

Abstract
Me-lex is a sequence-specific alkylating agent synthesized to preferentially (> 90%) generate N3-methyladenine (3-mA) in the minor groove of double-strand DNA, in A-T rich regions. In this paper we investigated the effect of XRCC1 deficiency in the processing of 3-mA adducts generated by Me-lex, through the molecular analysis of the Hprt mutations and the evaluation of cytogenetic end points such as sister chromatid exchanges (SCEs), micronuclei (MN) and nucleus fragmentation. EM-C11 cells, deficient in XRCC1 activity, showed a 2.5-fold higher sensitivity to the toxicity of Me-lex compared to the DNA repair proficient parental CHO-9 cells, but were not hyper mutable. The spontaneous mutation spectrum at the Hprt locus generated in EM-C11 cells revealed a high percentage of genomic deletions. After Me-lex treatment, the percentage of genomic deletions did not increase, but a class of mutations which appeared to target regulatory regions of the gene significantly increased (p=0.0277), suggesting that non-coding Hprt genomic sequences represent a strong target for the rare mutations induced by Me-lex. The number of SCEs per chromosome increased 3-fold above background in 50 µM Me-lex treated CHO-9 cells, while at higher Me-lex concentrations a sharp increase in the percentage of MN and fragmented nuclei was observed. In EM-C11 cells the background level of SCEs (0.939±0.182) was approximately 10-fold higher than in CHO-9 (0.129±0.027) and higher levels of multinucleated cells and MN were also found. In EM-C11, even low doses of Me-lex (25µM) led to a significant increase in genomic damage. These results indicate that XRCC1 deficiency can lead to genomic instability even in the absence of an exogenous genotoxic insult and low levels of Me-lex-induced lesions, i.e., 3-mA and/or a BER intermediate, can exacerbate this instability.
Keywords: Me-lex, mutation induction, Hprt, hamster cells, XRCC1, SCE, micronuclei
1. Introduction
Me-lex is a sequence-specific alkylating agent designed to preferentially (> 90%) generate N3-methyladenine (3-mA) in the minor groove of double-strand DNA mainly in A-T rich regions [1–4]. The interest in this molecule derives from the fact that 3-mA is a very cytotoxic lesion that exerts its action by blocking the progression of the DNA replication machinery [5–10], thus causing cell lethality. This implies that 3-mA generating compounds can be considered promising model molecules for the treatment of actively proliferating cancer cells.
In repair proficient cells, the removal of 3-mA is initiated by alkyladenine DNA glycosylase (AAG), which cleaves the glycoside bond connecting the adducted base to the deoxyribose sugar [11]. The resulting abasic site is recognized by an AP endonuclease that incises the phosphodiester bond 5’ to the lesion forming a strand break with a 3’ OH and a 5’ deoxyribosephosphate terminus. The removal of the abasic residue generates a nucleotide gap, which is filled in by the co-ordinate action of poly (ADP-ribose) polymerase (PARP), DNA polymerase β, XRCC1 and ligase I or III [11]. If encountered during replication, 3-mA may stall DNA replication due to the inability of replicating DNA polymerases to interact with the adducted N3 position of adenine in the DNA [12]. To overcome this block, DNA damage tolerance pathways, such as translesion synthesis (TLS), can be activated to allow survival, albeit with increased mutations [13].
Studies conducted in bacteria, yeast and mammalian cells have demonstrated that Me-lex toxicity is dependent on the DNA repair background. BER deficient Saccharomyces cerevisiae strains, lacking 3-methyladenine DNA glycosylase or both Apn1 and Apn2 AP endonucleases, are significantly more sensitive to Me-lex toxicity with respect to the parental strain [14]. Embryonic stem cells derived from Aag null mice are hypersensitive to killing by Me-lex, and unrepaired 3-mA induced SCEs, chromosome aberrations, S-phase arrest, p53 induction and apoptosis [15]. The cytotoxic potential of Me-lex has been demonstrated also in tumor cells. In human glioma cells Me-lex is cytotoxic and the deletion of AAG activity increases Me-lex toxicity, indicating that 3-mA is a lethal lesion [16]. In mismatch repair deficient leukemic cells, Me-lex has cytotoxic and clastogenic effects that are increased when PARP is inhibited by 3-aminobenzamide (3-AB) [17]. Interestingly, increased levels of XRCC1 were found both in cells treated with Me-lex and with Me-lex plus 3-AB [17], indicating an involvement of XRCC1 in the processing of 3-mA. Since these cancer cells are resistant to methylating agents inducing O6-MeG, such as temozolomide, Me-lex (alone or in combination with PARP inhibitors) could represent a pharmacological strategy for the treatment of tumors resistant to classical methylating agents [18,19].
In the perspective of using 3-mA generating compounds in cancer therapy, it is crucial to define their mutagenic potential, since currently used methylating agents (e.g., temozolomide and procarbazine for gliomas) induce pro-mutagenic DNA adducts and, consequently, may promote the development of secondary tumors derived from the treatment [20]. Me-lex mutagenicity has been addressed in yeast and hamster cells. In yeast, we have shown that Me-lex is poorly mutagenic in a wild-type background and its mutagenicity increases significantly in the absence of AP endonucleases while only slightly in the absence of 3-methyladenine DNA glycosylase [14]. This indicates that a deficiency in the removal of 3-mA is deleterious, but unrepaired AP sites (likely derived from 3-mA) are even more mutagenic. Both in repair proficient and deficient yeast strains, AT-targeted base pair substitutions were the predominant type of mutation [3,14]. Using the same approach, we have also recently demonstrated an involvement of yeast TLS polymerases Polζ, REV1 [13] and Polrη [21] in the fixation of mutations derived from Me-lex induced lesions. Our data indicate that the by-pass of Me-lex induced lesions is a multi DNA-polymerases process that is most effective when all three yeast TLS polymerases are present.
In mammalian cells, Me-lex is cytotoxic but poorly mutagenic, resulting only in a 7-fold increase (relative to background) of the Hprt mutation frequency. However, in the mutation spectrum at the Hprt locus, there was a high percentage of genomic deletions [22], while base pair substitutions in the Hprt coding region represented only 25% of the mutagenic events. This unexpected result led us to hypothesize that a large proportion of the rare mutations in mammalian cells may result from the processing of 3-mA within A/T rich sequences in non-coding regions of the Hprt gene. Due to the specific reactivity of Me-lex towards nucleophilic sites in the DNA minor groove, the possible clustering of these lesions in A/T rich sequences could represent a strong impediment to the progression of DNA replication forks, thus promoting genomic rearrangements and/or large deletions [22].
