

Abstract
Cycloaddition reactions are attractive strategies for rapid formation of molecular complexity in organic synthesis as multiple bonds are formed in a single process. To this end, several research groups have been actively involved in the development of catalytic methods to activate readily accessible π-components to achieve cycloadditions. However, the use of C-N π-components for the formation of heterocycles by these processes is less well developed. It has been previously demonstrated that the combination of different isocyanates with two alkynes yields pyridones of several types by metal-catalyzed [2+2+2] cycloadditions. The potential of this chemistry has been extended to alkenes as C-C π-components, allowing the formation of sp3-stereocenters. In this tutorial review directed towards [n+2+2] cycloaddition of heterocumulenes, alkynes and alkenes, the recent advances in catalytic asymmetric synthesis of indolizidine, quinolizidine and azocine skeletons are discussed.
1. Introduction
Recent advances in transition metal-catalyzed cycloadditions have made these transformations among the most efficient methods to assemble polycyclic frameworks. Efforts in this field have mainly focused on carbocycle synthesis involving various modes of cycloadditions, including [2+1],1 [4+2]2 and [5+2]3 among others. Three-component cycloadditions, such as the Pauson-Khand reaction ([2+2+1])4 and the alkyne cyclotrimerization ([2+2+2])5, are also common examples. The formation of heterocycles by these processes is less well developed, with aziridinations6 and 1,3-dipolar cycloadditions7 being prominent exceptions.8 Given the demonstrated importance of nitrogen heterocycles in biology and medicine, the stereoselective synthesis of these moieties constitutes an area of intense research. This review will focus on rhodium-catalyzed [n+2+2] cycloadditions of alkenes, alkynes and isocyanates to form different bicyclic N-heterocycles.
Isocyanates are readily accessible from the corresponding carboxylic acids using the Curtius rearrangment or other methods and are primed to react with a variety of reagents.9 Their modest basicity at the nitrogen atom makes them compatible with transition metals. Several research groups have demonstrated that isocyanates are competent 2π-components in metal-catalyzed cycloadditions with two alkynes to form pyridones. In the 1970's, Hong and Yamazaki first reported the metal-catalyzed [2+2+2] cycloaddition of isocyanates (and carbodiimides) with two equivalents of alkyne to form pyridone isomers.10 However, most of the early and important advances in this field come from Hoberg's pioneering work using nickel as a catalyst for different transformations involving heterocumulenes and C-C π-components (eq. 1).11 Interestingly, using stoichiometric nickel, they were able to isolate metallacycles of the general form A from the oxidative cyclization of alkenes and heterocumulenes (isocyanates and carbon dioxide). Based on this precedent, they proposed mechanisms for their catalytic transformations, including the catalytic synthesis of 2-pyridones from two alkynes and one isocyanate.
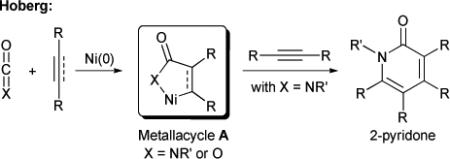 |
(1) |
By tethering two of the three components, Vollhardt was able to overcome the problem associated with the formation of multiple pyridone regioisomers in the cobalt-catalyzed [2+2+2] cycloadditions (eq. 2).12 Itoh has shown the cycloaddition of various tethered diynes and isocyanates using a ruthenium(II) catalyst (eq. 3),13 while Louie has found that nickel(II) complexes modified with a carbene or a phosphorous ligand are also competent catalysts in this chemistry.14 Takahashi has developed a highly regioselective, fully intermolecular method to incorporate three different π-components for the synthesis of pyridones (and other heterocycles), but uses stoichiometric amounts of zirconium and nickel.15
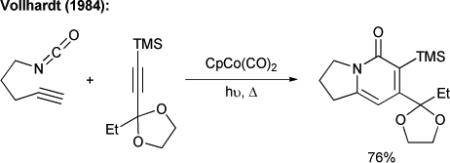 |
(2) |
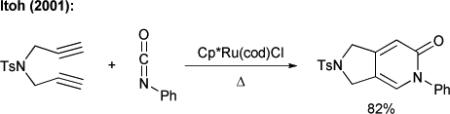 |
(3) |
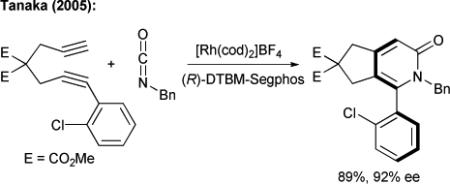 |
(4) |
The potential of metal-catalyzed [2+2+2] cycloadditions with isocyanates has been illustrated by Tanaka with the synthesis of biaryl systems with control of axial chirality using a cationic rhodium(I) complex (eq. 4).16 Except for Tanaka's example, all reported [2+2+2] cycloadditions involving isocyanates allow only for the synthesis of achiral bicyclic pyridones. The use of two alkynes in the previous transformations is an inherent limitation, affording cycloadducts lacking sp3-stereocenters and flexibility for further chemical transformations towards complex molecule syntheses.
It would be a clear benefit if alkenes could participate in [2+2+2] cycloadditions with isocyanates since they would introduce sp3-stereocenters in the product. The significance of a catalytic, enantioselective transformation combining isocyanates, alkynes and alkenes lies in the ability to access complex biologically active molecules in few steps from readily available starting material. Indolizidine and quinolizidine skeletons are ubiquitous natural product scaffolds with wide diversity in their substitution pattern, stereochemistry and biological activity.17 Alkaloids ranging from structurally simple indolizidine 209D, lasubine I and II, to more complex ones such as halichlorine, lythrine, secu'amamine A, cylindricine C and FR901483, all contain such ring systems (Fig. 1).
Fig. 1.

