

Abstract
Mutations in LMNA, which encodes A-type nuclear lamins, cause disorders of striated muscle that have as a common feature dilated cardiomyopathy. We have demonstrated an abnormal activation of both the extracellular signal-regulated kinase (ERK) and the c-Jun N-terminal kinase (JNK) branches of the mitogen-activated protein kinase signaling cascade in hearts from LmnaH222P/H222P mice that develop dilated cardiomyopathy. We previously showed that pharmacological inhibition of cardiac ERK signaling in these mice delayed the development of left ventricle dilatation and deterioration in ejection fraction. In the present study, we treated LmnaH222P/H222P mice with SP600125, an inhibitor of JNK signalling. Systemic treatment with SP600125 inhibited JNK phosphorylation, with no detectable effect on ERK. It also blocked increased expression of RNAs encoding natriuretic peptide precursors and proteins involved in the architecture of the sarcomere that occurred in placebo-treated mice. Furthermore, treatment with SP600125 significantly delayed the development of left ventricular dilatation and prevented decreases in cardiac ejection fraction and fibrosis. These results demonstrate a role for JNK activation in the development of cardiomyopathy caused by LMNA mutations. They further provide proof of principle for JNK inhibition as a novel therapeutic option to prevent or delay the cardiomyopathy in humans with mutations in LMNA.
Keywords: Cardiomyopathy, lamin, MAP kinase, JNK, Emery-Dreifuss muscular dystrophy
1. Introduction
Mutations in LMNA encoding A-type nuclear lamins are responsible for at least 3 severe diseases involving striated muscles: autosomal Emery-Dreifuss muscular dystrophy [1], limb girdle muscular dystrophy type 1B [2] and dilated cardiomyopathy type 1A [3]. A common feature of these disorders is dilated cardiomyopathy, which is characterized by an age of onset generally in the third decade of life and frequently associated with progressive conduction system disease leading to implantation of defibrillators [4]. Affected individuals eventually develop heart failure, for which there is currently no curative treatment, ultimately necessitating cardiac transplantation.
Identification of LMNA mutations in patients with dilated cardiomyopathy raised intriguing questions about the relationship between A-type nuclear lamins and dilated cardiomyopathy, since lamins are not known to contribute in force transmission or generation in cardiomyocytes. The link between cardiomyopathy and abnormalities in A-type lamins is poorly understood and only a few hypotheses have been raised concerning pathophysiology. We have recently reported an abnormal activation of the extracellular signal-regulated kinase (ERK) and the c-Jun N-terminal kinase (JNK) branches of the mitogen-activated protein kinase (MAP kinase) signalling cascade in hearts of Lmna H222P knock-in mice, a model of autosomal Emery-Dreifuss muscular dystrophy [5]. Male LmnaH222P/H222P mice develop left ventricular (LV) dilatation and depressed contractile function starting at approximately 8-10 weeks of age and invariably develop LV dilatation and decreased cardiac contractility at 16 weeks of age with death typically occurring between 16 to 36 weeks [6]. On the basis of our observations that ERK and JNK are activated in these mice prior to the onset of clinically detectable cardiomyopathy, as well as our demonstration that lamin A variants that cause striated muscle disease activate both of these protein kinases when expressed in cultured cells, we hypothesized that abnormal activation of ERK and JNK plays a primary pathogenic role in the development of cardiomyopathy [5]. Recently, we showed that blocking ERK signalling in male LmnaH222P/H222P mice, using a small molecule inhibitor of extracellular signal-regulated kinase kinase (MEK) that activates ERK, induced normal LV diameters and cardiac ejection fraction (EF) at an age of 16 weeks, when placebo-treated mice had significant abnormalities in these parameters [7]. In the present study, we sought to determine if pharmacological inhibition of JNK signaling would similarly prevent or delay the development of dilated cardiomyopathy in LmnaH222P/H222P mice. To test this hypothesis, we treated LmnaH222P/H222P mice with SP600125, an inhibitor of JNK.
2. Methods
2.1. Mice
LmnaH222P/H222P mice were generated and genotyped as previously described [6]. Genotyping was performed by polymerase chain reaction using genomic DNA isolated from tail clippings and oligonucleotides with sequences 5’-cagccatcacctctcctttg-3’ and 5’-agcaccagggagaggacagg-3’. Mice were fed a chow diet and housed in a disease-free barrier facility with 12h/12h light/dark cycles. The Institutional Animal Care and Use Committee at Columbia University Medical Center approved the use of animals and the study protocol. The investigation conforms with the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health (NIH Publication No. 85-23, revised 1996).
2.2. JNK inhibitor
The anthrapyrazolone inhibitor of JNK, SP600125 (Calbiochem), was dissolved in dimethyl sulfoxide (DMSO) (Sigma) at a concentration of 0.5 mg/ml and administered at a dose of 3 mg/kg/day for 5 days a week. The inhibitory activity of SP600125 is selective for all JNK isoforms (IC50 = 40 nM for JNK-1 and JNK-2 and 90 nM for JNK-3) [8]. The placebo consisted of DMSO alone and was delivered in the same volume. Placebo and SP600125 were administered by intraperitoneal injection using a 27 5/8-gauge syringe. Treatment was initiated when mice were 8 weeks of age and continued until 16 weeks of age.