The observation of a high level of genetic deletions in DNA repair proficient mammalian cells suggests that other enzymes (and/or other repair pathways) besides DNA glycosylases may have a role in the mutagenic processing of 3-mA (or a subsequent derivative) in genomic regions with high A/T content. During BER, AP sites arising spontaneously from the action of glycosylases are enzymatically converted into single strand breaks (SSBs). DNA intermediates with a strand break are potentially deleterious since they can lead to double strand breaks (DSBs) and recombination. XRCC1 plays a crucial role in the coordination of BER and SSB repair (SSBR), through its interaction with several key enzymes involved at different steps of these pathways (i.e., Polβ, PARP, Ligase III) [23,24]. In addition to protein-protein interactions, XRCC1 can bind different DNA substrates, such as gapped and nicked DNA [25] and AP sites [26]. For these different activities, it has been proposed that XRCC1 could optimize the passage of DNA substrates from one enzyme to the next in the pathway as a scaffold that co-localizes the proteins through its different interacting domains [27].
XRCC1 deficient cell lines are hypersensitive to monofunctional alkylating agents and exhibited roughly a 10-fold higher level of spontaneous SCEs [28]. The EM-C11 cell line, isolated on the basis of its high sensitivity to killing by ethyl methansulfonate (EMS), has a defect in the rejoining of SSBs after exposure to X-rays and EMS [28]. The molecular characterization of Hprt mutants isolated in EM-C11 cells following EMS or methyl methanesulfonate (MMS) treatments revealed a higher incidence of deletions compared to their parental repair proficient CHO-9 cells, suggesting that XRCC1deficiency may lead to the formation of genomic deletions following exposure to alkylating agents [29,30].
In order to determine the role of XRCC1 in the cytotoxicity and mutagenicity of Me-lex, the spontaneous and the Me-lex induced mutation spectra at the Hprt locus were obtained in the EM-C11 cell line. In addition, cytogenetic assays were conducted in EM-C11 cells, and in its parental CHO-9 cell line, in order to investigate the ability of Me-lex to induce SCEs, micronuclei (MN) and other types of genomic damage.
2. Materials and Methods
2.1. Hazardous procedures
Me-lex should be considered a toxic compound, and was handled accordingly.
2.2. Compounds
Reagents of the highest purity were purchased from Sigma (St. Louis, MO, USA) or Aldrich (Milwaukee, WI, USA) unless otherwise stated. Me-lex was prepared as previously described [1].
2.3. Cell culture conditions
The Chinese hamster ovary CHO-9 and EM-C11 cell lines (obtained from M. Zdzienicka, Leiden University Medical Center, NL) were grown in Ham’s F10:D-MEM (GIBCO, Invitrogen, Milano, Italy) 1:1, supplemented with 10% fetal calf serum (Euroclone, Milano, Italy), 100 IU/ml penicillin and 0,1 mg/ml streptomycin (MP Biomedicals, Irvine), in a humidified incubator at 37 °C with 5% CO2. In order to reduce pre-existing spontaneous mutants, cells were treated with HAT medium (Ham’s F10:D-MEM 1:1 supplemented with 10−4 M hypoxanthine, 10−6 M aminopterin and 10−5 M thymidine, Sigma Aldrich, Milano, Italy).
2.4. Mutation experiments
Independent populations starting from 1000 cells were isolated and one population was used for each experiment. For each treatment, Me-lex stock solution (10 mM) was prepared by dissolving 1 mg of Me-lex in 20.7 µl DMSO and 200 µl EtOH. Survival and mutagenicity were evaluated up to 150 µM Me-lex. Mutagenicity experiments were performed as described by Russo et al. [22]. For the isolation of Hprt mutants, 12×106 EM-C11 cells were exposed to 75 µM Me-lex. Soon after treatment, the cultures were split into 30 parallel subcultures to ensure the growth of independent mutants. Simultaneously, the spontaneous mutant frequencies were determined in order to calculate the level of mutant induction. Spontaneous Hprt mutants were isolated and analyzed in parallel with those induced by Me-lex. The mutant frequency was expressed as the number of mutants per 105 clone-forming cells.
2.5. Molecular analysis of Hprt mutants
The molecular analysis of Hprt mutants was performed as described [22]. Briefly, total RNA was isolated from 2×106 cells of each mutant (RNeasy Mini Kit, Qiagen, Italy). The Hprt cDNA was synthesized in 20 µl volumes containing 1 µg RNA (Reverse-iT 1st strand Synthesis kit, AB Gene). Approximately 20% of the cDNA product was used as template for PCR to amplify the entire Hprt coding sequence or, when the complete Hprt cDNA sequence was not obtained, the 5’- and the 3’-ends of the coding sequence to verify the presence of shorter cDNAs [22]. PCR products were gel purified and sequenced (BMR Genomics, CRIBI, Padova, Italy). In order to analyze mutants missing one (or more) exon(s) from the Hprt cDNA, a crude cell lysate was used as source of genomic DNA for amplification of single exons using primers lying in the introns flanking the missing exons. A multiplex PCR-based amplification of all nine exons was carried out with primers described by Xu et al., [31]. PCR products were displayed by electrophoresis on 4% agarose gel.
2.6. MN assay
The day before treatment, cells were seeded in 35-mm dishes with coverslips. Cells were cultured at densities of 2.5–10×104 cells/coverslip (about 6×103 cells/cm2), using conditions ensuring that cells were actively growing after 1 h treatment. At the appropriate harvest time, cells were rinsed and exposed to a hypotonic shock in situ using Iskandar solution (0.9% NaCl/75 mM KCl; 9:1). Cells were then fixed with MeOH/HOAc (3:1) and stained with Giemsa. All slides including those of positive and solvent controls were coded before analysis and scored blindly for the evaluation of MN by two independent observers. The criteria used for identifying MN fulfil those recommended by the HUMN work [32]: (1) area < 1/3 the main nucleus area; (2) no overlapping with the nucleus (distinct borders); and (3) same aspect as the chromatin.
2.7. Measurement of SCE levels
Cells seeded onto 20×20 coverslips and treated as above, were cultured in the presence of 50 µM BUdR for two cell cycle periods in the dark, and pulsed with colcemid 0.1 µg/mL for the last 2 h. Cells were then treated in situ with 75 mM KCl for 20 min and subsequently fixed with MeOH/HOAc (3:1) for 15 min. To visualize SCEs, FPG technique was employed [33]. Briefly, dried slides were incubated with 5 µg/mL of Hoechst 33258 in Sorensen’s buffer (pH 6.8) for 15 min, rinsed with the same buffer and irradiated with black light (UVP Upland UVGL-15 lamp, 365 nm, arranged at a distance of 6 cm above each specimen) for 30 min. After incubation in Sorensen’s buffer at 60 °C for 1 h, slides were stained with 2.5 % Giemsa solution in the same buffer. Coded slides were scored blindly by three independent observers. Twenty metaphases per each experimental point were scored.
3. Results
3.1. XRCC1deficiency increases Me-lex cytotoxicity but not mutagenicity
XRCC1deficient EM-C11 cells were treated with increasing Me-lex concentrations to determine the survival curve and the mutation frequency (MF) at the Hprt locus. Results were compared to those obtained in the CHO-9 parental cell line [22]. As shown in Figure 1, based on LD50 values, EM-C11 cells were 2.5-fold more sensitive to Me-lex compared to the parental CHO-9 cells. A low mutability of EM-C11 cells was found (Table 1) since the MF increased significantly over the background only at 100 µM Me-lex (p=0.0357), a dose that reduced the viability to less than 10%, and did not increase even when cell survival was below 1% (Table 1). When compared to the parental repair proficient CHO-9, the EM-C11 cells did not show a significantly higher mutability following Me-lex exposure (Table 1). Thus, the XRCC1deficiency affected the cytotoxicity but not the mutation frequency induced by Me-lex.