Some indolizidine- and quinolizidine-based natural products
In light of potentially providing a general and efficient route to many indolizidine and quinolizidine alkaloid natural products,18 we have focused on developing metal-catalyzed [n+2+2] three-component cycloadditions of alkenes, alkynes and C-N π-components, such as isocyanates and carbodiimides. Our vision relies on the catalytic generation of metallacycle M1 from an oxidative cyclization of a heterocumulene and a C-C π-component, which allows the insertion of an alkene or an alkyne to form a heterocycle after reductive elimination of M2 (Scheme 1).
Scheme 1.

General scheme for metal-catalyzed generation of heterocycles
Initial investigations focused on the use of isocyanates bearing tethered alkenes in the presence of exogenous alkynes (Scheme 2). The goal is to achieve this transformation in a fully intermolecular fashion using different heterocumulenes. Both strategies allow the formation of three new σ-bonds from three π-components and up to four stereocenters, providing a useful methodology for the asymmetric synthesis of indolizidine and quinolizidine natural products.
Scheme 2.

Intra- and intermolecular strategies for [2+2+2] cycloadditions with heterocumulenes
2. Development of the Rhodium-Catalyzed [2+2+2] Cycloaddition of Alkenyl Isocyanates and Alkynes: Discovery of a CO Migration Process
In our initial catalyst screen for the three-component cycloaddition of alkenes, alkynes and isocyanates, we found that the combination of 5 mol % of bis(ethylene)-rhodium(I) chloride dimer ([Rh(C2H4)2Cl]2) and 10 mol % of tris(4-methoxyphenyl)phosphine provides an active catalyst for this transformation.19 Reactions of alkenyl isocyanate 1a with different symmetrical internal aryl alkynes (tolanes) in the presence of the rhodium(I) catalyst afford indolizinone adducts in excellent yields (Scheme 3). Unexpectedly, the products formed are vinylogous amides 3a, resulting from an apparent carbonyl migration,20 rather than the anticipated lactams 2a. This unusual rhodium-catalyzed cycloaddition consists of two C-C and two C-N bond forming events and results in a fragmentation of the isocyanate unit.
Scheme 3.

Rh-catalyzed [2+2+2] cycloaddition of alkenyl isocyanates and internal symmetrical alkynes
This [2+2+2] cycloaddition tolerates a wide range of symmetrical internal aryl alkynes. Neutral and electron-rich tolanes substituted at either the para- or meta-position readily undergo the desired cycloaddition. However, electron-withdrawing substituted tolanes appear to be less reactive. This protocol can also be used to build the quinolizinone framework 3b (n = 2) using isocyanate 1b. Switching from tolanes to symmetrical alkyl alkynes has a profound effect on the reaction, as the lactam/vinylogous amide product selectivity is reversed, furnishing lactams 2a-b selectively (Scheme 3).19 However, this first generation catalyst proved to be of limited scope, since reactions with terminal alkynes often proceed sluggishly and with poor isolated yields.
3. Enantioselective Rhodium-Catalyzed [2+2+2] Cycloaddition of Alkenyl Heterocumulenes with Terminal Alkynes
As terminal alkynes are the most readily available alkynyl substrates, developing an efficient [2+2+2] cycloaddition between alkenyl heterocumulenes and terminal alkynes is highly desirable. The lack of desired reactivity using the previous catalytic system is attributed to the known competitive rhodium-catalyzed dimerization of terminal alkynes under the reaction conditions.21 In fact, when the terminal alkyne 4 is subjected to the reaction conditions in the absence of alkenyl isocyanate, dimerized products 5 and 6 are obtained in 50% yield in a 4:1 ratio favoring the head-to-tail product 5 (eq. 5).22
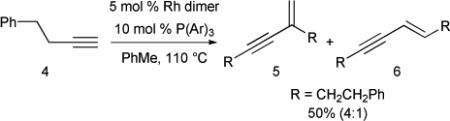 |
(5) |
3.1. Isocyanates as C-N π-Components
3.1.1. Aliphatic Terminal Alkynes
Attempts to improve the reaction of aliphatic terminal alkynes with alkenyl isocyanates led to the discovery of rhodium(I)-phosphoramidite complexes as more efficient catalysts (Fig. 2).23 By employing TADDOL-derived monodentate phosphoramidite ligand (−)-T1, cycloadditions with alkyl acetylenes proceed smoothly to afford lactams 7a with excellent product selectivities (up to >20:1) and good enantioselectivities (Scheme 4).24 Enantioselective synthesis of lactam-type indolizinones bearing a quaternary substituted stereocenter (7b) is also possible using 1,1-disubstituted alkenyl isocyanates in combination with ligand (−)-T4.25
Fig. 2.