2.3. Thansthoracic echocardiography
At 16 weeks of age, mice were anesthetized with 1.5% isoflurane in O2 and placed on a heating pad (37°C). Echocardiography was performed using a Visualsonics Vevo 770 ultrasound with a 30 MHz transducer applied to the chest wall. Cardiac ventricular dimensions and EF were measured in 2D mode and M-mode 3 times for the number of animals indicated. A “blinded” echocardiographer (J.S.), unaware of the genotype or treatment, performed the examinations and interpreted the results.
2.4. Histopathological analysis
Mice were sacrificed at 16 weeks of age after being examined by echocardiography and freshly removed hearts were fixed in 4% formaldehyde for 48 hours, embedded in paraffin, sectioned at 5 μm and stained with Gomori’s trichrome or hematoxylin and eosin. Representative stained sections were photographed using a Microphot SA (Nikon) light microscope attached to a Spot RT Slide camera (Diagnostic Instruments). Images were processed using Adobe Photoshop CS (Adobe Systems).
2.5. Protein extraction and immunoblotting
Heart tissue was homogenized in extraction buffer as previously described [7]. Protein samples were subjected to SDS-PAGE, transferred to nitrocellulose membranes and blotted with primary antibodies against JNK (Santa-Cruz), phosphorylated JNK (No 9251, Cell Signaling), phosphorylated c-Jun (No Sc-822, Santa-Cruz), ERK1/2 (No Sc-94, Santa-Cruz) and phosphorylated ERK1/2 (No 9101, Cell Signaling). Secondary antibodies were horseradish peroxidate conjugated (Amersham). Recognized proteins were visualized by enhanced chemiluminescence (ECL, Amersham). The signal generated using antibody against GAPDH was used as internal control to normalize the amounts of protein between immunoblots.
2.6. Quantitative real-time RT-PCR analysis
Total RNA was extracted using the Rneasy isolation kit (Qiagen) as previously described [7]. cDNA was synthesized using Superscript first strand synthesis system according to the manufacturer’s instructions (Invitrogen) on total RNA. For each replicate in each experiment, RNA from tissue samples of different animals was used. Primers were designed correspond to mouse RNA sequences using Primer3 (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi) for Nppa (forward 5’-gcttccaggccatattggag-3’, reverse 5’-ccctgcttcctcagtctgct-3’), Nppb (forward 5’-ggaccaaggcctcacaaaag-3’, reverse 5’-tacagcccaaacgactgacg-3’), Myl4 (forward 5’-cccaagcctgaagagatgag-3’, reverse 5’-agacaacagctgctccacct-3’), Myl7 (forward 5’-tcaaggaagccttcagctgc-3’, reverse 5’-cggaacacttaccctcccg-3’), Myh7 (forward 5’-tgcagcagttcttaaccac-3’, reverse 5’-tcgaggcttctggaagttgt-3’) and JunD (forward 5’-atgtgcacgaaaatggaaca-3’, reverse 5’-cctgacccgaaaagtagctg-3’). The real-time RT-PCR reaction contained iQ SYBR green super mix (Bio-Rad), 200 nM of each primer and 0.2 μl of template in a 25 μl reaction volume. Amplification was carried out using the MyiQ Single-Color Real-Time PCR Detection System (Bio-Rad) with an initial denaturation at 95°C for 2 min followed by 50 cycles at 95°C for 30 s and 62°C for 30 s. Relative levels of mRNA expression were calculated using the ΔΔCT method [7]. Individual expression values were normalized by comparison with Gapdh mRNA (forward 5’-tgcaccaccaactgcttag-3’, reverse 5’-ggatgcagggatgatgttc-3’).
2.7. Statistical analysis
Results of immunoblots, real-time RT-PCR and fibrosis quantification were compared using a Student unpaired t-test. Comparisons of the echocardiographic parameters between DMSO-treated LmnaH222P/H222P and Lmna+/+ mice and between SP600125-treated and DMSO-treated LmnaH222P/H222P were performed using a Student unpaired t-test; to validate these results, a non-parametric test (Mann-Whitney) was performed and concordance checked. Statistical analyses were performed using GraphPad Prism software.
3. Results
3.1. Effect of SP600125 on JNK activity
Systemic administration of SP600125 to LmnaH222P/H222P mice partially blocked phosphorylation of JNK in hearts as shown by immunoblot (Fig. 1A). At 3 mg/kg/day (5 times a week), we did not detect inhibition of phosphorylation of ERK in the hearts (Fig. 1A). Quantification of the immunoblot signals for JNK showed that DMSO-treated LmnaH222P/H222P mice had a 2.5-fold increase of phopshorylated JNK expression compared to Lmna+/+ mice but LmnaH222P/H222P treated with SP600125 had a significantly reduced level of phosphorylated JNK similar to Lmna+/+ mice (Fig. 1A). Phosphorylation of the downstream target, c-Jun, was also significantly reduced by SP600125 (Fig. 1B), as well as the expression of JunD mRNA (Fig. 1C), confirming the efficacy of the small molecule inhibitor in heart at the given dose.
