α, contain highly oncogenic activating PI3K gene mutations (E545K in MCF-7, H1047R in T47D), and are routinely used as contrasting models for studies of drug resistance to the anti-estrogens toremifene and tamoxifen [11-13]. Herein we describe an unusual PI3K/Akt-sensitive MEK-independent circuit of EGF-dependent ERK activation in T47D cells, which may underlie higher survival rates and reported anti-estrogen resistance of these cells in comparison with anti-estrogen-sensitive MCF7 cells.
MATERIALS AND METHODS
Chemicals and antibodies
Insulin was obtained from Sigma-Aldrich (St. Louis, MO), 1,2-Dioleoyl-sn-glycerol – from Cayman Chemical (Ann Arbor, MI) and other growth factors were purchased from PeproTech Inc. (Rocky Hill, NJ). The stock solutions of inhibitors listed in Table S1 were prepared in dimethyl sulphoxide (DMSO) (Sigma-Aldrich). List of specific antibodies used in this study and their commercial sources are indicated in Table S2. All other common chemicals, solvents and reagents were of highest grade available from various commercial sources.
Cell lines and culture conditions
T47D (ATCC No. HTB-133) cells were cultured in a complete RPMI-1640 media with L-glutamine and 25mM HEPES (Mediatech Inc., Manassas, VA) supplemented with 10% fetal bovine serum (FBS) (Gemini Bio-Products, West Sacramento, CA), 20 μg/ml bovine insulin (Sigma-Aldrich) and penicillin-streptomycin solution (100 μg/ml each) (Mediatech Inc.). MCF7 (ATCC No. HTB-22), BT-474 (ATCC No. HTB-20) and CAPAN-1 (ATCC No. HTB-79) cells were grown in a complete DMEM/F-12 media (Mediatech Inc.) containing 10% FBS and 1% penicillin-streptomycin solution. All cells were cultivated in a humidified 5% CO2 incubator at 37°C. Cells were grown for 4–5 days and after reaching confluency were harvested by exposure to 0.25% Trypsin–EDTA solution (GIBCO) and then passed into new T-75 tissue culture flasks (Denville Scientific, Metuchen, NJ).
Cell stimulation and protein extraction
For ligand-response studies, cells were plated in 60×15 cm dishes (Denville Scientific, Metuchen, NJ) and grown until they reached 80% confluency. The cells were starved overnight in appropriate FBS/insulin-free media, (if required) preincubated with fixed concentrations of inhibitors or DMSO vehicle alone, stimulated with indicated ligand(s) for indicated time intervals at 37°C and lysed with the lysis buffer. The preparation of total protein extracts, electrophoresis and Multistrip Western blotting procedures were performed as described previously [6, 14]. Briefly, cell lysates were subjected to LDS-PAGE. Separated proteins were electrotransferred onto nitrocellulose membranes. For semiquantitative immunoblot analyses, the membranes previously blocked with 5% bovine serum albumin (BSA) solution were probed with specific primary and corresponding secondary antibodies (dilutions are indicated in Table S2). Signals of protein bands were detected by enhanced chemiluminescence system (Pierce Biotechnology/Thermo Fisher Scientific, Rockford, IL) and quantified using KODAK Image Station 440CF software. Kinetic curves and charts were plotted in SigmaPlot. Obtained signal values were normalized to the appropriate loading control.
Cell transfection
Half an hour or less before transfection T47D cells were trypsinized and resuspended in antibiotics-free complete media. 8×105 cells per sample were aliquoted into Eppendorf tubes and centrifuged at 90 g for 10 min at room temperature. Supernatant was removed and cell pellet was resuspended in 100 μl of Nucleofector solution V (Lonza, Basel, Switzerland) containing siRNA. The responses of cells transfected with 200 nM of MEK1 and MEK2 siRNA were compared to those transfected with siCONTROL scrambled Non-Targeting siRNA (all from Dharmacon, Lafayette, CO). The following validated MEK siRNA sequences were used: 5′-AAGCAACUCAUGGUUCAUGCUUU-3′ for MEK1 and 5′-AAGAAGGAGAGCCUCACAGCAUU-3′ for MEK2 [15]. The responses of cells transfected with 50 nM of validated PIK3R1, PIK3CA, AKT1, AKT2, PBK/TOPK, BLVRA, FER or RIPK2 siRNA were compared to those transfected with AllStar negative control siRNA (all from Qiagen, Valencia, CA). Cell suspensions containing siRNA were electroporated using the X-005 program on Amaxa’s Nucleofector II device (Lonza Cologne AZ, Basel, Switzerland). Immediately after electroporation, 0.5 ml of the pre-equilibrated antibiotics-free complete media was added to the cuvette and cell suspension was gently transferred into 6-well plates (final volume 2.0 ml media per well). Cells were allowed to attach for 6 hours before the addition of penicillin-streptomycin solution. Total proteins were isolated from cells harvested 72 hours post-transfection. Approximately 70% ± 5% suppression of target proteins was achieved.
Cell viability assay
Cells were serum-starved overnight in T-75 flask. Equal amounts of serum-starved cells were seeded into 96-well plates at a density of 5×103 cells/well and maintained in serum-free medium with or without inhibitors for an hour before the addition of 2 nM EGF. The plates were further incubated for 72 hours at 37°C. Three hours prior to the designated time point, 20 μl of AlamarBlue (AB) (Invitrogen, Carlsbad, CA) was directly added to the medium of the test wells resulting in a final vol/vol concentration of 10%. AB contains resazurin, a non-fluorescent indicator dye, which is converted to bright red–fluorescent resorufin via the reduction reactions of metabolically active cells. The amount of fluorescence produced is proportional to the number of living cells. As negative control AB was added to the serum-free medium without cells, whereas cells grown in complete media served as a positive control. After incubation with AB at 37°C, the fluorescence of test, positive and negative control wells was read with Synergy HT microplate reader (BioTek Instruments, Winooski, VT) using the excitation/emission wavelengths of 530/590 nm filter settings. Negative control values were averaged and subtracted from the fluorescence values obtained from each test well. Results were presented as percentage increase in cell proliferation compared with control groups for the same incubation time. Each bar is mean ± SD of 12 well replicates.
RESULTS AND DISCUSSION
Differential sensitivity of T47D and MCF7 breast cancer cells to MEK/ERK and PI3K/Akt signaling in response to EGF
We examined the interaction between cell survival (PI3K/Akt) and mitogenic (Ras/MAPK) signal transduction pathways in EGF-responsive T47D and MCF7 breast cancer cell lines (Fig. 2). For this purpose, serum-starved T47D or MCF7 cells were pretreated with the specific small molecule inhibitors of MEK1/2 (U0126), PI3K (wortmannin), PDK1 (OSU-03012) or Akt1/2/3 (Akt-VIII), followed by stimulation with 1 nM EGF for varying time intervals. To assess the kinetics of ERK and Akt activation and compare it in control (untreated cells) and inhibitor-treated cells, Multistrip Western blotting analysis was performed using anti-phospho-p44/42 MAPK antibodies recognizing dually phosphorylated ERK1 (Thr202/Tyr204) and ERK2 (Thr204/Tyr187) isoforms and anti-phospho-Akt antibodies detecting endogenous levels of Akt1, Akt2 and Akt3 isoforms if they are phosphorylated on Ser473 residues.
