

Abstract
Nuclear factor-kappaB (NF-κB) activation occurs following ischemic preconditioning (IPC) in brain. However, the upstream signaling messengers and down-stream targets of NF-κB required for induction of IPC remain undefined. In a previous study, we demonstrated that epsilon protein kinase c (εPKC) was a key mediator of IPC in brain. Activation of εPKC induced cyclooygenase-2 (COX-2) expression and conferred ischemic tolerance in the neuronal and hippocampal slice models. Here, we hypothesized that IPC-mediated COX-2 expression was mediated by NF-κB. We tested this hypothesis in mixed cortical neuron/astrocyte cell cultures. To simulate IPC or ischemia, cell cultures were exposed to 1 or 4 h of oxygen–glucose deprivation, respectively. Our results demonstrated translocation of p65 and p50 subunits of NF-κB into nucleus following IPC or εPKC activation. NF-κB inhibition with pyrrolidine dithiocarbamate (10 μM) abolished IPC or εPKC activator-mediated neuroprotection indicating that NF-κB activation was involved in ischemic tolerance. In parallel studies, inhibition of either εPKC or the extracellular signal-regulated kinase (ERK 1/2) pathway reduced IPC-induced NF-κB activation. Finally, inhibition of NF-κB blocked IPC-induced COX-2 expression. In conclusion, we demonstrated that IPC-signaling cascade comprises εPKC activation→ERK1/2 activation→NF-κB translocation to nucleus→COX-2 expression resulting in neuroprotection in mixed neuronal culture.
Keywords: Cerebral ischemia, Ischemic tolerance, Epsilon protein kinase C, Extracellular signal-regulated kinase (ERK1/2), Neuroprotection, Mixed cortical neuron/astrocyte cell cultures
Introduction
Ischemic preconditioning (IPC) is an intrinsic neuroprotective mechanism invoked by sublethal ischemic insults prior to a subsequent lethal ischemic insult. The resulting state of ischemic tolerance has gained attention as a potentially robust prophylactic approach with the potential for clinical use in humans having a high risk for pathological cerebrovascular events and for patients anticipating neurosurgical procedures. More importantly, therapies based on endogenous neuroprotective mechanisms may as well have less intense and fewer side effects. Therefore, the main emphasis in the field of IPC is to better understand signaling mechanisms of initiation, induction, and maintenance.
The induction of IPC requires activation of several transcription factors and expression of genes that confer cell resistance against subsequent stress conditions. Numerous signal transduction pathways have been proposed to play a role in induction of brain tolerance by IPC [1-6]. We have identified that epsilon protein kinase C (εPKC) is one of the pieces of the IPC-mediated signaling pathways [1, 7-10]. We proposed that εPKC orchestrates cell viability by initiating signal transduction pathways that are favorable for cell survival after IPC in brain [11, 12]. Recently, we demonstrated that activated εPKC subsequently increased cyclooxygenase 2 (COX-2) expression in mixed cortical neuronal cultures and organotypic hippocampal slice culture models of cerebral ischemia [8]. Cyclooxygenase-2 is the rate-limiting enzyme for prostaglandin (PG) synthase, catalyzing the conversion of arachidonic acid to prostaglandin H2 [13].
COX-2 expression is regulated by transcription factor nuclear factor kappa B (NF-κB) in various tissues [14, 15]. Nuclear factor-κB is a family of closely related protein dimers that bind to a common sequence motif in DNA called the κB site. The most usual form of NF-κB is a heterodimer of p65 and p50, which normally exists in the cytoplasm in a dormant form, bound to one of the members of inhibitory proteins called IκBα, IκBβ, and IκBε [16, 17]. Several stimuli promote dissociation of IκBα through phosphorylation, ubiquitinylation, and its ultimate degradation in the proteasomes. This process unmasks the nuclear localization sequence of NF-κB, facilitating its nuclear entry, binding to κB regulatory elements and activation of transcription of target genes [16, 18]. A dual role for NF-κB has been suggested in neurodegenerative diseases [19-21]. A recent report proposed that NF-κB was neuroprotective after focal cerebral ischemia in mice, in part, by activation of the Akt signaling mechanism [22]. Activation of the NF-κB was also identified as a key event in brain tolerance after IPC [23]. However, the upstream and down-stream signaling cascade of NF-κB activation after ischemic preconditioning neuroprotection remains undefined. Thus, in the current study we hypothesized that the NF-κB activation is dependent on εPKC and extracellular signal-regulated kinase (ERK 1/2) signaling pathways and required for COX-2 expression after IPC.
Materials and Methods
All animal procedures were carried out in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health and were approved by the Animal Care and Use Committee of the University of Miami. According to these guidelines, efforts were made to minimize the number of animals and their discomfort.
Preparation of Mixed Cortical Neuron/Astrocyte Cell Cultures
For mixed cortical neuron/astrocyte cell cultures, astrocytes were prepared from neonatal rat as described previously [8]. Sprague–Dawley neonatal (1–2 days old) rats were anesthetized by intraperitoneal injection of ketamine (1.0 mg/pup). Animals were decapitated and the brains quickly removed. The cerebral cortices of the neonates were isolated and dissociated astrocytes were plated with minimum essential medium (MEM) (Invitrogen, Carlsbad, CA, USA) containing 10% fetal bovine serum (FBS), 10% equine serum, 2 mM glutamine, and 1 μg/ml epidermal growth factor (Sigma, St. Louis, MO, USA) in three cortical hemisphere were dissociated and distributed equally to the 24-well plate. After 10 days, 19-day pregnant Sprague–Dawley rats were anesthetized by halothane and embryos were quickly removed and decapitated to isolate cortical neurons. The cerebral cortices of the embryos were isolated and dissociated cortical neurons were placed on a confluent monolayer of astrocytes cultured for 10 days to generate co-culture with MEM containing 5% FBS, 5% equine serum, and 2 mM glutamine. The mixed cortical neuron/astrocyte cells were used after 10–11 days in vitro.
