

Abstract
Plasmodium falciparum, in addition to scavenging essential fatty acids from its intra- and intercellular environments, possesses a functional complement of type II fatty acid synthase (FAS) enzymes targeted to the apicoplast organelle. Recent evidence suggests that products of the plasmodial FAS II system may be critical for the parasite's liver-to-blood cycle transition, and it has been speculated that endogenously generated fatty acids may be precursors for essential cofactors, such as lipoate, in the apicoplast. β-Ketoacyl-acyl carrier protein (ACP) synthase III (pfKASIII or FabH) is one of the key enzymes in the initiating steps of the FAS II pathway, possessing two functions in P. falciparum: the decarboxylative thio-Claisen condensation of malonyl-ACP and various acyl coenzymes A (acyl-CoAs; KAS activity) and the acetyl-CoA:ACP transacylase reaction (ACAT). Here, we report the generation and characterization of a hybrid Lactococcus lactis strain that translates pfKASIII instead of L. lactis fabH to initiate fatty acid biosynthesis. The L. lactis expression vector pMG36e was modified for the efficient overexpression of the plasmodial gene in L. lactis. Transcriptional analysis indicated high-efficiency overexpression, and biochemical KAS and ACAT assays confirm these activities in cell extracts. Phenotypically, the L. lactis strain expressing pfKASIII has a growth rate and fatty acid profiles that are comparable to those of the strain complemented with its endogenous gene, suggesting that pfKASIII can use L. lactis ACP as substrate and perform near-normal function in L. lactis cells. This strain may have potential application as a bacterial model for pfKASIII inhibitor prescreening.
The rapid replication of Plasmodium falciparum in infected hosts requires a continuous supply of lipids. As lipids are abundant in blood serum and hepatic tissues, it was initially presumed that plasmodia would likely not possess innate fatty acid biosynthesis machinery and instead rely on salvaging lipids from host organisms (16). This assumption was questioned upon the sequencing of the P. falciparum genome (3), which revealed that plasmodia possess an intact endogenous type II fatty acid biosynthesis pathway (FAS II) for de novo synthesis of fatty acids (Fig. 1) (6, 10, 12). Enzymes of the FAS II system of P. falciparum were subsequently determined to be targeted to the apicoplast organelle via bipartite leader peptides, suggesting a critical role in parasite growth and development (6, 12, 16a).
