

Abstract
Objective
To determine whether HLA-B27 misfolding and the unfolded protein response (UPR) results in cytokine dysregulation, and whether this is associated with Th1 and/or Th17 activation in HLA-B27/human beta-2-microglobulin (hβ2m) transgenic rats (HLA-B27 transgenic), an animal model of spondyloarthropathy.
Methods
Cytokine expression in LPS-stimulated macrophages was analyzed in the presence and absence of a UPR induced by chemical agents or HLA-B27 upregulation. Cytokine expression in colon tissue and in cells purified from the lamina propria was determined by real time reverse-transcriptase polymerase chain reaction, and differences in Th1 and Th17 CD4+ T cell populations were quantified with intracellular cytokine staining.
Results
IL-23 was found to be synergistically upregulated by LPS in macrophages undergoing a UPR induced either by pharmacologic agents or HLA-B27 misfolding. IL-23 was also increased in the colon of HLA-B27 transgenic rats concurrent with the development of intestinal inflammation, and IL-17, a downstream target of IL-23, exhibited robust upregulation in a similar temporal pattern. IL-23 and IL-17 transcripts were localized to CD11+ antigen presenting cells and CD4+ T cells, respectively, from the colonic lamina propria. Colitis was associated with a 6-fold expansion of CD4+ IL-17-expressing T cells.
Conclusion
The IL-23/IL-17 axis is strongly activated in the colon of HLA-B27 transgenic rats with spondyloarthropathy-like disease. HLA-B27 misfolding and UPR activation in macrophages can result in enhanced induction of the pro-Th17 cytokine, IL-23. These results suggest a possible link between HLA-B27 misfolding and immune dysregulation in this animal model with implications for human disease.
Keywords: Rodent, MHC, inflammation
Introduction
Spondyloarthropathies encompass a group of heterogeneous immune-mediated inflammatory diseases with overlapping clinical manifestations that can include gastrointestinal inflammation, axial and peripheral arthritis, and uveitis. Although these are complex genetic diseases and susceptibility genes are likely to vary, many are strongly linked to the MHC-encoded class I allele, HLA-B27.
Expression of HLA-B27 and human β2m (hβ2m) in rats (HLA-B27 transgenic) results in an inflammatory disease resembling human spondyloarthropathies (1), thus providing a model to investigate the role of this allele (2). CD4+ T cells have been implicated in the pathogenesis of disease in rats, and overexpression of IFN-γ, TNF-α, and IL-12p40 in the gastrointestinal tract has suggested that colitis is predominantly a Th1-mediated process (1). Furthermore, elimination of CD8αβ T cells does not prevent disease (3), suggesting that canonical recognition of HLA-B27-peptide complexes is not necessary. Despite progress in defining cellular requirements for disease, upstream events responsible for pathogenesis, and in particular the relationship between HLA-B27 and pathogenic CD4+ T cells, remain unclear.
The propensity of HLA-B27 to misfold (4, 5) has been associated with disease in transgenic rats (6). Upregulation of HLA-B27 in rat macrophages enhances the accumulation of misfolded heavy chains, resulting in endoplasmic reticulum (ER) stress and activation of the unfolded protein response (UPR) (7, 8). The UPR maintains ER homeostasis, initially by dampening the flux of protein into this organelle, and then by expanding its capacity to fold, secrete, and/or degrade protein (9). However, depending on the magnitude and duration of ER stress, and the type of cell that is affected, the UPR can result in apoptosis. The UPR has been implicated in the pathogenesis of a number of protein misfolding diseases, in part through cell death. We recently found that XBP1, a transcription factor induced by UPR activation, mediates synergistic type I IFN induction in cells exposed to certain pattern recognition receptor (PRR) agonists (‘UPR-PRR synergy’) (10), and there is increasing recognition that the UPR plays a role in immune modulation with potential links to inflammatory disease pathogenesis (11).
Here, we report that IL-23p19 is an additional target gene of UPR-PRR synergy. The active IL-23 cytokine is comprised of two subunits, IL-23p19 and IL-12/23p40, and plays a key role in driving memory CD4+ T cells (‘Th17′) to produce pro-inflammatory cytokines including IL-17 (12). This prompted further examination of colitis in HLA-B27 transgenic rats, where we found a striking upregulation of IL-17 and expansion of IL-17-producing CD4+ T cells. Taken together these results demonstrate activation of the IL-23/IL-17 axis in an HLA-B27-mediated disease model, and suggest a novel paradigm linking protein misfolding, ER stress, and UPR activation with inflammatory disease.
Materials and Methods
Animals
Wild type (WT) C57BL/6 mice (Jackson Laboratory, Bar Harbor, ME) were housed in the barrier facility at Cincinnati Children's Research Foundation (CCRF). HLA-B*2705/hβ2m transgenic (HLA-B27 transgenic) rats on the F344 background (33-3 line) (1), and WT control F344 rats were purchased from Taconic (Germantown, NY) and housed in the conventional animal facility at CCRF. All HLA-B27 transgenic rats were hemizygous for the 33-3 locus, which contains 55 copies of the HLA-B27 transgene and 66 copies of the hβ2m transgene. Both transgenes are genomic clones and contain promoter regions enabling regulation by interferons. All experiments were performed in accord with protocols approved by the CCRF Institutional Animal Care and Use Committee.
Reagents
L929 cells (CCL-1) (ATCC, Manassas, VA) were used to prepare cell culture supernatants (containing M-CSF). Thapsigargin (TPG) and S. enteridis lipopolysaccharide (LPS) (Sigma-Aldrich, St. Louis, MO) were used at final concentrations of 1 μM and 10 ng/ml, respectively. Recombinant rat IFN-γ (R&D Systems, Minneapolis, MN) was used at a concentration of 100 U/ml.
Bone marrow-derived macrophage culture and RNA preparation
Mouse bone marrow (BM) macrophages were derived using CMG 12-14-conditioned media (provided by D. Williams) containing M-CSF (10), and rat BM macrophages were generated using L929-conditioned media (7) as described previously. Mature macrophages were plated at a density of 3-5 × 106 per well in 12 well plates for experiments. For RNA isolation TRIzol Reagent (Invitrogen, Carlsbad, CA) was added directly to cells followed by RNA extraction.