Scheme of Oxygen/Glucose Deprivation Injury
To mimic ischemic injury, we exposed cells to oxygen–glucose deprivation (OGD) for 4 h. To simulate ischemic preconditioning, cell cultures were exposed to OGD for a short period of 1 h, 48 h prior to ischemia. For OGD, cell cultures were washed twice with glucose-free HBSS (pH 7.4) of the following constitution: 1.26 mM CaCl2·2H2O, 5.37 mM KCl, 0.44 mM KH2PO4, 0.49 mM MgCl2, 0.41 mM MgSO4, 136.9 mM NaCl2, 4.17 mM NaHCO3, 0.34 mM Na2HPO4, and 10 mM HEPES (Sigma, St. Louis, MO, USA). The cell cultures were then transferred into an anaerobic chamber (PROOX model 110, BioSpherix, Ltd. Redfield, NY, USA) which was placed in a water-jacketed incubator gassed with 95% N2/5% CO2 at 37°C. The chamber was sealed and incubated for either 1 h (preconditioning) or 4 h (ischemic insult). Following OGD, the cell cultures were transferred to their respective normal culture media and placed back into the incubator.
Assessment of Cell Death of Mixed Cortical Neuron/Astrocyte Cell Cultures
To determine assessment of cell death, cytotoxicity was measured by lactate dehydrogenase (LDH) released for 48 h into culture medium as described by Koh and Choi [24]. Maximal neuronal LDH release was measured in the neuronal cultures exposed to NMDA (500 μM; 48 h; maximal neuronal death). LDH release was measured by absorbance at 340 nm using a microplate reader (Molecular Devices, Sunnyvale, CA, USA). Values were expressed relative to LDH measurement from maximal neuronal LDH.
Preparation of Cytosolic and Nuclear Extracts
To visualize nuclear expression of p65 and p50 subunit of NF-κB, nuclear extracts of neurons were prepared by using similar procedures described previously [25]. Cells were harvested, washed twice with ice-cold phosphate-buffered saline (PBS), and lysed in lysis buffer containing 1% Nonidet P-40, 20 mM Tris (pH8.0) 137 mM NaCl, 0.5 mM EDTA, 10% glycerol, 10 mM sodium pyrophosphate, 10 mM sodium fluoride, 1μg/ml aprotinin, 10μg/ml leupeptin, 1 mM vanadate, and 1 mM phenylmethylsulfonyl fluoride for 15 min on ice [8]. Cells were vortexed vigorously for 15 s, and then centrifuged at 14,000 rpm for 10 min. The supernatant was used for COX-2 expression. Nuclear pellet was resuspended in buffer B (10 mM HEPES, pH8, 2 mM MgCl2, 10 mM KCl, 0.5 mM DTT, 1μg/ml aprotinin, 10μg/ml leupeptin, 1 mM vanadate, and 1 mM phenylmethylsulfonyl fluoride) and centrifuged at 14,000 rpm for 10 min. The resulting pellet was incubated with the extraction buffer (20 mM HEPES, pH8, 400 mM NaCl, 0.5 mM EDTA, 2 mM MgCl2, 1 mM DTT, 1μg/ml aprotinin, 10μg/ml leupeptin, 1 mM vanadate, and 1 mM phenylmethylsulfonyl fluoride) for 30 min on ice. Following the centrifugation at 15,000 rpm for 15 min, the supernatant was used as nuclear extract. Protein samples (15–20 μg) of nuclear or (50–80 μg) of cytosolic extracts were used for Western blot analysis.
Western Blot Analysis
Equal amounts of proteins were subjected to 8% SDS-PAGE and the separated proteins were electrophoretically transferred to a nitrocellulose membrane (Bio Rad Laboratories, Hercules, CA, USA). The blot was blocked with 5% non-fat dried milk, incubated overnight at 4°C with the antibody of COX-2 (1:1,000, Cayman Chemicals, Ann Arbor, MI, USA), β-actin (1:5,000, Sigma St. Louis, MO, USA), p65, p50, and a nuclear marker, Lamin B (1:500, Santa Cruz Biotechnology, Santa Cruz, CA, USA), Lamin B was present in nuclear fraction and not detected in cytosolic extract. Then, incubation was followed by horseradish peroxidase-conjugated-specific secondary antibody for 1 h at room temperature. The immunoreactive bands were revealed by ECL western blotting detection reagents (Amersham, Buckinghamshire, England). Western blot images were digitized at eight-bit precision by means of a CCD camera (eight to 12 bit, Xillix Technologies Corporation, Vancouver, Canada) equipped with a 55 mm Micro-Nikkor lens (Nikon, Japan). The camera was interfaced to an advanced image-analysis system (MCID Model M2, Imaging Research, Inc., St. Catherines, Ontario, Canada). The digitized immunoblots were subjected to densitometric analysis using MCID software.
Immunocytochemistry
Mixed cortical neuron/astrocyte cell cultures were grown onto a cover glass in a 24-well plate. At 15 and 30 min of reperfusion after 1 h of IPC, cells were fixed with 4% paraformaldehyde in phosphate buffer (10 mM, pH7.4). After washing with triton X-100 in PBS (0.4% ; PBS-T), cells were blocked in 10% goat serum, 10% horse serum, and 10% bovine serum albumin (BSA) in PBS-T and incubated with primary antibody against p50 (1:1,000, Cell Signaling Technology, Danvers, MA, USA), neuron nuclear (NeuN, a neuronal-specific marker) and glial fibrillary acidic protein (GFAP, an astrocytes marker; 1:100 and 1:400, Chemican, Temeculal, CA, USA) in 5% goat serum, 5% horse serum, and 10% BSA in PBS-T. Incubations were performed for 24 h at 4°C. After several washes in PBS-T, cells were incubated with biotinylated anti-rabbit and rhodamine-labeled anti-mouse secondary antibodies (1:500, Vector, Burlingame, CA, USA) for 1 h at room temperature. After several washes in PBS-T, cells were incubated with the fluorescent secondary antibodies, fluorescein-labeled anti-rabbit (Santa Cruz Biotechnology) and rhodamine-labeled anti-mouse (Santa Cruz Biotechnology) for 30 min at room temperature. Cells were mounted on slides using Prolong Antifade Kit (Molecular Probes, OR, USA) and were visualized by laser scanning microscopy (LSM; Carl Zeiss Inc., Germany). The images of cells were analyzed using an LSM 5 image browser.