Fig. 1.
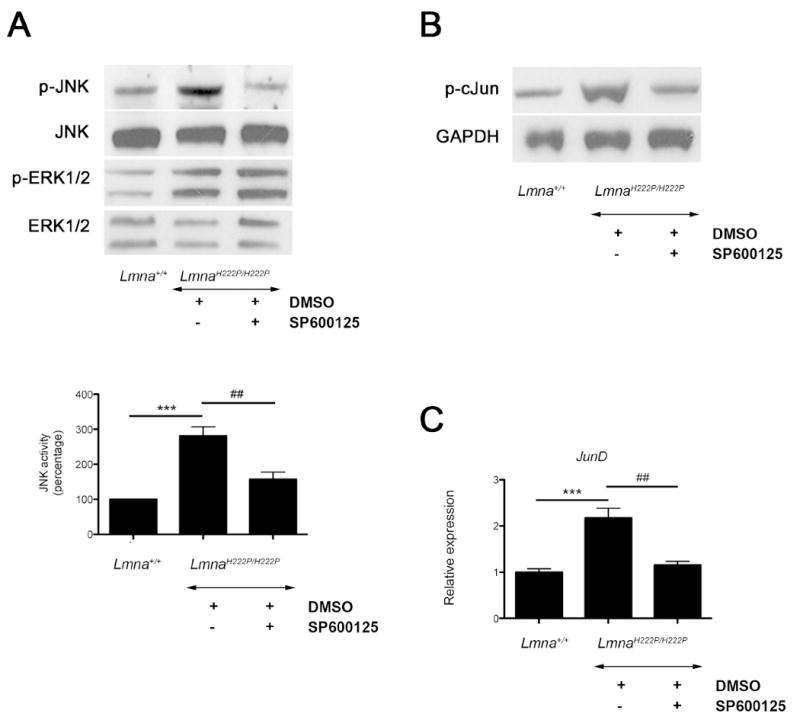
Treatment of male LmnaH222P/H222P mice with SP600125 inhibits phosphorylation of JNK signaling. (A) Representative immunoblots using antibodies against phophorylated JNK (p-JNK), total JNK, phosphorylated ERK1/2 (p-ERK1/2) and total ERK1/2 to probe proteins extracted from hearts from LmnaH222P/H222P mice treated with SP600125 or DMSO. Blots of proteins extracted from hearts of Lmna+/+ mice are shown for comparison. Data in bar graphs show quantification of phosphorylated JNK compared with total JNK measured by scanning immunoblots and using ImageJ64 software. Values are means ± standard errors for n=5 samples from different animals per group. Comparison between DMSO-treated LmnaH222P/H222P and Lmna+/+ mice was performed using Student unpaired t-test; ***P<0.0005. Comparison between SP600125-treated and DMSO-treated LmnaH222P/H222P was performed using Student unpaired t-test; ‡‡P<0.005. (B) Representative immunoblots using antibody against phosphorylated cJun (p-cJun) to probe proteins extracted from hearts from LmnaH222P/H222P mice treated with SP600125 or DMSO. Blot of proteins extracted from hearts of Lmna+/+ mice is shown for comparison. Antibody against GAPDH is used as a loading control. (C) Quantitative real-time RT-PCR showing the expression of mRNA from JunD. Results from hearts from Lmna+/+ mice are shown for comparison. Bars indicate the fold over-expression of the indicated mRNA calculated by the ΔΔCT method. Values are means ± standard errors for n=5 samples from different animals per group. Reactions were performed in triplicate for each different RNA sample. Comparison between DMSO-treated LmnaH222P/H222P and Lmna+/+ mice was performed using Student unpaired t-test; ***P<0.0005. Comparison between SP600125-treated and DMSO-treated LmnaH222P/H222P was performed using Student unpaired t-test; ‡‡P<0.005.
3.2. Effect of SP600125 on cardiac expression of natriuretic peptides and myosin
A feature of dilated cardiomyopathy is the upregulation of natriuretic peptides [9]. The upregulation of genes involved in sarcomere organization also occurs in dilated cardiomyopathy [5]. In hearts from male LmnaH222P/H222P mice, expression of NppA and NppB mRNAs encoding natriuretic peptides precursors were significantly increased compared to Lmna+/+ mice (Fig. 2A). Similarly, the expression of Myl7 and Myl4 mRNAs encoding myosin light chain and the expression of Myh7 mRNA encoding myosin heavy chain were significantly increased compared to Lmna+/+ mice (Fig. 2B). SP600125-treated LmnaH222P/H222P mice had a significantly decreasd cardiac expression of Myl7, Myl4, Myh7, NppA and NppB compared to DMSO-treated LmnaH222P/H222P mice (Fig. 2A and Fig. 2B).
Fig. 2.