Quantitative real time reverse-transcriptase polymerase chain reaction (RT-PCR) and semi-quantitative RT-PCR
Total RNA was reverse transcribed using oligo (dT) primers and the Superscript one-step RT-PCR system (Invitrogen). Real time PCR was performed using SYBR Green I and an iCycler™ (BioRad, Hercules, CA). For all samples, target gene expression was normalized to β-actin. XBP1 splicing was determined on reverse transcribed mRNA samples by amplifying across the region of XBP1 containing the splice site, separating PCR products on 4% agarose gels (Cambrex Bioscience, Rockland, ME), and measuring the relative amounts of unspliced and spliced cDNA using a PhosphorImager (Amersham Biosciences, Piscataway, NJ) and ImageQuant software. XBP1 splicing is expressed as the percent of the total PCR product that is spliced (7). Oligonucleotide primer sequences are available upon request.
Colon isolation and fractionation
Colon tissue was obtained from cohorts of WT and HLA-B27 transgenic rats at times indicated in the figure legends. Colon was dissected free from connective tissue, washed in sterile PBS and transected longitudinally to remove fecal matter. Samples were then used immediately for isolation of lamina propria (LP) cells or stored overnight in RNAlater® (Ambion) at 4°C prior to lysis in TRIzol Reagent for RNA isolation. To isolate LP cells, colon sections were placed in sterile CMF medium (Ca2+/Mg2+-free Hanks Balanced Salt Solution (HBSS), HEPES-bicarbonate buffer, and 2% FCS) (13). Tissue was then cut into 0.5 cm sections and placed into fresh CMF at 4°C. Samples were washed with multiple rounds of inversion in fresh CMF until supernatants were clear. Tissue was then vortexed for 15 seconds in fresh CMF/FCS/EDTA media (CMF with 10% FCS, 5 mM EDTA, and 100 mg/ml gentamicin). Supernatants were removed and remaining tissue was subjected to multiple additional rounds of vortexing in fresh media until supernatants were clear. The tissue that remained after vortexing was placed into 60 ml of complete RPMI supplemented with 10% FCS, 300 U/ml collagenase (Sigma-Aldrich) and 0.25 mg/ml Type II-O trypsin inhibitor (Sigma-Aldrich). After shaking the tissue samples at 250 rpm at 37°C for 2 hours, the supernatant was filtered through a cell strainer, and cells were collected by centrifugation. Cells were then either lysed in TRIzol Reagent for RNA isolation (LP fraction) or used for purification of LP leukocyte subsets by FACS (FACSVantage SE Cell Sorter, BD Biosciences, San Jose, CA). For purification of LP leukocyte subsets by FACS, cells were stained with the following mAb: APC conjugated OX35 (anti-CD4; BD Biosciences), PE conjugated OX42 (anti-CD11b/c; BD Biosciences) and biotinylated R73 (anti-αβTCR; BioLegend, San Diego, CA). A streptavidin-PE/Cy7 conjugated secondary antibody (BioLegend) was used to label biotinylated R73. Sorted cells were then lysed in TRIzol Reagent for RNA isolation.
Intracellular cytokine staining
Lymphocytes were isolated from LP cell suspensions on discontinuous Percoll gradients of 75% and 40% by centrifugation at 600 g for 20 min at room temperature. The interface between the layers was collected and washed with HBSS supplemented with 5% FCS, and resuspended for counting. Cells were then stimulated with PMA (EMD Bioscience, Gibbstown, NJ) (200 μg/ml) and ionomycin (EMD Bioscience) (10 mM) in the presence of brefeldin A (Sigma-Aldrich) (10 mg/ml) for 6 hours at 37°C. For flow cytometry, the following antibodies were used (BD Biosciences): CD4 PerCP, CD3 APC, IFN-γ PE and IL-17 PE (anti-mouse IL-17 Ab that cross reacts with rat IL-17) and used according the manufacturers protocols. Labeled cells were analyzed using a FACSCalibur and CellQuest Pro software (BD Biosciences).
Statistical analysis
Statistical analysis was performed using a Student's t-test. A value of P <0.05 was considered significant. The confidence interval (CI) was determined using a significance level of 0.05 comparing a mean and standard deviation from three separate experiments.
Results
Synergistic induction of IL-23 by LPS during UPR activation
Previously we found that IFN-β is synergistically upregulated by certain PRR agonists (via TLR4, TLR3, and MDA5) in macrophages undergoing a UPR (10). To identify similarly affected transcripts, we performed a microarray analysis of mouse BM macrophages pretreated with TPG for 1 hour, followed by LPS for an additional 3 hours. TPG inhibits ER Ca2+ ATPase activity, causing Ca2+ depletion and impaired protein folding, resulting in ER stress and robust UPR activation. LPS induced a number of cytokine transcripts that were minimally affected by TPG alone, and TPG induced a classical ER stress response. In addition, LPS treatment of TPG-primed cells resulted in dramatic synergistic upregulation of IL-23p19 as well as IFN-β mRNA. TNF-α was induced 4-5 fold more with the combination of LPS and TPG, while most other cytokines were not differentially affected ((10) and unpublished observations). We confirmed and quantified synergistic induction of IL-23p19 by LPS in cells undergoing a UPR using rat BM macrophages and real time RT-PCR. The combination of ER stress and LPS resulted in a striking increase in IL-23p19 transcripts compared to LPS alone (∼20-fold) or TPG alone (Figure 1A). UPR activation in TPG treated cells is documented by upregulation of BiP transcripts and activation of XBP1 splicing (Figure 1A). LPS alone had no effect on BiP expression or XBP1 splicing, but in TPG-treated cells appears to slightly exacerbate the UPR (Figure 1A). We also found synergistic upregulation of IL-12p35 transcripts in rat macrophages over LPS alone (∼10-fold), and a modest increase (∼2-fold) in IL-12/23p40 mRNA (Figure 1A).
Figure 1.