Figure 1. Survival curve in EMC −11 and CHO-9 cell lines after Me-lex treatment.

The survival curve in EMC −11 (♦) and the parental CHO-9 (□) cells after Me-lex treatment are reported. Each point represents the mean ± s.d. of 5 independent experiments.
Table 1.
Cell survival and Hprt mutation frequency induced by Me-lex in EM-C11 and CHO-9
| Me-lex dose | Survival | Mut Freq | |
|---|---|---|---|
| Survival CHO-9a | Mut | ||
| Freq CHO-9a | |||
| (µM) | % | (×10−5)b | |
| % | (×10−5)b | ||
| 0 | 100 | 0.5 ± 0.2 | |
| 100 | 0.4 ± 0.3 | ||
| 25c | 89.2 ± 11.4 | 1.6 ± 0.4 | N.S. |
| 50c | 57.8 ± 10.1 | 1.8 ± 0.8 | N.S. |
| 75c | 19.2 ± 8.7 | 2.7 ± 0.9 | N.S. |
| 100 | 6.1 ± 3.1 | 2.8 ± 1.5 | |
| p= 0.0357 | 80.0 ± 11.7 | 1.1 ± | |
| 0.7 | |||
| 150 | 0.4 ± 0.5 | 2.8 ± 2.2 | p= |
| 0.0357 | 40.8 ± 18.7 | 1.6 ± 0.9 | |
| 200 | |||
| 13.3 ± 8.2 | 2.5 ± 1.4 | ||
| 250 | |||
| 2.9 ± 2.3 | 0.9 ± 0.9 | ||
From Russo et al., 2009
The Hprt mutation frequency is expressed as the number of 6-TG-resistant clones per 105 clone-forming cells. Each point represents the mean ± s.d. of 5 independent experiments for survival and 3 experiments for mutation induction. Non-parametric Mann-Whitney test was used for statistical analysis.
For these doses, only two repetitions could be used for statistical analysis and the comparison was not statistically significant (N.S.).
3.2. XRCC1 deficiency affects the frequency of genomic deletions in untreated EM-C11 cells
A spontaneous mutation spectrum was generated in order to characterize the mutations arising as a result of XRCC1deficiency per se. Ten independent EM-C11 sub-populations starting from 1000 cells were isolated and, for each sub-population, the spontaneous Hprt MF (sMF) was determined and Hprt mutant clones isolated and characterized (Table 2). Low sMFs were found in every sub-population with an average of 0.13×10−5 ± 0.07. Two sub-populations were further expanded to simultaneously isolate Me-lex induced and spontaneous mutants.
Table 2.
Spontaneous Hprt mutation spectrum in EM-C11
| Mutant/(s) | cDNA producta | Position | Type of mutation | Target | ||
|---|---|---|---|---|---|---|
| sequenceb | Aminoacid change | N° of | ||||
| (exon) | (5’ | |||||
| >3’) | (or cDNA change) | mutants | ||||
| Base pair changes | ||||||
| EG-sp2/3 | full length | 118 (2) | GC>TA | CCT | ||
| CAT GGA GTG ATT | Gly>STOP | 2 | ||||
| EG-8/-10 | full length | 118 (2) | GC>TA | CCT CAT | ||
| GGA GTG ATT | Gly>STOP | 2c | ||||
| EG-spl A/1B | full length | 139 (3) | GC>TA | AGG | ||
| ACT GAA AGA CTT | Glu>STOP | 2 | ||||
| EB-17 | full length | 533 (8) | AT>GC | CAG ACT | ||
| TTT GTT GG | Phe>Ser | 1c | ||||
| EC-7 | full length | 595 (8) | AT>TA | GAG TAC | ||
| TTC AGG GAT | Phe>Ser | 1c | ||||
| EN-13 | full length | 614 (9) | AT>GC | AAT CAT | ||
| ATT TGT GTC | Ile>Thr | 1c | ||||
| Frameshift mutations | ||||||
| EN-sp17 | very low, full length | 207–212 (3) | +G | GTG CTG | ||
| AAG GGG GGC TAT | 1 | |||||
| EC-13 | 112/113 | −C | TTT ATT | |||
| CCT CAT GGA | 1c | |||||
| Splice mutations | ||||||
| EN-sp9 | very low, short cDNA | Splice junctions not sequenced | ||||
| Skipping ex 2+3 | 1 | |||||
| EA-6/−9 | short cDNAs | All exons present on gDNA | ||||
| Skipping exon 2/2+3 | 2c | |||||
| Genomic Deletions | ||||||
| EN-spl/5/10 | short cDNA | Deletion of exon 2+3 | ||||
| 3 | ||||||
| EH-5 | Deletion exs 6–8 on gDNA | |||||
| Skipping ex 6–8/Ins 58 bp | 1c | |||||
| EG-sp4/13 | no cDNA | Deletion of all Hprt exons | ||||
| 2 | ||||||
| EF-8/−10 | no cDNA | Deletion of all Hprt exons | ||||
| 3c | ||||||
| EH-3 | ||||||
| EL-1 | no cDNA | Deletion ex 7–8 on gDNA. Low amplification of | ||||
| other Hprt exons | 1c | |||||
| EL-6 | no cDNA | Deletion ex 7–8 on gDNA | ||||
| 1c | ||||||
| Mutations in regulatory regions | ||||||
| EN-sp 15/19/20 | no cDNA | All exons present on gDNA | ||||
| 3 | ||||||
| Other mutations | ||||||
| EN-sp13 | longer cDNA | Duplication of exons 2+3 | ||||
| 1 | ||||||
cDNA amplifications of the full lenght, the 5'- (ex 1–4) and the 3'- (ex 4–8) half of the Hprt coding sequence were performed for each mutant (see M&M). Short cDNA refers to PCR fragments lacking the internal part of the hprt coding sequence.
Non transcribed strand gDNA = genomic DNA
Spontaneous mutants isolated from EM-C11 independent populations before treatment
The analysis of spontaneous or Me-lex induced Hprt mutations was first performed on cDNA level by PCR amplification [34,35]. The mutants that generated short cDNA amplicons or that did not give a PCR product were further analyzed by sequencing the intron/exon boundaries and by multiplex PCR [22]. Mutants with all Hprt exons present in genomic DNA but which produced either very low amounts of shorter cDNA or no cDNA products were classified as mutants with “mutations in regulatory regions”.
In the spontaneous spectrum (Table 2), 9 mutants carried base pair changes, 6 GC-targeted and 3 AT-targeted. Two mutants had frame shifts and 3 mutants showed the skipping of exons 2 and 2+3. The spontaneous spectrum showed a high number of genomic deletions encompassing at least two Hprt exons. Three mutants did not give a cDNA product even though all exons were present at the genomic level and 1 mutant produced a longer cDNA due to a duplication of exons 2+3.