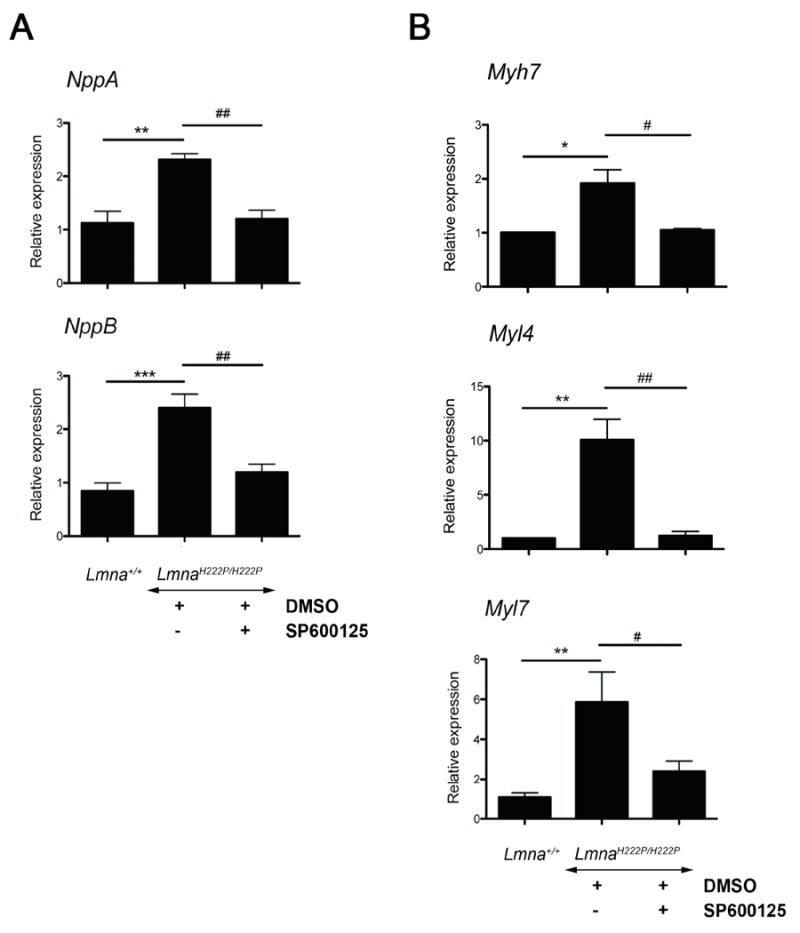
Treatment of male LmnaH222P/H222P mice with SP600125 inhibits expression of natriuretic peptides and myosin chains. Quantitative real-time RT-PCR showing the expression of (A) mRNAs from NppA and NppB genes encoding natriuretic peptide precursors A and B, respectively and (B) mRNAs from Myh7 encoding myosing heavy chain and Myl4 and Myl7 encoding myosin light chain. Results from hearts from Lmna+/+ mice are shown for comparison. Bars indicate the fold over-expression of the indicated mRNA calculated by the ΔΔCT method. Values are means ± standard errors for n=5 samples from different animals per group. Reactions were performed in triplicate for each different RNA sample. Comparison between DMSO-treated LmnaH222P/H222P and Lmna+/+ mice was performed using Student unpaired t-test; *P<0.05, **P<0.005, ***P<0.0005. Comparison between SP600125-treated and DMSO-treated LmnaH222P/H222P was performed using Student unpaired t-test; ‡P<0.05, ‡‡P<0.005.
3.3. Effect of JNK inhibition on cardiac function
Histopathological analysis of hearts at 16 weeks of age showed that DMSO-treated LmnaH222P/H222P mice had an increase in fibrotic tissue compared to Lmna+/+ mice (P<0.0005) (Fig. 3A). As assessed by quantification of collagen staining, SP600125-treated LmnaH222P/H222P mice had a statistically significant decrease of fibrosis compared to DMSO-treated LmnaH222P/H222P (P<0.05) (Fig. 3A). Hence, treatment with SP600125 prevents cardiac fibrosis in LmnaH222P/H222P mice. To confirm the degree of fibrosis, we further determined the expression of Col1a1 and Co1a2 encoding type I collagen by quantitative real-time RT-PCR. At 16 weeks of age, DMSO-treated LmnaH222P/H222P mice had a significantly increased expression of both genes in the heart compared to Lmna+/+ mice (Fig. 3B). In DMSO-treated LmnaH222P/H222P mice, Col1a1 and Col1a2 mRNAs were increased by 3-fold compared to Lmna+/+ mice (Fig. 3B). Treatment with SP600125 significantly lowered the expression of both Col1a1 and Col1a2 mRNAs in the heart of LmnaH222P/H222P mice compared to the DMSO-treated LmnaH222P/H222P mice (Fig. 3B).
Fig. 3.