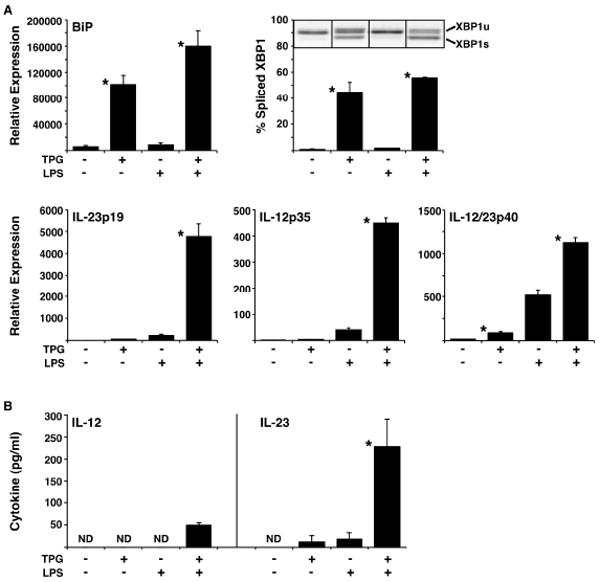
LPS-stimulated IL-23 production by macrophages is augmented by UPR activation. A, The UPR was induced in BM macrophages from WT rats by 1 μM TPG for 4 hours, with or without the addition of 10 ng/ml of LPS for the final 3 hours. Expression of BiP, IL-23p19, IL-12p35, and IL-12/23p40 mRNA was quantified by real time PCR normalized to β-actin. XBP1 splicing was determined by electrophoresis of RT-PCR products. Representative image of unspliced (upper band; XBP1u) and spliced (lower band; XBP1s) XBP1 PCR products are shown. Bars represent the mean of biological triplicates and are representative of at least three separate experiments. B, The UPR was induced in mouse BM macrophages by 1 μM TPG for 2 hours. This was followed by a 7 h washout period, after which cells were treated without or with 10 ng/ml of LPS for 24 hours. Supernatants were collected, and levels of IL-12 and IL-23 measured by ELISA. Bars represent the mean of triplicates and are representative of at least three separate experiments. Error bars represent standard error of the mean and statistical significance was determined using a standard t-test, where p <0.05 is considered significant as indicated by the asterisks (*).
To determine whether stressed macrophages produce more IL-23 and/or IL-12, cells were pulsed with TPG, allowed to recover, and then stimulated without or with LPS for 24 h. These experiments were performed using mouse macrophages since there are no antibodies available to measure rat IL-23, and the anti-mouse IL-23 antibodies we have tested do not cross-react. We found a striking increase in IL-23 in the supernatants of LPS-stimulated cells previously pulsed with TPG (∼10-fold), compared to either LPS or TPG alone (Figure 1B). Stressed macrophages also produce more IL-12, but the amount is much lower, and just above the level of detection (30 pg/ml) (Figure 1B). Similar results were obtained after pulsing cells with Tunicamycin, which causes UPR activation by inhibiting glycosylation of nascent ER proteins (unpublished observations). The similar effect of these diverse agents shows that generalized ER stress increases IL-23 production induced by a TLR4 agonist.
IL-23p19 expression in HLA-B27 transgenic rat macrophages
To determine whether HLA-B27 misfolding can augment LPS-induced IL-23p19 expression, we examined BM macrophages. Cells were first incubated without or with IFN-γ to upregulate MHC class I expression, followed by LPS. HLA-B27 upregulation exacerbates misfolding and activates the UPR in rat macrophages (7, 8, 10) as evidenced by BiP induction and increased XBP1 mRNA splicing (Figure 2A). This is accompanied by a several fold increase in IL-23p19 induction by LPS in HLA-B27 transgenic macrophages compared to WT cells (Figure 2B, right). In contrast, IL-12p35 and IL-12/23p40 induction appears to be minimally affected by HLA-B27 upregulation. Note that IFN-γ priming results in an increase in LPS-induced IL-12p35 and IL-12/23p40 (14), while it has little effect on IL-23p19, at least in WT cells (Figure 2B). Interestingly, LPS exacerbated UPR activation in IFN-γ-treated HLA-B27-expressing macrophages (Figure 2A, right), and actually caused low-level BiP upregulation and XBP1 splicing in the absence of IFN-γ after 3-4 hours of exposure (Figure 2A, left). Induction of IL-6, TNF-α, and IFN-γ mRNA was no different in HLA-B27-expressing macrophages (unpublished observations). Taken together these results suggest that ER stress caused by HLA-B27 misfolding is sufficient to augment the induction of IL-23p19.
Figure 2.
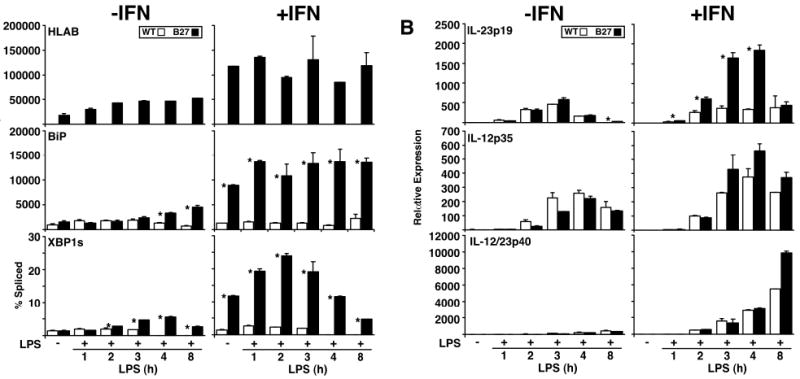
LPS-induced IL-23p19 expression is augmented in the presence of a UPR in HLA-B27 transgenic rat macrophages. BM macrophages from WT and HLA-B27 transgenic (B27) rats were treated without or with 100 U/ml of recombinant rat IFN-γ for 20 h prior to stimulation with LPS for the number of hours shown. RNA expression for the indicated targets was quantified by real time PCR using β-actin for normalization, except for XBP1, which is shown as percent of spliced (XBP1s) XBP1 PCR products. A, Relative expression of HLA-B, BiP and percent spliced XBP1 mRNA. B, Relative expression of IL-23p19, IL-12p35 and IL-12/23p40 mRNA in BM macrophages. Bars indicate the mean of triplicate biological replicates and are representative of at least five separate experiments. Error bars represent standard error of the mean. The asterisk (*) indicates differences that are statistically significant (p < 0.05).
Activation of the IL-23/IL-17 axis in HLA-B27 transgenic rat colon
Based on the observation that HLA-B27 expressing macrophages undergoing a UPR can become polarized to overexpress IL-23, we were interested in determining the relative expression of this and related cytokines in the colon. HLA-B27 transgenic rats (33-3 transgene locus) develop colitis shortly after weaning at 4 weeks of age, so we compared colon tissue from cohorts of transgenic and WT rats at two-week intervals from 2 to 12 weeks of age. Histologic evidence of colitis was first apparent at 6 weeks of age (Figure 3A) with an epithelial growth response with some loss of goblet cells, followed by progressive mononuclear cell infiltration of the mucosal and submucosal layers. This is coincident with increased expression of HLA-B27 and the UPR marker, BiP. IL-23p19 transcripts were also elevated at 6 weeks, paralleling the earliest histologic changes, with persistent elevations seen after 10 weeks (Figure 3B). IL-12/23p40 paralleled IL-23p19, whereas IL-12p35 expression was similar in WT and transgenic rats until 8-10 weeks when expression in HLA-B27 transgenic animals dropped off (Figure 3B). The increase in IL-23p19 was associated with a striking and persistent increase in IL-17 transcripts, as well as a smaller elevation of IFN-γ (Figure 3C). We confirmed increases in TNF-α, IL-1, and IL-6, and the lack of any change in TGF-β, as reported previously by others (1). These data demonstrate activation of the IL-23/IL17 axis in parallel with the development of inflammation in the colon of HLA-B27 transgenic rats in addition to confirming previous evidence of an apparent Th1 response.
