

Abstract
Extracellular β-NAD is known to elevate intracellular levels of calcium ions, inositol 1,4,5-trisphate and cAMP. Recently, β-NAD was identified as an agonist for P2Y1 and P2Y11 purinergic receptors. Since β-NAD can be released extracellularly from endothelial cells (EC), we have proposed its involvement in the regulation of EC permeability. Here we show, for the first time, that endothelial integrity can be enhanced in EC endogenously expressing β-NAD-activated purinergic receptors upon β-NAD stimulation. Our data demonstrate that extracellular β-NAD increases the transendothelial electrical resistance (TER) of human pulmonary artery EC (HPAEC) monolayers in a concentration-dependent manner indicating endothelial barrier enhancement. Importantly, β-NAD significantly attenuated thrombin-induced EC permeability as well as the barrier-compromising effects of Gram-negative and Gram-positive bacterial toxins representing the barrier-protective function of β-NAD. Immunofluorescence microscopy reveals more pronounced staining of cell-cell junctional protein VE-cadherin at the cellular periphery signifying increased tightness of the cell-cell contacts after β-NAD stimulation. Interestingly, inhibitory analysis (pharmacological antagonists and receptor sequence specific siRNAs) indicates the participation of both P2Y1 and P2Y11 receptors in β-NAD-induced TER increase. B-NAD-treatment attenuates the lipopolysaccharide (LPS)-induced phosphorylation of myosin light chain indicating its involvement in barrier protection. Our studies also show the involvement of cAMP-dependent protein kinase A and EPAC1 pathways as well as small GTPase Rac1 in β-NAD-induced EC barrier enhancement. With these results, we conclude that β-NAD regulates the pulmonary EC barrier integrity via small GTPase Rac1- and MLCP- dependent signaling pathways.
Keywords: P2Y1 and P2Y11 receptors, Gs protein, Gq protein, cAMP, HPAEC, P2Y antagonists, EPAC1, Rac1
Introduction
The vascular endothelium is a semi-selective diffusion barrier between the plasma and interstitial fluid and is critical to vessel wall homeostasis. Inflammatory factor-induced barrier dysfunction of the endothelium is associated with cytoskeletal remodeling, disruption of cell-cell contacts and the formation of paracellular gaps (Dudek and Garcia, 2001). Reorganization of the endothelial cytoskeleton leads to alteration in cell shape and provides a structural basis for increase of vascular permeability, which has been implicated in the pathogenesis of various diseases (Dudek and Garcia, 2001; Garcia and Schaphorst, 1995; Lum and Malik, 1996). Disruption of the endothelial barrier occurs during inflammatory diseases such as acute lung injury (ALI) and acute respiratory distress syndrome (ARDS), with an overall mortality rate of 30–40% (Ware and Matthay, 2000), and results in the movement of fluid and macromolecules into the interstitium and pulmonary air spaces causing pulmonary edema (Pugin et al., 1999). Some naturally occurring substances such as sphingosine-1-phosphate (Dudek et al., 2007), ATP (Kolosova et al., 2005), angiopoietin-1 (Baffert et al., 2006) and cAMP (Fukuhara et al., 2005) are known to enhance the EC barrier function. These findings raise the possibility that sphingosine-1-phosphate, or ATP, or angiopoietin-1 may link vasculogenesis with the formation of stable EC barrier and these results indicate the potential role of these mediators in reducing endothelial permeability.
The EC are connected to each other by a complex set of junctional proteins that comprise tight junctions (TJs), adherent junctions (AJs) and gap junctions (GJs). Endothelial AJs contain vascular endothelial (VE)-cadherin as the major structural protein responsible for homophilic binding and adhesion of adjacent cells. VE-cadherin is essential for proper assembly of AJs and development of normal endothelial barrier function (Mehta and Malik, 2006). Ectopic expression of a cadherin mutant lacking VE-cadherin extracellular domain in dermal EC (Venkiteswaran et al., 2002) resulted in a leaky junctional barrier indicating the significance of VE-cadherin. Although the precise mechanisms of the regulation of junctional assembly by VE-cadherin have not been identified, actin-binding proteins appear to be crucial (Mehta and Malik, 2006). Lampugnani et al showed that transfection of VE-cadherin cDNA in EC from VE-cadherin-null murine embryos induced actin cytoskeleton rearrangement and activated Rho family GTPase Rac1 (Lampugnani et al., 2002). Likewise, in another study, engagement of VE-cadherin was shown to activate Rac1 (Noren et al., 2001) suggesting an important role of VE-cadherin in recruiting Rac1 during cytoskeletal reorganization.
During vascular injury, lysed cells are a source of extracellular nucleotides. Additionally, vascular EC is also regulated by extracellular nucleotides released from platelets. β-nicotinamide adenine dinucleotide (β-NAD) is a coenzyme found in all living cells. In metabolism, β-NAD is involved in redox reactions, carrying electrons from one reaction to another and these electron transfer reactions are the main known function of β-NAD. It is also used in other cellular processes, notably as a substrate of enzymes that add or remove chemical groups from proteins, and in posttranslational modifications. β-NAD has been demonstrated as a cytokine targeting human polymorphonuclear granulocytes (Bruzzone et al., 2006) and a rapid increase of cAMP was observed followed by exposure to extracellular β-NAD (Bruzzone et al., 2006). Present in nanomolar to sub-micromolar concentrations in human serum (De Flora et al., 2004), β-NAD, released extracellularly from the injured cells (Krebs et al., 2005; Scheuplein et al., 2009), could be involved in various signaling mechanisms. Recently, Mutafova-Yambolieva et al (Mutafova-Yambolieva et al., 2007) and Moreschi et al (Moreschi et al., 2006) showed that β-NAD is an agonist of human P2Y1 and P2Y11 receptor respectively. Interestingly, the signaling properties of P2Y receptors characterized so far suggested that P2Y11 is the only receptor that increases both IP3 and cAMP levels by virtue of its dual coupling to heterotrimeric proteins, Gq and Gs (Moreschi et al., 2006). Despite these findings, the effect of β-NAD on vascular barrier function has not been investigated. Therefore, in the present article, we delineated the effect of β-NAD on EC permeability and demonstrated that β-NAD displayed a potent barrier protection via activation of P2Y1 and P2Y11 receptors followed by PKA-, EPAC1-, Rac1- and MLCP-dependent cytoskeletal modifications.