Phosphoramidite ligands used in the Rh-catalyzed [n+2+2] cycloadditions of heterocumulenes
Scheme 4.

Enantioselective Rh-catalyzed [2+2+2] cycloaddition of alkenyl isocyanates and aliphatic terminal alkynes
On the other hand, vinylogous amide products 8a are favored when biaryl ligand (R)-L1 is used, illustrating that the catalyst exerts significant control over product formation in this type of cycloaddition (Scheme 4).26 It is noteworthy that the cycloaddition proceeds in a highly regioselective manner since 7a, 7b and 8a are always isolated as single regioisomers (for discussions on the regioselectivity of these cycloadditions, see sections 3.3 and 4.2). Interestingly, it has been observed that for any given cycloaddition, the minor product (for example, vinylogous amides 8 in the case of the reactions with ligands (−)-T1 and (−)-T4) is always of opposite absolute configuration.
To validate this new methodology, we undertook the total synthesis of indolizidine (−)-209D (Scheme 5).26 The key intermediate 5-hexyl indolizinone 9 can be prepared conveniently by the cycloaddition protocol in one step and is suitable for scale-up. This constitutes the shortest enantioselective synthesis of 209D reported to date.27
Scheme 5.

Total synthesis of (−)-209D
3.1.2. Aromatic Terminal Alkynes Part I
In contrast to the lactam selectivity observed for alkyl acetylenes with TADDOL-derived phosphoramidites, electron-rich terminal aryl alkynes provide the vinylogous amides 12 predominantly; this is presumably due to the steric and electronic differences between alkyl and aryl groups (Scheme 6).24 The cycloadditions generally proceed cleanly to furnish cycloadducts 12a in high yields and enantioselectivities. Electron-withdrawing substituted aryl acetylenes also participate readily in the cycloaddition, with the product selectivity gradually favoring lactam with increasing EWG strength. For example, substitution of the p-OMe for a p-CF3 leads to the formation of the lactam product in 2.5:1 ratio (entry 1 vs entry 2 in the table of Scheme 6). This preference is rationalized by electronic factors as will be discussed in section 3.3. Heteroaryl acetylenes including both free and protected indoles also undergo the cycloaddition efficiently as shown in Scheme 6.
Scheme 6.

Enantioselective Rh-catalyzed [2+2+2] cycloaddition of alkenyl isocyanates and aromatic terminal alkynes
Asymmetric synthesis of quinolizinones can also be achieved as demonstrated by the rapid total synthesis of (+)-lasubine II (Scheme 7).24 However, [2+2+2] cycloadditions involving longer olefin tethers (or conducted in more concentrated solutions) are accompanied by varying amounts of pyridones, such as 14, suggesting that the tethered alkene is the last 2π-component to be incorporated.22
Scheme 7.