3.3. Molecular analysis of Me-lex induced Hprt mutants in EM-C11 cells
Hprt-deficient mutants were isolated at 75 µM Me-lex, a dose inducing almost a 5-fold increase of the MF over background (see Table 1). Due to the low mutagenic but high cytotoxic potential of Me-lex, we decided not to use higher Me-lex concentrations, which would have greatly reduced the possibility of isolating mutants. Besides, treatments from 75 µM up to 150 µM of Me-lex resulted in approximately the same MF. Fifty-six Me-lex induced Hprt mutants were isolated at 75 µM Me-lex.
In the Me-lex induced mutation spectrum (Table 3), 7 mutants carried AT-targeted base pair changes, 2 had frameshift mutations involving A/T bases and 9 carried mutations that generated exon skipping. The mutation responsible for exon skipping was identified for 7 mutants, 6 of which presented a mutated G at the donor or acceptor splice site. In all these cases, a very AT-rich sequence was adjacent to the site of mutation suggesting a Me-lex sequence selective induction. Genomic deletions were found in 11 mutants, including three-base deletions, single exon deletions, and deletion of all Hprt exons. In two cases, namely EG-13B and EG-21B, the deletion likely extended far from exons 4 or 5, respectively, since no cDNA products were detected. This result suggests that introns 4 and 5 probably contained A/T rich sequences important for Hprt mRNA processing or stability. The largest class of mutations comprised a heterogeneous group of 16 mutants (defined as “mutations in regulatory regions”) that produced low amounts, if any, of shorter length cDNAs that were better characterized using nested primers for the presence of the 5’-end, or the 3’-end of the Hprt coding sequence. However, all Hprt exons were present at genomic level. One mutant (EN-23A) carried a deletion of 5 base pairs in exon 2 that induced partial skipping of exon 2 resulting in the amplification of both the full-length and the shorter cDNA. Ten out of the 56 Me-lex induced Hprt mutants carried mutations that had been already observed in the spontaneous spectrum. Those mutants, being about 18% of all mutants isolated after Me-lex treatment, represent the portion of mutants that are expected to be of spontaneous origin based upon the 5-fold increase of the MF after Me-lex, and were not considered Me-lex induced. Therefore, they were excluded from further analysis.
Table 3.
Me-lex induced Hprt mutation spectrum in EM-C11
| Mutant/(s) | cDNA producta | Position | Type of mutation | Target | ||
|---|---|---|---|---|---|---|
| sequenceb | Aminoacid change N° of | |||||
| (exon) | (5’ > 3’) | (or | ||||
| cDNA change) | mutants | |||||
| Base pair changes | ||||||
| EN-15A | full length | 110 (2) | TA>CG | GTG TTT | ||
| ATT CCT CAT | Ile>Thr | 1 | ||||
| EN-11A | full length | 296 (3) | TA>GC | GTA GAT | ||
| TTT ATC AGA | Phe>Cys | 1 | ||||
| EN-20C | full length | 437 (6) | TA>GC | CAA ACT | ||
| CTG CTT TCC | Leu>Arg | 1 | ||||
| EN-25C | full length | 466 (6) | AT>TA | AAC CCC | ||
| AAA ATG GTT | Lys>STOP | 1 | ||||
| EN-5A | full length | 479 (6) | TA>AT | GTT AAG | ||
| GTT GCA AGC | Val>Asp | 1 | ||||
| EN-6B | full length | 543 (8) | TA>GC | GTT GGA | ||
| TTT GAA ATT | Phe>Leu | 1 | ||||
| EG-18B | full length | 545 (8) | AT>GC | GGA TTT GAA | ||
| ATT CCA | Glu>Gly | 1 | ||||
| Frameshift mutations | ||||||
| EN-29A | low, full length | 294–297 (3) | −T | CT GTA GAT | ||
| TTT ATC AG | 1 | |||||
| EN-19B | low, full length | 408–409 (6) | +AA | AG GAC ATA | ||
| ATT GAC AC | 1 | |||||
| Splice mutations | ||||||
| EN-28A | short cDNA | In 3: +1 | GC>AT | TACTGT g | ||
| taagtataatt | Skipping ex 3 | 1 | ||||
| EN-2B/−14C | short cDNA | In 3: −1 | GC>CG | ttttttaacta g | ||
| AATGAT | Skipping ex 4 | 5 | ||||
| /−17B/−21A | ||||||
| /−27C | ||||||
| EN-30B | short cDNA | In6: +2 | TA>GC | TGCAAGg t | ||
| atgtatgcc | Skipping ex 6 | 1 | ||||
| EN-3C/−7B | short cDNA | Splice junctions not sequenced | ||||
| Skipping ex 2 | 2 | |||||
| All exons present in gDNA | ||||||
| Genomic deletions | ||||||
| EN-1C | full length | 81–83 (2) | -CTA | AAT CAC | ||
| TAT GTC GAG | -Tyr | 1 | ||||
| EG-3A/−30A | full length | 565–567 (6) | -GTT | TTT GTT GTT | ||
| GGA TAT | -Val | 2 | ||||
| EN-10A | low, two short cDNAs | Deletion of exon 3 | ||||
| 1 | ||||||
| EG-13B | no cDNAs | Deletion of exon 4 | ||||
| 1 | ||||||
| EG-21B | no cDNAs | Deletion of exon 5 | ||||
| 1 | ||||||
| EG-2B/−8B | no cDNAs | Deletion of all Hprt exons | ||||
| 5 | ||||||
| /−24C/−27B | ||||||
| /−28C | ||||||
| Mutations in regulatory regions | ||||||
| EN-12A/−24A | no cDNAs | All exons present on gDNA | ||||
| 2 | ||||||
| EN-22B | very low, short cDNA | All exons present on gDNA | ||||
| Skipping ex 4 | 1 | |||||
| No mutations found at splice junctions | ||||||
| EG-4A/−5C | low ex (1–4) | All exons present on gDNA | ||||
| 8 | ||||||
| /−16A/−20B | ||||||
| /−21A/−22C | ||||||
| /−25A/−26C | ||||||
| EG-6B | Low gDNA ex 1. All other exons present on | |||||
| gDNA | 1 | |||||
| EG-9A | low cDNA ex (3–9) | All exons present on gDNA | ||||
| 1 | ||||||
| EG-12A/−19B | no cDNAs | All exons present on gDNA | ||||
| 3 | ||||||
| /−29A | ||||||
| Other mutations | ||||||
| EN-23A | two cDNA: full length, | Deletion (28–32) in exon 2 | tttttcag | |||
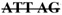 T GAT T GAT |
1 | |||||
| short | ||||||
| Skipping ex 2 | ||||||
cDNA amplifications of the full length, the 5'- (ex 1–4) and the 3'- (ex 4–8) half of the Hprt coding sequence were performed for each mutant (see M&M). Short cDNA refers to PCR fragments lacking the internal part of the hprt coding sequence.