Material and Methods
Materials
All the reagents were obtained from Sigma-Aldrich (St. Louis, MO) unless otherwise indicated. Mouse monoclonal VE-cadherin antibody was purchased from BD Biosciences (San Diego, CA). Rabbit polyclonal antibodies against P2Y1 and P2Y11 receptors were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). siPORT Amine transfection reagent was obtained from Ambion (Austin, TX). P2Y1-, P2Y11-, EPAC1- and Rac1-specific siRNAs were purchased from Santa Cruz Biotechnology. TRIzol was obtained from Invitrogen (Carlsbad, CA). P2Y1- and P2Y11-specific antagonists were obtained from Tocris (Ellisville, MO). PKA inhibitor, H89, was purchased from Calbiochem (San Diego, CA). Phospho-MLC-specific antibodies were purchased from Cell Signaling (Beverly, MA). G-LISA kit was obtained from Cytoskeleton Inc. (Denver, CO).
Cell Culture
Human pulmonary artery EC (HPAEC) and EBM-2 complete medium were purchased from Lonza (Allendale, NJ). HPAEC were cultured according to the manufacturer’s protocol and utilized at early (3–6) passages.
Measurement of endothelial monolayer electrical resistance
The barrier properties of EC monolayers were characterized using a highly sensitive electrical cell-substrate impedance sensing (ECIS) instrument to measure transendothelial electrical resistance (TER) as described previously (Birukova et al., 2004a; Kolosova et al., 2005). The TER data was normalized to the initial voltage.
Immunofluorescence microscopy
Immunostaining was performed as described previously (Kolosova et al., 2005). The DNA-binding, fluorescent dye 7-amino-actinomycin D (7AAD) was used to stain cell nuclei (Rabinovitch et al., 1986). The percentage of total cell surface area occupied by VE-cadherin-positive cell-cell junctions was quantitatively determined using Zeiss Microscope quantification Software.
Semi-quantitative RT-PCR analysis
To compare the amounts of P2Y1 and P2Y11 mRNAs, the total RNA (1.0 μg) isolated from HPAEC was subjected to PCR in 25-μl reaction mixture using reagents from Superscript One Step RT-PCR kit (Invitrogen, Carlsbad, CA). 18S ribosomal RNA 184 bp fragment (internal control for normalization) was amplified using 50 nM primers from TaqMan Gold RT-PCR Core Reagents Kit (Applied Biosystems, Foster City, CA). To amplify 134 bp fragment of Homo sapiens P2Y1 cDNA (Accession No. NM_002563.2), the following primers were used: forward, 5′-TATTCATCATCGGCTTCCTGGGCA-3′; reverse, 5′-AGCGGCATCTCCGTGTACATGTTCAA-3′; and probe, 5′-AGCGGCATCTCCGTGTACATGTTCAA-3′. For the amplification of 189 bp fragment of Homo sapiens P2Y11 cDNA (Accession No. NM_002566.4), the following primers were used: forward, 5′-CTCCTATGTGCCCTACCACATCA-3′; reverse, 5′-AGCTTTGCAGACATAGCCCAGGCCA-3′; and probe, 5′-AGCTTTGCAGACATAGCCCAGGCCA-3′. For the amplification of 391 bp fragment of Homo sapiens EPAC1 (Accession No. NM_001098351), the following primers were used: forward, 5′-TTGTTGTCAACCCACACGAAGTGC-3′; reverse, 5′-GAGGCCAAACATGACGGCAAAGAA-3′. The final concentration of all primers used was 200 nM. The PCR products were analyzed by agarose gel electrophoresis.
RT-PCR Analysis of Expression of mRNA Transcripts
The presence of specific mRNA transcripts for P2Y1, P2Y11, and EPAC1 was evaluated by RT-PCR. Total RNA was prepared from HPAEC using TRIzol. For RT-PCR analysis, 1 μg total RNA was reverse transcribed using a RNA-PCR kit (Gene-Amp; Applied Biosystems, Foster City, CA) according to the manufacturer’s protocol. PCR was performed using 1.0 μmol each of sense and antisense primers, 2.5 U of AmpliTaq DNA polymerase (Applied Biosystems), and the following cycling conditions: 94°C for 0.5 minutes; 35 cycles of 94°C for 1 minute, 60°C for 1 minute, and 72°C for 1 minute; 1 cycle of 72°C for 5 minutes. The PCR products were analyzed by agarose gel electrophoresis.
P2Y1 and P2Y11 receptor antagonists study
HPAEC were pretreated with receptor-specific antagonists, MRS2279 or NF157, for 30 min, and then challenged with 50 μM β-NAD. TER was registered throughout to examine the barrier enhancement induced by β-NAD in the presence or absence of the antagonists.
Depletion of endogenous mRNA using siRNA approach
To deplete the mRNA content of endogenous P2Y1, P2Y11 or EPAC1, the cells were treated with respective siRNA duplexes, which guide sequence-specific degradation of the homologous mRNA. A non-specific, scrambled siRNAs were used as a control treatment. HPAEC were plated on 60-mm dishes to yield 60–70% confluence, and transfection of siRNAs was performed using siPORT Amine transfection reagent according to the manufacturer’s protocol. Briefly, cells were serum-starved for 1 hr followed by incubation with 20 nM of target siRNA (or scrambled siRNA) for 6 hrs in serum-free media. Then media with serum was added (1% serum final concentration) for 42 hrs before biochemical experiments, ECIS and/or functional assays were conducted. To estimate the efficiency of mRNA depletion, 48 hrs later, the cells were lysed in TRIzol and specific mRNA depletion was analyzed by RT-PCR. For TER measurement, cells were plated to yield 60–70% confluence in electrode wells and transfected with siRNA as described previously (Kolosova et al., 2005).
Immunoblotting and G-LISA
Protein extracts were separated by SDS-PAGE, transferred to nitrocellulose membrane and probed with specific antibodies as previously described (Kolosova et al., 2005). Horseradish peroxidase-conjugated goat anti-rabbit IgG antibody (Santa Cruz Biotechnology, Santa Cruz, CA) was used as the secondary antibody, and immunoreactive proteins were detected using enhanced chemiluminescence detection kit (ECL) according to the manufacturer’s protocol (Amersham, Little Chalfont, UK). For quantification, immunoblot data were analyzed using NIH Image 1.63 software. Active Rac1 was determined using G-LISA Rac activation assay according to the manufacturer’s recommendations (Cytoskeleton Inc., Denver, CO).
Statistical analysis
All measurements are presented as the mean ± SEM of at least 3 independent experiments. To compare results between groups, a 2-sample Student t test was used. For comparison among groups, 1-way ANOVA was performed. Differences were considered statistically significant at p<0.05. We routinely use this analysis in our studies (Kolosova et al., 2005).