Total synthesis of (+)-lasubine II
Enantioselective synthesis of vinylogous amide-type indolizinones bearing a quaternary substituted stereocenter (12b) is also possible using ligand (−)-T3 (Scheme 6).25 In this particular case, the ratio of lactam to vinylogous amide products obtained remains independent of the alkene substitution (entries 2-4 in the table of Scheme 6). However, with sterically bulky alkenyl substituents, an increase in pyridone byproduct and a corresponding decrease in vinylogous amide yields are observed. The increase in steric demand about the alkene presumably decreases the rate of coordination and/or insertion of the alkene to the extent that intermolecular insertion of another alkyne is competitive.
Based on the previous observations concerning pyridone formation with longer tether chains and bulkier alkenes, a highly regioselective rhodium-catalyzed intermolecular [2+2+2] cycloaddition of terminal alkynes with a variety of isocyanates to provide 2- and 4-pyridones has been developed (eq. 6).28 Again, an apparent carbonyl migration is responsible for the formation of the 4-pyridones (17).
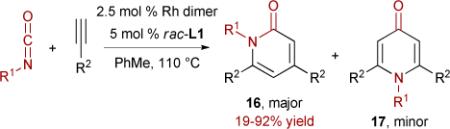 |
(6) |
Before we discuss the synthesis of lactam-type products 11 using aromatic terminal alkynes, a discussion on the current understanding of the reaction mechanism is appropriate.
3.2. Mechanistic Discussion
The current mechanistic understanding is illustrated in Scheme 8.22 While several rhodium(I) complexes are possible, only one seems to be productive (C1). Based on Wakatsuki's calculations on the coordination of alkynes to cobalt,29 the preferred orthogonal orientation of both the alkyne and the isocyanate in C1 appears to be under ligand control (vide infra). Partitioning between lactam and vinylogous amide products occurs during the first step (path A vs path B). Oxidative cyclization of C1 to form M3 via a C-N bond formation first generates a species in which the olefin cannot reach the rhodium(III) to insert because of a prohibitively strained bridged geometry in the transition state. CO extrusion to form M4 followed by re-insertion leads to M5, in which the olefin is now in a proximal relationship to the metal. Alkene insertion and reductive elimination afford the vinylogous amide product.
Scheme 8.

Current mechanistic understanding for the Rh-catalyzed [2+2+2] cycloadditions
The oxidative cyclization of C1 occuring with a C-C bond formation generates metallacycle M7. Subsequent olefin insertion and reductive elimination lead to the lactam product. Based on the pyridone formation with longer tethers (or higher concentration of alkyne), an alternative mechanism involving intramolecular oxidative cyclization of the alkenyl isocyanate to form M9 is less likely (path C). This mechanism also suggests that both products would have the same absolute configuration (which is not the case) since stereoinduction would occur prior to CO migration.
3.3 Lactam vs Vinylogous Amide Selectivity
Although the observation of pyridone byproducts has shed some light on the mechanism, a crucial question remains: what dictates the selectivity between the formation of metallacycles M3 and M7, leading respectively to the vinylogous amide and the lactam products. Results using the TADDOL-derived phosphoramidite ligands reveal that the electronic and steric contributions of the alkyne have a profound effect in determining the product selectivity. In addition, the steric environment of the ligand seems to have a strong influence on product selectivity (cf. Scheme 4, (−)-T1 vs (R)-L1).
As discussed previously, both TADDOL- and BINOL-derived phosphoramidites seem to establish a preorganization of the alkyne and the isocyanate moiety in the rhodium(I) complex C1 before the product discriminating oxidative cyclization step (Fig. 3).22 More precisely, the positioning of the bigger substituents (R for the alkyne and NR' for the isocyanate) on the same side is dictated by the steric environment of the ligand, which hinders one face of the rhodium(I) complex C1. This observation is borne out in the crystal structures of Rh(cod)Cl•(−)-T3 and Rh(cod)Cl•(R)-L2.
Fig. 3.

Rhodium-ligand crystal structures
Cycloadditions of alkyl alkynes and alkenyl isocyanates with TADDOL-derived phosphoramidites afford selectively the lactam products (cf. Scheme 4) while aryl alkynes provide vinylogous amide products (cf. Scheme 5). In both cases, mitigating factors reduce product selectivities: branched aliphatic alkynes provide considerable amounts of vinylogous amide adducts and extremely electron-deficient aryl alkynes provide lactams as the major products. It is thus clear that there are elements of both steric and electronic control that affect product distribution.
The product selectivities reported with electron-donating (EDG) and electron-withdrawing (EWG) aryl groups (cf. Scheme 5) are consistent with Stockis and Hoffmann's theoretical treatise of metallacycle formation.30 The authors argue that the kinetic regioselectivity results from the positioning of the π-systems with the largest coefficients in the LUMO (in other words, the most δ+ atom) β to the metal (Scheme 9). This preference arises from an interaction between a filled s-type orbital on the metal with the empty LUMO's of the two π-components, which results in a lower tansition state for the formation of the metallacycle. In our case, the isocyanate LUMO coefficient, resident largely on the central carbon, overwhelms the much smaller LUMO on the aliphatic terminal alkyne, leading to metallacycle M7 (via TS2a) and the resulting lactam product.22 This electronic control can be overriden by steric demand. Larger R-groups prefer to be oriented distal to the metal and its sterically demanding phosphoramidite ligand (TS1a over TS2a), leading to M3. Aryl acetylenes provide vinylogous amides product largely controlled by steric factors. With electron-releasing substituents on the arene, an electronic contribution due to a larger LUMO coefficient on the alpha carbon reinforces the steric selectivity (TS1a') leading to the vinylogous amide product. With strongly electron-withdrawing groups on the arene, sterics can be partially overriden by the electronic control (TS2a').
Scheme 9.