Non transcribed strand gDNA = genomic DNA
3.4. Me-lex induces SCEs, MN and nuclear fragmentation
Previous results in CHO-9 cells led us to hypothesize that the processing of 3-mA within A/T rich sequences in non-coding regions of the Hprt gene could result in recombination and genomic deletions [22]. To further investigate this issue, we performed a genome-wide analysis of Me-lex genotoxicity through the determination of SCEs and MN in both CHO-9 and EM-C11 cell lines.
In CHO-9 cells, SCE induction was evaluated at a range of 25–100 µM Me-lex concentrations, which induced low levels of mortality (80–90% of cell survival) [22]. The number of SCEs per chromosome increased 3-fold from mock to 50 µM Me-lex treated cells (Figure 2A). At 100 µM Me-lex, a high percentage of fragmented nuclei and a very low number of metaphases were counted (see below), making the evaluation of SCEs unreliable. MMS, an alkylating agent known to induce SCEs and chromosomal aberrations [36,37], was used as positive control at equitoxic (sub-lethal) concentrations [30]. In MMS-treated cells a dose dependent induction of SCE up to 250 µM was found (Figure 2B), demonstrating the robustness of the assay. Me-lex preferentially (> 90%) generates 3-mA and at equimolar concentrations of Me-lex and MMS, the induction of other SCE-inducing lesions, e.g., 7-mG and 6-mG, is comparable. Since the yield of 3-mA is approximately 40-fold higher with Me-lex treatment than MMS [2], these results are consistent with 3-mA (or its derived BER intermediate) being responsible for the induction of SCEs.
Figure 2. SCE, MN and nuclear fragmentation induced by Me-lex and MMS in CHO-9 cells.

The number of SCE per chromosome induced in CHO-9 cells is reported for Me-lex (A) and MMS (B). The number of MN, fragmented nuclei and apoptotic cells, over 1000 scored cells, are reported for Me-lex (C) and MMS (D) treated CHO-9 cells.
The induction of MN [38,39], as well as the appearance of fragmented and apoptotic nuclei were also investigated. As shown in Figure 2C, 100 µM Me-lex induced a sharp increase in the percentage of MN and fragmented nuclei, and the appearance of apoptotic nuclei. In MMS-treated cells, the percentage of MN increased at 100 µM (14.7 ‰) but did not reach the level detected for Me-lex even at the highest MMS concentration tested (i.e., 500 µM MMS; 28.2 ‰) (Figure 2D).
The same end points were also explored in EM-C11 cells. This cell line is known to have a high background level of SCEs and chromosomal aberrations [28]. In agreement with published data [28], in mock-treated EM-C11 cells about a 10-fold higher level of SCEs (0.939 ± 0.182) than in CHO-9 cells (0.129 ± 0.027) was found. In addition, the levels of multinucleated cells and MN were significantly higher in EMC-11 than in CHO-9 cells (Figure 3A). In Me-lex treated EM-C11 cells, aberrant mitosis and a high level of fragmented nuclei were detected at very low doses (Figure 3B). These results indicate that XRCC1deficiency may lead to genomic instability in the absence of an exogenous genotoxic insult, and that in this cellular environment even low levels of Me-lex induced lesions, i.e., 3-mA (or its derivatives), are sufficient to generate a large amount of genomic damage.
Figure 3. Cytogenetic analysis of Me-lex treated EMC −11 cells.

A) Multinucleated cells and MN spontaneously induced in CHO-9 and EM-C11 cells. B) Multinucleated cells, aberrant mitosis and fragmented nuclei in EM-C11 cells before and after Me-lex treatment. Insert in the top right of panel B shows a magnification of a micronucleated cell. MultiN = multinucleated cells; AbMi = aberrant mitoses; AbMe = aberrant metaphase; MN = micronucleus; FN = fragmented nucleus; G = ghost.
4. Discussion
The effect of XRCC1deficiency in the processing of 3-mA adducts generated by Me-lex was explored through the molecular analysis of Hprt mutations and the determination of cytogenetic end points, including SCEs, MN and nucleus fragmentation in Me-lex treated EM-C11 and the parental DNA repair proficient CHO-9 cells.
EM-C11 cells exposed to Me-lex showed a 2.5-fold higher sensitivity to toxicity than their DNA repair proficient counterpart (CHO-9), but they were not hypermutable. A comparable level of mutability, despite their different sensitivities, has been already reported for these two cell lines following treatment with EMS [29] and MMS [30]. Our results strongly suggest that unrepaired 3-mA (or a derived AP site) is a lethal lesion and that XRCC1 is involved in its repair, probably by facilitating the processing of BER intermediates.
In order to characterize the mutations arising as a result of XRCC1 deficiency per se, a spontaneous mutation spectrum at the Hprt locus was generated in EM-C11 cells. When compared with the spontaneous mutation spectrum obtained in the parental CHO-9 cell line ([22] and unpublished data), the two spectra appear significantly different (p=0.0016; Cariello’s test [40]) especially in terms of genomic deletions (p=0.0054; Fisher’s exact test) (Table 4). These results clearly indicated that XRCC1 deficiency per se causes genomic instability leading to deletions. After Me-lex treatment, mutations in regulatory regions reached a level of 35% with respect to the 10.5% in the spontaneous spectrum (p=0.0277 Fisher’s exact test). This class of mutations was defined in a previous study [22] as mutations that produced low levels and/or short Hprt cDNAs without affecting the genomic sequence of Hprt exons. The mutations responsible for the formation of short or unstable mRNAs are likely localized in non-coding regulatory regions of the Hprt gene [22]. Regulatory regions may contain large stretches of A/T-repeats that constitute the preferential target for Me-lex binding and methylation [3]. Interestingly, within the whole Hprt mouse genomic sequence, A/T-runs in introns are abundant (http://www.ensembl.org).
Table 4.