FIG. 1.
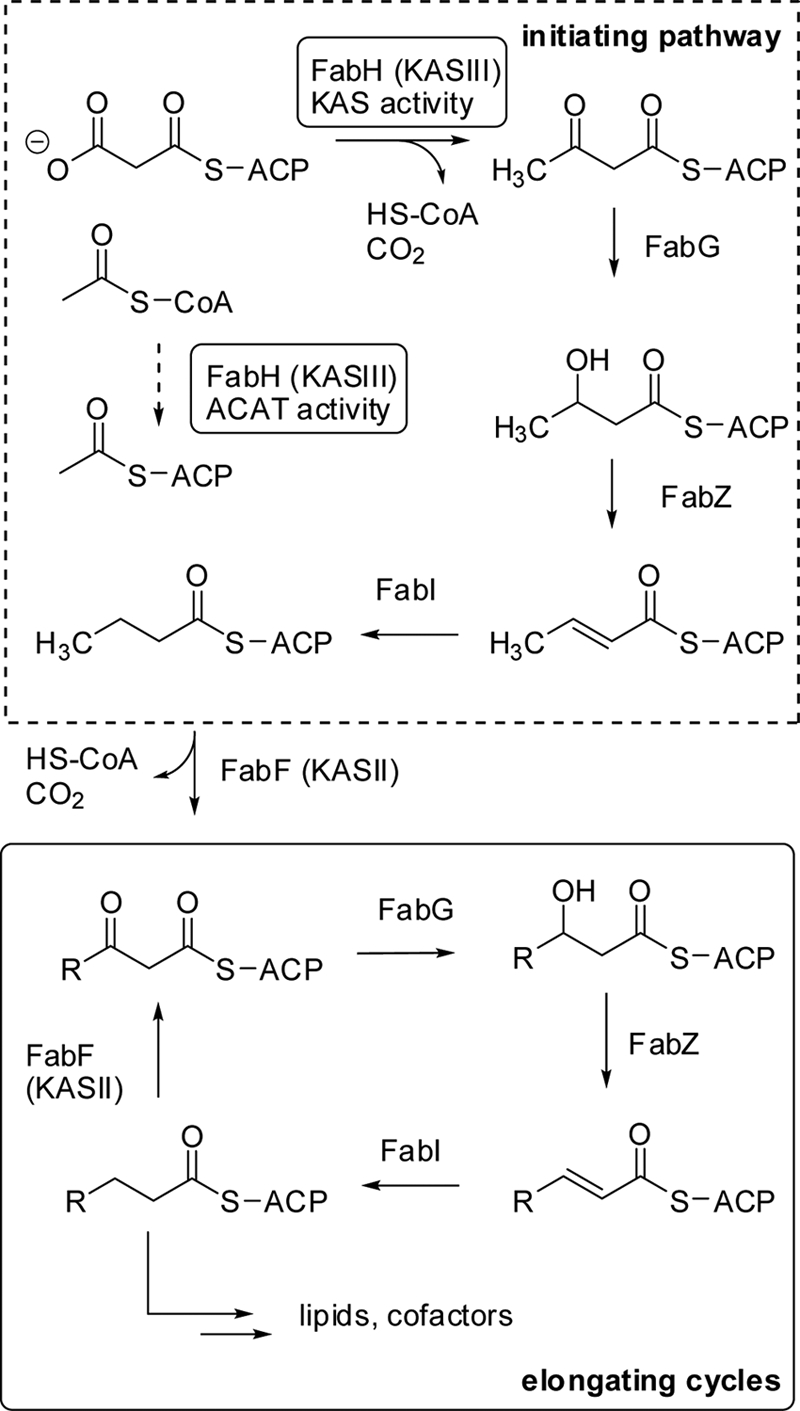
The initiating step of de novo fatty acid biosynthesis is mediated by β-ketoacyl-ACP synthase III (KASIII or FabH), which selectively condenses acetyl-CoA and malonyl-ACP. Malonyl-ACP is generated via malonyl-CoA:ACP transferase (MCAT or FabD [not shown]). The product of pfKASIII is further elongated via the iterative action of β-ketoacyl-ACP synthase II (KASII or FabF), β-ketoacyl-ACP reductase (FabG), β-hydroxyacyl-ACP dehydratase (FabZ), and enoyl-ACP reductase (FabI) activities.
The importance of FAS II enzymes in plasmodial development in infected hosts remains unclear. A number of compounds with FAS II enzyme inhibitory activity and in vitro blood stage antiplasmodial assay results have been reported, including inhibitors of FabI and FabH (pfKASIII in P. falciparum) (6, 8). However, recent data demonstrate that genetic knockouts of fatty acid biosynthetic genes encoding FabI, FabF, and FabZ, each generating an ultimate compromise of the FAS II pathway, have little or no effect on the blood stage replication of various plasmodia (15, 22). These data suggest that inhibitors of FabI and FabH likely exert their activity in the blood stage via an alternative mechanism. A similar FAS II independence in a blood context has been observed in the bacterial pathogen Staphylococcus aureus (1), questioning the utility of FAS II-targeting drugs as a general therapeutic strategy.
Unlike S. aureus, P. falciparum propagates via several distinct developmental stages. Recent detailed genetic analysis of the plasmodial FAS II pathway suggests that it may indeed be critical for pathogenicity. Notably, several FAS II-deficient Plasmodium yoelii strains have been shown to be largely incompetent in the developmental transition from liver stage trophozoite into erythrocytic merozoites (15, 22). Consequently, it has been speculated that interrupting this key stage of the infective life cycle may have therapeutic potential.