Fig. 2. Time courses of EGF-induced ERK and Akt activation in T47D and MCF7 breast cancer cells in the presence of MEK/ERK or PI3K/Akt inhibitors.

Serum-starved T47D (A) or MCF7 (B) cells were either left untreated (control, black circles) or treated with wortmannin (200 nM, 30 min; black triangles down), Akt-VIII inhibitor (5 μM, 1 h; grey circles), OSU-03012 (50 μM, 1 h; grey squares) or U0126 (10 μM, 30 min; white circles) before stimulation with EGF (1 nM) for the indicated time intervals (min). Equal amounts of total cell lysates were resolved by NuPAGE and subjected to Multistrip Western blotting. Immunoblots (IB) were probed with antibodies against phospho-p44/42 MAPK (ERK1/2) (Thr202/Tyr204 and Thr204/Tyr187) and p44/42 MAPK (ERK1/2) (loading control) or phospho-Akt (Ser473) and Akt (loading control). Each graph represents the intensity of protein activation in arbitrary units (AU) plotted as the ratio between signals of phosphorylated and non-phosphorylated protein forms.
In both cell lines, EGF caused robust phosphorylation of ERK1/2, but its activation patterns were notably distinct, being sustained in T47D cells (Fig. 2A) and more transient in MCF7 cells (Fig. 2B). PI3K and PDK1 were involved in the positive regulation of Ras/MAPK signaling to a greater extent in MCF7 than in T47D cells. In both cell lines, wortmannin attenuated ERK1/2 phosphorylation levels much stronger than Akt-VIII inhibitor, which indicates that wortmannin inhibits additional positive regulators of ERK that lay upstream of Akt, e.g. PH-domain containing adaptor protein GAB1, PDK-1 and PI3K-activated PAK.
MEK inhibition by U0126 increased Akt activation in both cell lines (Figs. 2A-B, lower panels), which is consistent with previous reports in other cellular systems upon EGF treatment [6, 16]. Surprisingly, in U0126-pretreated T47D cells, ERK1/2 remained substantially phosphorylated, whereas its phosphorylation was completely abrogated in MCF7 cells (Fig. 2A-B, upper panels).
EGF-induced ERK phosphorylation in T47D cells depends only partially on MEK activity
To explore a possible mechanism of ERK1/2 activation, under conditions where MEK is inhibited, we determined the detailed kinetics of ERK1/2 phosphorylation on T47D cells, stimulated with saturating (1 nM), mid-range (0.1 nM) and low (0.01 nM) EGF doses in the presence or absence of 10 μM U0126. As shown in Fig. 3A, ERK1/2 phosphorylation in MEK inhibited cells was time-dependent, starting after 5 min and reaching the saturation by 30 min, then slowly declined with a temporal pattern that is independent of EGF dose. Similar, albeit substantially weaker, U0126-resistant ERK1/2 phosphorylation was observed in 1 nM EGF-treated BT-474 breast cancer cells (Fig. 1S, upper panel). By contrast, treatment with 10 μM U0126 completely abrogated EGF-induced ERK1/2 phosphorylation in pancreatic cancer CAPAN-1 cells (Fig. 1S, lower panel), and has also been reported to prevent MEK1/2 signaling to ERK1/2 in a variety of other EGF-responsive epithelial cells [6, 17-19].
Fig. 3. A. Effects of MEK inhibitor U0126 on ERK phosphorylation upon treatment with different doses of EGF (0.01, 0.1, 1 nM).

Serum-starved cells were incubated with U0126 (10 μM) for 30 min before stimulation with indicated EGF doses for the indicated time intervals (min). Equal amounts of total cell lysates were resolved by NuPAGE and subjected to Multistrip Western blotting. Immunoblots (IB) were probed with anti-phospho-p44/42 MAPK (ERK1/2) (labeled as p-ERK). B. Effects of structurally distinct MEK inhibitors on ERK phosphorylation in T47D cells. Cells were preincubated with PD 098059 (50 μM) or PD 198306 (200 nM) for 30 min and stimulated with EGF (1 nM) for the indicated time intervals (min). C. The effect of suppression of MEK protein levels on ERK phosphorylation in the presence or absence of MEK inhibitor. T47D cells were transfected with either 200 nM of Scrambled (Scr) or MEK1/2 siRNA as described in “Materials and Methods”. Serum-starved cells were pretreated with U0126 (10 μM, 30 min) and stimulated with 1 nM EGF for 30 min. Suppression levels of MEK1/2 were assessed by probing immunoblots with anti-MEK1/2 antibody. GAPDH levels were used as loading controls.
The behavior of phospho-ERK1/2 in the presence of U0126 was similar when immunoassayed with antibodies that detect a) either individually or dually phosphorylated ERK1/2 b) only monophosphorylated Thr residues of ERK1/2 (Fig. 2S) or c) all phosphorylated Tyr residues (pY20) in ERK1/2 immunoprecipitates (data not shown). These findings confirm that the U0126-resistant ERK species are active, since the phosphorylation of both Thr and Tyr residues is required to attain full ERK1/2 activity.
Drug sensitivities can vary between cell lines due to differences in inhibition constants, different cell penetration, metabolic degradation or excretion of the inhibitor [20-22]. To verify that the effect was not caused by non-specific properties of U0126, which is also reported to inhibit MEK5 signaling to ERK5 [23], T47D cells were treated with two other structurally unrelated MEK inhibitors PD 098059 and PD 198306. At concentrations over 10-50 fold higher than their IC50, these inhibitors failed to prevent EGF-induced ERK1/2 activation (Fig. 3B). To further test the interpretation that there is a MEK-independent ERK phosphorylation, T47D cells were transfected with small interfering RNA (siRNA) specifically targeting MEK1 and MEK2 or scrambled control siRNA (Scr siRNA) for 72 hours and treated with 1 nM EGF for an additional 30 min in the presence or absence of 10 μM U0126 (Fig. 3C). Quantification of the blots showed, that while MEK1/2 expression was suppressed by 70% in MEK1/2 siRNA transfected cells compared to Scr siRNA, phospho-ERK1/2 protein levels were reduced by only ~20%. By comparison, ERK1/2 phosphorylation in cells treated with U0126 alone was only ~30% less than in untreated control cells. GAPDH levels did not show significant differences between MEK1/2- and Scr siRNA-transfected groups.
Taken together, these data suggest that initial ERK1/2 activation at early time points is exclusively mediated by MEK, but at later times MEK’s contribution becomes less, since the MEK-independent pathway accounts for more than 40% of remaining ERK1/2 phosphorylation.