Statistical Analysis
Data were expressed as the mean±SEM. An analysis of variance followed by Dunnett’s multiple comparison tests was used for statistical comparison. Results from the densitometric analysis were analyzed by a two-tailed Student’s t test. In all cases, a p value less than 0.05 was considered statistically significant.
Experimental Design
The mixed cortical neuron/astrocyte cell cultures were divided into five major groups, as follows :
Control: after 10 days in vitro, cell death was measured by the LDH assay and cells were lysed with lysis buffer for western blot analysis
Ischemia (4 h of oxygen–glucose deprivation): cell cultures were exposed to sham ischemic preconditioning, and 48 h later, 4 h of OGD was induced. At 48 h following OGD, cell death was measured by the LDH assay.
Ischemic preconditioning: cell cultures were exposed to ischemic preconditioning (1 h of OGD), and 48 h of reperfusion later, 4 h of OGD was induced followed by the LDH assay. Cells were lysed with lysis buffer at different times for Western blot analysis.
Ischemic preconditioning + drug treatment: cell cultures were treated by PD98059 (a MAPK-K inhibitor,10 μM, Sigma, St. Louis, MO, USA) or εPKC specific inhibitory Tat-conjugated peptide (εV1-2, 100 nM, KAI Pharmaceuticals Inc., 270 Littlefield Ave, South San Francisco, CA 94080, USA; [26]) during 1 h of IPC and 48 h of reperfusion. Pyrrolidine dithiocarbamate (PDTC, NF-κB inhibitor, 10 μM, Sigma, St. Louis, MO, USA) was administrated to cell cultures for 48 h of reperfusion after 1 h of IPC just prior to OGD. At 48 h following OGD, cell death was measured by the LDH assay. Cells were lysed with lysis buffer at the indicated times for Western blot analysis.
Pharmacological preconditioning (PPC): cell cultures were treated by εPKC specific activating Tat-conjugated peptide (ψεRACK at 100 nM, KAI Pharmaceuticals Inc., 270 Littlefield Ave, South San Francisco, CA 94080, USA) for 1 and 48 h of reperfusion later, 4 h of OGD was induced followed by the LDH assay. Cells were lysed with lysis buffer at the indicated times for Western blot analysis.
Results
Ischemic Preconditioning Induced Nuclear Translocation of NF-κB
First, we tested the hypothesis that NF-κB is activated by IPC in mixed cortical neurons. To test this hypothesis, cell cultures were preconditioned (1 h OGD) and collected at different time points for Western blot analysis. Our data indicated that preconditioning mediated translocation of p65 and p50 subunit of NF-κB to the nucleus at 15 min and lasted for 2 h (Fig. 1a, b, and c). Immunohistochemistry showed neurons as well as astrocytes with positive immuno-reactivity for p50 subunit of NF-κB at 30 min of reperfusion after IPC.
Fig. 1.
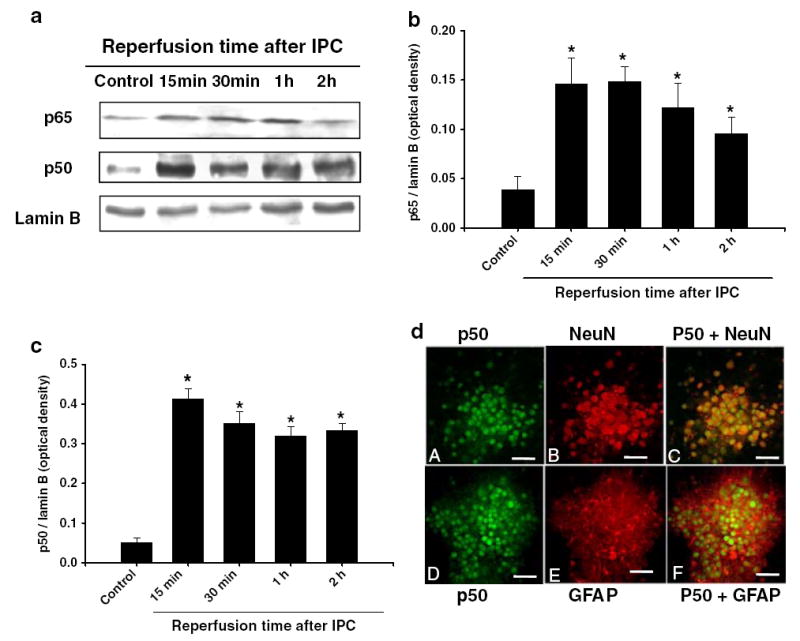
Ischemic preconditioning induced p65 and p50 translocation to the nucleus: a cells were lysed immediately after 1 h of IPC and at 15 min, 30 min, 1 h, and 2 h of reperfusion after 1 h of PC. Western blotting for p65 was carried out with nuclear protein extracts and the membrane was re-probed with the p50 antibody or nuclear lamin B antibody. b and c Histogram depicted densitometric analysis of Western blots of p65 or p50 in nuclear protein extracts compared with lamin B, respectively, *p<0.05 compared with control (n=5). d Confocal microscopic images of mixed cortical neuron/astrocyte cell cultures depicting co-localization of immunoreactivities for neuronal-specific antibody, NeuN (red) or astrocyte-specific antibody GFAP (red) and p50. At 30 min of reperfusion after IPC, NeuN (arrows; a, b, c) and GFAP (d, e, f) positive cells expressed p50. Bar:20μM
To test the hypothesis that NF-κB activation is required for IPC-induced neuroprotection, an NF-κB inhibitor (PDTC, 10 μM), was applied during IPC. Forty-eight hours later, cells were exposed to lethal OGD and neuronal cell death was measured (Fig. 2). Results showed that IPC reduced the neuronal death by 30% as compared to OGD (50±1.2% vs. 80±1.8% of maximal cell death; IPC + OGD vs. OGD, respectively, p<0.05, n=20) and PDTC treatment during IPC significantly abolished IPC-induced neuroprotection (50±1.2% vs. 65.7±2.5% of maximal cell death: IPC + OGD vs. IPC + PDTC + OGD, p<0.05, n=20). These results suggested that NF-κB activation was required for the IPC-induced neuroprotection against OGD injury.