A lynchpin enzyme in the plasmodial FAS II cycle is the initiating β-ketoacyl-acyl carrier protein (ACP) synthase III (pfKASIII or FabH) (Fig. 1), which primarily catalyzes the decarboxylative condensation of malonyl-acyl carrier protein (mal-pfACP) and acetyl coenzyme A (acetyl-CoA; KAS activity) and secondarily catalyzes acetyl-CoA:pfACP transacylase activity (ACAT activity) (10, 19). To study the function of pfKASIII in the context of an intact type II FAS pathway in vivo, we endeavored to insert the encoding gene into bacterial fabH knockout strains. Our initial studies of fabH deletions in B type E. coli were complicated by the propensity of these strains to readily acquire bypass mutations, a property that has also been previously observed by J. E. Cronan and coworkers (personal communication). As L. lactis fabH mutants that do not display the propensity to form similar bypass mutations have been reported (5), perhaps due to this organism's more compact genome (2.3 Mb; GenBank accession number AE005176), we have developed a pfKASIII expression vector for L. lactis and used it to complement an L. lactis ΔfabH strain (5). Transcriptional analysis demonstrates high copy number of pfKASIII-encoding mRNA in the transformed L. lactis strain. While the ΔfabH strain is dependent upon exogenously added fatty acids for growth, the pfKASIII-complemented strain grew robustly in media lacking fatty acids. Consistent with this phenotype, significant pfKASIII activity of cell extracts of chimeric strains was observed along with ACAT activity relative to that of the knockout strain. Analysis of membrane fatty acids of wild-type and chimeric strains indicates that membrane composition is largely unaltered relative to that of L. lactis complemented with its wild-type fabH.
Together, these data suggest that the L. lactis-Plasmodium chimera contains an intact FAS II system dependent upon pfKASIII for the initiation of de novo fatty acid biosynthesis. The chimera was further evaluated as a bioassay strain for FAS II inhibitor prescreening.
MATERIALS AND METHODS
Materials.
All chemicals were obtained from Sigma-Aldrich unless otherwise indicated. Restriction enzymes were obtained from New England Biolabs, and M17 medium was purchased from Difco (catalog number 218561).
Strains, plasmids, and culture conditions.
L. lactis CL112 possesses a 36-bp in-frame deletion in the active-site-encoding region of chromosomal fabH in L. lactis IL1403. Escherichia coli TOP10 cells were from Invitrogen (catalog number 553001A). All E. coli strains were grown in LB medium according to standard protocols. L. lactis strains were maintained in GM17 medium (M17 medium plus 0.5% glucose) or SGM17 medium (M17 medium plus 0.5% glucose plus 0.5 M sucrose) with or without added sodium oleate or bovine serum fatty acids, as necessary.
Synthesis and modification of the gene encoding pfKASIII.
The codon-optimized 972-bp P. falciparum fabH gene was synthesized based on the deposited protein sequence (GenBank accession number AF038929; protein identification number AAC63960.1) by DNA2.0 Inc., and restriction sites for cloning into expression vector pMG36e (SalI and HindIII) (14) were subsequently appended to the synthetic gene along with a stop codon and ribosomal binding site via PCR by using the primers pfKAS-36e-s2 and pfKAS-36e-a (Table 1).
TABLE 1.