MEK-independent ERK activation is triggered only by ErbB family ligands
The results presented above suggest that in EGF-stimulated T47D cells a significant pool of ERK becomes activated via MEK-independent mechanism. To examine whether this mechanism is ErbB receptor family-specific, T47D cells were stimulated with TGF-α, HRG-β and ligands that are important for mammary tumor progression (insulin (INS), IGF-1, PDGF, FGF, prolactin (PRL), VEGF). The left panel in Fig. 4 shows that ERK1/2 responses to INS and IGF-1 are weaker compared to EGF, TGF-α and HRG-β (which were equipotent in stimulating ERK1/2). While U0126 abrogated ERK1/2 activation in response to IGF-1 and INS, the cells that were stimulated with the EGF family peptides continued to signal downstream of MEK and increase phospho-ERK1/2 levels. Moreover, neither a very high dose of IGF-1 (50 nM), nor PDGF, FGF, PRL, VEGF or FBS were able to induce ERK1/2 phosphorylation in the presence of U0126 (Fig. 4, right panel). This can imply that the proto-oncogene ErbB2 receptor, the preferred heterodimerization partner of EGFR [24], prolongs the duration of MEK/ERK response [25] and accounts for the ERK resistance to MEK inhibition, as reported previously [26]. Theoretically, in this case, the cell lines expressing higher levels of ErbB2 and showing sustained ERK kinetics (e.g. BT-474 or CAPAN-1) would be even more resistant to U0126 treatment. This is not what we observed (Fig. 1S). Moreover, the inhibition of ErbB2 with a selective ATP-competitive inhibitor tyrphostin (AG-825, 5 μM) did not alter the levels of phospho-ERK1/2 in the presence of U0126 (data not shown).
Fig. 4. Effects of various growth factors on p-ERK resistance to MEK inhibition by U0126.

Serum-starved T47D cells were incubated with U0126 (10 μM) for 30 min before stimulation with EGF (1 nM), TGF-α (10 nM), HRG-β (1 nM), IGF-1 (10 nM) and insulin (50 nM) for the indicated time intervals (min) (left panel) or with EGF (1 nM), PDGF-BB (1 nM), FGF-basic (10 nM), prolactin (PRL, 10 nM), VEGF (1 nM), IGF-1 (50 nM) and 5% FBS for 30 min (right panel). Equal amounts of total cell lysates were resolved by NuPAGE and subjected to Western blotting. Immunoblots (IB) were probed with antibodies against phospho-ERK1/2 (Thr202/Tyr204), phospho-Akt1/2/3 (Ser473) or GAPDH (loading control).
U0126-resistant ERK activation depends on kinase(s) located downstream of PI3K/Akt
To distinguish the kinase(s) that could be potentially involved in an alternative route of ERK activation in EGF-stimulated T47D cells, we applied a set of widely used small molecule inhibitors and measured the relative amounts of phosphorylated ERK1/2 by immunoblotting at various time points (Fig. 5A, upper panel). Total levels of ERK1/2 were constant (not shown). The ratios between phospho- and total ERK signal values were plotted as a graph (Fig. 5A, bottom panel).
Fig. 5. MEK-independent ERK activation depends on kinase(s) located downstream of PI3K.

A. Serum-starved T47D cells were either left untreated (Control, blot strip (1); black circles) or treated with U0126 alone (10 μM, 30 min; blot strip (2); white circles) or in combination with SQ22536 (50 μM, 1 h; blot strip (3); grey circles), Gö6850 (5 μM, 1 h; blot strip (4); black squares), Gö6983 (5 μM, 1 h; blot strip (5); white squares), Su6656 (10 μM, 1 h; blot strip (6); grey squares), wortmannin (WT) (200 nM, 30 min; blot strip (7); black diamonds), OSU-03012 (50 μM, 1 h; blot strip (8); white diamonds) and Akt-VIII inhibitor (5 μM, 1 h; blot strip (9); grey diamonds) before stimulation with EGF (1 nM) for the indicated time intervals (min). Total cell lysates were subjected to Multistrip Western blotting for ERK activation as described previously. The graph show the intensity of ERK activation in arbitrary units (AU) plotted as the ratio between phospho-ERK1/2 and total ERK1/2 signal. B. T47D cells were transfected with either 50 nM of PIK3R1 (p85-α regulatory subunit of PI3K), PIK3CA (p110-α catalytic subunit of PI3K), Akt1, Akt2 siRNA or with AllStar negative control siRNA as described in “Materials and Methods”. Serum-starved cells were pretreated with U0126 (10 μM, 30 min) and stimulated with 1 nM EGF for 30 min. The ratios between phospho-ERK1/2 and total ERK1/2 signals are shown as a percentage of siRNA control. Values represent a mean ± SD of three signal measurements.
EGFR-dependent cross-activation of G-protein coupled receptors could activate cAMP-dependent protein kinase A (PKA), involved in the activation of ERK-specific phosphatases PTP-SL and MKPs [27] (Fig. 1B). To inhibit cAMP production, and thus, PKA activity, we used the adenylate cyclase inhibitor SQ22536 (Fig. 5A). Compared to the U0126-only treated cells (blot strip 2), cAMP inhibition slightly increased ERK1/2 phosphorylation at 10-20 and then at 90-120 minutes (blot strip 3), indicating that there is no tight regulatory relationship between cAMP-dependent signal transducers and ERK1/2 when MEK is inactive.
To test the direct action of PKC isozymes on ERK1/2 activation, mixtures of U0126 and selective inhibitors of multiple PKC isoforms Gö6850 (blot strip 4) or Gö6983 (blot strip 5) were applied, but none of these combinations significantly changed ERK1/2 phosphorylation dynamics. The concentrations we used in our studies (5 μM of Gö6983 and Gö6850) far exceeded IC50 values (see Table S1), abolishing the activities of most PKC isoforms, except for PKC-μ , which functions at the Golgi compartment [28]. Although the expression of constitutively active PKCμ activates c-Raf leading to ERK1/2 phosphorylation, this effect entirely depends on MEK activity [29]. Furthermore, ERK1/2 phosphorylation, which was induced by cell stimulation with 1,2-Dioleoyl-sn-glycerol (DiC8), an analog of the PKC-activating secondary messenger DAG, was eliminated by U0126 treatment (Fig. 3S).
Stimulation of T47D cells with EGF results in the activation of tyrosine kinase c-Src, which generally contribute to the MAPK signaling upstream of MEK [30]. In addition, there are reports of c-Src-mediated regulation of ERK1/2 activity through the inactivation of PP2A phosphatase [31] (Fig. 1B). However, in our experimental conditions, c-Src inhibition by Su6656 (Fig. 5A, blot strip 6) or PP2 (5 μM, data not shown) did not attenuate ERK1/2 phosphorylation, exluding the role of c-Src for ERK1/2 activation downstream of MEK.