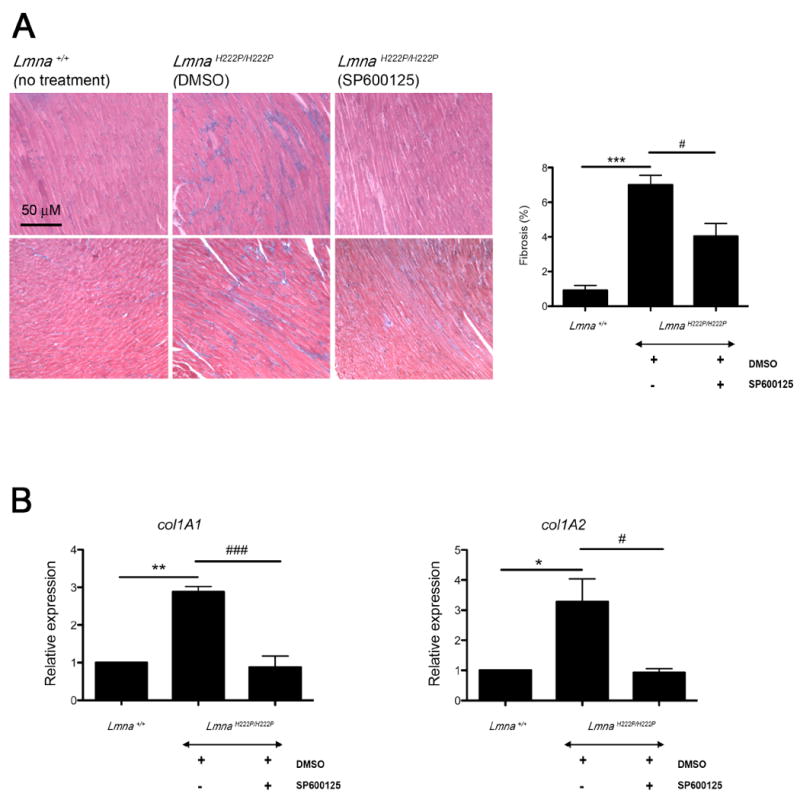
Treatment with SP600125 prevents cardiac fibrosis in LmnaH222P/H222P mice. (A) Representative heart tissue sections from LmnaH222P/H222P mice treated with SP600125 or DMSO stained with Gomori’s Trichrome are shown. Heart tissue section from Lmna+/+ mice is shown for comparison. Scale bar: 50 μm. Bar graph represents the quantification of fibrotic area in hearts from mice. Micrographs (n=3) for each heart were processed (JMicroVision software) and blue-staining fibrotic tissue was quantified (ImageJ64 software). Bars indicate the percentage of fibrosis in heart from Lmna+/+ and LmnaH222P/H222P mice treated with SP600125 or DMSO. Values are means ± standard errors for n=3 mice per group. Comparison between DMSO-treated LmnaH222P/H222P and Lmna+/+ mice was performed using Student unpaired t-test; ***P<0.0005. Comparison between SP600125-treated and DMSO-treated LmnaH222P/H222P was performed using Student unpaired t-test ‡P<0.05. (B) Effect of SP600125 inhibitor on cardiac expression of collagens genes in LmnaH222P/H222P mice. Bars indicate the expression of Col1a1 and Col1a2 in heart from Lmna+/+ and LmnaH222P/H222P mice treated with SP600125 or placebo (DMSO). Values are means ± standard errors for n=3 hearts. Comparison between DMSO-treated LmnaH222P/H222P and Lmna+/+ mice was performed using Student unpaired t-test; *P<0.05, **P<0.005. Comparison between SP600125-treated and DMSO-treated LmnaH222P/H222P mice was performed using Student unpaired t-test; ‡P<0.05, ‡‡‡P<0.0005.
We recently reported alterations in nuclear morphology, including abnormal elongation of nuclei, in cardiomyocytes of LmnaH222P/H222P mice [7]. At 16 weeks of age, DMSO-treated LmnaH222P/H222P mice had a significant increase in nuclear length, compared to Lmna+/+ mice (Fig. 4A). Cardiomyocyte nuclei in SP600125-treated LmnaH222P/H222P mice had an overall length that was similar to those in Lmna+/+ mice (Fig. 4A). Mean lengths of cardiomyocyte nuclei in placebo-treated LmnaH222P/H222P mice were significantly longer than in Lmna+/+ mice and SP600125-treated LmnaH222P/H222P mice (Fig. 4B).
Fig. 4.