Bacteria strains, plasmids, and PCR primers
| Strain, plasmid, or primer | Genotype, sequence, or properties | Reference or source |
|---|---|---|
| Strains | ||
| E. coli | ||
| TOP10 | Used as a cloning strain; F−mcrA Δ(mrr-hsdRMS-mcrBC) φ80lacZΔM15 ΔlacX74 nupG recA1 araD139 Δ(ara-leu)7697 galE15 galK16 rpsL (Strr) endA1 λ− | Invitrogen |
| BL21 Star(DE3) | F−ompT hsdSB (rB−mB−) gal dcm rne131 (DE3) | Invitrogen |
| L. lactis | ||
| IL1403 | Wild type | 5 |
| CL112 | IL1403 with a 36-bp in-frame L. lactis fabH deletion (ΔfabH) | 5 |
| YD037 | CL112 carrying pYD37; Emr | This study |
| YD038 | CL112 carrying pYD38; Emr | This study |
| Plasmids | ||
| pMAL-c2x | Maltose-binding protein expression vector | New England Biolabs |
| pSTP1 | pMAL-c2x with P. falciparum acp | 10 |
| pSTP4 | pMAL-c2x with P. falciparum mcat | 10 |
| pMAL-cHT | pMAL-c2x with encoding TEV cleavage site and histidine tag | 9 |
| pSTP5 | pMAL-cHT with P. falciparum fabH | 10 |
| pSPr022 | pMAL-cHT with P. falciparum acp | This study |
| pMG36e | pGKV432 promoter/pWV01 ori; Emr; expression vector in L. lactis | 14 |
| pYD37 | pMG36e with P. falciparum fabH | This study |
| pYD38 | pMG36e with L. lactis fabH | This study |
| Primers | ||
| pfACP-cHT-s | 5′-GGTGGTGAATTCAGCTCTTTAAAAAGTACTTTTGATG-3′ | This study |
| pfACP-cHT-a | 5′-GGTGGTGTCGACTTATTGCTTATTATTTTTTTCTATATAATC-3′ | This study |
| pfKAS-36e-s2 | 5′-ATGTCGACCTAATGATTAACTTTATAAGGAGGAAAAACATATGGGAGGCAAGATTATC-3′ | This study |
| pfKAS-36e-a | 5′-GAATTCAAGCTTTTAGTATTTTAAAATAACAC-3′ | This study |
| ll-FabH-s | 5′-ATGTCGACCTAATGATTAACTTTATAAGGAGGAAAAACATATGACTTTTGCGAAAATTACG-3′ | This study |
| ll-FabH-a | 5′-TTCCTGCAGTTATAAATTAATAATTGCTGTACC-3′ | This study |
| llKASRealF | 5′-ACGGGTTCAAGCTCAAGT-3′ | This study |
| llKASRealR | 5′-AATGACAAGCCCTCGTTG-3′ | This study |
| pfKASRealF | 5′-TCTTATCGTAGGGTCTGATGC- 3′ | This study |
| pfKASRealR | 5′-CGGCATCCCCAAATAGAA-3′ | This study |
| llpheSRealF | 5′-CCCGAAAGCAAAACCAGA-3′ | This study |
| llpheSRealR | 5′-GGAAAAGGCTGTAACGTCTG-3′ | This study |
L. lactis electrocompetent cell preparation.
L. lactis strains were inoculated via colony or glycerol stock into medium GM17O (M17 medium plus 0.5% glucose plus 50 μg/ml sodium oleate) and incubated with shaking at 30°C to an optical density at 600 nm (OD600) of 0.5 to 0.8 (∼40 h). A total of 600 μl of this seed culture was inoculated into 30 ml of GM17O with 0.5% glycine supplementation. After growing at 30°C to an OD600 of 0.6 to 0.8 (3 to 4 h), the cells were incubated on ice for 20 min and harvested by centrifugation (4°C at 3,000 × g for 20 min). Following two washes in ice-cold sterile 0.5 M sucrose-10% glycerol, the cells were gently resuspended in 1/100 culture volume of 0.5 M sucrose-10% glycerol, aliquoted (50 μl/Eppendorf tube), and stored at −80°C.
Ligation of pfKASIII-encoding gene and pMG36e vector and transformation into L. lactis CL112.