Despite the relatively weak action of the PI3K inhibitor wortmannin, the PDK1 inhibitor OSU-03012 or Akt1/2/3 inhibitor Akt-VIII on ERK activation when MEK was active (Fig. 2), the combination of each agent with U0126 resulted in the dramatic suppression of ERK1/2 phosphorylation (Fig. 5A, blot strips 7-9). Out of the three inhibitors, phospho-ERK1/2 was particularly sensitive to wortmannin treatment.
To further verify the requirement of PI3K and Akt for MEK-independent ERK activation, we transfected T47D cells either with siRNA against Akt1, Akt2 (Akt 3 isoform is not expressed in these cells), catalytic or regulatory subunits of PI3K or with negative control siRNA. 72 hours post-transfection, cells were pretreated with MEK inhibitor U0126 and stimulated with 1 nM EGF for 30 minutes. As a result of these treatments, ERK phosphorylation levels in Akt1/2- or PI3K-downregulated cells dropped by an additional 40 to 60% in comparison with control cells (Fig. 5B).
These findings provide evidence that the protein(s), responsible for MEK-independent ERK1/2 activation, functions downstream of PI3K, thereby supporting the hypothesis, that in some cellular systems ERK activation is mediated by an alternative, cell type and growth factor-specific MEK- and PKC-independent, but PI3K-sensitive pathway [32-34].
MEK-independent ERK activation is not mediated by p38 MAPK and GSK-3 kinases and does not depend on PP2A or Cdc25 phosphatase activity
Since Akt negatively regulates p38 MAPK, which, in turn, activates PP2A and MAPK phosphatases (MKPs) that are able to directly dephosphorylate ERK1/2 (Fig. 1B), we investigated whether MEK-independent ERK phosphorylation could be mediated by p38 MAPK and its targets. For this purpose, T47D cells were treated with wortmannin alone or its combination with U0126, p38 MAPK inhibitor PD 169316 and both inhibitors (Fig. 6A). Inhibition of PI3K and subsequent donwregulation of Akt activity by wortmannin significantly increased basal and EGF-induced p38 MAPK phosphorylation levels (Fig. 6A, left panel), implying the existence of negative regulation between Akt and p38 MAPK.
Fig. 6. MEK-independent ERK phosphorylation is not mediated by p38 MAPK (A), PP2A (B) or GSK-3 (C).

A. Left panel. Serum-starved T47D cells were either left untreated (Control) or treated with wortmannin (WT) (200 nM, 30 min) before stimulation with EGF (1 nM) for the indicated time intervals (min). Total cell lysates were subjected to Multistrip Western blotting with anti-phospho-p38 MAPK (Thr180/Tyr182) antibodies. Right panel. Serum-starved T47D cells were treated with either wortmannin alone (200 nM, 30 min; WT) or with wortmannin plus PD169316 (5 μM, 60 min), U0126 (10 μM, 30 min) and their combination (U0126 + PD 169316 + WT) before stimulation with EGF (1 nM) for the indicated time intervals (min). Total cell lysates were subjected to Multistrip Western blotting for ERK activation as described previously. B. Similarly, T47D cells were treated with U0126 in the presence or absence of endothall (2 μM, 60 min) before the stimulation with EGF (1 nM) for the indicated time intervals (min) and ERK activation was measured. C. T47D cells were treated with Akt-VIII (5 μM, 1 h), SB 216763 (7 μM, 1 h) or their combination in the presence or absence of U0126 (10 μM, 30 min) before the stimulation with EGF (1 nM) for 30 min. ERK and Akt activation was measured.
Assuming that p38 MAPK negatively regulates GSK-3 and that MEK-independent ERK activation occurs exclusively through the GSK-3- and p38 MAPK-controlled phosphatases (Fig. 1B), but not through an unknown kinase, the combined cell treatment either with Akt inhibitor or wortmannin plus U0126 should collectively result in the downregulation of ERK activity. In this case the inhibition of p38 MAPK and PP2A phosphatase should upregulate ERK phosphorylation levels. However, in the presence of wortmannin, p38 MAPK inhibition by PD 169316 and successive PP2A/MKPs deactivation did not enhance phospho-ERK1/2, rather it decreased ERK1/2 activation at early time points (2-5 min) (Fig. 6A, right panel). Furthermore, upon combined MEK and PI3K inhibition, when ERK1/2 phosphorylation was near the basal level, PD 169316 treatment did not significantly increase phospho-ERK1/2.
However, we could not exclude the presence of a p38 MAPK-independent mechanism of PP2A activation. To test this hypothesis, we used endothall (IC50 = 90 nM for PP2A, IC50 = 5 μM for PP1), a specific and more selective inhibitor of PP2A than okadaic acid (IC50 = 0.2-1 nM for PP2A, IC50 = 3 nM for PP1). At 2 μM concentration, which fully inhibits PP2A activity, endothall did not change ERK1/2 activation at 5 or 30 minutes after EGF stimulation in the absence of MEK activity (Fig. 6B).
At the same time, the failure of p38 MAPK inhibition to enhance phospho-ERK1/2 could indicate the absence of p38-MAPK—GSK-3 inhibitory feedback. To assess the role of GSK-3 in MEK-independent ERK activation, T47D cells were pretreated with Akt-VIII, GSK-3 inhibitor SB 216763 or their combination in the presence or absence of U0126. Although Akt inhibition decreased U0126-resistant ERK1/2 phosphorylation 30 minutes post-EGF, SB 216763 treatment did not change the remaining phospho-ERK1/2 levels (Fig. 6C). Similar results were observed with another GSK-3 inhibitor SB 415286 (data not shown).
Increased expression of oncogenic cell cycle-regulatory dual-specificity phosphatase Cdc25A is frequently observed in human cancers, especially those with activating PI3K/Akt mutations and concomitant decreased activity of GSK-3 [35]. Hence, PI3K inhibition by wortmannin could inactivate Cdc25A. According to literature data, Cdc25A inhibition or/and endogenous suppression by siRNA causes prolonged and enhanced ERK phosphorylation in response to EGF even in the presence of mutated MEK [36]. However, T47D cell treatment with a selective inhibitor of the CDC25 phosphatase family (NSC 95397, 5 μM) in the presence of wortmannin and U0126 did not alter the U0126-resistant ERK response to EGF stimulation (data not shown).
These data suggest that neither p38 MAPK nor GSK-3 participate in MEK-independent ERK1/2 activation, which does not require PP2A and Cdc25 phosphatase activities either.