Figure 3.
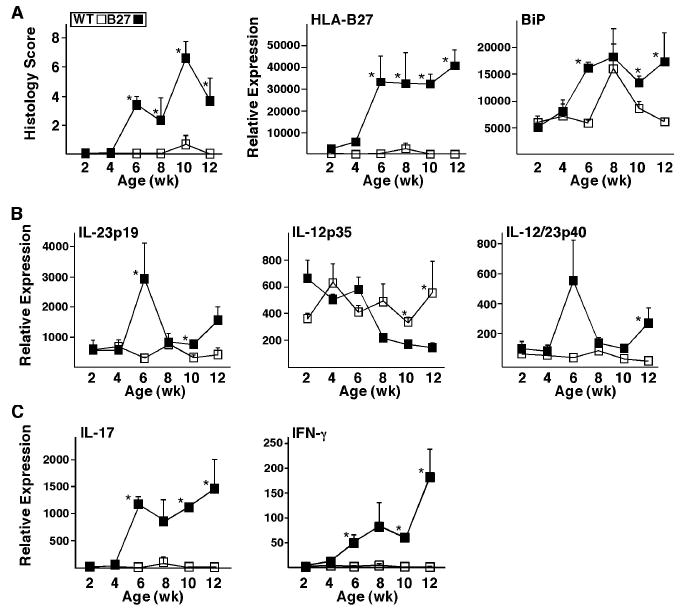
IL-23/IL-17 axis activation correlates with disease onset and progression in rat colon tissue. Histological assessment was performed and RNA was isolated from colon tissue of WT and HLA-B27 transgenic (B27) rats at 2, 4, 6, 8, 10 and 12 weeks of age. Transcript levels were analyzed by real time PCR and normalized to β-actin. A, Histological scores and relative expression of HLA-B27 and BiP in colon tissue. B, Relative expression of IL-23p19, IL-12p35 and IL-12/23p40 mRNA in colon tissue. C, Relative expression of IL-17 and IFN-γ mRNA in colon tissue. Data points represent the mean of measurements from 3 animals performed in duplicate. Error bars represent standard error of the mean. The asterisk (*) indicates differences that are statistically significant (p < 0.05).
Cellular localization of Th17 and Th1 cytokines in HLA-B27 transgenic rat colon
To further explore the cellular sources of cytokines we performed a preliminary experiment, fractionating colon tissue from HLA-B27 transgenic rats with active colitis and WT controls into intestinal epithelial cells, intraepithelial lymphocytes, and lamina propria cells. The majority of IL-17 mRNA was found in lamina propria cells, where transcripts were ∼200-fold greater in cells from HLA-B27 transgenic animals (unpublished observations). We then examined different leukocyte subpopulations from the lamina propria. Briefly, cells stained with antibodies against CD4, TCRαβ, and CD11b/c (CD11), were first separated based on forward and side scatter into populations enriched for antigen presenting cells (APCs containing macrophages and dendritic cells) and lymphocytes. The majority of cells in the lymphocyte population were TCRαβ positive (TCR+), while a smaller proportion in the APC-enriched population was CD11 positive (CD11+) (Figure 4A). Lamina propria cells from the lymphocyte gate were then sorted into three fractions based on CD4 and TCR staining (Figure 4A). There was a 6-fold expansion of CD4+/TCR+ T cells (5.5 vs. 0.9% of total) in the lamina propria of HLA-B27 transgenic compared to WT rats (Figure 4A, right panel). Lamina propria cells from the APC-enriched gate were sorted into CD11+ and CD11- fractions (Figure 4A). There was an approximate 4-fold expansion of CD11+ cells in HLA-B27 transgenic colon compared to WT, but little difference in the CD11- population.
Figure 4.
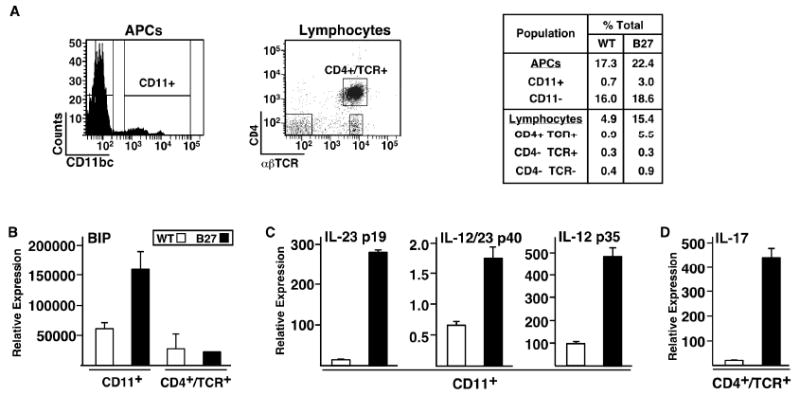
Localization of IL-17 expression to CD4+/TCRαβ+ cells in HLA-B27 transgenic rat colonic lamina propria. Lamina propria cells were isolated from WT and HLA-B27 transgenic rat colon and further subdivided using FACS with antibodies against the indicated antigens. RNA was isolated from the sorted populations and analyzed by real time PCR to quantify transcript levels. A, Populations designated APCs and Lymphocytes were identified based on forward and side scatter characteristics. These populations were subdivided based on cell surface staining for the indicated antigens. B, C, and D, Transcript levels for the indicated genes comparing populations of either CD11+ cells or CD4+/TCR+ cells from WT and HLA-B27 transgenic rats. Data points represent the mean of 2-3 samples. Error bars represent standard error of the mean.
Using these lamina propria-derived leukocyte populations, we looked for evidence of UPR activation by quantifying BiP mRNA expression (Figure 4B). BiP transcripts were elevated approximately 2.5-fold in CD11+ cells from HLA-B27 transgenic rats, whereas BiP expression in CD4+ T cells was unchanged (Figure 4B), consistent with cell type specificity of UPR activation (8).
The majority of IL-12 and IL-23 subunit mRNA transcripts were found in the CD11+ fraction (unpublished observations), where IL-23p19, IL-12p35, and IL-12/23p40 mRNAs were more highly expressed in CD11+ cells from HLA-B27 transgenic rats (Figure 4C). The greatest difference was seen for IL-23p19 (19-fold), whereas IL-12/23p40 and IL-12p35 transcripts were increased approximately 2.5 and 5-fold, respectively, over WT controls (Figure 4C).