Both the gene encoding pfKASIII and the vector pMG36e were digested with SalI/HindIII, and the gel-purified DNA fragments were ligated using T4 DNA ligase (NEB). Individual ligation reactions were desalted by sodium acetate/isopropanol precipitation, and the resulting DNA pellets were resuspended in sterile water prior to transformation. Electroporation was performed using a Bio-Rad Gene Pulser Xcell with a PC module. Competent cell suspensions were mixed with desalted ligation reaction mixtures (1 to 10 μl, in water), transferred to an ice-cooled electroporation cuvette (2-mm electrode gap), and exposed to a single electrical pulse (3.0 kV, 25 μF, 200 Ω, 5 ms). Immediately following the discharge, the suspension was mixed with 1 ml of SGM17OMC(E) broth (M17 medium plus 0.5% glucose, 0.5 M sucrose, 50 μg/ml sodium oleate, 20 mM MgCl2, 2 mM CaCl2, and 50 ng/ml erythromycin) and incubated on ice for 5 min. The cells were then incubated with shaking at 30°C for 2 h. A total of 100 to 150 μl was spread onto SGM17OE agar (M17 medium plus 0.5% glucose, 0.5 M sucrose, 50 μg/ml sodium oleate, 5 μg/ml erythromycin, and 2% agar) and incubated at 30°C. Colonies appeared after 24 to 48 h. One of the erythromycin-resistant transformants, harboring the pfKASIII expression plasmid pYD37, was designated YD037 and used in the following studies.
Cloning and modification of the L. lactis fabH gene.
The L. lactis fabH gene (GenBank accession number NC_002662) was amplified from the genomic DNA of L. lactis IL1403 by PCR by the use of the primers ll-FabH-s and ll-FabH-a (Table 1). The resulting 978-bp DNA fragment and pMG36e were restricted with SalI/PstI and purified by gel electrophoresis. Ligation reactions were transformed into the fabH knockout strain CL112 as described above. The resulting strain, L. lactis YD038, which carries the L. lactis fabH expression plasmid pYD38, was used in the following studies.
Confirmation of the L. lactis transformants via PCR.
The integrity of the inactivated fabH gene in the constructed L. lactis strain YD037 was confirmed by PCR and sequencing. PCR was performed on purified genomic DNA (Promega Wizard kit, number A1120) with primers ll-FabH-s and ll-FabH-a (Table 1). Sequenced PCR products demonstrated that the fabH gene lacks the 36 bp deleted in the fabH knockout and that this genotype is stable in YD037. The presence of insert in plasmids pYD37 and pYD38 in L. lactis strains YD037 and YD038 was similarly confirmed by PCR and DNA sequencing. Plasmid DNA was purified from both strains, and PCR was performed with primers pfKAS-36e-s2 and pfKAS-36e-a (for YD037) or ll-FabH-s and ll-FabH-a (for YD038). The PCR products were sequenced to demonstrate that both strains contain the expected plasmids.
RT-PCR analysis of L. lactis strains.
Total mRNA was purified from log-phase cultures of L. lactis IL1403 (wild type), CL112 (ΔfabH mutant), YD037 (pfKASIII-complemented strain), and YD038 (L. lactis FabH-complemented strain) by using the RNeasy mini kit from Qiagen (catalog number 74104). cDNA was generated from each mRNA preparation by using reverse transcriptase. Reverse transcriptase PCR (RT-PCR) analysis was performed by the Molecular Genetics Core at Vanderbilt University, using an ABI PRISM7700 sequence detection system. Primers were designed based on the sequences of the codon-optimized gene encoding pfKASIII (P. falciparum 3D7) and L. lactis FabH (L. lactis IL1403). A housekeeping gene, pheS of L. lactis IL1403 (GenBank accession number NC_002662; encoding phenylalanine tRNA synthetase) (11), was used as a reference, and the L. lactis fabH transcription level in IL1403 (compared with that of pheS) was used as a calibrator. RT-PCR primers llKASRealF, llKASRealR, pfKASRealF, pfKASrealR, llpheSRealF, and llpheSRealR are described in Table 1. SYBR green was used to monitor DNA synthesis, and the cycle points at which the fluorescence crossed the detection threshold (CT values) were measured. Three parallel experiments of each sample were performed, and the data were analyzed using the comparative CT method of relative quantification (13). The relative amount of target, normalized to an endogenous reference and relative to a calibrator, is given by the following equation: expression fold value = 2−ΔΔCT, where ΔΔCT = ΔCTq − ΔCTcb, where q corresponds to the target PCR product and cb is the calibrator product; ΔCT = CTX − CTR, the difference in threshold cycles for target and reference.