Fig. 2.
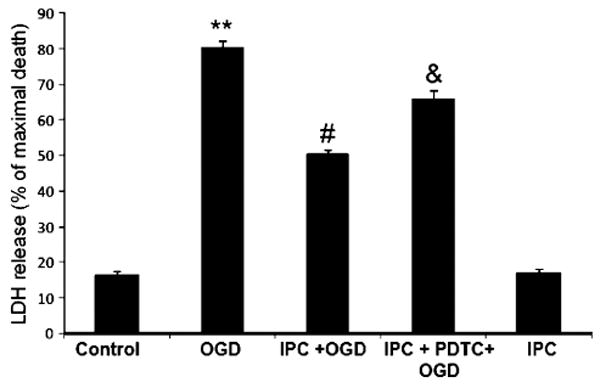
NF-kB inhibition reduced ischemic preconditioning-induced neuroprotection: histogram representing cell death measured by LDH release at 48 h of reperfusion after OGD (4 h). Note that neuroprotection was lost when cells were exposed to NF-kB inhibitor after IPC. **p<0.01 compared with control, #p<0.05 compared with OGD, and &p<0.05 compared with PC (n=20)
εPKC Mediated NF-κB Activation Following Ischemic Preconditioning
εPKC plays a key role in ischemic preconditioning [8-10]. Here we examined the role of εPKC as an upstream regulator of NF-κB in IPC signaling. To characterize this signaling pathway, we inhibited εPKC activation (εPKC inhibitor; εV1-2, 100 nM) during IPC. Inhibition of εPKC significantly reduced translocation of NF-κB p65 subunit to the nucleus as compared with IPC (2.0±0.3 vs. 0.8±0.4 optical density: IPC vs. IPC + εPKC inhibitor, p<0.05 compared with IPC, n=5, Fig. 3). Furthermore, we tested the hypothesis that activation of εPKC required NF-κB activation. The preconditioning mediated by εPKC-specific activating peptide (ψεRACK, 100 nM, pharmacological preconditioning) induced translocation of p65 and p50 subunit of NF-κB to the nucleus at 15 min and lasted for 1 h (Fig. 4a). To characterize whether NF-κB activation is necessary for εPKC preconditioning, cells were exposed to NF-κB inhibitor (PDTC; 10 μM) during εPKC preconditioning. Results of LDH assay showed that εPKC preconditioning significantly reduced neuronal death from OGD injury (47±1.3% vs. 80±1.8% of maximal cell death; PPC + OGD vs. OGD, p<0.05, n=20, Fig. 4b) and PDTC abolished εPKC-induced neuroprotection significantly (47±1.2% vs. 65.7±2.5% of maximal cell death: PPC + OGD vs. PPC + PDTC + OGD, p<0.05, n=20, Fig. 4b). These results suggested that εPKC activation could be an upstream signaling pathway for NF-κB activation leading to neuroprotection.
Fig. 3.
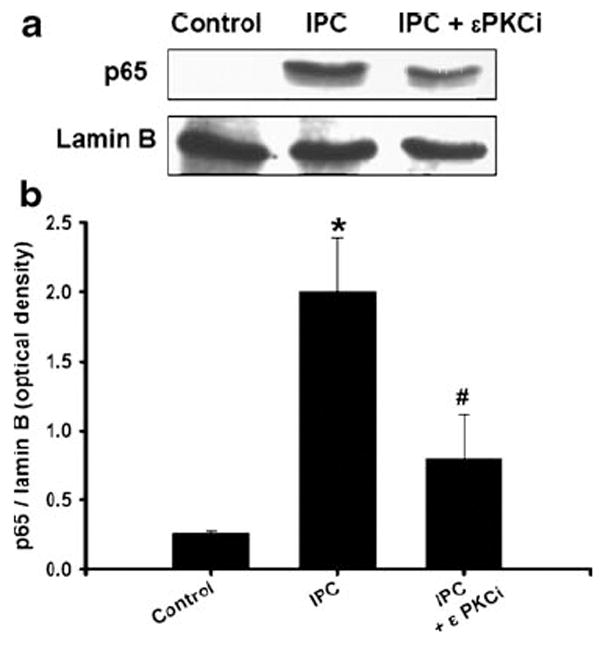
Epsilon PKC activation is required for p65 translocation after IPC: a inhibition of epsilon PKC-reduced PC-induced translocation of p65 to nucleus. Cells were treated with epsilon PKC inhibitor (εPKCi was εV1-2, 100 nM) or vehicle (tat carrier peptide) during IPC and reperfusion. Cells were isolated at 1 h of reperfusion after IPC (1 h of OGD). Western blotting for p65 was carried out and the membrane was re-probed with lamin B antibody. b Histogram depicted densitometric analysis of Western blots of p65 in nuclear protein extracts compared with lamin B, respectively. *p<0.05 compared with control, #p<0.05 compared with IPC
Fig. 4.