RESULTS
Extracellular β-NAD increases transendothelial electrical resistance and affects endothelial cell-cell junctions
In order to explore a possibility for β-NAD to regulate endothelial monolayer integrity, the effectors were used in the TER assay (Fig. 1). In the first set of experiments we studied a dose-dependent effect of β-NAD on quiescent HPAEC monolayers (Fig. 1A). Data obtained indicate a positive effect of micromolar concentrations of β-NAD on endothelial barrier function. β-NAD-treated HPAEC underwent changes in distribution of cell-cell junctional proteins, which could be demonstrated by immunofluorescence microscopy. For example, VE-cadherin, a major component of endothelial adherens junctions, was more pronounced at the cellular periphery, presumably at cell-cell contacts (Fig. 2A). The calculated percentage of total cell surface area occupied by VE-cadherin-positive cell-cell junctions confirmed that β-NAD induced a significant increase in the surface area of cell-cell interfaces as a percentage of total cell surface area (Fig. 2B). Taken together, our data (Fig. 2) clearly signify the role of β-NAD in the control of vascular permeability and maintaining a restrictive endothelial barrier.
Figure 1.

Extracellular β-NAD enhances barrier function of HPAEC monolayers and increases VE-cadherin presentation in cell-cell contacts. (A) Dose-dependent TER response of β-NAD. HPAEC were challenged with increasing concentration of β-NAD (10–100 μM). Data are representative of several independent experiments (minimum of three) (*p < 0.05 compared with control). (B) Immunofluorescence staining of VE-cadherin in quiescent and β-NAD-stimulated HPAEC monolayers. The cells were treated with 50 μM β-NAD for 30 min, then fixed and immunostained using VE-cadherin antibody. Appreciably more VE-cadherin was recruited to the area of cell-cell junctions after β-NAD treatment. Arrows indicate overlapping edges of neighboring cells. (C) Quantification of the surface area of the cell-cell interface. The percentage of total cell-surface area occupied by VE-cadherin-positive cell-cell junctions was calculated for 20 cells in each group. The graph demonstrates that β-NAD induced a significant increase in cell-cell interface surface area as a percentage of total cell surface area (*p < 0.05 compared with control). The box and Whiskers plot show the means (lines at the box centers, 17.42% and 61.03% for control and β-NAD-treated cells respectively).
Figure 2.
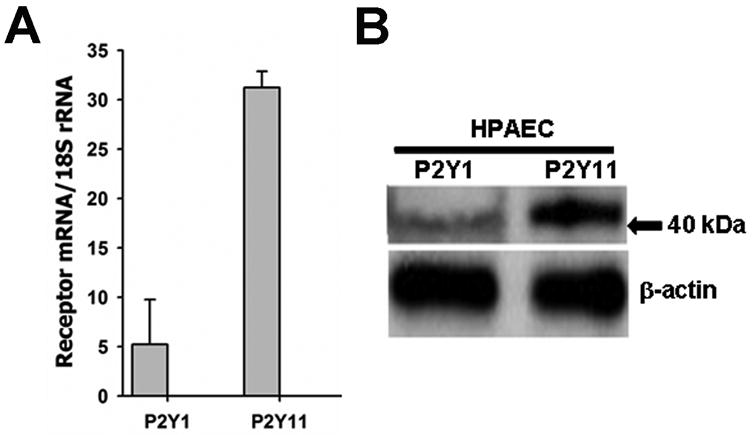
Expression of β-NAD-activated purine receptors P2Y1 and P2Y11 on mRNA and protein levels in HPAEC. (A) The receptor mRNA expressions were examined by Real-Time RT-PCR. Data were calculated relative to internal housekeeping gene (18S rRNA) and are expressed as fold change compared to control ± SEM (n=4). (B) The cell lysates obtained from HPAEC were analyzed by SDS-PAGE followed by immunoblotting using rabbit polyclonal antibodies against P2Y1 and P2Y11. Position of 40 kDa protein marker is shown by arrow. Immunoblotting of β-actin was used as a loading control.
Expression of β-NAD-activated P2Y receptors in HPAEC and their role in β-NAD-induced TER increase
Recent studies have demonstrated that extracellular β-NAD may interact with and activate two P2Y purine receptors, namely P2Y1 (Mutafova-Yambolieva et al., 2007) and P2Y11 (Moreschi et al., 2006). To evaluate the expression levels of these receptors in HPAEC, we have carried out semi-quantitative Real-Time RT-PCR analysis. Our data indicate that HPAEC express both of these receptors (Fig. 2A) and the mRNA levels of P2Y11 receptor appears to be higher than P2Y1 receptor. Immunoblotting experiments with receptor specific antibodies indicate that HPAEC express both P2Y1 and P2Y11 receptor proteins (Fig. 2B). To reveal a possible involvement of either of them in HPAEC TER increase, we have employed two different experimental approaches: (1) specific inhibition of the receptors by selective receptor antagonists and (2) specific depletion using siRNAs.
In first set of the experiments, we used the classical pharmacological approach with receptor-specific antagonists. As shown in Fig. 3A, a treatment of HPAEC with either P2Y1 antagonist (MRS2279) or P2Y11 antagonist (NF157) attenuated the β-NAD-induced TER increase, suggesting the involvement of both of these receptors in the enhancement of TER response. However, P2Y11 inhibition by NF157 attenuated the β-NAD-induced TER increase more significantly than P2Y1 inhibition by MRS2279. Data obtained may reflect the difference in the receptor expression levels and indicate the major role of highly expressed P2Y11 in HPAEC TER increase.
Figure 3.

Inhibitory analysis (selective antagonists and siRNA-mediated depletion) of the involvement of P2Y1 and P2Y11 receptors in β-NAD-stimulated TER increase. (A) HPAEC were pretreated with either 10 μM MRS2279 (P2Y1 antagonist) or 1 μM NF157 (P2Y11 antagonist) for 30 min prior β-NAD stimulation and used in ECIS assay. Data are representative of several independent experiments (minimum of three) (*p < 0.05 compared with control). (B) RT-PCR analysis of the expression of P2Y1 and P2Y11 mRNAs in the cells treated with scrambled and receptor-specific siRNA. siRNA approach was proved to be very effective in the depletion of P2Y1 and P2Y11 expression. Expression of hypoxanthine-guanine phosphoribosyltransferase (HPRT) was used as a loading control. (C, D) Depletions of P2Y11 (C) or P2Y1 (D) receptors negatively affect β-NAD-mediated TER response. HPAEC plated in ECIS arrays were transfected with respective scrambled (nsRNA) and siRNA as described in Materials and Methods. 48 hrs after transfection, the cells were used in ECIS assay in the presence or absence of β-NAD. Time-points of β-NAD addition are indicated by arrows. Data are representative of several independent experiments (minimum of three) (*p < 0.05 compared with control).