Growth of L. lactis YD037 and YD038.
Determination of the growth rates of the L. lactis strains were performed as follows. Three microliters of glycerol stock was inoculated into 3 ml of GM17 medium containing 0 or 5 μg/ml erythromycin and/or 0 or 50 μg/ml sodium oleate. The seed cultures were incubated at 30°C for 20 h and then inoculated into 30 ml of fresh GM17 medium containing 0 or 50 μg/ml sodium oleate and 0 or 5 μg/ml erythromycin, resulting in final OD600 values ranging from 0.01 to 0.03. The cultures were incubated with shaking at 30°C, and 100-μl samples were withdrawn for measurements of OD600 every hour. Relative growth curves were rendered based on these data.
Expression and purification of pfACP, pfMCAT, and pfKASIII.
A truncated form of pfACP (amino acids S57 to Q137, with the putative signal/transit peptide removed) was subcloned from a previously described plasmid (pSTP1) (10). After digestion with EcoRI and SalI, pfACP was ligated into the expression vector, pMAL-cHT, which has been modified to allow for in vivo cleavage of the MBP fusion partner from the recombinant protein, exposing a His6 tag (9). This construct, pSPr022, was transformed into BL21 Star (DE3) cells (Invitrogen), which already contained plasmids pRK586 (encoding tobacco etch virus [TEV] protease [4]) and pRIL isolated from BL21-CodonPlus (DE3) cells (Stratagene). Cell cultures were grown at 37°C to an OD600 of 0.6. The cultures were then transferred to 20°C and induced with 0.4 mM isopropyl-β-d-thiogalactopyranoside (IPTG) for 10 h. Cells were harvested by centrifugation and stored at −20°C. The frozen cell pellets were resuspended in lysis buffer (20 mM phosphate [pH 7.5], 200 mM NaCl, 1 mg/ml lysozyme, 2.5 μg/ml DNase I, and 1 mM phenylmethylsulfonyl fluoride [PMSF]) and sonicated. The resulting lysate was centrifuged to remove the insoluble fraction, and the supernatant was loaded onto a 5-ml HisTrap FF chelating column (GE Healthcare). Protein bound to the column was eluted using imidazole. Fractions containing pfACP (identified by SDS-PAGE) were dialyzed against 20 mM phosphate (pH 7.5) and 10 mM dithiothreitol (DTT) overnight at 4°C. pfACP was further purified using a 5-ml HiTrap Q HP column (GE Healthcare) equilibrated with 20 mM phosphate (pH 6.5) and 10 mM DTT. Fractions containing pfACP were concentrated and stored at −80°C. pfMCAT was purified as described by Prigge et al. (10), using the truncated construct of pfMCAT encoding residues 104 to 408 (pSTP4). pfKASIII was purified as described by Prigge et al. (10), using the truncated construct of pfKASIII encoding residues 50 to 371 (pSTP5).
Total protein preparation.