Steric and electronic contributions to rationalize the partitioning between the lactam and vinylogous amide product
3.4. Carbodiimides as C-N π-Components
In an effort to selectively access lactam-type products using aromatic acetylenes, we envisoned that a cycloaddition employing carbodiimides should favor TS2b by placing the bulky imido moiety farther away from the rhodium(I) center during the first oxidative cyclization step (Scheme 10). Although one may envision two different carbodiimide complexes in the ground state, only C2a is productive, leading to TS2b. We suggest that C2b would afford a metallacycle bearing the bulky NAr moiety proximal to the Rh and is thus disfavored.
Scheme 10.

Proposed approach to the synthesis of bicyclic amidines
We were pleased to see the successful application of this strategy using ligand (−)-T5 to offer a novel entry to the asymmetric synthesis of bicyclic amidines of type 18a (Scheme 11).31 An isocyanide (R-N=C) migration as minor process during the cycloaddition accounts for the formation of products 19. Cycloadditions with alkyl acetylenes are also possible, generating bicyclic amidines 18b with high efficiency.
Scheme 11.

Enantioselective Rh-catalyzed [2+2+2] cycloaddition of alkenyl carbodiimides and terminal alkynes
4. Enantioselective Rhodium-Catalyzed [2+2+2] Cycloaddition of Alkenyl Isocyanates with Internal Alkynes
The previous work on the rhodium-catalyzed [2+2+2] cycloaddition of terminal alkynes leads to an extension of the reaction scope by the introduction of internal alkynes.
4.1. Symmetrical Aryl Alkynes (Tolanes)
The advent of Guiphos ((R)-L2)26 proved to be a turning point for the expansion of the alkyne scope to symmetrical aryl alkynes, the product selectivity favoring the vinylogous amide adducts.32 However, low to moderate yields (15 to 65%) and large variations in enantioselectivities (62 to 91% ee) were observed between different tolanes. We have proposed that a second alkyne acts as spectator ligand on the rhodium(III) metallacycle in an octahedral geometry before olefin insertion (Fig. 4). Partitioning between several diastereomeric metallacycles, with the two more reasonable being M11 and M12, might account for these variations.
Fig. 4.

Proposed Rh(III)-complexes for the enantioselective [2+2+2] cycloaddition of alkenyl isocyanates and tolanes
Simple kinetic experiments reveal a first order dependence on alkyne for the cycloaddition. This is consistent with an oxidative cyclization occuring through a four-coordinate intermediate such as C1 (cf. Scheme 8). Therefore, the effect of the coordination of a second alkyne on enantioselectivity must occur after the oxidative cyclization (C1 to M3), most likely during the olefin insertion (M5 to M6).
These observations led to the discovery that additives can be used to improve the enantioselectivity of the [2+2+2] cycloaddition of alkenyl isocyanates and tolanes, with methyl nicotinate (20) being optimum (Scheme 12).32 The data suggest that the additive affects the environment of the rhodium(III) intermediate, and thus the enantioselectivity, by replacing the second alkyne as the spectator ligand in the reaction (see M13 in Fig. 4), prior to olefin insertion.
Scheme 12.

Enantioselective Rh-catalyzed [2+2+2] cycloaddition of alkenyl isocyanates and tolanes
A series of tolanes are compatible in this reaction. For example cycloadducts 21a and 21b are isolated with 93% ee and 92% ee respectively, compared to 84% and 76% ee without any additive. Importantly, the large variation in ee seen in reactions without additive is eliminated when the cycloaddition is conducted in the presence of 20.
4.2 Internal Unsymmetrical Alkynes
Insertion of unsymmetrical alkynes in metal-catalyzed [2+2+2] cycloadditions is not without precedent. However, most reports only apply to carbocycle synthesis33 and only a few concern heterocycle formation.34 As one can imagine, the main challenge in both cases is to achieve regioselective insertion.
From the previous results, it seems clear that the nature of the alkyne as well as the ligand have a profound influence on the product selectivity and the regioselectivity in the [2+2+2] cycloaddition with alkenyl isocyanates (cf. Schemes 4 and 5). However, all the above studies concern terminal alkynes with a very large difference in both steric bulk and electronic contributions. These inherent differences are not necessarily prevalent in internal alkynes.
Alkynoates have been shown to undergo metal-catalyzed [2+2] and [2+2+2] cycloadditions in a regioselective fashion.35 We have demonstrated that the large electronic difference between esters and alkyl or aryl functionalities leads to high regioselection in the rhodium-catalyzed cycloaddition with mono- and 1,1-substituted alkenyl isocyanates (Scheme 13, 26 and 27).36 While all reactions proceed in excellent yields, there is a marked increase in enantioselectivity when disubstituted alkenes are used (26 vs 27). The scope of the reaction has also been extended to other carbonyl groups (not shown). Exploiting electronic control of the alkyne, it is possible to reverse the regioselectivity of insertion by using electron-donating substituents on the internal alkynes, such as an alkoxy group (27 vs 28). Smaller electronic difference, such as aryl versus alkyl, also allows for the reaction to proceed with high regioselectivity (29).
Scheme 13.