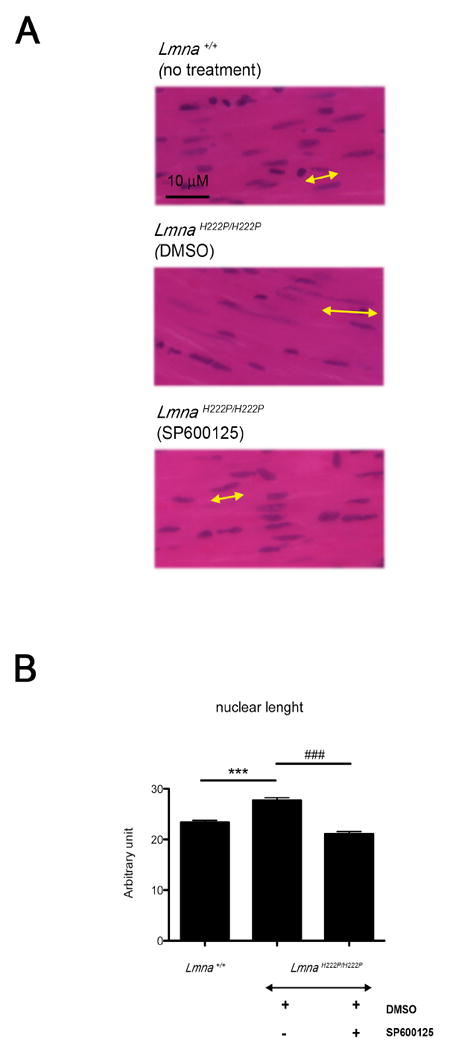
Treatment with SP600125 prevents abnormal elongation of cardiomyocyte nuclei in LmnaH222P/H222P mice. (A) Histological analysis of cross-sections of hearts from LmnaH222P/H222P mice treated with SP600125 or placebo (DMSO). Heart from Lmna+/+ mice was used for comparison. Sections are stained with hematoxylin and eosin. Yellow lines with arrowheads demonstrate the measurement of nuclear length. Scale bar: 10 μm. (B) Quantification of nuclear elongation in cardiomyocytes from mice. Cardiomyocyte nuclei are measured along the yellow lines with arrowheads. Bars indicate the length of cardiomyocyte nuclei in the indicated hearts. Values are means ± standard errors for n=230 cardiomyocytes. Comparison between DMSO-treated LmnaH222P/H222P and Lmna+/+ mice was performed using Student unpaired t-test; ***P<0.0005. Comparison between SP600125-treated and DMSO-treated LmnaH222P/H222P mice was performed using Student unpaired t-test; ‡‡‡P<0.0005.
Compared with Lmna+/+ mice, LmnaH222P/H222P mice treated with DMSO had significant increases in LV end-diastolic and end-systolic diameters and decreases in interventricular septal diameter, EF and fractional shortening (Table 1), consistent with what has been described in previous studies [6,7]. We then analyzed the effects of the JNK inhibitor compared to the placebo on echocardiographic parameters. The analysis was performed with heart rates similar between the two groups (358.47 ± 81.75 beats/min for DMSO-treated LmnaH222P/H222P mice and 355 ± 80.77 beats/min for SP600125-treated LmnaH222P/H222P mice). When treated with SP600125, LmnaH222P/H222P mice had approximately 5% smaller mean LV end-diastolic diameter, although the difference compared to placebo-treatment mice did not reach statistical significance. When treated with SP600125, LmnaH222P/H222P mice had a 15% smaller mean LV end-systolic diameter compared to DMSO-treated LmnaH222P/H222P mice (P<0.05). Systemic treatment with SP600125 had a positive effect on cardiac function leading to an EF approximately 20% higher than in DMSO-treated LmnaH222P/H222P mice (P<0.005). Hence, treatment with SP600125 for 8 weeks prevented or delayed the development of significant cardiac contractile dysfunction in LmnaH222P/H222P mice.
Table 1.
Echocardiographic data at 16 weeks of age for Lmna+/+ mice and LmnaH222P/H222P mice treated with DMSO or SP600125
| Genotype (Treatment) | n | LVEDD (mm) | LVESD (mm) | IVSD (mm) | EF (%) | FS (%) |
|---|---|---|---|---|---|---|
| Lmna+/+ (No Treatment) | 13 | 3.35 ± 0.09 | 2.00 ± 0.09 | 0.72 ± 0.02 | 76.82 ± 2.01 | 44.21 ± 1.77 |
| LmnaH222P/H222P (DMSO) | 15 | 3.59 ± 0.09* | 2.68 ± 0.14*** | 0.65 ± 0.03* | 56.89 ± 2.87*** | 29.70 ± 1.87*** |
| LmnaH222P/H222P (SP600125) | 13 | 3.46 ± 0.07 | 2.30 ± 0.12‡ | 0.72 ± 0.02‡ | 69.24 ± 2.66‡‡ | 38.70 ± 2.05‡‡ |
LVEDD, left ventricular end-diastolic diameter; LVESD, left ventricular end-systolic diameter; IVSD, interventricular septum diameter; EF, ejection fraction; FS, fractional shortening. Values are means ± standard errors.
Comparison between DMSO-treated LmnaH222P/H222P and Lmna+/+ mice was performed using Student unpaired t-test;
P<0.05,
P<0.005,
P<0.0005.
Comparison between SP600125-treated and DMSO-treated LmnaH222P/H222P was performed using Student unpaired t-test and results validated by a non-parametric test (Mann-Whitney);
P<0.05,
P<0.005.
4. Discussion
Our results show that abnormal activation of the stress-induced JNK signalling pathway contributes to the pathogenesis of cardiomyopathy caused by mutations in LMNA encoding A-type lamins. It remains unclear how A-type lamins with amino acid substitutions activate JNK signalling. Some investigators have hypothesized that alterations in response to stress may underlie the development of cardiac disease caused by LMNA mutation [10]. Abnormal responses to stress in cardiomyocytes with abnormalities in A-type lamins could therefore potentially have an impact on activation of JNK. This hypothesis remains to be tested.