In order to confirm the results obtained by inhibitory analysis, we have individually depleted P2Y1 and P2Y11 receptor using receptor-specific siRNAs and the depletion of both P2Y1 and P2Y11 receptor mRNAs was confirmed by RT-PCR analysis (Fig. 3B). Our ECIS data (Fig. 3C, D) indicate that depletion of either P2Y1 or P2Y11 attenuated the β-NAD-induced HPAEC TER increase. However, the effect of depletion of the P2Y11 receptor on TER response was much more profound (Fig. 3C). The control siRNA with scrambled sequence failed to attenuate the β-NAD-induced TER increase, demonstrating the sequence-specific depletion. Taken together, the results shown in Fig. 3 strongly suggest an involvement of both, P2Y1 and PY11, receptors in β-NAD-induced HPAEC TER increase.
Effects of β-NAD on thrombin-, lipopolysaccharide (LPS)- or pneumolysin (PLY)-induced HPAEC barrier dysfunction
To evaluate EC barrier-protective functions of β-NAD, we analyzed the effect of β-NAD treatment on HPAEC challenged with various barrier-disruptive factors, such as protease thrombin, Gram-negative bacterial toxin LPS or Gram-positive bacterial toxin PLY. Data obtained in these experiments are presented in Fig. 4. Thrombin, a protease activated on the surface of injured endothelium, stimulates protease-activated receptors (PARs) coupled to heterotrimeric G12/13, Gq/11 and Gi proteins which, in turn, stimulate PLCβ, PKCα and RhoA pathways and inhibit AC (Knezevic et al., 2007; Rebecchi and Pentyala, 2000). This can eventually lead to activation of MLC kinase and inhibition of MLC phosphatase, stress fiber formation and EC barrier dysfunction. Our data indicate that β-NAD-dependent cell signaling can antagonize thrombin-activated cascades. Simultaneous treatment of the cells with thrombin and β-NAD significantly attenuated the thrombin-induced EC permeability, demonstrating the barrier-protective effect of β-NAD (Fig. 4A). LPS, a component of the outer membrane of Gram-negative bacteria, act as an endotoxin and elicit a strong immune response. LPS has been used as a model endotoxin to induce barrier disruption in HPAEC (Bai et al., 2009; Koh et al., 2007; Kolosova et al., 2008). It has been demonstrated that LPS-treatment of human umbilical vein EC (HUVEC) decreased the activity of myosin light chain (MLC) phosphatase (MLCP), resulting in an increase in MLC phosphorylation followed by cell contraction and an increase in EC permeability. To evaluate the protective role of β-NAD in LPS-induced HPAEC barrier disruption, we performed TER measurement assay in the cell monolayers. As shown in Fig. 4B, HPAEC exposed to LPS (100 nM) caused a significant and sustained decrease in HPAEC TER, which reflects a significant EC barrier dysfunction (~60% decrease in TER from the baseline). However, added to LPS, β-NAD significantly attenuated the LPS-induced barrier disruption (~30% decrease in TER from base line). These differences in TER values are significant as the protection sustained for several hours. Treatment with β-NAD alone caused a significant initial increase in TER which is in full agreement with Fig. 1A. Similar results were also obtained when HPAEC exposed to the PLY (125 ng/ml) (Fig. 4C). PLY is a pore-forming protein with multiple effects on eukaryotic cells (Marriott et al., 2008). One of the effects characteristic for PLY-treated cells includes cytoskeletal reorganization due to elevation of intracellular Ca2+ followed by activation of Rho/Rho-kinase pathway (Iliev et al., 2007). In order to test protective properties of β-NAD, we treated HPAEC with either PLY alone or in a mixture with 50 μM β-NAD, while measured TER response. Data shown in Fig. 4C demonstrate a rapid and dramatic loss of the monolayer integrity after PLY addition (~80% decrease in TER from the baseline). In contrast, PLY added to the cells in a mixture with β-NAD, failed to produce such drastic effect (~30% decrease in TER from the baseline) (Fig. 4C), likely, because of rapid activation of interfering pathways leading to inhibition of RhoA and activation of MLCP.
Figure 4.
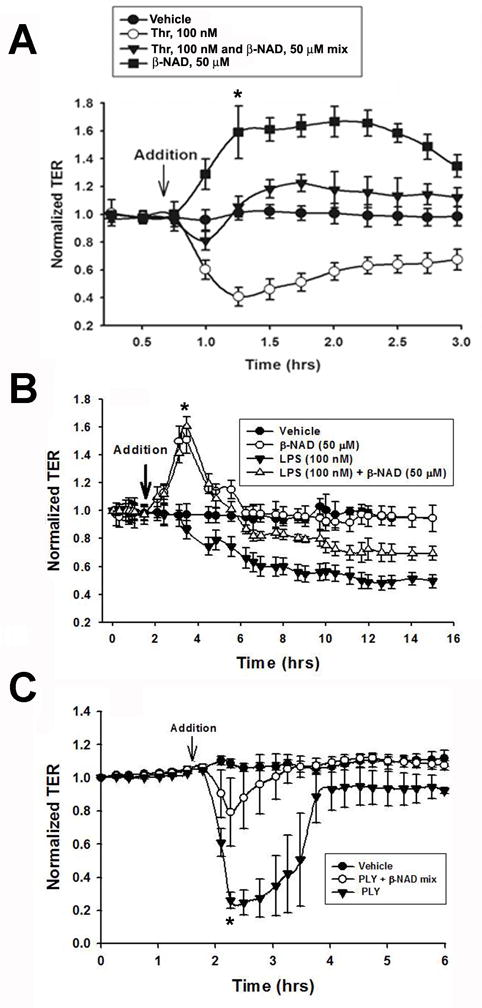
Extracellular β-NAD protects HPAEC monolayers from barrier-disruptive effects of thrombin and Gram-negative and Gram-positive bacterial toxins, lipopolysaccharide (LPS) and pneumolysin (PLY). HPAEC plated in ECIS arrays were challenged with either 100 nM thrombin (A) or 100 nM LPS (B) or 31.2 ng/ml PLY (C). The challengers were added either alone or in mixture with 50 μM β-NAD. β-NAD consistently prevented HPAEC barrier dysfunction in the cells treated with thrombin or PLY and significantly protected barrier integrity in the cells treated with LPS. Data are representative of several independent experiments (minimum of three) (*p < 0.05 compared with control).