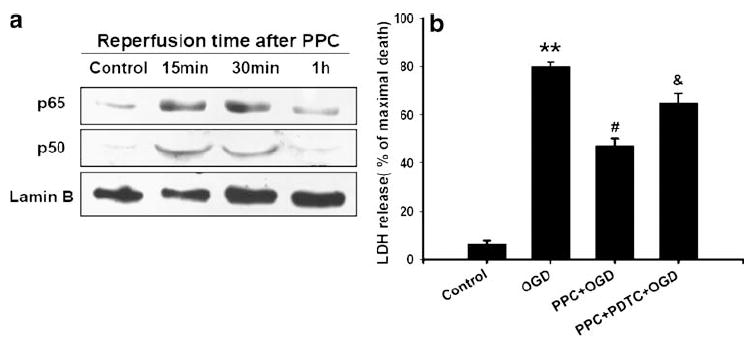
a Epsilon PKC activation mimicked IPC and induced nuclear translocation of p65 and p50: cells were lysed at 15 min, 30 min, and 1 h after εPKC activator (ψεRACK, 100 nM) treatment. Western blotting for p50 was carried out and the membrane was re-probed for p65 and lamin B antibodies. b Histogram depicted densitometric analysis of Western blots of p65 or p50 in nuclear protein extracts compared with lamin B, respectively, *p<0.05 compared with control (n=5)
ERK1/2 Activation Mediated NF-κB Activation Following Ischemic Preconditioning
Previously, we demonstrated that IPC-signaling-induced εPKC activation leads to phosphorylation of ERK1/2 [8]. Here, we examined whether blockade of ERK1/2 phosphorylation reduced IPC-induced translocation of NF-κB p65 subunit to the nucleus. We inhibited ERK1/2 activation (mitogen-activated protein kinase–kinase (MAPK-K) inhibitor, PD98059, 10 μM) during IPC and reperfusion in cell cultures. Inhibition of ERK1/2 completely inhibited IPC-induced p65 translocation to nucleus at 30 min of reperfusion as compared to IPC group (p<0.05, n=5, Fig. 5).
Fig. 5.
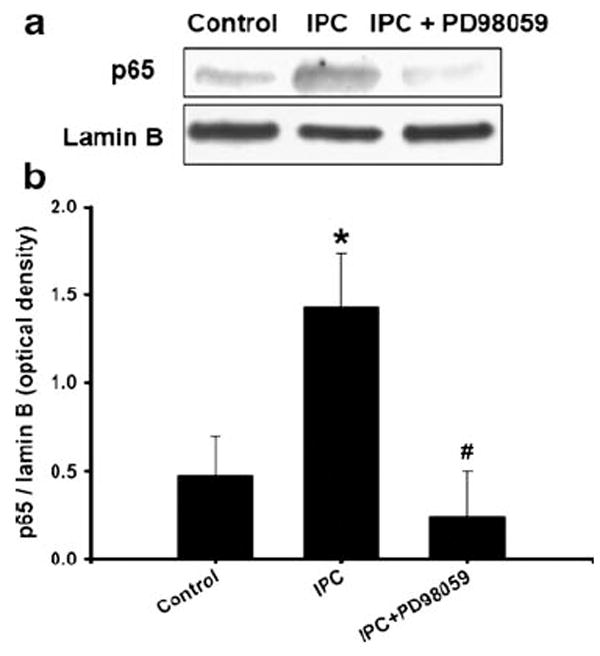
Inhibition of ERK1/2 activation blocked IPC-induced p65 translocation to nucleus: a cells were treated with ERK1/2 inhibitor PD98059 (MAPK-K inhibitor, 10 μM) or vehicle during IPC and reperfusion. Cells were isolated at 1 h of reperfusion after IPC (1 h of OGD). Western blotting for p65 was carried out and the membrane was re-probed with lamin B antibody. b Histogram depicted densitometric analysis of Western blots of p65 in nuclear protein extracts compared with lamin B, respectively, *p<0.05 compared with control (n=5), #p<0.01 compared with OGD, and &p<0.05 compared with PPC + OGD
NF-κB Activation Regulated Cyclooxygenase-2 (COX-2) Expression Following Ischemic Preconditioning
To determine whether NF-κB activation is involved in IPC-induced COX-2 expression in neuronal cell cultures, cells were treated with NF-κB inhibitor during IPC. Our results demonstrated that NF-κB inhibition significantly reduced IPC-induced COX-2 expression (0.6±0.2 vs. 0.15±0.02 optical density: IPC vs. IPC + PDTC, p<0.05 compared with IPC, p<0.05 compared with IPC, n=5, Fig. 6). Taken together, COX-2 up-regulation requires NF-κB activation via εPKC and ERK1/2-dependent signaling pathways following ischemic preconditioning in neuronal cultures.
Fig. 6.
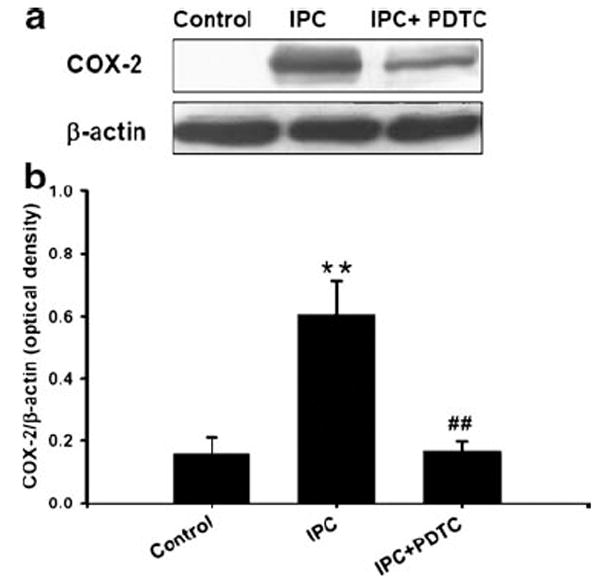
Inhibition of NF-kB reduced preconditioning-induced cyclooygenase-2 (COX-2) expression: a cells were treated with NF-kB inhibitor (PDTC, 1 μM) or vehicle during 24 h of reperfusion after 1 h of IPC. Cells were lysed at 24 h of reperfusion and analyzed by Western blotting with COX-2 antibody. The membrane was re-probed with β-actin antibody. Histogram depicted densitometric analysis of Western blots of COX-2 compared with β-actin. **p<0.01 compared with control, #p<0.05 compared with PC (n=3). b Histogram depicted densitometric analysis of Western blots of COX-2 and β-actin, *p<0.05 compared with control (n=5)
Discussion
In the present study, we demonstrated that NF-κB activation was required for induction of IPC, that upstream pathways were εPKC and ERK1/2, and finally that NF-κB regulated cyclooxygenase-2 expression after IPC in mixed cortical neuronal/astrocyte cell cultures. Taken together, these results suggested an IPC-signaling cascade comprised of εPKC activation → MAPK-K → ERK1/2 → NF-κB → COX-2 resulting in neuroprotection against subsequent lethal OGD stress in mixed cortical neuronal/astrocyte cell cultures.