We have demonstrated that partial pharmacological inhibition of JNK in vivo, using SP600125, prevents significant cardiomyopathy in male LmnaH222P/H222P mice at an age when placebo-treated controls have detectable cardiac dysfunction. We recently reported that the partial pharmacological blockade of ERK signalling for the same duration in LmnaH222P/H222P mice of the same age similarly prevents cardiomyopathy [7]. Data in our previous study [7] and our current results show that inhibiting either the ERK or the JNK branches of the MAP kinase signalling cascade prevents the re-expression of fetal genes such as those encoding myosins, the up-regulation in expression of natriuretic peptides, LV dilatation and decreased cardiac contractility. We have also shown that the JNK inhibitor prevents onset of cardiac fibrosis in 16 week-old LmnaH222P/H222P mice.
This preclinical study in mice assessed primary (LV dilatation, EF) and secondary (expression of natriuretic factors) endpoints that are used in many human clinical heart failure trials. The measurements of LV function we used are strictly correlated to prognosis and in many human clinical trials their behaviour parallels changes in mortality with treatment [11]. For example, LV end-systolic volume is the major determinant of survival in human subjects after recovery from myocardial infarction and after coronary artery bypass grafting for impaired LV function [12,13]. While mortality is a reasonable endpoint in phase III clinical trials for advanced heart failure, it is rarely if ever used in the initial drug assessment phase or in treatment of subjects with early heart disease [11], as were both the case in our study. The effect of JNK inhibition at later treatment onset, after the development of decreased cardiac ejection fraction, is currently being evaluated in our laboratory, as many patients will be diagnosed at an advance stage. Nonetheless, our previous [7] and current results clearly provide proof-of-principle that specific inhibitors that target both JNK and ERK signalling could prevent or delay the onset of cardiomyopathy caused by LMNA mutations and indicate that additional studies are warranted. Future studies of the effects of ERK and JNK signalling pathway inhibitors on cardiac conduction defects could also be interesting, given that early conduction abnormalities usually occur in human subjects with LMNA mutations.
JNK has been shown to play a central role in tissue remodelling through its ability to interact to AP-1 mediated transcription [14,15]. AP-1 function is regulated both through changes in the abundance of its Jun and Fos components and post-translational modification by phosphorylation [16]. Of interest, AP-1 modulates the regulation of type I collagen [17,18]. This is consistent with our observation that SP600125 decreased the expression of JunD mRNA as well as Col1a1 and Col1a2 mRNAs in hearts of LmnaH222P/H222P mice. We also showed that there was a markedly decreased amount of myocardial fibrosis in hearts of 16 week-old LmnaH222P/H222P mice treated with the JNK inhibitor. This anti-fibrotic action of SP600125 could be secondary to the beneficial effect on the cardiac structure and function.
Additional preclinical research should be performed before initiating clinical trials of ERK and JNK inhibition in human subjects with cardiomyopathy caused by LMNA mutations. To resolve the possible but unlikely issue that off-target effects are providing benefits on cardiomyopathy in LmnaH222P/H222P mice, these animals should be treated with other drugs in these classes, such as JNK inhibitors of different structure that recognize a different interaction site [19]. For ERK signalling, several compounds of different structures that act at various sites in the pathway are currently in clinical development and several have already been used in human subjects [20]. The effects of longer term ERK and JNK inhibition on various tissues also needs to be examined in experimental animals, especially on skeletal muscle that is often simultaneously affected in individuals with LMNA mutations that cause cardiomyopathy [1,2].
Acknowledgments
This work was supported by Grants from the National Institutes of Health [AR048997] and Muscular Dystrophy Association [MDA4287].
Footnotes
Conflict of Interest H.J.W. and A.M. are inventors on a pending PCT patent application on methods for treating and/or preventing cardiomyopathies by ERK and JNK inhibition filed by the Trustees of Columbia University in the City of New York.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bonne G, Di Barletta MR, Varnous S, Becane HM, Hammouda EH, Merlini L, Muntoni F, Greenberg CR, Gary F, Urtizberea JA, Duboc D, Fardeau M, Toniolo D, Schwartz K. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat Genet. 1999;21:285–288. doi: 10.1038/6799. [DOI] [PubMed] [Google Scholar]