The vast majority of IL-17 transcripts were in the CD4+ T cell fraction. Although we did not specifically isolate CD8+ or γδ T cells or neutrophils, which can express IL-17 (15, 16), the fractions expected to contain these cell types (i.e. CD4-/αβTCR+, CD11+) expressed at least 10-fold lower levels of IL-17 mRNA than CD4+ T cells (unpublished observations). Comparing genotypes, IL-17 transcripts were 20-fold greater in CD4+ T cells from HLA-B27 transgenic rats compared to WT controls (Figure 4D), suggesting Th17 activation.
Expansion of Th17 cells in HLA-B27 transgenic rat colon
To determine whether Th17 expansion contributes to the large increase in IL-17 transcripts in HLA-B27 transgenic rats we performed intracellular cytokine staining. Lamina propria lymphocytes were isolated from the colon of HLA-B27 transgenic rats with active colitis and WT controls, and stained for CD3, CD4, IL-17 and IFN-γ. After gating on CD3+ cells, we examined IL-17 and IFN-γ expression in CD4+ cells. This revealed a striking increase in CD4+/IL-17+ (Th17) cells (Figure 5A) and a smaller increase in CD4+/IFN-γ+ Th1 cells (Figure 5B) in HLA-B27 transgenic compared to WT controls. Figure 5C shows the average from 3 independent experiments where Th17 cells are expanded 6.3-fold (CI 3.1), and Th1 cells 3.4-fold (CI 1.3), in HLA-B27 transgenic rats.
Figure 5.
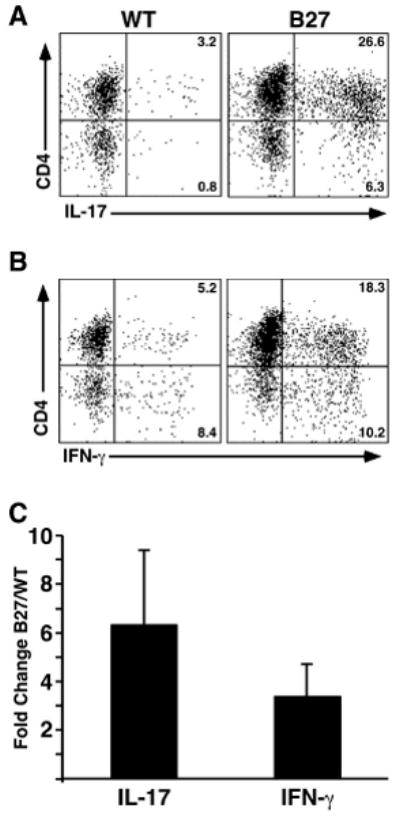
Expansion of CD4+ T cells expressing IL-17 in colonic lamina propria from HLA-B27 transgenic rats. Lamina propria lymphocytes were isolated from WT and HLA-B27 transgenic colon on a percoll gradient. Cells were analyzed for cell surface antigen and intracellular cytokines using FACS. Cells were gated on CD3+ and compared for cytokine production. A, Percentage of CD4+/IL-17+ and CD4-/IL-17+ cells in WT and HLA-B27 transgenic rats. B, Percentage of CD4+/IFN-γ+ and CD4-/IFN-γ+ cells in WT and HLA-B27 transgenic rats. C, Average fold-change in percentage of IL-17-expressing CD4+ T cells (IL-17; CD4+/IL-17+) and IFN-γ-expressing CD4+ T cells (IFN-γ; CD4+/IFN-γ+) populations between WT and HLA-B27 transgenic rats. Data shown are representative of three separate experiments using at least two animals per genotype and error bars represent standard error of the mean. Th17 cells are increased ∼6.3-fold (CI 3.1), and Th1 ∼3.4-fold (CI 1.3).
Discussion
HLA-B27 transgenic rats exhibit striking Th17 expansion and activation in the colon that is temporally related to the development of colitis. At the cellular level, macrophages undergoing a UPR induced either by HLA-B27 misfolding or exposure to pharmacologic inducers of ER stress, are polarized to produce more IL-23 in response to LPS. Taken together, these data suggest that the IL-23/IL-17 axis may play a role in the pathogenesis of spondyloarthritis-like disease in HLA-B27 transgenic rats, and demonstrate a potential link between HLA-B27 misfolding and immune dysregulation.
In recent years, the IL-23/IL-17 axis and CD4+ Th17 cells have gained widespread attention for their role in immune-mediated inflammatory diseases in rodent models (17-20) and several human diseases including inflammatory bowel disease (IBD) (21). IL-17 is also overexpressed in subjects with HLA-B27-associated spondyloarthropathies (22-24), and polymorphisms in the IL23R gene are associated with susceptibility to ankylosing spondylitis (25, 26). These findings provide strong support for involvement of the IL-23/IL-17 axis in human spondyloarthritis as well as in the rat model.
Th17 cells are important regulators of intestinal homeostasis, and are present in healthy lamina propria at much higher frequency than in peripheral tissues (27). They also have the capacity to become pathogenic under the influence of increased local expression of IL-23 (28-30). The increase in IL-23 subunit expression found in the colon of HLA-B27 transgenic rats occurs at least as early as the increase in IL-17 (6 weeks; Figure 3C). We also found increased expression of IFN-γ in the colon (Figure 3), and evidence of Th1 expansion and activation (Figure 5). These early changes either precede or are coincident with, the development of diarrhea, which typically begins between 6 and 9 weeks of age. IFN-γ overexpression has been demonstrated previously along with increases in IL-1α, IL-1β, TNF-α, IL-6, MIP-2 and iNOS (1, 31-33). These data have been interpreted in support of colitis being a Th1-mediated process. However, this type of cytokine profile (i.e. IL-17 in addition to IFN-γ, TNF-α, IL-6, and IL-1β) is also seen in mouse models of inflammatory bowel disease shown to be driven by IL-23 (16). Our studies demonstrate that colitis in HLA-B27 transgenic rats is associated with prominent Th17 expansion and activation, and are consistent with the idea that it could be driven by excess local production of IL-23 in the lamina propria. In support of this, the striking inflammatory phenotype of IL-23p19 transgenic mice includes gastrointestinal inflammation (17).