Comparison among Hprt mutation spectra in EM-C11 and CHO-9 cell lines
| EM-C11 | |||
|---|---|---|---|
| CHO-9a | |||
| Type of Hprt mutations | Spontaneous | Me-lex | |
| induced | Spontaneous | Me-lex induced | |
| n° (%) | n° (%) | ||
| n° (%) | n° (%) | ||
| Base pair changes | 9 (31%) | 7 | |
| (15%) | 16 (43%) | 10 (23%) | |
| AT-targeted | 3 (10%) | 7 | |
| (15%) | 12 (32%) | 7 (16%) | |
| AT>TA | 1 | 2 | |
| 1 | 2 | ||
| AT>GC | 2 | 2 | |
| 9 | 4 | ||
| AT > CG | 0 | 3 | |
| 2 | 2 | ||
| GC-targeted | 6 (21%) | 0 | |
| 4 (11%) | 3 (7%) | ||
| GC > AT | 0 | 0 | |
| 0 | 1 | ||
| GC > CG | 0 | 0 | |
| 1 | 0 | ||
| GC > TA | 6 | 0 | |
| 3 | 2 | ||
| Splice mutationsb | 3 (10%) | 9 | |
| (19%)c | 1 (3%) | 1(2%)c | |
| Frame shifts | 2 (7%) | 2 | |
| (4.3%) | 0 | 3 (7%) | |
| Genomic deletions | 11 (38%)d | 11 | |
| (24%) | 3 (8%)d | 21 (48%) | |
| Exon deletions | 6 (21%) | 6 | |
| (13%) | 3 | 9 (21%) | |
| Complete Hprt deletions | 5 (17%) | 5 | |
| (11%) | 0 | 12 (27%) | |
| Mutations in regulatory regions | 3 (10.5%)e | 16 | |
| (35%)e | 12 (32%) | 8 (18%) | |
| Other | 1 (3.5%) | 1 | |
| (2%) | 5 (14%) | 1 (2%) | |
| Total n° of mutations | 29 (100%) | 46 | |
| (100%) | 37 (100%) | 44 (100%) | |
From Russo et al., 2009 and data unpublished
This category includes splice mutants for which the mutation at splice junction was found
p=0.0153 (Fisher’s exact test)
p=0.0054 (Fisher’s exact test)
p=0.0277 (Fisher’s exact test)
The Me-lex induced mutation spectrum in EM-C11 cells differs from the one in CHO-9 cells (p=0.0157, Cariello’s test) (Table 4). The number of splice mutations in EM-C11 is significantly higher than in CHO-9 (p=0.0153; Fisher’s exact test). In these mutants, an A/T-rich sequence is adjacent to the site of base pair change suggesting a sequence specific methylation by Me-lex. A-tracts occur often in regulatory regions of many organisms [41] and it has been postulated that their ability to adopt unusual DNA conformation may be linked to their participation to regulatory processes. An unexpected result from our studies is the low level of genomic deletions found in EM-C11 vs. CHO-9 cells (24% vs 48%). Notably, in EM-C11, a high level of genomic deletions occurred without treatment (38%) and did not increase after Me-lex exposure (24%). Since XRCC1 deficient cells are spontaneously prone to accumulate genomic deletions as well as high levels of SCEs and MN, a Me-lex treatment, which further increases the overall burden of SSBs, may generate large genomic deletions detrimental for cell viability. Besides the presence of multinucleated cells, aberrant mitosis and fragmented nuclei were generated at very low Me-lex doses (25 µM). At 50 µM a lower number of multinucleated cells were counted but the mitotic index was also very low (not shown) making the determination of these cytogenetic endpoints not statistically reliable. It is likely that at the 75 µM Me-lex dose used to isolate mutants, only cells carrying less severe genomic damage could be recovered.
Comparison of the spontaneous and induced mutation spectra in EM-C11 cells and in Me-lex treated EM-C11 and CHO-9 cells indicate that in cells lacking XRCC1, Me-lex promotes the induction of mutations in non-coding Hprt regions crucial for the regulation and/or stability of mRNA processing. All together these data suggest that non-coding Hprt genomic sequences represent a good target for the rare pro-mutagenic lesions induced by Me-lex, and such a specificity is exacerbated by Xrcc1 gene dysfunction. These results could also be attributed to an intrinsic instability of these repetitive regions that may become even more unstable in a XRCC1 deficient cell.
The ability of Me-lex to induce recombination and genomic rearrangements [22] was confirmed through the analysis of SCE and MN induction in CHO-9 cells. The formation of SCEs has been correlated with recombination repair and the induction of point mutations, gene amplification and cytotoxicity [42]. Our results clearly show that even in a repair proficient background, i.e., CHO-9 cells, Me-lex induced SCEs in a dose-dependent manner. A comparable level of basal SCEs was found in both AAG proficient and AAG deficient ES cells [15], indicating that AAG deficiency per se does not affect spontaneous recombination. After Me-lex exposure, a significant increase of SCEs was found in Aag −/− cells providing evidence that 3-mA was the lesion responsible for SCE induction [15]. The appearance of MN, fragmented and apoptotic nuclei was also analysed. MN assay has emerged as one of the preferred methods for assessing chromosome damage because it enables both chromosome loss and chromosome breakage to be measured reliably [38,39]. Our results showed that sub-lethal Me-lex concentrations were able to induce a significant increase of MN, fragmented nuclei and apoptotic cells. When cells were treated with MMS, a lower induction of the same endpoints was observed, suggesting a direct role of A/T-targeted 3-mA lesions in chromosome damage induction. In a recent study conducted in yeast, a large number of unselected chromosomal aberrations arising from randomly induced DSBs in the entire Saccharomyces cerevisiae genome has been analysed. Interestingly, only repeat-associated DSBs could efficiently lead to chromosome aberrations, while DSBs in unique sequences were efficiently repaired through homologous recombination [43]. Because MN can only be expressed in cells that complete nuclear division, Me-lex concentrations that poorly affect the overall survival have to be used to perform this assay. Nevertheless, the cytogenetic analyses revealed a significant genotoxic effect of Me-lex that can explain its high cytotoxicity.
To reconcile all our data, we propose a model for Me-lex cytotoxicity and mutagenicity in mammalian cells. We emphasize that any consideration should take into account the specific reactivity of Me-lex towards A/T-rich sequences. In repair proficient cells, 3-mA is efficiently removed by AAG and processed through the subsequent steps of the BER pathway. If 3-mA (or one of its BER derivatives) is encountered during DNA replication it may stall DNA replication; in these circumstances, TLS can be involved and the lesion bypassed with the enhanced induction of base pair mutations (mainly A/T-targeted). However, the processing of 3-mA specifically localized in A/T-rich regions may promote the formation of clustered SSBs and even generate an irreversible block of DNA replication that stimulates homologous recombination. If we assume that regulatory regions are characterized by the presence of A/T-rich sequences, the presence of 3-mA could originate a high percentage of mutations that affect mRNA stability (mutations at regulatory regions and splice mutations). At low Me-lex doses (25–50µM) these events will promote DNA recombination, visualized as SCEs. As the level of 3-mA increases, double strand breaks may be generated, which would promote deletions, MN, fragmented nuclei and apoptosis. In a XRCC1 deficient background, cells have to manage the burden of SSBs spontaneously generated genome-wide. The presence of 3-mA or other repair intermediates from Me-lex exposure, in addition to spontaneous SSBs, would cause DNA replication forks to collapse resulting in extensive genomic damage even at a low Me-lex concentrations.
In summary, we have shown that XRCC1 deficiency increases Me-lex cytotoxicity and affects the type of mutations induced at the Hprt locus without increasing the Me-lex induced MF. Our results indicate that the processing of 3-mA, or one of its BER derivatives, is a complex process where DNA repair processes, besides BER, may participate. With this perspective, it will be important to explore other repair pathways, such as homologous recombination and nonhomologous end joining, that may contribute to the error-free repair of 3-mA and the modulation of Me-lex cytotoxicity.
Acknowledgments
This work was supported by the National Institute of Health, Grant RO1 CA29088 (to BG), and partially by the Associazione Italiana per la Ricerca sul Cancro (AIRC) to GF.