Enantioselective Rh-catalyzed [2+2+2] cycloaddition of alkenyl isocyanates and unsymmetrical internal alkynes
Interestingly, the use of alkynyl nitriles shows a possible influence of size on product selectivity. The nitrile is much smaller than an ester and this is likely the cause of a change in product selectivity in favor of the lactam adduct with excellent regioselectivity (30). In the absence of electron-donating or electron-withdrawing groups, steric control clearly takes place. In that particular case, the ligand seems to direct the orientation of both the isocyanate moiety and the alkyne, placing the larger substituent β to the carbonyl in the vinylogous amide product (31).
To rationalize the regioselectivity in these reactions, a simple steric model falls short. The successful insertion of many polarized alkynes suggests an electonic component to regioselectivity. In all cases, the stronger electron-donating group is placed β to the carbonyl in both the lactam and the vinylogous amide adducts (22 and 23). However, one cannot exclude sterics entirely, since the larger group also prefers to insert distal to the carbonyl (24 and 25).
5. Enantioselective Rhodium-Catalyzed [4+2+2] Cycloaddition of Dienyl Isocyanates with Terminal Alkynes
Formation of medium-sized rings has become increasingly important in organic synthesis due to their occurrence in a large number of biologically important natural products, including the manzamine alkaloids nakadomarin A and manzamine A (Fig. 5).37 Unlike the formation of commonsized rings, preparation of eight-membered rings has proven to be notoriously difficult due to entropic factors and high transannular strain.38 Fortunately, the use of transition metal-catalyzed cycloadditions allows one to overcome entropic factors through coordination of the substrates to a metal prior to reaction.39
Fig. 5.

Two manzamine alkaloids
Strategies such as [4+4], [6+2], [5+2+1] and [4+2+2] cycloadditions have all been elegantly used for the synthesis of various eight-membered carbocycles. The formation of eight-membered nitrogen-containing rings (azocines) has not been extensively explored, our group being the only one to report a successful approach. In fact, we provided a powerful solution by developing the first asymmetric synthesis of nitrogen-containing eight-membered rings by a rhodiumcatalyzed [4+2+2] cycloaddition.40
Traditionally, the bicyclo[6.3.0] ring systems are built stepwise including a ring closing metathesis to afford the eight-membered ring.37,41 Our strategy allows a new route to functionalized bicyclic azocines in one chemical step (Scheme 14). This enantioselective rhodium-catalyzed [4+2+2] cycloaddition of terminal alkynes and dienyl isocyanates affords azocine derivatives 33 in good yields and exceptional enantioselectivities. In this case, no product corresponding to a CO migration has been observed. Cycloaddition of isocyanates with substitution at the diene portion is also possible.
Scheme 14.

Enantioselective Rh-catalyzed [4+2+2] cycloaddition of dienyl isocyanates and terminal alkynes
Our current mechanistic hypothesis is outlined in Scheme 15. The terminal alkyne and the isocyanate moiety of 32 coordinate to the rhodium(I) leading to the formation of metallacycle M14 after oxidative cyclization. Coordination of the diene followed by migratory insertion and reductive elimination affords cycloadduct 33.
Scheme 15.