- 2.Muchir A, Bonne G, van der Kooi AJ, van Meegen M, Baas F, Bolhuis PA, de Visser M, Schwartz K. Identification of mutations in the gene encoding lamins A/C in autosomal dominant limb girdle muscular dystrophy with atrioventricular conduction disturbances (LGMD1B) Hum Mol Genet. 2000;9:1453–1459. doi: 10.1093/hmg/9.9.1453. [DOI] [PubMed] [Google Scholar]
- 3.Fatkin D, MacRae C, Sasaki T, Wolff MR, Porcu M, Frenneaux M, Atherton J, Vidaillet HJ, Spudich S, De Girolami U, Seidman JG, Seidman C, Muntoni F, Müehle G, Johnson W, McDonough B. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med. 1999;341:1715–1724. doi: 10.1056/NEJM199912023412302. [DOI] [PubMed] [Google Scholar]
- 4.Meune C, Van Berlo JH, Anselme F, Bonne G, Pinto YM, Duboc D. Primary prevention of sudden death in patients with lamin A/C gene mutations. N Engl J Med. 2006;354:209–210. doi: 10.1056/NEJMc052632. [DOI] [PubMed] [Google Scholar]
- 5.Muchir A, Pavlidis P, Decostre V, Herron AJ, Arimura T, Bonne G, Worman HJ. Activation of MAPK pathway links LMNA mutations to cardiomyopathy in Emery-Dreifuss muscular dystrophy. J Clin Invest. 2007;117:1282–1293. doi: 10.1172/JCI29042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arimura T, Helbling-Leclerc A, Massart C, Varnous S, Niel F, Lacene E, Fromes Y, Toussaint M, Mura AM, Keller DI, Amthor H, Isnard R, Mallisen M, Schwartz K, Bonne G. Mouse model carrying H222P-lmna mutation develops muscular dystrophy and dilated cardiomyopathy similar to human striated muscle laminopathies. Hum Mol Genet. 2005;14:155–169. doi: 10.1093/hmg/ddi017. [DOI] [PubMed] [Google Scholar]
- 7.Muchir A, Shan J, Bonne G, Lehnart SE, Worman HJ. Inhibition of extracellular signal-regulate kinase signaling to prevent cardiomyopathy caused by mutation in the gene encoding A-type lamins. Hum Mol Genet. 2009;18:241–247. doi: 10.1093/hmg/ddn343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bennett BL, Sasaki DT, Murray BW, O’Leary EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, Bhagwat SS, Manning AM, Anderson DW. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci USA. 2001;98:13681–13686. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mizuno Y, Yoshimura M, Harada E, Nakayama M, Sakamoto T, Shimasaki Y, Ogawa H, Kugiyama K, Saito Y, Yasue H. Plasma levels of A- and B-type natriuretic peptides in patients with hypertrophic cardiomyopathy or idiopathic dilated cardiomyopathy. Am J Cardiol. 2000;86:1036–1040. doi: 10.1016/s0002-9149(00)01147-4. [DOI] [PubMed] [Google Scholar]
- 10.Muchir A, Worman HJ. The nuclear envelope and human disease. Physiology. 2004;19:309–314. doi: 10.1152/physiol.00022.2004. [DOI] [PubMed] [Google Scholar]
- 11.Zanolla L, Zardini P. Selection of endpoints for heart failure clinical trials. Eur J Heart Fail. 2003;5:717–723. doi: 10.1016/s1388-9842(03)00101-6. [DOI] [PubMed] [Google Scholar]
- 12.White HD, Norris RM, Brown MA, Brandt PW, Whitlock RM, Wild CJ. Left ventricular end-systolic volume as the major determinant of survival after recovery from myocardial infarction. Circulation. 1987;76:44–51. doi: 10.1161/01.cir.76.1.44. [DOI] [PubMed] [Google Scholar]
- 13.Hamer AW, Takayama M, Abraham KA, Roche AH, Kerr AR, Williams BF, Ramage MC, White HD. End-systolic volume and long-term survival after coronary artery bypass graft surgery in patients with impaired left ventricular function. Circulation. 1994;90:2899–2904. doi: 10.1161/01.cir.90.6.2899. [DOI] [PubMed] [Google Scholar]
- 14.Han H, Boyle D, Chang L, Bennett B, Karin M, Manning A, Firestein GS. c-Jun N-terminal kinase is required for metalloproteinase expression and joint destruction in inflammatory arthritis. J Clin Invest. 2001;108:73–81. doi: 10.1172/JCI12466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tibbles LA, Woodgett JR. The stress-activated protein kinase pathways. Cell Mol Life Sci. 1999;55:1230–1254. doi: 10.1007/s000180050369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Minden A, Karin M. Regulation and function of the JNK subgroup of MAP kinases. Biochim Byophys Acta. 1997;1333:85–104. doi: 10.1016/s0304-419x(97)00018-8. [DOI] [PubMed] [Google Scholar]
- 17.Ghosh AK. Factors involved in the regulation of type I collagen gene expression: implication in fibrosis. Exp Biol Med. 2002;227:301–314. doi: 10.1177/153537020222700502. [DOI] [PubMed] [Google Scholar]
- 18.Rossert J, Terraz C, Dupont S. Regulation of type I collagen genes expression. Nephrol Dial Transplant. 2000;15:66–68. doi: 10.1093/ndt/15.suppl_6.66. [DOI] [PubMed] [Google Scholar]
- 19.Stebbins JL, De SK, Machleidt T, Becattini B, Vazquez J, Kuntzen C, Chen LH, Cellitti JF, Riel-Mehan M, Emdadi A, Solinas G, Karin M, Pellecchia M. Identification of a new JNK inhibitor targeting the JNK-JIP interaction site. Proc Natl Acad Sci USA. 2008;105:16809–16813. doi: 10.1073/pnas.0805677105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wong KK. Recent developments in anti-cancer agents targeting the Ras/Raf/ MEK/ERK pathway. Recent Pat Anticancer Drug Discov. 2009;4:28–35. doi: 10.2174/157489209787002461. [DOI] [PubMed] [Google Scholar]