ACAT enzyme activity assays were conducted on total protein preparations generated by ammonium sulfate precipitation of cell extracts of IL1403 (wild type), CL112 (ΔfabH strain), and YD037 (complemented with pfKASIII) as follows. Ten microliters of glycerol stocks of the above-described strains was inoculated into 10 ml of GM17 medium (for CL112, 50 μg/ml sodium oleate was added) and cultured at 30°C for 20 h. The 10-ml seed cultures were inoculated into 500 ml of fresh GM17 medium (for CL112, 50 μg/ml sodium oleate was added). The 500-ml cultures were shaken at 30°C, and 100-μl aliquots were removed every hour for OD600 measurements. Relative growth curves were rendered, and cells were collected by centrifugation when the growth curves reached the mid-logarithmic phase. All subsequent steps were performed at 4°C unless otherwise noted. Cells were collected by centrifugation (3,000 × g for 20 min) and washed with 20 ml of 20 mM Tris-HCl (pH 7.5) buffer. Cells were resuspended in 10 ml of 20 mM Tris-HCl (pH 7.5) buffer with 0.2 mg/ml lysozyme, and protease inhibitor cocktail (Roche) was added according to the manufacturer's instructions. Cell suspensions were incubated at 37°C for 30 min for partial digestion of cell walls, and the cells were then disrupted by sonication in an ice bath (Fisher sonicator F550; 20% amplitude, 5 s on, 5 s pulsing, for 5 min). The cell-free extractions were obtained by centrifugation at 20,000 × g for 30 min. Cell extracts were precipitated with 75% saturated ammonium sulfate at 0°C. The precipitated protein was collected by centrifugation at 20,000 × g for 30 min, and the protein pellets were used for acetyl-CoA:ACP transacylase (ACAT) activity assays.
ACAT assay.
The ACAT assay was performed in a 96-well plate format as previously described by Lee et al. (6), with some minor modifications. Briefly, the assay is a radioactive 96-well plate assay that measures pfKASIII activity by detecting the transfer of the radiolabeled carbon in 14C-acetyl-CoA to the acyl carrier protein (pfACP), forming 14C-acetyl-ACP. Greiner black nonbinding plates were used for all reactions and Millipore MultiScreen HTS filter plates for filtration washing of unincorporated radiolabel. Optimal concentrations of pfKASIII enzyme (0.00314 mg/ml), 14C-acetyl-CoA (26 μM; 0.064 mCi/mMol per reaction), and pfACP (10 μl per reaction) and the reaction incubation time (20 min) were experimentally determined. To determine IL1403, CL112, and YD037 lysate activities in comparison to the lysate activity of the purified pfKASIII enzyme by using the ACAT assay, each lysate pellet was resuspended in 100 μl of buffer and titrated over 11 wells in duplicate. One well was used to determine the background cpm with no enzyme or lysate added.
The purified pfKASIII enzyme and three lysates (IL1403, CL112, and YD037) were all tested with a background (no enzyme) and concentrations above and below the 1× concentration of the enzyme or lysate. All reactions were performed in duplicate. Two microliters of purified pfKASIII enzyme was added to each well. When adding the enzyme or lysate in excess, we chose to add 3.5 μl and 7 μl at the 1× concentration. In order to obtain the serial dilution of enzyme and lysate, we used another plate to do a 2-fold serial dilution of the enzyme and lysate and then transferred 2 μl of all concentrations to the assay plate.
Whole-cell lysate KAS assays using pfACP and pfMCAT.
In order to test the KAS activity of the L. lactis IL1403, CL112, and YD37 strains described above, 200 μl of culture (at an OD600 of 1) per reaction were centrifuged and stored at −20°C. These cell pellets were resuspended in BugBuster (Novagen) and 1 mg/ml lysozyme and incubated for 10 min at room temperature. After this lysis step, the whole-cell lysate was diluted with reaction buffer (100 mM morpholineethanesulfonic acid [MES], 2 mM 2-mercaptoethanol) to a final volume of 10 μl per reaction, with a final concentration of 20% BugBuster. In order to test the KAS activity of these lysates, an MCAT reaction mixture was incubated at room temperature for 20 min (117.6 μM pfACP, 28.6 μM pfMCAT, and 1.5 mM malonyl-CoA in 100 mM MES [pH 6.5] and 2 mM 2-mercaptoethanol). A negative-control assay without pfACP was performed. After the MCAT incubation, 21.65 μM 1-14C-acetyl-CoA (specific activity of 60.4 mCi/mmol; Moravek Biochemicals) was added to the MCAT reactions. Two microliters was added to each whole-cell lysate reaction. For the complete KAS reaction, the final concentrations were 30 μM pfACP, 5 μM pfMCAT, 0.25 mM malonyl-CoA, and 3.6 μM 1-14C-acetyl-CoA. The control MCAT reaction mixture was identical to the complete reaction mixture except that it lacked pfACP. The KAS reactions were allowed to proceed at room temperature for 30 min. The reactions were quenched by adding 20% trichloroacetic acid (TCA) and precipitating labeled pfACP for 30 min at 4°C. The protein pellets were washed twice with 10% TCA, resuspended in 100 μl 100 mM NaOH. Each sample was dissolved in scintillation fluid, and 14C counts were obtained. The assay was performed in triplicate.