Numerous studies demonstrated an increased NF-κB activity under pathological conditions in which neurons were dying [27, 28]. In particular, cerebral ischemia resulted in an early activation of NF-κB in the hippocampal CA1 neurons in rat model [28, 29]. A delayed activation (several days after ischemia) of NF-κB was also observed in reactive glia and identified as the promoter of ischemic neuronal death. In contrast, studies using neuronal cultures provide evidence that the activation of NF-κB is neuroprotective against amyloid-peptide toxicity [30], excitotoxicity, or oxidative stress [31, 32]. In support of the neuroprotective role of NF-κB, Blondeau et al. [23], suggested that the NF-κB is at the crossing of neuronal cell death and survival pathways and is a crucial component of the signal transduction cascade of cerebral IPC. Overall, these studies point to a high degree of complexity in the manner in which NF-κB activation occurs following preconditioning and lethal ischemia. For example, a differential regulation of NF-κB occurs via tumor necrosis factor alpha after ischemic tolerance [33]. Furthermore, the time for NF-κB activation and the cell types in which this activation occurs are important determinants for post-ischemic cell viability. In the present study, we observed that NF-κB activation lasted for 1 h after IPC and occurred in neurons as well as in astrocytes. Thus, we conjecture that NF-κB activation for preconditioning neuroprotection must occur within an hour and last for a short time. In contrast, in lethal ischemia, activation of NF-κB might last for hours in neurons and for days in reactive glia, resulting in cell death.
In brain, activated NF-κB acted on genes for cytokine, cyclooxygenase-2, nitric oxide synthase, and apoptotic proteins (see reviews [34, 35]). Cerebrovascular inflammation plays a central role in the pathogenesis of cerebral ischemia [36, 37]. The important process in cerebrovascular inflammation is the induction of proinflammatory mediators such as cyclooxygenase-2 (COX-2) [38, 39]. COX-2 expression is required to mediate ischemic tolerance in the brain [8, 40]. Our previous study demonstrated that ischemic preconditioning-induced COX-2 expression by εPKC → MAPK-K → ERK1/2 signaling pathways and conferred neuroprotection against OGD injury. In the current study, we showed that the inhibition of NF-κB translocation to the nucleus after ischemic preconditioning reduced COX-2 expression, suggesting that NF-κB regulates COX-2 expression in preconditioned neurons and astrocytes. In parallel, our recent study demonstrated that the phosphorylation of signal transducers and activators of transcription-3 (STAT3) is crucial for COX-2 expression after preconditioning [41]. At this juncture it is imperative to emphasize the fact that IPC activates a complex cascade of signaling pathways. Interestingly, neuroprotective signaling pathways activated by IPC appeared to be activated simultaneously and converge to a point leading to protection of the cell. This is also apparent from our data as NF-κB inhibition (Fig. 2) partially reversed the protective effects of ischemic tolerance, while at the same time the degrees of nuclear translocation of p65 after εPKC or ERK inhibition were different, indicating that other mechanisms might be activated in parallel to the one we observed. Here, the point of convergence might be COX-2 in neuronal/astrocyte cultures.
Conclusions
Ischemic preconditioning increased the translocation of the p50 and p65 subunits of NF-κB into the nucleus, suggesting NF-κB activation after IPC in mixed cortical neuron/astrocyte cell cultures. Activation of NF-κB is regulated via epsilon PKC and ERK1/2 signaling pathways. Inhibition of NF-κB abolished COX-2 activation and ischemic tolerance. Thus, NF-κB activation is necessary for COX-2 expression leading to neuroprotection via ischemic preconditioning and its activation is mediated by εPKC and ERK1/2 activation.
Acknowledgments
This study was supported by PHS grants NS34773, NS054147, NS045676 (MAPP), AHA-National center # 0730089 N, the James and Esther King Biomedical Research Program, Florida Department of Health 07KN-10 (APR), and AHA Florida & Puerto Rico Affiliate grant 0525331B (MAPP/EJ).