Proposed mechanism for the Rh-catalyzed [4+2+2] cycloaddition of dienyl isocyanates
6. Conclusions
Metal-catalyzed asymmetric [m+n] and [m+n+o] cycloadditions has been intensively studied over the past three decades. However, the use of C-N π-components has proven to be more challenging. Recent achievements accomplished by our group in this field have demonstrated that the [n+2+2] cycloaddition of alkenyl isocyanates (and carbodiimides) with different alkynes provides a powerful option for the rapid formation of complex heterocycles in excellent yields and enantioselectivities. Moreover, extensive ligand optimizations led to a broader scope of heterocycles since one can control the selective formation of the bicyclic lactams or vinylogous amides, the latter arising from a CO migration process during the cycloaddition. To date, the synthetic utility of these transformations has been demonstrated by the rapid total syntheses of indolizidine (−)-209D and (+)-lasubine II. Additional studies on the reaction scope and mechanism to allow a fully intermolecular three-component cycloaddition, as well as the introduction of other heterocumulenes should provide a much more powerful tool for the synthesis of complex alkaloids and other heterocycles.
Footnotes
Part of the rapid formation of molecular complexity in organic synthesis themed issue.
Notes and references
- 1.Pfaltz A. Cyclopropanation. In: Beller M, Bolm C, editors. Transition Metals for Organic Synthesis. Vol. 1. Wiley-VCH; Weinheim: 2004. p. 157. [Google Scholar]; Lebel H, Marcoux J-F, Molinaro C, Charette AB. Chem. Rev. 2003;103:977. doi: 10.1021/cr010007e. [DOI] [PubMed] [Google Scholar]
- 2.Robinson JE. Rhodium(I)-Catalyzed [4+2] and [4+2+2] Carbocyclizations. In: Evans PA, editor. Modern Rhodium-Catalyzed Organic Reactions. Vol. 241. Wiley-VCH; Weinheim: 2005. [Google Scholar]
- 3.Wender PA, Gamber GG, Williams TJ. Rhodium(I)-Catalyzed [5+2], [6+2] and [5+2+1] Cycloadditions: New Reactions for Organic Synthesis. In: Evans PA, editor. Modern Rhodium-Catalyzed Organic Reactions. Vol. 263. Wiley-VCH; Weinheim: 2005. [Google Scholar]
- 4.Strbing D, Beller M. Pauson-Khand Reactions. In: Beller M, Bolm C, editors. Transition Metals for Organic Synthesis. Vol. 1. Wiley-VCH; Weinheim: 2004. p. 619. [Google Scholar]; Brummond KM, Kent JL. Tetrahedron. 2000;56:3263. [Google Scholar]
- 5.Galan BR, Rovis T. Angew. Chem. Int. Ed. 2009;48:2830. doi: 10.1002/anie.200804651. and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Möβner C, Bolm C. Catalyzed Asymmetric Aziridinations. In: Beller M, Bolm C, editors. Transition Metals for Organic Synthesis. Vol. 2. Wiley-VCH; Weinheim: 2004. p. 389. [Google Scholar]
- 7.Savizky RM, Austin DJ. Rhodium(II)-Catalyzed 1,3-Dipolar Cycloaddition Reactions. In: Evans PA, editor. Modern Rhodium-Catalyzed Organic Reactions. Vol. 433. Wiley-VCH; Weinheim: 2005. [Google Scholar]
- 8.Nakamura I, Yamamoto Y. Chem. Rev. 2004;104:2127. doi: 10.1021/cr020095i. [DOI] [PubMed] [Google Scholar]
- 9.For reviews on the synthesis of isocyanates and their reactions, see: Braunstein P, Nobel D. Chem. Rev. 1989;89:1927. Ozaki S. Chem. Rev. 1972;72:457.
- 10.Hong P, Yamazaki H. Tetrahedron Lett. 1977;18:1333. [Google Scholar]
- 11.Hoberg H, Bärhausen D, Mynott R, Schroth G. J. Organomet. Chem. 1991;410:117. and references therein. [Google Scholar]; Hoberg H, Ballesteros A, Sigan A, Jegat C, Milchereit A. Synthesis. 1991;395 and references therein. [Google Scholar]
- 12.Earl RA, Vollhardt KPC. J. Org. Chem. 1984;49:4786. and references therein. [Google Scholar]
- 13.Yamamoto Y, Kinpara K, Saigoku T, Takagishi H, Okuda S, Nishiyama H, Itoh K. J. Am. Chem. Soc. 2005;127:605. doi: 10.1021/ja045694g. and references therein. [DOI] [PubMed] [Google Scholar]
- 14.Duong HA, Cross MJ, Louie J. J. Am. Chem. Soc. 2004;126:11438. doi: 10.1021/ja046477i. [DOI] [PubMed] [Google Scholar]
- 15.Takahashi T, Tsai F-Y, Li Y, Wang H, Kondo Y, Yamanaka M, Nakajima K, Kotora M. J. Am. Chem. Soc. 2002;124:5059. doi: 10.1021/ja017507+. [DOI] [PubMed] [Google Scholar]