Synergistic downregulation of ERK phosphorylation and its downstream effectors by combined inhibition of MEK and PI3K activities
The comparison of phospho-ERK1/2 kinetics in the presence of U0126-only, wortmannin-only or their combination in T47D cells is shown in Fig. 7A. We found that the concurrent inhibition of PI3K/Akt and MEK kinases blocked ERK1/2 activation in a synergistic, but not additive manner after 5 minutes of EGF addition to the media (Fig. 7A, right panel). The inhibitory synergy was preserved over a wide range of wortmannin concentrations (50-1000 nM) (Fig. 4S) and occurred in both soluble and membrane subcellular fractions (Fig. 5S). Neither U0126 or 200 nM wortmannin (this dose completely inhibits the phosphorylation of PI3K’s substrate Akt, as shown in left panel of Fig. 7B) nor their combination reduced EGFR and Shc phosphorylation levels (Fig. 6S A-B). As expected, PI3K inhibition decreased phosphorylated c-Raf and MEK levels due to disruption of GAB-mediated positive feedbacks, but their total levels were unaffected (Fig. 7B, left panel, and Fig. 6S C). At the same time ERK1/2 phosphorylation (left panels in Figs. 7A and 7B) and its activity towards direct and indirect downstream targets were abolished (Fig. 7B, right panel).
Fig. 7. Synergistic downregulation of ERK phosphorylation and its downstream effectors by combined inhibition of MEK and PI3K activities.

A-B. Serum-starved T47D cells were either left untreated (Control) or treated with U0126 (10 μM, 30 min), wortmannin (WT) (200 nM, 30 min) or both before stimulation with EGF (1 nM) for the indicated time intervals (min) (A) or for 60 min (B) A. Total cell lysates were subjected to Multistrip Western blotting with anti-phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) and anti-p44/42 MAPK (Erk1/2) antibodies (left panel). The graph on the right panel shows the synergistic inhibitory effect of combined treatment with MEK and PI3K inhibitors on ERK activation at 30 min following EGF stimulation (grey bars) compared to estimated theoretical value of additive effect (white bar) ± SD (error bars) (n=3, p<0.05). B. Total cell lysates were subjected to immunoblotting with antibodies against phospho-c-Raf (Ser338), c-Raf, phospho-MEK1/2 (Ser217/221), MEK1/2, phospho-ERK1/2 (Thr202/Tyr204), ERK1/2, phospho-Akt1/2/3 (Ser473), Akt1/2/3, phospho-p90RSK (Ser380), phospho-Elk1 (Ser383), phospho-STAT3 (Ser727), phospho-Estrogen receptor-alpha (Ser118), c-Fos, c-Myc or GAPDH (loading control).
Cell treatment with single-agent U0126 did not significantly reduce c-Raf or MEK phosphorylation (Fig. 7B, left panel). U0126 inhibits catalytic MEK activity, when it has already been phosphorylated by upstream kinase c-Raf. Although U0126 itself should not change c-Raf and MEK phosphorylation status, we cannot exclude the effects emerging from ERK-mediated positive/negative feedbacks upstream of c-Raf and MEK that may influence phospho-MEK levels. Similarly, U0126 does not decrease, but rather increases Akt phosphorylation due to the ERK-mediated negative feedback on the GAB1-PI3K interaction (Fig. 1A).
As a readout of ERK1/2 activity we measured the expression levels of Ser380 phosphorylated p90 ribosomal S6 kinase (p90RSK) (see detailed time-course in Fig. 6S D) [37], Ser383 phosphorylated transcription factor Elk-1, and their dowstream target “immediate-early” gene c-Fos [38]. The phosphorylation of p90RSK and Elk-1 (Fig. 7B, right panel), was almost unaffected by U0126, which is consistent with moderate decrease in ERK activation at 60 min (Fig. 7B, left panel). In contrast, reduced c-Fos expression levels at 60 min perhaps is a consequence of U0126-mediated decrease in the amplitude of ERK phosphorylation at early time points, which can readily impede a threshold of c-Fos induction.
Previous studies have shown that ERK1/2-mediated phosphorylation of ER-α at Ser118, Ser104 and Ser106 residues stimulates ER-α activity in estrogen-independent manner [39]. Phosphorylation of ER-α at Ser118 was strongly reduced in T47D cells treated with the combination of U0126 and wortmannin (Fig. 7B, right panel).
We found that combined inhibition of MEK/ERK and PI3K/Akt signaling efficiently suppresses the phosphorylation of signal transducer and activator of transcription 3 (STAT3) on Ser727, known to modulate STAT3 transcriptional activity. STAT family transcription factors participate in oncogenesis through up-regulation of genes encoding apoptosis inhibitors and cell cycle regulators such as c-Myc. In correlation with the decrease in STAT3 phosphorylation, the expression levels of c-Myc were downregulated by the administration of both PI3K and MEK inhibitors (Fig. 7B, right panel). Similarly to c-Myc expression, the synergistic inhibition of S6RP phosphorylation on Ser235/Ser236 also reflected the activities of both PI3K/Akt and Ras/MAPK signaling pathways, which was confirmed by independent studies [40, 41].
These data demonstrate that complete inhibition of ERK activity can be achieved only by coinhibition of MEK and PI3K, but not by treatment with either agent alone.
Growth inhibition of EGF-stimulated T47D cells by Akt-VIII
Wortmannin is unstable in cell culture media if incubated for long time periods. Therefore, to assess the long-term effects of combined inhibition of PI3K/Akt and MEK/ERK signaling pathways on T47D cell growth, we used more stable cell-permeable quinoxaline compound Akt-VIII that potently and selectively inhibits Akt1/2/3 activities (see Table S1) and efficiently suppressed U0126-independent ERK phosphorylation as shown in Fig. 5A, upper panel, blot 9.
Figure 8 shows the effects of different Akt-VIII doses (1, 2 and 4 μM) on 2 nM EGF-induced proliferation of T47D and MCF7 cells grown for 72 hours in serum-free media containing U0126, Akt-VIII or their combination. Cells were then incubated with AlamarBlue, a redox indicator, which is reduced by reactions innate to cellular metabolism and, therefore, provides an indirect measure of viable cell number. Addition of U0126 decreased viable cell numbers by 15% and 58% in T47D and MCF7 cells, respectively. At the same time, increasing doses of Akt inhibitor retarded cell growth by 27%, 34% and 42% in T47D cells and by 34%, 44% and 78% in MCF7 cells. The combination of both U0126 and Akt inhibitors in the media caused additive decrease in MCF7 viable cell numbers, which displayed greater sensitivity for each of inhibitors. On the contrary, Akt-VIII and U0126 worked in a synergistic manner in preventing T47D cell proliferation. These results indicate that inactivation of Akt isoforms progressively sensitizes T47D cells to MEK inhibition.
Fig. 8. The effect of single or dual inhibition of Akt and MEK kinases on EGF-induced cell proliferation of T47D and MCF7 cells.