References
- 1.Dave KR, DeFazio RA, Raval AP, Torraco A, Saul I, Barrientos A, et al. Ischemic preconditioning targets the respiration of synaptic mitochondria via protein kinase C epsilon. J Neurosci. 2008;28(16):4172–4182. doi: 10.1523/JNEUROSCI.5471-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee TH, Yang JT, Ko YS, Kato H, Itoyama Y, Kogure K. Influence of ischemic preconditioning on levels of nerve growth factor, brain-derived neurotrophic factor and their high-affinity receptors in hippocampus following forebrain ischemia. Brain Res. 2008;1187:1–11. doi: 10.1016/j.brainres.2007.09.078. [DOI] [PubMed] [Google Scholar]
- 3.Miyawaki T, Mashiko T, Ofengeim D, Flannery RJ, Noh KM, Fujisawa S, et al. Ischemic preconditioning blocks BAD translocation, Bcl-xL cleavage, and large channel activity in mitochondria of postischemic hippocampal neurons. Proc Natl Acad Sci U S A. 2008;105(12):4892–4897. doi: 10.1073/pnas.0800628105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Orio M, Kunz A, Kawano T, Anrather J, Zhou P, Iadecola C. Lipopolysaccharide induces early tolerance to excitotoxicity via nitric oxide and cGMP. Stroke. 2007;38(10):2812–2817. doi: 10.1161/STROKEAHA.107.486837. [DOI] [PubMed] [Google Scholar]
- 5.Stevens SL, Ciesielski TM, Marsh BJ, Yang T, Homen DS, Boule JL, et al. Toll-like receptor 9: a new target of ischemic preconditioning in the brain. J Cereb Blood Flow Metab. 2008;28(5):1040–1047. doi: 10.1038/sj.jcbfm.9600606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yin W, Signore AP, Iwai M, Cao G, Gao Y, Johnnides MJ, et al. Preconditioning suppresses inflammation in neonatal hypoxic ischemia via Akt activation. Stroke. 2007;38(3):1017–1024. doi: 10.1161/01.STR.0000258102.18836.ca. [DOI] [PubMed] [Google Scholar]
- 7.Raval AP, Dave KR, DeFazio RA, Perez-Pinzon MA. epsilonPKC phosphorylates the mitochondrial K(+) (ATP) channel during induction of ischemic preconditioning in the rat hippocampus. Brain Res. 2007;1184:345–353. doi: 10.1016/j.brainres.2007.09.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim E, Raval AP, Defazio RA, Perez-Pinzon MA. Ischemic preconditioning via epsilon protein kinase C activation requires cyclooxygenase-2 activation in vitro. Neuroscience. 2007;145(3):931–941. doi: 10.1016/j.neuroscience.2006.12.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lange-Asschenfeldt C, Raval AP, Dave KR, Mochly-Rosen D, Sick TJ, Perez-Pinzon MA. Epsilon protein kinase C mediated ischemic tolerance requires activation of the extracellular regulated kinase pathway in the organotypic hippocampal slice. J Cereb Blood Flow Metab. 2004;24(6):636–645. doi: 10.1097/01.WCB.0000121235.42748.BF. [DOI] [PubMed] [Google Scholar]
- 10.Raval AP, Dave KR, Mochly-Rosen D, Sick TJ, Perez-Pinzon MA. Epsilon PKC is required for the induction of tolerance by ischemic and NMDA-mediated preconditioning in the organotypic hippocampal slice. J Neurosci. 2003;23(2):384–391. doi: 10.1523/JNEUROSCI.23-02-00384.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bright R, Sun GH, Yenari MA, Steinberg GK, Mochly-Rosen D. varepsilonPKC confers acute tolerance to cerebral ischemic reperfusion injury. Neurosci Lett. 2008;441(1):120–124. doi: 10.1016/j.neulet.2008.05.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perez-Pinzon MA, Dave KR, Raval AP. Role of reactive oxygen species and protein kinase C in ischemic tolerance in the brain. Antioxid Redox Signal. 2005;7(9–10):1150–1157. doi: 10.1089/ars.2005.7.1150. [DOI] [PubMed] [Google Scholar]
- 13.Smith WL, Garavito RM, DeWitt DL. Prostaglandin endoperoxide H synthases (cyclooxygenases)-1 and -2. J Biol Chem. 1996;271(52):33157–33160. doi: 10.1074/jbc.271.52.33157. [DOI] [PubMed] [Google Scholar]
- 14.Shinmura K, Xuan YT, Tang XL, Kodani E, Han H, Zhu Y, et al. Inducible nitric oxide synthase modulates cyclooxygenase-2 activity in the heart of conscious rabbits during the late phase of ischemic preconditioning. Circ Res. 2002;90(5):602–608. doi: 10.1161/01.res.0000012202.52809.40. [DOI] [PubMed] [Google Scholar]
- 15.Zhao TC, Taher MM, Valerie KC, Kukreja RC. p38 Triggers late preconditioning elicited by anisomycin in heart: involvement of NF-kappaB and iNOS. Circ Res. 2001;89(10):915–922. doi: 10.1161/hh2201.099452. [DOI] [PubMed] [Google Scholar]
- 16.Baeuerle PA, Baltimore D. I kappa B: a specific inhibitor of the NF-kappa B transcription factor. Science. 1988;242(4878):540–546. doi: 10.1126/science.3140380. [DOI] [PubMed] [Google Scholar]
- 17.Baeuerle PA, Baltimore D. A 65-kappaD subunit of active NF-kappaB is required for inhibition of NF-kappaB by I kappaB. Genes Dev. 1989;3(11):1689–1698. doi: 10.1101/gad.3.11.1689. [DOI] [PubMed] [Google Scholar]
- 18.Chen Z, Hagler J, Palombella VJ, Melandri F, Scherer D, Ballard D, et al. Signal-induced site-specific phosphorylation targets I kappa B alpha to the ubiquitin-proteasome pathway. Genes Dev. 1995;9(13):1586–1597. doi: 10.1101/gad.9.13.1586. [DOI] [PubMed] [Google Scholar]
- 19.Camandola S, Mattson MP. NF-kappa B as a therapeutic target in neurodegenerative diseases. Expert Opin Ther Targets. 2007;11(2):123–132. doi: 10.1517/14728222.11.2.123. [DOI] [PubMed] [Google Scholar]
- 20.Song YS, Lee YS, Narasimhan P, Chan PH. Reduced oxidative stress promotes NF-kappaB-mediated neuroprotective gene expression after transient focal cerebral ischemia: lympho-cytotrophic cytokines and antiapoptotic factors. J Cereb Blood Flow Metab. 2007;27(4):764–775. doi: 10.1038/sj.jcbfm.9600379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chan PH. Future targets and cascades for neuroprotective strategies. Stroke. 2004;35(11 Suppl 1):2748–2750. doi: 10.1161/01.STR.0000143325.25610.ac. [DOI] [PubMed] [Google Scholar]