Abbreviations
- Me-lex
{1-methyl-4-[1-methyl-4-(3-methoxysulfonylpropanamido) pyrrole-2-carboxamido]-pyrrole-2-carboxamido}propane
- XRCC1
(X-ray repair cross-complementing 1).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The authors declare that there are no conflicts of interest.
References
- 1.Zhang Y, Chen FX, Mehta P, Gold B. Groove- and sequence-selective alkylation of DNA by sulfonate esters tethered to lexitropsins. Biochemistry. 1993;32:7954–7965. doi: 10.1021/bi00082a017. [DOI] [PubMed] [Google Scholar]
- 2.Encell L, Shuker DE, Foiles PG, Gold B. The in vitro methylation of DNA by a minor groove binding methyl sulfonate ester. Chem Res Toxicol. 1996;9:563–567. doi: 10.1021/tx9501849. [DOI] [PubMed] [Google Scholar]
- 3.Kelly JD, Inga A, Chen FX, Dande P, Shah D, Monti P, Aprile A, Burns PA, Scott G, Abbondandolo A, Gold B, Fronza G. Relationship between DNA methylation and mutational patterns induced by a sequence selective minor groove methylating agent. J Biol Chem. 1999;274:18327–18334. doi: 10.1074/jbc.274.26.18327. [DOI] [PubMed] [Google Scholar]
- 4.Fronza G, Gold B. The biological effects of N3-methyladenine. J Cell Biochem. 2004;91:250–257. doi: 10.1002/jcb.10698. [DOI] [PubMed] [Google Scholar]
- 5.Dinglay S, Gold B, Sedgwick B. Repair in Escherichia coli alkB mutants of abasic sites and 3-methyladenine residues in DNA. Mutat Res. 1998;407:109–116. doi: 10.1016/s0921-8777(97)00065-7. [DOI] [PubMed] [Google Scholar]
- 6.Shah D, Kelly J, Zhang Y, Dande P, Martinez J, Ortiz G, Fronza G, Tran H, Soto AM, Marky L, Gold B. Evidence in Escherichia coli that N3-methyladenine lesions induced by a minor groove binding methyl sulfonate ester can be processed by both base and nucleotide excision repair. Biochemistry. 2001;40:1796–1803. doi: 10.1021/bi0024658. [DOI] [PubMed] [Google Scholar]
- 7.Engelward BP, Dreslin A, Christensen J, Huszar D, Kurahara C, Samson L. Repair-deficient 3-methyladenine DNA glycosylase homozygous mutant mouse cells have increased sensitivity to alkylation-induced chromosome damage and cell killing. EMBO J. 1996;15:945–952. [PMC free article] [PubMed] [Google Scholar]
- 8.Boiteux S, Huisman O, Laval J. 3-Methyladenine residues in DNA induce the SOS function sfiA in Escherichia coli. EMBO J. 1984;3:2569–2573. doi: 10.1002/j.1460-2075.1984.tb02175.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Larson K, Sahm J, Shenkar R, Strauss B. Methylation-induced blocks to in vitro DNA replication. Mutat Res. 1985;150:77–84. doi: 10.1016/0027-5107(85)90103-4. [DOI] [PubMed] [Google Scholar]
- 10.Plosky BS, Frank EG, Berry DA, Vennall GP, McDonald JP, Woodgate R. Eukaryotic Y-family polymerases bypass a 3-methyl-2'-deoxyadenosine analog in vitro and methyl methanesulfonate-induced DNA damage in vivo. Nucleic Acids Res. 2008;36:2152–2162. doi: 10.1093/nar/gkn058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA Repair and Mutagenesis. Washington, DC: AMS Press; 2006. [Google Scholar]
- 12.Patel PH, Kawate H, Adman E, Ashbach M, Loeb LA. A single highly mutable catalytic site amino acid is critical for DNA polymerase fidelity. J Biol Chem. 2001;276:5044–5051. doi: 10.1074/jbc.M008701200. [DOI] [PubMed] [Google Scholar]
- 13.Monti P, Ciribilli Y, Russo D, Bisio A, Perfumo C, Andreotti V, Menichini P, Inga A, Huang X, Gold B, Fronza G. Rev1 and Polzeta influence toxicity and mutagenicity of Me-lex, a sequence selective N3-adenine methylating agent. DNA Repair (Amst) 2008;7:431–438. doi: 10.1016/j.dnarep.2007.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Monti P, Campomenosi P, Ciribilli Y, Iannone R, Inga A, Shah D, Scott G, Burns PA, Menichini P, Abbondandolo A, Gold B, Fronza G. Influences of base excision repair defects on the lethality and mutagenicity induced by Me-lex, a sequence-selective N3-adenine methylating agent. J Biol Chem. 2002;277:28663–28668. doi: 10.1074/jbc.M203384200. [DOI] [PubMed] [Google Scholar]
- 15.Engelward BP, Allan JM, Dreslin AJ, Kelly JD, Wu MM, Gold B, Samson LD. A chemical and genetic approach together define the biological consequences of 3-methyladenine lesions in the mammalian genome. J Biol Chem. 1998;273:5412–5418. doi: 10.1074/jbc.273.9.5412. [DOI] [PubMed] [Google Scholar]
- 16.Bobola MS, Varadarajan S, Smith NW, Goff RD, Kolstoe DD, Blank A, Gold B, Silber JR. Human glioma cell sensitivity to the sequence-specific alkylating agent methyl-lexitropsin. Clin Cancer Res. 2007;13:612–620. doi: 10.1158/1078-0432.CCR-06-1127. [DOI] [PubMed] [Google Scholar]
- 17.Tentori L, Vernole P, Lacal PM, Madaio R, Portarena I, Levati L, Balduzzi A, Turriziani M, Dande P, Gold B, Bonmassar E, Graziani G. Cytotoxic and clastogenic effects of a DNA minor groove binding methyl sulfonate ester in mismatch repair deficient leukemic cells. Leukemia. 2000;14:1451–1459. doi: 10.1038/sj.leu.2401842. [DOI] [PubMed] [Google Scholar]
- 18.Tentori L, Forini O, Fossile E, Muzi A, Vergati M, Portarena I, Amici C, Gold B, Graziani G. N3-methyladenine induces early poly(ADP-ribosylation), reduction of nuclear factor-kappa B DNA binding ability, and nuclear up-regulation of telomerase activity. Mol Pharmacol. 2005;67:572–581. doi: 10.1124/mol.104.004937. [DOI] [PubMed] [Google Scholar]
- 19.Tentori L, Balduzzi A, Portarena I, Levati L, Vernole P, Gold B, Bonmassar E, Graziani G. Poly (ADP-ribose) polymerase inhibitor increases apoptosis and reduces necrosis induced by a DNA minor groove binding methyl sulfonate ester. Cell Death Differ. 2001;8:817–828. doi: 10.1038/sj.cdd.4400863. [DOI] [PubMed] [Google Scholar]
- 20.Allan JM, Travis LB. Mechanisms of therapy-related carcinogenesis. Nat Rev Cancer. 2005;5:943–955. doi: 10.1038/nrc1749. [DOI] [PubMed] [Google Scholar]