Role of actin cytoskeleton in β-NAD-dependent cytoskeletal rearrangement
During recent years the regulation of the assembly and organization of cytoskeletal structures has obtained a great deal of interest in cell biology. Rho family GTPases are important regulators of the actin cytoskeleton and consequently influence the shape and movement of the cells. One of the major functions of the Rho GTPases is reorganization of the actin cytoskeleton in response to various extracellular stimuli (Ridley et al., 1992; Wojciak-Stothard et al., 2006) and the GTP-bound form of Rac1 has several common downstream targets that regulate the actin cytoskeleton and advancing the motility of fibroblasts (Sells et al., 1997). On the other hand, in addition to the actin cytoskeleton, it has also been shown that Rho family GTPases, which are key regulators of cell migration, affect microtubules (Watanabe et al., 2004). Therefore, in the next set of experiments we attempted to identify the dynamic cytoskeletal component(s) (actin and/or microtubules) indispensable for a β-NAD-induced increase in TER. For these experiments, the cells were treated with cytoskeleton-disrupting agents prior to β-NAD stimulation. Using ECIS approach, we were able to evaluate the involvement of the actin and tubulin components of the cytoskeleton in β-NAD-stimulated endothelial barrier enhancement (Fig. 5A, B). First, the HPAEC monolayers were pretreated with the actin-depolymerizing agent, cytochalasin B, which produced a prompt attenuation in TER (Fig. 5A). Distinct from the protective effect observed for thrombin-induced barrier disruption (Fig. 4A), β-NAD treatment did not increase the TER when added after cytochalasin B. This data suggests a critical requirement for cytoskeletal rearrangement and an intact actin cytoskeleton in β-NAD-induced increase in HPAEC TER. Second, we used a classical microtubule-disrupting agent, nocodazole, which is known to compromise the EC barrier integrity (Birukova et al., 2004b; Verin et al., 2001). In contrast to the experiment with cytochalasin B, the result shown in Fig. 5B demonstrates that β-NAD treatment restored the EC barrier integrity disrupted by nocodazole. Taken together our data shown in Fig. 5A, B suggest a pivotal role of actin filaments in dynamic cytoskeleton rearrangement induced by β-NAD. Moreover, in the experiments with LPS-treated cells we were able to obtain data suggesting that β-NAD-dependent cell signaling might lead to regulation of the actin cytoskeleton via shifting the regulatory myosin light chain to dephosphorylated form (Fig. 5C) as previously was demonstrated for ATP-dependent EC barrier enhancement (Kolosova et al., 2005). Treatment of HPAEC with LPS caused a robust phosphorylation of MLC, which was significantly inhibited by β-NAD suggesting the involvement of MLCP in β-NAD-induced increase in TER (Fig. 5C).
Figure 5.
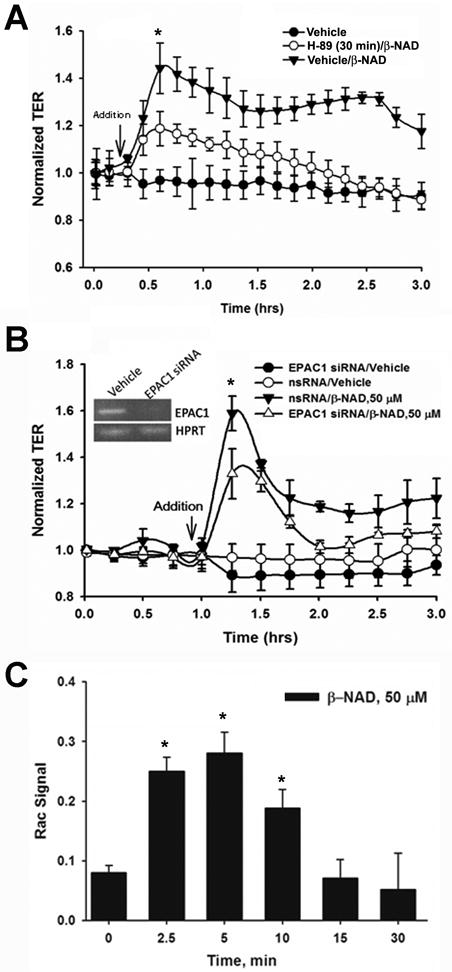
Molecular mechanisms of β-NAD-activated endothelial barrier enhancement in HPAEC. (A) β-NAD treatment activated cAMP-dependent PKA. HPAEC were pretreated with either vehicle or PKA-specific inhibitor, 5 μM H-89, for 30 min and then stimulated with 50 μM β-NAD in TER measurement assay. The inhibitor pretreatment significantly attenuated β-NAD-dependent increase in TER. (B) cAMP-activated EPAC1 is involved in β-NAD-activated TER response. HPAEC plated in ECIS arrays were transfected with either scrambled (nsRNA) or EPAC1-specific siRNA as described in Materials and Methods. The cells were stimulated with 50 μM β-NAD in TER measurement assay. Successful depletion of EPAC1 led to a significant decrease of β-NAD-activated TER response. (C) Downstream target of PKA/EPAC1 pathways, Rac1, is activated in HPAEC upon β-NAD stimulation. The cells treated with 50 μM β-NAD for time periods indicated were used in G-LISA assay as described in Materials and Methods to estimate the levels of activated Rac1. Data obtained demonstrate a rapid and dramatic elevation of Rac1-activity in β-NAD-stimulated cells. Data are representative of several independent experiments (minimum of three) (*p < 0.05 compared with control).
Signaling pathways responsible for β-NAD-induced enhancement of EC barrier function
To elucidate the signaling pathways involved in β-NAD-induced HPAEC TER increase, we studied the classical cAMP-activated protein kinase A (PKA) and the recently described nucleotide exchange protein directly activated by cAMP (EPAC) pathways. Since activation of P2Y11 receptor may lead to the well-documented Gαs-mediated pathway including direct stimulation of adenylate cyclase, elevation of cAMP levels and cAMP-dependent activation of PKA, we have performed a simple inhibitory test to confirm an activation of PKA and its participation in β-NAD-induced HPAEC barrier enhancement. For this test, we used H-89, an inhibitor of PKA activity, in ECIS experiments. HPAEC were pre-treated with H-89 for 30 min and then challenged with β-NAD and the effect of β-NAD-mediated barrier enhancement was determined using TER measurement. Our data (Fig. 6A) indicate that H-89 pre-treatment attenuated the β-NAD-induced HPAEC TER increase. Another cAMP-dependent signaling cascade, EPAC1/Rap1/Rac1 may also be involved in EC barrier protection (Birukova et al., 2007). To elucidate whether or not EPAC1 is also critical for β-NAD-inducible TER response, we depleted the expression of EPAC1 in HPAEC with the siRNA specific for EPAC1 and then the cells were challenged with β-NAD in TER assay. Our data showed that the HPAEC with depleted EPAC1 have markedly decreased TER response to β-NAD signifying the involvement of EPAC1 in β-NAD-induced TER response. Small GTPases of the Rho family regulate many aspects of cytoskeletal dynamics and three members of the family (Rac1, RhoA and Cdc42) have been studied extensively. Rho family GTPases control cell growth, cytokinesis, cell motility, trafficking and organization of the cytoskeleton (Jaffe and Hall, 2002). We hypothesized that Rac1 could be involved in β-NAD-induced increase in TER as a downstream target of the EPAC1 pathway. To prove this, we treated the HPAEC monolayers with β-NAD, and the cell lysates obtained at several time points of β-NAD stimulation were used to determine the levels of Rac1 activation by G-LISA assay. Data shown in Fig. 6C clearly demonstrate dramatic β-NAD-dependent activation of Rac1 at early time points (2.5 and 5 min) and the activity gradually returned to the basal values by 30 min. Time-dependent increase of Rac1 activation corroborates with the rapid increase in TER of HPAEC upon β-NAD treatment (Fig. 1A).