The extent to which IL-17 mediates intestinal inflammation in animal models remains unclear. In IL-10-deficient mice IL-17 blockade is only effective in suppressing intestinal inflammation if IL-6 is also neutralized (20), and in other T cell-dependent inflammatory bowel disease models inhibition of Th1 responses attenuates disease (16). In uveitis and encephalomyelitis both Th1 and Th17 cells mediate pathology (34, 35). In HLA-B27 transgenic rats, IL-10 administration reduced IFN-γ, IL-1β and TNF-α expression in the colon without affecting the severity of colitis (32), consistent with the idea that other cytokines such as IL-17 may be important. In addition, Th17 cells can secrete IL-22 and IL-21, which may contribute to their pathogenicity (12, 36). Thus, while IL-17 is pro-inflammatory and clearly responsible for pathology in some animal models of inflammatory bowel disease (16), we do not at this time know its relative importance in the context of other pro-inflammatory cytokines in HLA-B27 transgenic rats.
Interestingly, IFN-γ is an antagonist of Th17 development in mice (37, 38), and genetic ablation of IFNG results in Th17 expansion and exacerbates several types of Th17-mediated immunopathology (39). However, in humans with psoriasis, IFN-γ has been shown to induce Th17 T cells, in part through enhancing IL-23 expression, and promote their trafficking and function (40). In HLA-B27 transgenic but not WT rats, our data suggest that IFN-γ promotes IL-23 expression via HLA-B27-induced ER stress and UPR activation. In addition, preliminary studies suggest that a subset of Th17 CD4+ T cells in the colon of HLA-B27 transgenic rats also expresses IFN-γ (unpublished observations), consistent with evidence for IL-17/IFN-γ double positive T cells in humans (41). It will be important to further explore the balance and interplay between Th17 and Th1 T cells and cytokines in mediating inflammation in HLA-B27 transgenic rats.
The findings presented here are consistent with a model where HLA-B27 misfolding might promote a chronic inflammatory process such as colitis. Colonization of the gastrointestinal tract with commensal organisms results in a low level immune response including IFN-γ production (42), which is normally controlled (43). However, in HLA-B27 transgenic rats, IFN-γ could have a paradoxical effect by increasing HLA-B27 expression and generating ER stress, thus superimposing the UPR on macrophage activation. Macrophages may then become sensitized to pathogen-associated molecular patterns such as LPS that signal through pattern recognition receptors including the TLRs (Figure 6). Increased expression of IL-23 in response to microbial products would activate CD4+ Th17 cells to produce IL-17 leading to tissue specific inflammation and damage. Production of IFN-γ by Th1 and Th17 T cells could perpetuate HLA-B27 misfolding and UPR activation (Figure 6), particularly in the presence of other cytokines such as TNF-α that synergize with IFNs to increase class I expression. IFN-γ also primes antigen-presenting cells to produce more IL-12 (14). Consistent with this possibility, we find upregulation of IL-12 subunits (p35 and p40) in antigen-presenting cells isolated from the lamina propria (Figure 4). While there appears to be a shift in the Th17/Th1 balance associated with UPR activation and increased IL-23 expression, there is still considerable Th1 activation, which may play an important role in the inflammatory disease.
Figure 6.
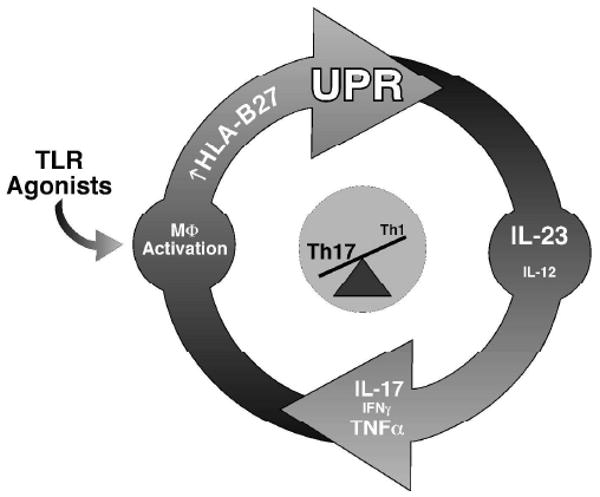
Proposed mechanism linking UPR activation as a consequence of HLA-B27 misfolding to activation of the IL-23/Il-17 axis. HLA-B27 upregulation may occur initially as a result of antigen presenting cell stimulation with TLR agonists from commensal microorganisms and/or low level IFN-γ production from innate immune cells such as NK cells. UPR activation is superimposed on macrophage activation because of HLA-B27 misfolding, resulting in greater IL-23 production in response to TLR agonists. This, in turn, stimulates Th17 cells to produce IL-17. Th1 activation, and/or double positive IL-17/IFN-γ producing cells, may help to sustain HLA-B27 expression thus perpetuating this cycle.
There are several other hypotheses to explain the role of HLA-B27 in disease (reviewed in (44)). Evidence that CD8αβ T cells are not required for spondyloarthritis-like disease in rats argues against antigen presentation as an initiating event (3). Dendritic cell dysfunction that could reduce tolerance to microbial flora has been reported (45), and cell surface dimers of HLA-B27 heavy chains have been hypothesized to modulate the immune response and lead to inflammation (46). Our studies do not rule out the involvement of alternative mechanisms, and it is conceivable that more than one mechanism is responsible.
The rats used for these studies did not develop arthritis, consistent with previous observations that this component of the inflammatory phenotype is rare in younger animals, particularly on the F344 background ((1) and unpublished observations). In future studies it will be important to examine the IL-23/IL-17 axis in arthritis, including the spondylitis phenotype that occurs when additional hβ2m is expressed in HLA-B27 transgenic rats (47). Overexpression of additional hβ2m was reported to curb HLA-B27 misfolding and caused a small reduction in BiP mRNA levels in splenocytes (47). However, these studies did not examine macrophages or the response to HLA-B27 upregulation, which is important for maximal UPR activation (7, 8).
Our results suggest a novel mechanism linking HLA-B27 misfolding and the generation of ER stress to augmented TLR4-mediated induction of IL-23p19 via activation of the UPR. This may sustain CD4+ Th17 cells and drive the production of IL-17 and IFN-γ from double positive T cells. The IL-23/IL-17 axis has been implicated in the pathogenesis of several immune-mediated inflammatory diseases in humans, including psoriasis and Crohn's disease, as well as animal models. Our results strongly support a role for this axis in the pathogenesis of colitis in HLA-B27 transgenic rats. Considering genetic studies implicating IL23R polymorphisms in susceptibility to ankylosing spondylitis, this work suggests a mechanism that might link HLA-B27 misfolding to the IL-23/IL-17 axis in humans, and should prompt further inquiry into the role of these cytokines in the pathogenesis of human spondyloarthritis.
Acknowledgments
We thank David P. Witte for analysis of the histopathology; Gerlinde Layh-Schmitt for critical evaluation of the manuscript, and Shuzhen Bai for technical assistance.