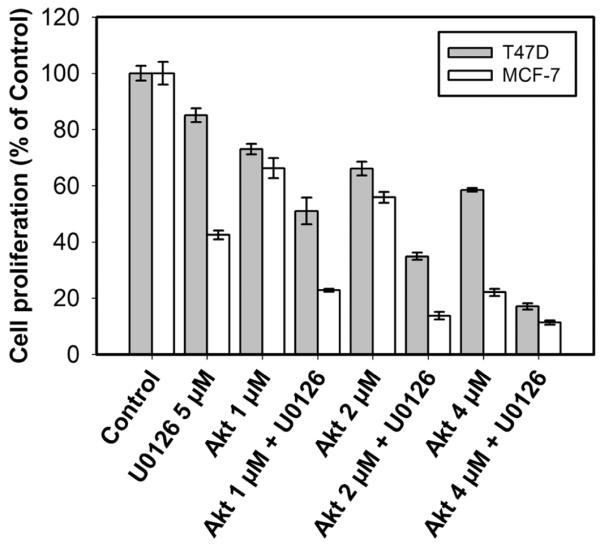
Equal amounts of serum-starved cells were plated into 96-well plates at a density of 25000 cells/ml and maintained in 2 nM EGF-containing medium without FBS in the presence or absence of U0126 (10 μM), different concentrations of Akt-VIII (1, 2 or 4 μM) or their mixtures for 72 hours. The numbers of viable cells were determined with AlamarBlue assay. Each data point is the mean ± SD of 12 replicates and is representative of three independent trials.
MEK-independent ERK activation depends on PBK/TOPK kinase
The effect of combined inhibition of MEK and PI3K/Akt implies the existence of a MEK-independent ERK1/2 activation mechanism, which could involve currently unidentified PI3K/Akt-inducible kinase(s). To the best of our knowledge, besides upstream kinases c-Raf and MEK1/2, the candidate list of kinases that have direct effects on ERK1/2 phosphorylation and can be directly or indirectly regulated by PI3K/Akt, includes only the following proteins: biliverdin reductase (BVR) [42] [43], PDZ-Binding Kinase/T-LAK Cell-Originated Protein Kinase (PBK/TOPK), receptor interacting protein 2 (RIP2) [44, 45] and Fer kinase [46].
Since selective inhibitors for these kinases are not commercially available, we used an siRNA approach to silence BVR, RIP2, Fer and TOPK gene expression and clarify whether any of them may account for MEK-independent ERK1/2 activation in T47D cells. The data in Fig. 9A show that upon MEK inhibition by U0126, ERK phosphorylation decreases by 62 % ± 10% in TOPK-siRNA-treated cells as compared to control cells. By contrast, downregulation of the BVR, RIP2 and FER kinases did not affect ERK activation in the presence of MEK inhibitor, indicating that TOPK is the most likely candidate kinase to provide an alternative ERK activation mechanism in T47D cells (Fig. 9B).
Fig. 9. A. Effects of siRNA-mediated suppression of TOPK, biliverdin reductase, Fer and RIP2 on EGF-induced ERK phosphorylation upon MEK inhibition.

T47D cells were transfected with either 50 nM of siRNA against indicated genes or AllStar negative control siRNA as described in “Materials and Methods”. Serum-starved cells were pretreated with U0126 (10 μM, 30 min) and stimulated with 1 nM EGF for 30 min. The ratios between phospho-ERK1/2 and total ERK1/2 signals are shown as a percentage of siRNA control. Values represent a mean ± SD of three signal measurements. B. Proposed mechanism of MEK-independent ERK activation downstream of PI3K/Akt involving TOPK or other unidentified kinases. Blunt-end arrows show some inhibitors used in this study.
PBK/TOPK is a novel MAPKK-like kinase [47], which is significantly upregulated in highly proliferative malignant cells, including colorectal and breast carcinomas [48, 49]. In fact, the levels of TOPK kinase are at least 2-fold higher in T47D than in MCF7 cells (data not shown). The regulatory connection between TOPK and PI3K/Akt may occur via Cdc2/cyclin B, which can be indirectly activated by Akt [50] and can phosphorylate TOPK on Thr-9 residue important for its enzymatic activity [47, 51]. Interaction between Raf and PBK/TOPK was shown by yeast two-hybrid screening analysis [52]. In EGFR signaling, an autocatalytic loop between TOPK and ERK has been demonstrated by the suppression of either TOPK or ERK2, which resulted in a decreased phosphorylation of ERK2 or TOPK, respectively [49]. Furthermore, p38 MAPK and ERK2 were good substrates of TOPK in vitro.
Further studies are required to determine whether TOPK co-localizes with ERK in cytoplasm and nucleus, and whether it can phosphorylate ERK or form a complex that protects ERK against phosphatase activity. It is also important to understand TOPK activation mechanism by upstream proteins, including PI3K and/or Akt.
CONCLUSION
In conclusion, we demonstrate that EGF induces a crosstalk between PI3K/Akt and Ras/MAPK signaling pathways, which significantly differs between ER-positive PI3K-mutant T47D and MCF7 breast cancer cells. Our data confirm the existence of a dynamic MEK-independent compensatory circuit for ERK activation in T47D cells, which is induced by EGF and other ligands of ErbB family receptors, but not through other receptor tyrosine kinases. Such a circuit is absent in MCF7 cells, thus making their growth more susceptible to inhibition by small molecule inhibitors. Our data suggest that MEK inhibitor-resistant ERK1/2 response is not mediated by phosphatases, but requires the contribution of PBK/TOPK or other currently unknown protein, which functions downstream of PI3K/Akt signaling pathway. Since the persistent activation of ERK’s targets promotes uncontrolled cell growth, survival, angiogenesis and invasion, thereby increasing the risk of breast cancer development and progression, the coinhibition of PI3K/Akt and MEK/ERK may have implications for breast cancer therapy [53-55]. Identifying key signaling molecules that are indispensable for ERK activation in T47D cells can be useful for understanding the molecular mechanisms by which cancer cells escape normal growth controls and acquire drug resistance.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the NIH Grant GM059570. W.K. and B.K. also acknowledge support by Science Foundation Ireland under Grant No. 06/CE/B1129.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
- Akt
- protein kinase B
- BVR
- biliverdin reductase
- c-Src
- cytosolic src avian sarcoma viral oncogen homolog