Figure 6.

Effect of cytoskeletal alterations on β-NAD-induced endothelial cell barrier protection. (A) HPAEC were pretreated with either vehicle or cytochalasin B for 30 min and then stimulated with 50 μM β-NAD in TER measurement assay. Actin depolymerization decreased TER and completely prevented the effect of β-NAD on TER. (B) HPAEC were pretreated with either vehicle or the microtubule-disrupting agent, nocodazole, for 30 min and then stimulated with 50 μM β-NAD. Disruption of microtubules decreased TER, but failed to alter β-NAD-induced increase in TER. Results are presented as mean ± SE and derived from three independent experiments (*p < 0.05 compared with control). (C) β-NAD treatment decreases myosin light chain (MLC) phosphorylation stimulated by LPS. HPAEC treated by either vehicle or LPS alone, or LPS/β-NAD mixture for 4 hrs were lysed and analyzed by SDS-PAGE followed by immunoblotting with anti-phosphoMLC antibody. Immunoblotting with anti-MLC antibody was used as a loading control. Data obtained indicate that barrier-protective mechanism of β-NAD may be realized via stimulation of MLC phosphatase activity, decreasing phosphoMLC levels and preventing, therefore, actin stress fiber formation.
DISCUSSION
The major finding of the present study is that β-NAD significantly increases the TER of pulmonary EC in a dose-dependent manner (Fig. 1A) and attenuates the thrombin-, LPS-, and PLY-induced EC barrier disruption (Fig. 4). β-NAD induces rearrangement of VE-cadherin suggesting tightening of cell-cell contacts leading to barrier enhancement (Fig. 1B). These results demonstrate for the first time β-NAD as a putative novel extracellular nucleotide in the regulation of endothelial permeability.
Although β-NAD structure is similar with those of the classic ligands of purine receptors, ATP and ADP, it was only recently demonstrated to be a ligand of purine receptors (Moreschi et al., 2006; Mutafova-Yambolieva et al., 2007). Interactions of β-NAD can bind to only two purine receptors, P2Y1 and P2Y11. Such selectivity indicated that extracellular β-NAD could be an attractive, physiologically relevant agent for positive regulation of endothelial barrier function, since these receptors are coupled only to heterotrimeric Gs and Gq proteins. Indeed, Gs protein is a well-known direct activator of AC, and elevation of cAMP levels in endothelial cells essentially leads to an enhancement of barrier integrity. Activation of heterotrimeric Gq protein followed by direct activation of the phospholipase Cβ and, therefore, elevation of inositol 1,4,5-triphosphate (IP3) and diacylglycerol (DAG) levels (Rebecchi and Pentyala, 2000). These two second messengers stimulate, in turn, intracellular calcium elevation, activation of PKC and/or PKG pathways. Cross-talks of these pathways may eventually lead to either negative or positive consequences for the barrier function (Knezevic et al., 2007; Moldobaeva et al., 2006). In contrast, ATP and ADP interact with four different P2Y receptors and can also activate Gi protein, an inhibitor of AC, via binding to P2Y13 (von Kugelgen, 2006). Therefore, a signaling cascade dependent upon P2Y13 activation should undoubtedly have a negative effect on endothelial barrier function. Taking all of this into account, we have explored, for the first time, a possibility that β-NAD may serve as effectors of endothelial integrity. The experiments with stimulated HPAEC monolayers revealed that β-NAD is a strong positive regulator of endothelial integrity (Fig. 1A). Besides, HPAEC were an adequate model for this study because these cells expressed both β-NAD-activated purine receptors (Fig. 2) and we were able to evaluate their involvement in β-NAD-dependent barrier enhancement. Interestingly, inhibitory analysis based on receptor-selective antagonists and sequence-specific siRNAs showed that both P2Y1 and P2Y11 receptors are involved in β-NAD-induced EC response (Fig. 3). β-NAD-activated receptors stimulate cAMP synthesis followed by activation of two cAMP-dependent pathways, PKA and EPAC1/Rac1 (Fig. 6). Both of them likely led to actin cytoskeleton rearrangement via RhoA/Rho-kinase inhibition and activation of MLCP. The actin component of cytoskeleton played an indispensable role in the HPAEC monolayer integrity enhancement, while microtubules were not involved in the TER response induced by β-NAD (Fig. 5). Taken together, our experimental data summarizing in Fig. 7 suggest that activation of both P2Y receptors lead to actin reorganization and barrier protection/enhancement, although via at least two different signaling pathways. We were able to study in detail signaling pathways activated by heterotrimeric protein Gs. We have demonstrated that in HPAEC treated with β-NAD, Gαs-induced stimulation of AC leads to two cAMP-dependent pathways, PKA and EPAC1 followed by rapid and dramatic activation of Rac1. Birukova et al (Birukova et al., 2007) demonstrated cross-talk between these cAMP-dependent pathways, which must increase an efficiency of actin reorganization, since both effectors (PKA and EPAC1) can directly or indirectly inhibit RhoA and activate Rac1. Thus, β-NAD is a novel and very efficient regulator of endothelial integrity. We were able to prove this idea in the experiments with various EC barrier-disruptive factors such as thrombin, and the bacterial toxins LPS and PLY (Fig. 4).
Figure 7.
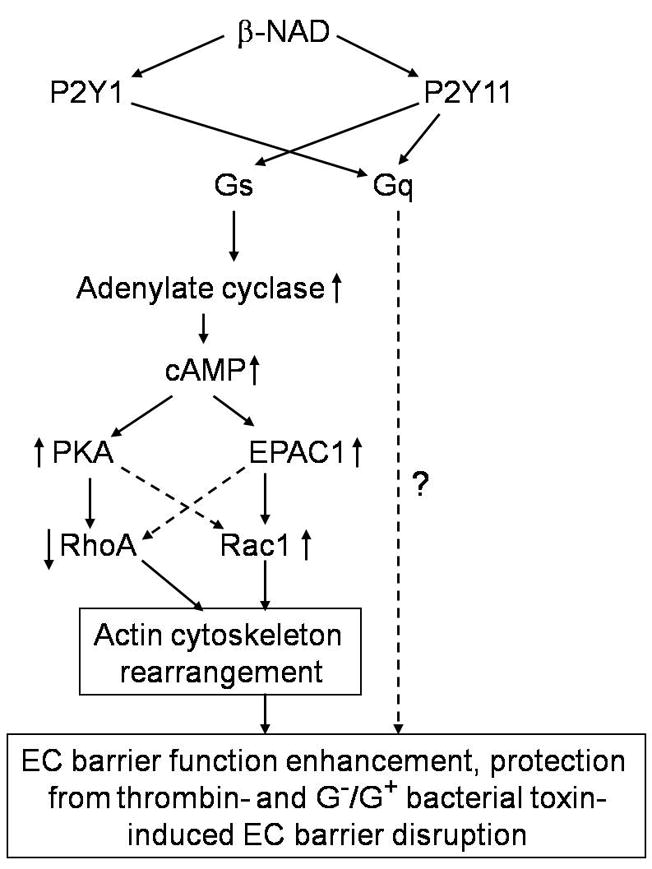
Schematic representation of putative EC barrier-protective molecular mechanisms activated by extracellular β-NAD.