Abbreviations used in this paper
- β2m
β2-microglobulin
- BM
bone marrow
- ER
endoplasmic reticulum
- hβ2m
human β2m
- UPR
unfolded protein response
- TPG
thapsigargin
- XBP1
X box binding protein-1
Footnotes
This work was supported by National Institutes of Health grants R01 AR46177 and AR48372. M.J.T. was supported by a Functional Genomics Fellowship granted through the University of Cincinnati College of Medicine. D.P.S. was supported by NIH Training Grant T32 AR07594, and J.A.S. was supported by an Arthritis Foundation Postdoctoral Fellowship.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Taurog JD, Maika SD, Satumtira N, Dorris ML, McLean IL, Yanagisawa H, et al. Inflammatory disease in HLA-B27 transgenic rats. Immunol Rev. 1999;169:209–223. doi: 10.1111/j.1600-065x.1999.tb01317.x. [DOI] [PubMed] [Google Scholar]
- 2.Smith JA, Marker-Hermann E, Colbert RA. Pathogenesis of ankylosing spondylitis: current concepts. Best Pract Res Clin Rheumatol. 2006;20(3):571–91. doi: 10.1016/j.berh.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 3.May E, Dorris ML, Satumtira N, Iqbal I, Rehman MI, Lightfoot E, et al. CD8ab T cells are not essential to the pathogenesis of arthritis or colitis in HLA-B27 transgenic rats. J Immunol. 2003;170:1099–1105. doi: 10.4049/jimmunol.170.2.1099. [DOI] [PubMed] [Google Scholar]
- 4.Dangoria NS, DeLay ML, Kingsbury DJ, Mear JP, Uchanska-Ziegler B, Ziegler A, et al. HLA-B27 misfolding is associated with aberrant intermolecular disulfide bond formation (dimerization) in the endoplasmic reticulum. J Biol Chem. 2002;277:23459–23468. doi: 10.1074/jbc.M110336200. [DOI] [PubMed] [Google Scholar]
- 5.Antoniou AN, Ford S, Taurog JD, Butcher GW, Powis SJ. Formation of HLA-B27 homodimers and their relationship to assembly kinetics. J Biol Chem. 2004;279(10):8895–902. doi: 10.1074/jbc.M311757200. [DOI] [PubMed] [Google Scholar]
- 6.Tran TM, Satumtira N, Dorris ML, May E, Wang A, Furuta E, et al. HLA-B27 in transgenic rats forms disulfide-linked heavy chain oligomers and multimers that bind to the chaperone BiP. J Immunol. 2004;172(8):5110–9. doi: 10.4049/jimmunol.172.8.5110. [DOI] [PubMed] [Google Scholar]
- 7.Turner MJ, Sowders DP, DeLay ML, Mohapatra R, Bai S, Smith JA, et al. HLA-B27 misfolding in transgenic rats is associated with activation of the unfolded protein response. J Immunol. 2005;175(4):2438–48. doi: 10.4049/jimmunol.175.4.2438. [DOI] [PubMed] [Google Scholar]
- 8.Turner MJ, Delay ML, Bai S, Klenk E, Colbert RA. HLA-B27 up-regulation causes accumulation of misfolded heavy chains and correlates with the magnitude of the unfolded protein response in transgenic rats: Implications for the pathogenesis of spondylarthritis-like disease. Arthritis Rheum. 2007;56(1):215–23. doi: 10.1002/art.22295. [DOI] [PubMed] [Google Scholar]
- 9.Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–89. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 10.Smith JA, Turner MJ, DeLay ML, Klenk EI, Sowders DP, Colbert RA. Endoplasmic reticulum stress and the unfolded protein response are linked to synergistic IFN-beta induction via X-box binding protein 1. Eur J Immunol. 2008;38(5):1194–203. doi: 10.1002/eji.200737882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Todd DJ, Lee AH, Glimcher LH. The endoplasmic reticulum stress response in immunity and autoimmunity. Nat Rev Immunol. 2008;8(9):663–674. doi: 10.1038/nri2359. [DOI] [PubMed] [Google Scholar]
- 12.Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol. 2007;25:821–52. doi: 10.1146/annurev.immunol.25.022106.141557. [DOI] [PubMed] [Google Scholar]
- 13.Lefrancois L, Lycke N. Isolation of mouse small intestinal intraepithelial lymphocytes, Peyer's patch, and lamina propria cells. In: Coligan JE, Bierer BE, Margulies DH, Shevach EM, Strober W, editors. Curr Protoc Immunol. New York: John Wiley & Sons; 2001. p. 3.3.19. [DOI] [PubMed] [Google Scholar]
- 14.Hayes MP, Wang J, Norcross MA. Regulation of interleukin-12 expression in human monocytes: selective priming by interferon-gamma of lipopolysaccharide-inducible p35 and p40 genes. Blood. 1995;86(2):646–50. [PubMed] [Google Scholar]
- 15.Happel KI, Dubin PJ, Zheng M, Ghilardi N, Lockhart C, Quinton LJ, et al. Divergent roles of IL-23 and IL-12 in host defense against Klebsiella pneumoniae. J Exp Med. 2005;202(6):761–9. doi: 10.1084/jem.20050193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hue S, Ahern P, Buonocore S, Kullberg MC, Cua DJ, McKenzie BS, et al. Interleukin-23 drives innate and T cell-mediated intestinal inflammation. J Exp Med. 2006;203(11):2473–83. doi: 10.1084/jem.20061099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wiekowski MT, Leach MW, Evans EW, Sullivan L, Chen SC, Vassileva G, et al. Ubiquitous transgenic expression of the IL-23 subunit p19 induces multiorgan inflammation, runting, infertility, and premature death. J Immunol. 2001;166(12):7563–70. doi: 10.4049/jimmunol.166.12.7563. [DOI] [PubMed] [Google Scholar]
- 18.Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421(6924):744–8. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 19.Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201(2):233–40. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yen D, Cheung J, Scheerens H, Poulet F, McClanahan T, McKenzie B, et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest. 2006;116(5):1310–6. doi: 10.1172/JCI21404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tesmer LA, Lundy SK, Sarkar S, Fox DA. Th17 cells in human disease. Immunol Rev. 2008;223:87–113. doi: 10.1111/j.1600-065X.2008.00628.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wendling D, Cedoz JP, Racadot E, Dumoulin G. Serum IL-17, BMP-7, and bone turnover markers in patients with ankylosing spondylitis. Joint Bone Spine. 2007;74(3):304–5. doi: 10.1016/j.jbspin.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 23.Singh R, Aggarwal A, Misra R. Th1/Th17 cytokine profiles in patients with reactive arthritis/undifferentiated spondyloarthropathy. J Rheumatol. 2007;34(11):2285–90. [PubMed] [Google Scholar]