- DAG
- diacyl glycerol
- DiC8
- 1,2-Dioleoyl-sn-glycerol
- EGF
- epidermal growth factor
- EGFR
- epidermal growth factor receptor
- ERK
- extracellular signal-regulated kinase
- ER-α
- estrogen receptor alpha
- FBS
- fetal bovine serum
- FGF
- fibroblast growth factor
- GAB
- Grb2-associated binder
- GSK-3
- glycogen synthase kinase-3
- HRG-β
- heregulin beta
- IB
- immunoblotting
- IGF-1
- insulin-like growth factor 1
- IRS
- insulin receptor substrate
- MAPK
- mitogen-activated protein kinase
- MEK
- mitogen-activated protein kinase kinase
- MKPs
- MAPK phosphatases
- p70S6K
- p70 ribosomal S6 kinase
- p90RSK
- p90 ribosomal S6 kinase
- PAK
- p21-activated kinase
- PBK/TOPK
- PDZ-Binding Kinase/T-LAK Cell-Originated Protein Kinase
- PDGF
- platelet-derived growth factor
- PDK1
- phosphoinositide-dependent protein kinase-1
- PH
- pleckstrin homology
- PI3K
- phosphatidylinositol-3-kinase
- PKA
- protein kinase A
- PKC
- protein kinase C
- PP2A
- protein phosphatase 2A
- PRL
- prolactin
- RIP2
- receptor interacting protein 2
- S6RP
- ribosomal S6 protein
- SHC
- Src homolohy and collagen domain protein
- siRNA
- small interfering RNA
- SOS
- Son of Sevenless
- STAT3
- signal transducer and activator of transcription 3
- TGF-α
- transforming growth factor alpha
- VEGF
- vascular endothelial growth factor
- WT
- wortmannin
REFERENCES
- [1].Janne PA, Gray N, Settleman J. Nat Rev Drug Discov. 2009 doi: 10.1038/nrd2871. [DOI] [PubMed] [Google Scholar]
- [2].Stern DF. Exp Cell Res. 2003;284(1):89–98. doi: 10.1016/s0014-4827(02)00103-9. [DOI] [PubMed] [Google Scholar]
- [3].Yarden Y, Sliwkowski MX. Nat Rev Mol Cell Biol. 2001;2(2):127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- [4].Mora A, Komander D, van Aalten DM, Alessi DR. Semin Cell Dev Biol. 2004;15(2):161–170. doi: 10.1016/j.semcdb.2003.12.022. [DOI] [PubMed] [Google Scholar]
- [5].Jorissen RN, Walker F, Pouliot N, Garrett TP, Ward CW, Burgess AW. Exp Cell Res. 2003;284(1):31–53. doi: 10.1016/s0014-4827(02)00098-8. [DOI] [PubMed] [Google Scholar]
- [6].Kiyatkin A, Aksamitiene E, Markevich NI, Borisov NM, Hoek JB, Kholodenko BN. J Biol Chem. 2006;281(29):19925–19938. doi: 10.1074/jbc.M600482200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Borisov N, Aksamitiene E, Kiyatkin A, Legewie S, Berkhout J, Maiwald T, Kaimachnikov NP, Timmer J, Hoek JB, Kholodenko BN. Mol Syst Biol. 2009;5:256. doi: 10.1038/msb.2009.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Potti A, Dressman HK, Bild A, Riedel RF, Chan G, Sayer R, Cragun J, Cottrill H, Kelley MJ, Petersen R, Harpole D, Marks J, Berchuck A, Ginsburg GS, Febbo P, Lancaster J, Nevins JR. Nat Med. 2006;12(11):1294–1300. doi: 10.1038/nm1491. [DOI] [PubMed] [Google Scholar]
- [9].Hoeflich KP, O’Brien C, Boyd Z, Cavet G, Guerrero S, Jung K, Januario T, Savage H, Punnoose E, Truong T, Zhou W, Berry L, Murray L, Amler L, Belvin M, Friedman LS, Lackner MR. Clin Cancer Res. 2009;15(14):4649–4664. doi: 10.1158/1078-0432.CCR-09-0317. [DOI] [PubMed] [Google Scholar]
- [10].Mirzoeva OK, Das D, Heiser LM, Bhattacharya S, Siwak D, Gendelman R, Bayani N, Wang NJ, Neve RM, Guan Y, Hu Z, Knight Z, Feiler HS, Gascard P, Parvin B, Spellman PT, Shokat KM, Wyrobek AJ, Bissell MJ, McCormick F, Kuo WL, Mills GB, Gray JW, Korn WM. Cancer Res. 2009;69(2):565–572. doi: 10.1158/0008-5472.CAN-08-3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].De Larco JE, Wuertz BR, Rosner KA, Erickson SA, Gamache DE, Manivel JC, Furcht LT. Am J Pathol. 2001;158(2):639–646. doi: 10.1016/S0002-9440(10)64005-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hollestelle A, Elstrodt F, Nagel JH, Kallemeijn WW, Schutte M. Mol Cancer Res. 2007;5(2):195–201. doi: 10.1158/1541-7786.MCR-06-0263. [DOI] [PubMed] [Google Scholar]
- [13].Toffanin S, Daidone MG, Miodini P, De Cecco L, Gandellini P, Cappelletti V. Int J Oncol. 2008;33(4):791–798. [PubMed] [Google Scholar]
- [14].Kiyatkin A, Aksamitiene E. Methods Mol Biol. 2009;536:149–161. doi: 10.1007/978-1-59745-542-8_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ussar S, Voss T. J Biol Chem. 2004;279(42):43861–43869. doi: 10.1074/jbc.M406240200. [DOI] [PubMed] [Google Scholar]
- [16].Yu CF, Liu ZX, Cantley LG. J Biol Chem. 2002;277(22):19382–19388. doi: 10.1074/jbc.M200732200. [DOI] [PubMed] [Google Scholar]
- [17].Karihaloo A, O’Rourke DA, Nickel C, Spokes K, Cantley LG. J Biol Chem. 2001;276(12):9166–9173. doi: 10.1074/jbc.M009963200. [DOI] [PubMed] [Google Scholar]
- [18].Jiang Q, Zhou C, Bi Z, Wan Y. J Ocul Pharmacol Ther. 2006;22(2):93–102. doi: 10.1089/jop.2006.22.93. [DOI] [PubMed] [Google Scholar]
- [19].Frey MR, Golovin A, Polk DB. J Biol Chem. 2004;279(43):44513–44521. doi: 10.1074/jbc.M406253200. [DOI] [PubMed] [Google Scholar]
- [20].Liu D, Liu Z, Jiang D, Dackiw AP, Xing M. J Clin Endocrinol Metab. 2007;92(12):4686–4695. doi: 10.1210/jc.2007-0097. [DOI] [PubMed] [Google Scholar]
- [21].Leboeuf R, Baumgartner JE, Benezra M, Malaguarnera R, Solit D, Pratilas CA, Rosen N, Knauf JA, Fagin JA. J Clin Endocrinol Metab. 2008;93(6):2194–2201. doi: 10.1210/jc.2007-2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Duncia JV, Santella JB, 3rd, Higley CA, Pitts WJ, Wityak J, Frietze WE, Rankin FW, Sun JH, Earl RA, Tabaka AC, Teleha CA, Blom KF, Favata MF, Manos EJ, Daulerio AJ, Stradley DA, Horiuchi K, Copeland RA, Scherle PA, Trzaskos JM, Magolda RL, Trainor GL, Wexler RR, Hobbs FW, Olson RE. Bioorg Med Chem Lett. 1998;8(20):2839–2844. doi: 10.1016/s0960-894x(98)00522-8. [DOI] [PubMed] [Google Scholar]