In summary, we demonstrated that β-NAD is protective against thrombin, LPS-and PLY- induced EC barrier dysfunction via cAMP-activated PKA and EPAC1/Rac1-dependent actin cytoskeleton rearrangement. The results from these studies can give important information about mechanisms of EC barrier protection and can thus contribute to the development of alternative therapeutic strategies for ALI/ARDS.
Acknowledgments
Contract grant sponsor: NIH; Contract grant number; HL083327 (ADV) and HL067307 (ADV) and MCG PSRP (NSU).
References
- Baffert F, Le T, Thurston G, McDonald DM. Angiopoietin-1 decreases plasma leakage by reducing number and size of endothelial gaps in venules. Am J Physiol Heart Circ Physiol. 2006;290(1):H107–118. doi: 10.1152/ajpheart.00542.2005. [DOI] [PubMed] [Google Scholar]
- Bai JW, Deng WW, Zhang J, Xu SM, Zhang DX. [Protective effect of myosin light-chain kinase inhibitor on acute lung injury] Zhongguo Wei Zhong Bing Ji Jiu Yi Xue. 2009;21(4):215–218. [PubMed] [Google Scholar]
- Birukova AA, Smurova K, Birukov KG, Kaibuchi K, Garcia JG, Verin AD. Role of Rho GTPases in thrombin-induced lung vascular endothelial cells barrier dysfunction. Microvasc Res. 2004a;67(1):64–77. doi: 10.1016/j.mvr.2003.09.007. [DOI] [PubMed] [Google Scholar]
- Birukova AA, Smurova K, Birukov KG, Usatyuk P, Liu F, Kaibuchi K, Ricks-Cord A, Natarajan V, Alieva I, Garcia JG, Verin AD. Microtubule disassembly induces cytoskeletal remodeling and lung vascular barrier dysfunction: role of Rho-dependent mechanisms. J Cell Physiol. 2004b;201(1):55–70. doi: 10.1002/jcp.20055. [DOI] [PubMed] [Google Scholar]
- Birukova AA, Zagranichnaya T, Fu P, Alekseeva E, Chen W, Jacobson JR, Birukov KG. Prostaglandins PGE(2) and PGI(2) promote endothelial barrier enhancement via PKA- and Epac1/Rap1-dependent Rac activation. Exp Cell Res. 2007;313(11):2504–2520. doi: 10.1016/j.yexcr.2007.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruzzone S, Moreschi I, Guida L, Usai C, Zocchi E, De Flora A. Extracellular NAD+ regulates intracellular calcium levels and induces activation of human granulocytes. Biochem J. 2006;393(Pt 3):697–704. doi: 10.1042/BJ20051302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Flora A, Zocchi E, Guida L, Franco L, Bruzzone S. Autocrine and paracrine calcium signaling by the CD38/NAD+/cyclic ADP-ribose system. Ann N Y Acad Sci. 2004;1028:176–191. doi: 10.1196/annals.1322.021. [DOI] [PubMed] [Google Scholar]
- Dudek SM, Camp SM, Chiang ET, Singleton PA, Usatyuk PV, Zhao Y, Natarajan V, Garcia JG. Pulmonary endothelial cell barrier enhancement by FTY720 does not require the S1P1 receptor. Cell Signal. 2007;19(8):1754–1764. doi: 10.1016/j.cellsig.2007.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek SM, Garcia JG. Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol. 2001;91(4):1487–1500. doi: 10.1152/jappl.2001.91.4.1487. [DOI] [PubMed] [Google Scholar]
- Fukuhara S, Sakurai A, Sano H, Yamagishi A, Somekawa S, Takakura N, Saito Y, Kangawa K, Mochizuki N. Cyclic AMP potentiates vascular endothelial cadherin-mediated cell-cell contact to enhance endothelial barrier function through an Epac-Rap1 signaling pathway. Mol Cell Biol. 2005;25(1):136–146. doi: 10.1128/MCB.25.1.136-146.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia JG, Schaphorst KL. Regulation of endothelial cell gap formation and paracellular permeability. J Investig Med. 1995;43(2):117–126. [PubMed] [Google Scholar]
- Iliev AI, Djannatian JR, Nau R, Mitchell TJ, Wouters FS. Cholesterol-dependent actin remodeling via RhoA and Rac1 activation by the Streptococcus pneumoniae toxin pneumolysin. Proc Natl Acad Sci U S A. 2007;104(8):2897–2902. doi: 10.1073/pnas.0608213104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe AB, Hall A. Rho GTPases in transformation and metastasis. Adv Cancer Res. 2002;84:57–80. doi: 10.1016/s0065-230x(02)84003-9. [DOI] [PubMed] [Google Scholar]
- Knezevic N, Roy A, Timblin B, Konstantoulaki M, Sharma T, Malik AB, Mehta D. GDI-1 phosphorylation switch at serine 96 induces RhoA activation and increased endothelial permeability. Mol Cell Biol. 2007;27(18):6323–6333. doi: 10.1128/MCB.00523-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh H, Tasaka S, Hasegawa N, Yamada W, Shimizu M, Nakamura M, Yonemaru M, Ikeda E, Adachi Y, Fujishima S, Yamaguchi K, Ishizaka A. Protective role of vascular endothelial growth factor in endotoxin-induced acute lung injury in mice. Respir Res. 2007;8:60. doi: 10.1186/1465-9921-8-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolosova IA, Mirzapoiazova T, Adyshev D, Usatyuk P, Romer LH, Jacobson JR, Natarajan V, Pearse DB, Garcia JG, Verin AD. Signaling pathways involved in adenosine triphosphate-induced endothelial cell barrier enhancement. Circ Res. 2005;97(2):115–124. doi: 10.1161/01.RES.0000175561.55761.69. [DOI] [PubMed] [Google Scholar]
- Kolosova IA, Mirzapoiazova T, Moreno-Vinasco L, Sammani S, Garcia JG, Verin AD. Protective effect of purinergic agonist ATPgammaS against acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2008;294(2):L319–324. doi: 10.1152/ajplung.00283.2007. [DOI] [PubMed] [Google Scholar]