- 22.Song YS, Narasimhan P, Kim GS, Jung JE, Park EH, Chan PH. The role of Akt signaling in oxidative stress mediates NF-kappaB activation in mild transient focal cerebral ischemia. J Cereb Blood Flow Metab. 2008;28(12):1917–1926. doi: 10.1038/jcbfm.2008.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blondeau N, Widmann C, Lazdunski M, Heurteaux C. Activation of the nuclear factor-kappaB is a key event in brain tolerance. J Neurosci. 2001;21(13):4668–4677. doi: 10.1523/JNEUROSCI.21-13-04668.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koh JY, Choi DW. Quantitative determination of glutamate mediated cortical neuronal injury in cell culture by lactate dehydrogenase efflux assay. J Neurosci Methods. 1987;20(1):83–90. doi: 10.1016/0165-0270(87)90041-0. [DOI] [PubMed] [Google Scholar]
- 25.Wang HL, He CY, Chou AH, Yeh TH, Chen YL, Li AH. Polyglutamine-expanded ataxin-7 decreases nuclear translocation of NF-kappaB p65 and impairs NF-kappaB activity by inhibiting proteasome activity of cerebellar neurons. Cell Signal. 2007;19(3):573–581. doi: 10.1016/j.cellsig.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 26.Chen L, Wright LR, Chen CH, Oliver SF, Wender PA, Mochly-Rosen D. Molecular transporters for peptides: delivery of a cardioprotective epsilonPKC agonist peptide into cells and intact ischemic heart using a transport system, R(7) Chem Biol. 2001;8(12):1123–1129. doi: 10.1016/s1074-5521(01)00076-x. [DOI] [PubMed] [Google Scholar]
- 27.Grilli M, Goffi F, Memo M, Spano P. Interleukin-1beta and glutamate activate the NF-kappaB/Rel binding site from the regulatory region of the amyloid precursor protein gene in primary neuronal cultures. J Biol Chem. 1996;271(25):15002–15007. doi: 10.1074/jbc.271.25.15002. [DOI] [PubMed] [Google Scholar]
- 28.Clemens JA, Stephenson DT, Smalstig EB, Dixon EP, Little SP. Global ischemia activates nuclear factor-kappa B in forebrain neurons of rats. Stroke. 1997;28(5):1073–1080. doi: 10.1161/01.str.28.5.1073. discussion 1080-1. [DOI] [PubMed] [Google Scholar]
- 29.Ueno T, Sawa Y, Kitagawa-Sakakida S, Nishimura M, Morishita R, Kaneda Y, et al. Nuclear factor-kappa B decoy attenuates neuronal damage after global brain ischemia: a future strategy for brain protection during circulatory arrest. J Thorac Cardiovasc Surg. 2001;122(4):720–727. doi: 10.1067/mtc.2001.115917. [DOI] [PubMed] [Google Scholar]
- 30.Barger SW, Horster D, Furukawa K, Goodman Y, Krieglstein J, Mattson MP. Tumor necrosis factors alpha and beta protect neurons against amyloid beta-peptide toxicity: evidence for involvement of a kappa B-binding factor and attenuation of peroxide and Ca2+ accumulation. Proc Natl Acad Sci U S A. 1995;92(20):9328–9332. doi: 10.1073/pnas.92.20.9328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goodman Y, Mattson MP. Ceramide protects hippocampal neurons against excitotoxic and oxidative insults, and amyloid beta-peptide toxicity. J Neurochem. 1996;66(2):869–872. doi: 10.1046/j.1471-4159.1996.66020869.x. [DOI] [PubMed] [Google Scholar]
- 32.Mattson MP. Cellular actions of beta-amyloid precursor protein and its soluble and fibrillogenic derivatives. Physiol Rev. 1997;77(4):1081–1132. doi: 10.1152/physrev.1997.77.4.1081. [DOI] [PubMed] [Google Scholar]
- 33.Ginis I, Jaiswal R, Klimanis D, Liu J, Greenspon J, Hallenbeck JM. TNF-alpha-induced tolerance to ischemic injury involves differential control of NF-kappaB transactivation: the role of NF-kappaB association with p300 adaptor. J Cereb Blood Flow Metab. 2002;22(2):142–152. doi: 10.1097/00004647-200202000-00002. [DOI] [PubMed] [Google Scholar]
- 34.Carroll JE, Hess DC, Howard EF, Hill WD. Is nuclear factor-kappaB a good treatment target in brain ischemia/reperfusion injury? NeuroReport. 2000;11(9):R1–R4. [PubMed] [Google Scholar]
- 35.Mattson MP, Camandola S. NF-kappaB in neuronal plasticity and neurodegenerative disorders. J Clin Invest. 2001;107(3):247–254. doi: 10.1172/JCI11916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.del Zoppo G, Ginis I, Hallenbeck JM, Iadecola C, Wang X, Feuerstein GZ. Inflammation and stroke: putative role for cytokines, adhesion molecules and iNOS in brain response to ischemia. Brain Pathol. 2000;10(1):95–112. doi: 10.1111/j.1750-3639.2000.tb00247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Vries HE, Kuiper J, de Boer AG, Van Berkel TJ, Breimer DD. The blood-brain barrier in neuroinflammatory diseases. Pharmacol Rev. 1997;49(2):143–155. [PubMed] [Google Scholar]
- 38.McKay LI, Cidlowski JA. Molecular control of immune/inflammatory responses: interactions between nuclear factor-kappa B and steroid receptor-signaling pathways. Endocr Rev. 1999;20(4):435–459. doi: 10.1210/edrv.20.4.0375. [DOI] [PubMed] [Google Scholar]
- 39.Nogawa S, Forster C, Zhang F, Nagayama M, Ross ME, Iadecola C. Interaction between inducible nitric oxide synthase and cyclooxygenase-2 after cerebral ischemia. Proc Natl Acad Sci U S A. 1998;95(18):10966–10971. doi: 10.1073/pnas.95.18.10966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gendron TF, Brunette E, Tauskela JS, Morley P. The dual role of prostaglandin E(2) in excitotoxicity and preconditioning-induced neuroprotection. Eur J Pharmacol. 2005;517(1–2):17–27. doi: 10.1016/j.ejphar.2005.05.031. [DOI] [PubMed] [Google Scholar]
- 41.Kim EJ, Raval AP, Perez-Pinzon MA. Preconditioning mediated by sublethal oxygen-glucose deprivation-induced cyclooxygenase-2 expression via the signal transducers and activators of transcription 3 phosphorylation. J Cereb Blood Flow Metab. 2008;28(7):1329–1340. doi: 10.1038/jcbfm.2008.26. [DOI] [PMC free article] [PubMed] [Google Scholar]