- [23].Mody N, Leitch J, Armstrong C, Dixon J, Cohen P. FEBS Lett. 2001;502(1-2):21–24. doi: 10.1016/s0014-5793(01)02651-5. [DOI] [PubMed] [Google Scholar]
- [24].Olayioye MA. Breast Cancer Res. 2001;3(6):385–389. doi: 10.1186/bcr327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Spencer KS, Graus-Porta D, Leng J, Hynes NE, Klemke RL. J Cell Biol. 2000;148(2):385–397. doi: 10.1083/jcb.148.2.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Yip-Schneider MT, Klein PJ, Wentz SC, Zeni A, Menze A, Schmidt CM. J Pharmacol Exp Ther. 2009;329(3):1063–1070. doi: 10.1124/jpet.108.147306. [DOI] [PubMed] [Google Scholar]
- [27].Pursiheimo JP, Kieksi A, Jalkanen M, Salmivirta M. FEBS Lett. 2002;521(1-3):157–164. doi: 10.1016/s0014-5793(02)02864-8. [DOI] [PubMed] [Google Scholar]
- [28].Hausser A, Link G, Bamberg L, Burzlaff A, Lutz S, Pfizenmaier K, Johannes FJ. J Cell Biol. 2002;156(1):65–74. doi: 10.1083/jcb.200110047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hausser A, Storz P, Hubner S, Braendlin I, Martinez-Moya M, Link G, Johannes FJ. FEBS Lett. 2001;492(1-2):39–44. doi: 10.1016/s0014-5793(01)02219-0. [DOI] [PubMed] [Google Scholar]
- [30].Kassenbrock CK, Hunter S, Garl P, Johnson GL, Anderson SM. J Biol Chem. 2002;277(28):24967–24975. doi: 10.1074/jbc.M201026200. [DOI] [PubMed] [Google Scholar]
- [31].Hu X, Wu X, Xu J, Zhou J, Han X, Guo J. BMC Neurosci. 2009;10:74. doi: 10.1186/1471-2202-10-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kinkl N, Sahel J, Hicks D. J Biol Chem. 2001;276(47):43871–43878. doi: 10.1074/jbc.M105256200. [DOI] [PubMed] [Google Scholar]
- [33].Saeki Y, Hazeki K, Hazeki O, Ui M, Itoh K, Matsumoto M, Toyoshima K, Akedo H, Seya T. Int J Mol Med. 2000;6(2):155–160. doi: 10.3892/ijmm.6.2.155. [DOI] [PubMed] [Google Scholar]
- [34].Pizon V, Baldacci G. Oncogene. 2000;19(52):6074–6081. doi: 10.1038/sj.onc.1203984. [DOI] [PubMed] [Google Scholar]
- [35].Kang T, Wei Y, Honaker Y, Yamaguchi H, Appella E, Hung MC, Piwnica-Worms H. Cancer Cell. 2008;13(1):36–47. doi: 10.1016/j.ccr.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wang Z, Zhang B, Wang M, Carr BI. J Cell Physiol. 2005;204(2):437–444. doi: 10.1002/jcp.20297. [DOI] [PubMed] [Google Scholar]
- [37].Dalby KN, Morrice N, Caudwell FB, Avruch J, Cohen P. J Biol Chem. 1998;273(3):1496–1505. doi: 10.1074/jbc.273.3.1496. [DOI] [PubMed] [Google Scholar]
- [38].Karin M. J Biol Chem. 1995;270(28):16483–16486. doi: 10.1074/jbc.270.28.16483. [DOI] [PubMed] [Google Scholar]
- [39].Thomas RS, Sarwar N, Phoenix F, Coombes RC, Ali S. J Mol Endocrinol. 2008;40(4):173–184. doi: 10.1677/JME-07-0165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Lee T, Yao G, Nevins J, You L. PLoS Comput Biol. 2008;4(2):e1000013. doi: 10.1371/journal.pcbi.1000013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Lehman JA, Gomez-Cambronero J. Biochem Biophys Res Commun. 2002;293(1):463–469. doi: 10.1016/S0006-291X(02)00238-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Lerner-Marmarosh N, Miralem T, Gibbs PE, Maines MD. Proc Natl Acad Sci U S A. 2008;105(19):6870–6875. doi: 10.1073/pnas.0800750105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zeng R, Yao Y, Han M, Zhao X, Liu XC, Wei J, Luo Y, Zhang J, Zhou J, Wang S, Ma D, Xu G. J Am Soc Nephrol. 2008;19(2):380–387. doi: 10.1681/ASN.2006111194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Hindley A, Kolch W. J Cell Sci. 2002;115(Pt 8):1575–1581. doi: 10.1242/jcs.115.8.1575. [DOI] [PubMed] [Google Scholar]
- [45].Navas TA, Baldwin DT, Stewart TA. J Biol Chem. 1999;274(47):33684–33690. doi: 10.1074/jbc.274.47.33684. [DOI] [PubMed] [Google Scholar]
- [46].Salem Y, Shpungin S, Pasder O, Pomp O, Taler M, Malovani H, Nir U. Cell Signal. 2005;17(3):341–353. doi: 10.1016/j.cellsig.2004.08.001. [DOI] [PubMed] [Google Scholar]
- [47].Gaudet S, Branton D, Lue RA. Proc Natl Acad Sci U S A. 2000;97(10):5167–5172. doi: 10.1073/pnas.090102397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Park JH, Lin ML, Nishidate T, Nakamura Y, Katagiri T. Cancer Res. 2006;66(18):9186–9195. doi: 10.1158/0008-5472.CAN-06-1601. [DOI] [PubMed] [Google Scholar]
- [49].Zhu F, Zykova TA, Kang BS, Wang Z, Ebeling MC, Abe Y, Ma WY, Bode AM, Dong Z. Gastroenterology. 2007;133(1):219–231. doi: 10.1053/j.gastro.2007.04.048. [DOI] [PubMed] [Google Scholar]
- [50].Katayama K, Fujita N, Tsuruo T. Mol Cell Biol. 2005;25(13):5725–5737. doi: 10.1128/MCB.25.13.5725-5737.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Matsumoto S, Abe Y, Fujibuchi T, Takeuchi T, Kito K, Ueda N, Shigemoto K, Gyo K. Biochem Biophys Res Commun. 2004;325(3):997–1004. doi: 10.1016/j.bbrc.2004.10.133. [DOI] [PubMed] [Google Scholar]
- [52].Yuryev A, Wennogle LP. Genomics. 2003;81(2):112–125. doi: 10.1016/s0888-7543(02)00008-3. [DOI] [PubMed] [Google Scholar]
- [53].Ali S, Coombes RC. J Mammary Gland Biol Neoplasia. 2000;5(3):271–281. doi: 10.1023/a:1009594727358. [DOI] [PubMed] [Google Scholar]
- [54].Milde-Langosch K, Roder H, Andritzky B, Aslan B, Hemminger G, Brinkmann A, Bamberger CM, Loning T, Bamberger AM. Breast Cancer Res Treat. 2004;86(2):139–152. doi: 10.1023/B:BREA.0000032982.49024.71. [DOI] [PubMed] [Google Scholar]
- [55].Bowman T, Garcia R, Turkson J, Jove R. Oncogene. 2000;19(21):2474–2488. doi: 10.1038/sj.onc.1203527. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.