- Krebs C, Adriouch S, Braasch F, Koestner W, Leiter EH, Seman M, Lund FE, Oppenheimer N, Haag F, Koch-Nolte F. CD38 controls ADP-ribosyltransferase-2-catalyzed ADP-ribosylation of T cell surface proteins. J Immunol. 2005;174(6):3298–3305. doi: 10.4049/jimmunol.174.6.3298. [DOI] [PubMed] [Google Scholar]
- Lampugnani MG, Zanetti A, Breviario F, Balconi G, Orsenigo F, Corada M, Spagnuolo R, Betson M, Braga V, Dejana E. VE-cadherin regulates endothelial actin activating Rac and increasing membrane association of Tiam. Mol Biol Cell. 2002;13(4):1175–1189. doi: 10.1091/mbc.01-07-0368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum H, Malik AB. Mechanisms of increased endothelial permeability. Can J Physiol Pharmacol. 1996;74(7):787–800. doi: 10.1139/y96-081. [DOI] [PubMed] [Google Scholar]
- Marriott HM, Mitchell TJ, Dockrell DH. Pneumolysin: a double-edged sword during the host-pathogen interaction. Curr Mol Med. 2008;8(6):497–509. doi: 10.2174/156652408785747924. [DOI] [PubMed] [Google Scholar]
- Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86(1):279–367. doi: 10.1152/physrev.00012.2005. [DOI] [PubMed] [Google Scholar]
- Moldobaeva A, Welsh-Servinsky LE, Shimoda LA, Stephens RS, Verin AD, Tuder RM, Pearse DB. Role of protein kinase G in barrier-protective effects of cGMP in human pulmonary artery endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2006;290(5):L919–930. doi: 10.1152/ajplung.00434.2005. [DOI] [PubMed] [Google Scholar]
- Moreschi I, Bruzzone S, Nicholas RA, Fruscione F, Sturla L, Benvenuto F, Usai C, Meis S, Kassack MU, Zocchi E, De Flora A. Extracellular NAD+ is an agonist of the human P2Y11 purinergic receptor in human granulocytes. J Biol Chem. 2006;281(42):31419–31429. doi: 10.1074/jbc.M606625200. [DOI] [PubMed] [Google Scholar]
- Mutafova-Yambolieva VN, Hwang SJ, Hao X, Chen H, Zhu MX, Wood JD, Ward SM, Sanders KM. Beta-nicotinamide adenine dinucleotide is an inhibitory neurotransmitter in visceral smooth muscle. Proc Natl Acad Sci U S A. 2007;104(41):16359–16364. doi: 10.1073/pnas.0705510104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noren NK, Niessen CM, Gumbiner BM, Burridge K. Cadherin engagement regulates Rho family GTPases. J Biol Chem. 2001;276(36):33305–33308. doi: 10.1074/jbc.C100306200. [DOI] [PubMed] [Google Scholar]
- Pugin J, Verghese G, Widmer MC, Matthay MA. The alveolar space is the site of intense inflammatory and profibrotic reactions in the early phase of acute respiratory distress syndrome. Crit Care Med. 1999;27(2):304–312. doi: 10.1097/00003246-199902000-00036. [DOI] [PubMed] [Google Scholar]
- Rabinovitch PS, Torres RM, Engel D. Simultaneous cell cycle analysis and two-color surface immunofluorescence using 7-amino-actinomycin D and single laser excitation: applications to study of cell activation and the cell cycle of murine Ly-1 B cells. J Immunol. 1986;136(8):2769–2775. [PubMed] [Google Scholar]
- Rebecchi MJ, Pentyala SN. Structure, function, and control of phosphoinositide-specific phospholipase C. Physiol Rev. 2000;80(4):1291–1335. doi: 10.1152/physrev.2000.80.4.1291. [DOI] [PubMed] [Google Scholar]
- Ridley AJ, Paterson HF, Johnston CL, Diekmann D, Hall A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 1992;70(3):401–410. doi: 10.1016/0092-8674(92)90164-8. [DOI] [PubMed] [Google Scholar]
- Scheuplein F, Schwarz N, Adriouch S, Krebs C, Bannas P, Rissiek B, Seman M, Haag F, Koch-Nolte F. NAD+ and ATP released from injured cells induce P2X7-dependent shedding of CD62L and externalization of phosphatidylserine by murine T cells. J Immunol. 2009;182(5):2898–2908. doi: 10.4049/jimmunol.0801711. [DOI] [PubMed] [Google Scholar]
- Sells MA, Knaus UG, Bagrodia S, Ambrose DM, Bokoch GM, Chernoff J. Human p21-activated kinase (Pak1) regulates actin organization in mammalian cells. Curr Biol. 1997;7(3):202–210. doi: 10.1016/s0960-9822(97)70091-5. [DOI] [PubMed] [Google Scholar]
- Venkiteswaran K, Xiao K, Summers S, Calkins CC, Vincent PA, Pumiglia K, Kowalczyk AP. Regulation of endothelial barrier function and growth by VE-cadherin, plakoglobin, and beta-catenin. Am J Physiol Cell Physiol. 2002;283(3):C811–821. doi: 10.1152/ajpcell.00417.2001. [DOI] [PubMed] [Google Scholar]
- Verin AD, Birukova A, Wang P, Liu F, Becker P, Birukov K, Garcia JG. Microtubule disassembly increases endothelial cell barrier dysfunction: role of MLC phosphorylation. Am J Physiol Lung Cell Mol Physiol. 2001;281(3):L565–574. doi: 10.1152/ajplung.2001.281.3.L565. [DOI] [PubMed] [Google Scholar]
- von Kugelgen I. Pharmacological profiles of cloned mammalian P2Y-receptor subtypes. Pharmacol Ther. 2006;110(3):415–432. doi: 10.1016/j.pharmthera.2005.08.014. [DOI] [PubMed] [Google Scholar]
- Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342(18):1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Wang S, Noritake J, Sato K, Fukata M, Takefuji M, Nakagawa M, Izumi N, Akiyama T, Kaibuchi K. Interaction with IQGAP1 links APC to Rac1, Cdc42, and actin filaments during cell polarization and migration. Dev Cell. 2004;7(6):871–883. doi: 10.1016/j.devcel.2004.10.017. [DOI] [PubMed] [Google Scholar]
- Wojciak-Stothard B, Tsang LY, Paleolog E, Hall SM, Haworth SG. Rac1 and RhoA as regulators of endothelial phenotype and barrier function in hypoxia-induced neonatal pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2006;290(6):L1173–1182. doi: 10.1152/ajplung.00309.2005. [DOI] [PubMed] [Google Scholar]