- 21.Monti P, Traverso I, Casolari L, Menichini P, Inga A, Ottaggio L, Russo D, Iyer P, Gold B, Fronza G. Mutagenicity of N3-methyladenine: a multi-translesion polymerase affair. Mutat Res. 2010;683:50–56. doi: 10.1016/j.mrfmmm.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Russo D, Fronza G, Ottaggio L, Monti P, Inga A, Iyer P, Gold B, Menichini P. High frequency of genomic deletions induced by Me-lex, a sequence selective N3-adenine methylating agent, at the Hprt locus in Chinese hamster ovary cells. Mutat Res. 2009;671:58–66. doi: 10.1016/j.mrfmmm.2009.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thompson LH, West MG. XRCC1 keeps DNA from getting stranded. Mutat Res. 2000;459:1–18. doi: 10.1016/s0921-8777(99)00058-0. [DOI] [PubMed] [Google Scholar]
- 24.Caldecott KW. XRCC1 and DNA strand break repair. DNA Repair (Amst) 2003;2:955–969. doi: 10.1016/s1568-7864(03)00118-6. [DOI] [PubMed] [Google Scholar]
- 25.Mani RS, Karimi-Busheri F, Fanta M, Caldecott KW, Cass CE, Weinfeld M. Biophysical characterization of human XRCC1 and its binding to damaged and undamaged DNA. Biochemistry. 2004;43:16505–16514. doi: 10.1021/bi048615m. [DOI] [PubMed] [Google Scholar]
- 26.Nazarkina ZK, Khodyreva SN, Marsin S, Lavrik OI, P J. Radicella XRCC1 interactions with base excision repair DNA intermediates. DNA Repair (Amst) 2007;6:254–264. doi: 10.1016/j.dnarep.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 27.Beernink PT, Hwang M, Ramirez M, Murphy MB, Doyle SA, Thelen MP. Specificity of protein interactions mediated by BRCT domains of the XRCC1 DNA repair protein. J Biol Chem. 2005;280:30206–30213. doi: 10.1074/jbc.M502155200. [DOI] [PubMed] [Google Scholar]
- 28.Zdzienicka MZ, van der Schans GP, Natarajan AT, Thompson LH, Neuteboom I, Simons JW. A Chinese hamster ovary cell mutant (EM-C11) with sensitivity to simple alkylating agents and a very high level of sister chromatid exchanges. Mutagenesis. 1992;7:265–269. doi: 10.1093/mutage/7.4.265. [DOI] [PubMed] [Google Scholar]
- 29.Op het Veld CW, Zdzienicka MZ, Vrieling H, Lohman PH, van Zeeland AA. Molecular analysis of ethyl methanesulfonate-induced mutations at the hprt gene in the ethyl methanesulfonate-sensitive Chinese hamster cell line EM-C11 and its parental line CHO9. Cancer Res. 1994;54:3001–3006. [PubMed] [Google Scholar]
- 30.Op het Veld CW, Jansen J, Zdzienicka MZ, Vrieling H, van Zeeland AA. Methyl methanesulfonate-induced hprt mutation spectra in the Chinese hamster cell line CHO9 and its xrcc1-deficient derivative EM-C11. Mutat Res. 1998;398:83–92. doi: 10.1016/s0027-5107(97)00243-1. [DOI] [PubMed] [Google Scholar]
- 31.Xu Z, Yu Y, Gibbs RA, Caskey CT, Hsie AW. Multiplex DNA amplification and solid-phase direct sequencing for mutation analysis at the hprt locus in Chinese hamster cells. Mutat Res. 1993;288:237–248. doi: 10.1016/0027-5107(93)90090-3. [DOI] [PubMed] [Google Scholar]
- 32.Fenech M, Chang WP, Kirsch-Volders M, Holland N, Bonassi S, Zeiger E. HUMN project: detailed description of the scoring criteria for the cytokinesis-block micronucleus assay using isolated human lymphocyte cultures. Mutat Res. 2003;534:65–75. doi: 10.1016/s1383-5718(02)00249-8. [DOI] [PubMed] [Google Scholar]
- 33.Perry P, Wolff S. New Giemsa method for the differential staining of sister chromatids. Nature. 1974;251:156–158. doi: 10.1038/251156a0. [DOI] [PubMed] [Google Scholar]
- 34.Menichini P, Inga A, Fronza G, Iannone R, Degan P, Campomenosi P, Abbondandolo A. Defective splicing induced by 4NQO in the hamster hprt gene. Mutat Res. 1994;323:159–165. doi: 10.1016/0165-7992(94)90028-0. [DOI] [PubMed] [Google Scholar]
- 35.Inga A, Iannone R, Degan P, Campomenosi P, Fronza G, Abbondandolo A, Menichini P. Analysis of 4-nitroquinoline-1-oxide induced mutations at the hprt locus in mammalian cells: possible involvement of preferential DNA repair. Mutagenesis. 1994;9:67–72. doi: 10.1093/mutage/9.1.67. [DOI] [PubMed] [Google Scholar]
- 36.Kaina B. Mechanisms and consequences of methylating agent-induced SCEs and chromosomal aberrations: a long road traveled and still a far way to go. Cytogenet Genome Res. 2004;104:77–86. doi: 10.1159/000077469. [DOI] [PubMed] [Google Scholar]
- 37.Wyatt MD, Pittman DL. Methylating agents and DNA repair responses: Methylated bases and sources of strand breaks. Chem Res Toxicol. 2006;19:1580–1594. doi: 10.1021/tx060164e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fenech M. The in vitro micronucleus technique. Mutat Res. 2000;455:81–95. doi: 10.1016/s0027-5107(00)00065-8. [DOI] [PubMed] [Google Scholar]
- 39.Aardema MJ, M K-V. Basel, Switzerland: Marcel Dekker; 2001. The in vitro micronucleus assay. [Google Scholar]
- 40.Cariello NF, Piegorsch WW, Adams WT, Skopek TR. Computer program for the analysis of mutational spectra: application to p53 mutations. Carcinogenesis. 1994;15:2281–2285. doi: 10.1093/carcin/15.10.2281. [DOI] [PubMed] [Google Scholar]
- 41.Haran TE, Mohanty U. The unique structure of A-tracts and intrinsic DNA bending. Q Rev Biophys. 2009;42:41–81. doi: 10.1017/S0033583509004752. [DOI] [PubMed] [Google Scholar]
- 42.Wilson DM, 3rd, Thompson LH. Molecular mechanisms of sister-chromatid exchange. Mutat Res. 2007;616:11–23. doi: 10.1016/j.mrfmmm.2006.11.017. [DOI] [PubMed] [Google Scholar]
- 43.Argueso JL, Westmoreland J, Mieczkowski PA, Gawel M, Petes TD, Resnick MA. Double-strand breaks associated with repetitive DNA can reshape the genome. Proc Natl Acad Sci U S A. 2008;105:11845–11850. doi: 10.1073/pnas.0804529105. [DOI] [PMC free article] [PubMed] [Google Scholar]