- 24.Agarwal S, Misra R, Aggarwal A. Interleukin 17 Levels Are Increased in Juvenile Idiopathic Arthritis Synovial Fluid and Induce Synovial Fibroblasts to Produce Proinflammatory Cytokines and Matrix Metalloproteinases. J Rheumatol. 2008;35:515–519. [PubMed] [Google Scholar]
- 25.Burton PR, Clayton DG, Cardon LR, Craddock N, Deloukas P, Duncanson A, et al. Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat Genet. 2007;39(11):1329–1337. doi: 10.1038/ng.2007.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rueda B, Orozco G, Raya E, Fernandez-Sueiro JL, Mulero J, Blanco FJ, et al. The IL23R Arg381Gln non-synonymous polymorphism confers susceptibility to ankylosing spondylitis. Ann Rheum Dis. 2008;67:1451–4. doi: 10.1136/ard.2007.080283. [DOI] [PubMed] [Google Scholar]
- 27.Maloy KJ. The Interleukin-23 / Interleukin-17 axis in intestinal inflammation. J Intern Med. 2008;263(6):584–90. doi: 10.1111/j.1365-2796.2008.01950.x. [DOI] [PubMed] [Google Scholar]
- 28.Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126(6):1121–33. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 29.Mangan PR, Harrington LE, O'Quinn DB, Helms WS, Bullard DC, Elson CO, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441(7090):231–4. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 30.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24(2):179–89. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 31.Rath HC, Herfarth HH, Ikeda JS, Grenther WB, Hamm TE, Balish E, et al. Normal luminal bacteria, especially bacteroides species, mediate chronic colitis, gastritis, and arthritis in HLA-B27/human b2 microglobulin transgenic rats. J Clin Invest. 1996;98:945–953. doi: 10.1172/JCI118878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bertrand V, Quere S, Guimbaud R, Sogni P, Chauvelot-Moachon L, Tulliez M, et al. Effects of murine recombinant interleukin-10 on the inflammatory disease of rats transgenic for HLA-B27 and human beta 2-microglobulin. Eur Cytokine Netw. 1998;9(2):161–70. [PubMed] [Google Scholar]
- 33.Qian BF, Tonkonogy SL, Hoentjen F, Dieleman LA, Sartor RB. Dysregulated luminal bacterial antigen-specific T-cell responses and antigen-presenting cell function in HLA-B27 transgenic rats with chronic colitis. Immunology. 2005;116(1):112–21. doi: 10.1111/j.1365-2567.2005.02206.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luger D, Silver PB, Tang J, Cua D, Chen Z, Iwakura Y, et al. Either a Th17 or a Th1 effector response can drive autoimmunity: conditions of disease induction affect dominant effector category. J Exp Med. 2008;205(4):799–810. doi: 10.1084/jem.20071258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kroenke MA, Carlson TJ, Andjelkovic AV, Segal BM. IL-12- and IL-23-modulated T cells induce distinct types of EAE based on histology, CNS chemokine profile, and response to cytokine inhibition. J Exp Med. 2008;205(7):1535–41. doi: 10.1084/jem.20080159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zheng Y, Danilenko DM, Valdez P, Kasman I, Eastham-Anderson J, Wu J, et al. Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature. 2007;445(7128):648–51. doi: 10.1038/nature05505. [DOI] [PubMed] [Google Scholar]
- 37.Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6(11):1133–41. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6(11):1123–32. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 39.Chu CQ, Swart D, Alcorn D, Tocker J, Elkon KB. Interferon-gamma regulates susceptibility to collagen-induced arthritis through suppression of interleukin-17. Arthritis Rheum. 2007;56(4):1145–51. doi: 10.1002/art.22453. [DOI] [PubMed] [Google Scholar]
- 40.Kryczek I, Bruce AT, Gudjonsson JE, Johnston A, Aphale A, Vatan L, et al. Induction of IL-17+ T cell trafficking and development by IFN-gamma: mechanism and pathological relevance in psoriasis. J Immunol. 2008;181(7):4733–41. doi: 10.4049/jimmunol.181.7.4733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Annunziato F, Cosmi L, Santarlasci V, Maggi L, Liotta F, Mazzinghi B, et al. Phenotypic and functional features of human Th17 cells. J Exp Med. 2007;204(8):1849–61. doi: 10.1084/jem.20070663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rhee SJ, Walker WA, Cherayil BJ. Developmentally regulated intestinal expression of IFN-gamma and its target genes and the age-specific response to enteric Salmonella infection. J Immunol. 2005;175(2):1127–36. doi: 10.4049/jimmunol.175.2.1127. [DOI] [PubMed] [Google Scholar]
- 43.Mowat AM, Donachie AM, Parker LA, Robson NC, Beacock-Sharp H, McIntyre LJ, et al. The role of dendritic cells in regulating mucosal immunity and tolerance. Novartis Found Symp. 2003;252:291–302. doi: 10.1002/0470871628.ch22. discussion 302-5. [DOI] [PubMed] [Google Scholar]
- 44.Colbert RA. The immunobiology of HLA-B27: variations on a theme. Curr Mol Med. 2004;4(1):21–30. doi: 10.2174/1566524043479293. [DOI] [PubMed] [Google Scholar]
- 45.Hacquard-Bouder C, Chimenti MS, Giquel B, Donnadieu E, Fert I, Schmitt A, et al. Alteration of antigen-independent immunologic synapse formation between dendritic cells from HLA-B27-transgenic rats and CD4+ T cells: selective impairment of costimulatory molecule engagement by mature HLA-B27. Arthritis Rheum. 2007;56(5):1478–89. doi: 10.1002/art.22572. [DOI] [PubMed] [Google Scholar]
- 46.Kollnberger S, Bird LA, Sunm MY, Retiere C, Braud VM, McMichael A, et al. Cell surface expression and immune receptor recogntion of HLA-B27 homodimers. Arth Rheum. 2002;46:2972–2982. doi: 10.1002/art.10605. [DOI] [PubMed] [Google Scholar]
- 47.Tran TM, Dorris ML, Satumtira N, Richardson JA, Hammer RE, Shang J, et al. Additional human beta(2)-microglobulin curbs HLA-B27 misfolding and promotes arthritis and spondylitis without colitis in male HLA-B27-transgenic rats. Arthritis Rheum. 2006;54(4):1317–1327. doi: 10.1002/art.21740. [DOI] [PubMed] [Google Scholar]