- 16.Tanaka K, Takahashi Y, Suda T, Hirano M. Synlett. 2008;1724 [Google Scholar]; Tanaka K, Wada A, Nogushi K. Org. Lett. 2005;7:4737. doi: 10.1021/ol052041b. [DOI] [PubMed] [Google Scholar]
- 17.Daly JW. J. Med. Chem. 2003;46:445. doi: 10.1021/jm0204845. [DOI] [PubMed] [Google Scholar]; Daly JW, Spande TF, Garraffo HM. J. Nat. Prod. 2005;68:1556. doi: 10.1021/np0580560. [DOI] [PubMed] [Google Scholar]
- 18.For reviews of recent syntheses, see: Michael JP. Nat. Prod. Rep. 2008;25:139. doi: 10.1039/b612166g. and references therein.
- 19.Yu RT, Rovis T. J. Am. Chem. Soc. 2006;128:2782. doi: 10.1021/ja057803c. [DOI] [PubMed] [Google Scholar]
- 20.For selected CO migration references on rhodium(I) species, see: Gonsalvi L. Organometallics. 2003;22:1047. Gonsalvi L, Adams H, Sunley GJ, Ditzel E, Haynes A. J. Am. Chem. Soc. 2002;124:13597. doi: 10.1021/ja0176191.; Cavallo L, Sola M. J. Am. Chem. Soc. 2001;123:12294. doi: 10.1021/ja016468z.
- 21.Lee C-C, Lin Y-C, Liu Y-H, Wang Y. Organometallics. 2005;24:136. and references therein. [Google Scholar]
- 22.Dalton DM, Oberg KM, Yu RT, Lee EE, Perreault S, Oinen ME, Pease ML, Malik G, Rovis T. doi: 10.1021/ja905065j. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.For reviews on the use of phosphoramidite ligands, see: Jerphagnon T, Renaud J-L, Bruneau C. Tetrahedron: Asymmetry. 2004;15:2101.; Feringa B. Acc. Chem. Res. 2000;33:346. doi: 10.1021/ar990084k.
- 24.Yu RT, Rovis T. J. Am. Chem. Soc. 2006;128:12370. doi: 10.1021/ja064868m. [DOI] [PubMed] [Google Scholar]
- 25.Lee EE, Rovis T. Org. Lett. 2008;10:1231. doi: 10.1021/ol800086s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu RT, Lee EE, Malik G, Rovis T. Angew. Chem. Int. Ed. 2009;48:2379. doi: 10.1002/anie.200805455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reddy PG, Baskaran S. J. Org. Chem. 2004;69:3093. doi: 10.1021/jo035258x. and references therein. [DOI] [PubMed] [Google Scholar]
- 28.Oberg KM, Lee EE, Rovis T. Tetrahedron. 2009;65:5056. doi: 10.1016/j.tet.2009.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wakatsuki Y, Nomura O, Kitaura K, Morokuma K, Yamazaki H. J. Am. Chem. Soc. 1983;105:1907. [Google Scholar]
- 30.Stockis A, Hoffmann R. J. Am. Chem. Soc. 1980;102:2952. [Google Scholar]
- 31.Yu RT, Rovis T. J. Am. Chem. Soc. 2008;130:3262. doi: 10.1021/ja710065h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oinen ME, Yu RT, Rovis T. submitted. [Google Scholar]
- 33.Zhang D, Liu Z, Yum EK, Larock RC. J. Org. Chem. 2007;72:251. doi: 10.1021/jo0620563. [DOI] [PubMed] [Google Scholar]; Wender PA, Gamber GG, Hubbard RD, Zhang L. J. Am. Chem. Soc. 2002;124:2876. doi: 10.1021/ja0176301. and references therein. [DOI] [PubMed] [Google Scholar]
- 34.Boren BC, Narayan S, Rasmussen L, Zhang L, Zhao H, Lin Z, Jia G, Fokin VV. J. Am. Chem. Soc. 2008;130:8923. doi: 10.1021/ja0749993. [DOI] [PubMed] [Google Scholar]; Duong HA, Louie J. J. Organomet. Chem. 2005;690:5098. [Google Scholar]; Earl RA, Vollhardt KPC. J. Am. Chem. Soc. 1983;105:6991. and references therein. [Google Scholar]
- 35.Evans PA, Lai KW, Sawyer JR. J. Am. Chem. Soc. 2005;127:12466. doi: 10.1021/ja053123y. [DOI] [PMC free article] [PubMed] [Google Scholar]; Shibata T, Takami K, Kawachi A. Org. Lett. 2006;8:1343. doi: 10.1021/ol060055r. [DOI] [PubMed] [Google Scholar]
- 36.Keller Friedman R, Rovis T. J. Am. Chem. Soc., ASAP [Google Scholar]
- 37.For two selected syntheses of manzamine alkaloids, see: Young IS, Kerr M. J. Am. Chem. Soc. 2007;129:1465. doi: 10.1021/ja068047t. Humphrey JM, Liao Y, Ali A, Rein T, Wong Y-L, Chen H-J, Courtney A,K, Martin SF. J. Am. Chem. Soc. 2002;124:8584. doi: 10.1021/ja0202964.
- 38.Illuminati G, Mandolini L. Acc. Chem. Res. 1981;14:95. [Google Scholar]; Engler EM, Andose JD, Schleyer P. v. R. J. Am. Chem. Soc. 1973;95:8005. [Google Scholar]
- 39.Yet L. Chem. Rev. 2000;100:2963. doi: 10.1021/cr990407q. [DOI] [PubMed] [Google Scholar]
- 40.Yu RT, Keller Friedman R, Rovis T. submitted. [Google Scholar]
- 41.Michaut A, Rodriguez J. Angew. Chem. Int. Ed. 2006;45:5740. doi: 10.1002/anie.200600787. [DOI] [PubMed] [Google Scholar]