Fatty acid analysis.
L. lactis strains IL1403, CL112 (ΔfabH), YD037, and YD038 were streaked onto GM17 agar with or without sodium oleate (50 μg/ml) to obtain single colonies, and these single colonies were used for fatty acid methyl ester (FAME) analysis. The fatty acid analysis was performed by the Bacterial Strain-Identification and Mutant Analysis Service (BSI-MAS) at Auburn University, AL. Single colonies were subjected to an extraction procedure which involves disrupting the cell walls and membranes, methylating the fatty acids, and extracting them with an organic solvent. This collection of FAMEs was then analyzed on a fully automated gas chromatographic analytical system, and a comprehensive analysis of the fatty acid composition was then obtained. Each set of data was obtained from three independent experiments.
Isolation of fatty acids from bovine serum.
Serum fatty acid extraction was performed by the method of Yin et al. (20). Ten milliliters of bovine serum (Gibco; catalog number 16170-078) was diluted with 10 ml of 0.9% NaCl and extracted with 40 ml of chloroform-methanol (2/1 [vol/vol]). The organic layer was separated and dried in vacuo. The residue was reconstituted in 1 ml methanol, and 1 ml of 1 M aqueous potassium hydroxide was added to hydrolyze phospholipids. The mixture was incubated at 37°C for 30 min, was acidified to pH 3 with 1 M HCl, and extracted with heptane. The organic layer was dried in vacuum and dissolved in dimethyl sulfoxide (DMSO) to the desired concentration (2 to 20 mg/ml).
Antibiotic assays.
Twenty microliters of glycerol stocks of strain YD037 was mixed with 20 ml of GM17 broth and GM17 broth supplemented with either 60 μg/ml bovine serum fatty acid or 20 μg/ml sodium oleate. One-hundred-microliter aliquots of cell dilutions were transferred into 96-well plates containing the following concentration ranges of antibiotics: 0 to 20 μg/ml cerulenin, 0 to 0.8 μg/ml vancomycin, 0 to 38 μg/ml rifampin, and 0 to 80 μg/ml kanamycin. Plates were incubated at 30°C for 16 h, and the absorption at 600 nm for each well was measured to determine the percentage of growth inhibition.
RESULTS
Generation of P. falciparum fabH- and L. lactis fabH-complemented strains.
L. lactis CL112 (ΔfabH) was previously generated from L. lactis IL1403, a genomically sequenced strain, by a 36-bp in-frame deletion of the region encoding the FabH active site (5). Notably, L. lactis CL112 requires supplementation with long-chain unsaturated fatty acids (e.g., oleic acid) for growth.
As the preferred codon usage of P. falciparum differs substantially from that of L. lactis, the pfKASIII-encoding gene was chemically synthesized using L. lactis-preferred codons. A 972-bp DNA fragment encoding 323 amino acids lacking the apicoplast targeting amino-terminal leader peptide was synthesized based on the amino acid sequence of pfKASIII (19) (GenBank accession number AF038929; protein identification number AAC63960.1). The codon-optimized gene sequence has been deposited into GenBank under accession number GQ861515.
The L. lactis expression plasmid vector pMG36e was originally designed to clone a target gene as an in-frame fusion with an uncharacterized L. lactis subsp. cremoris Wg2 chromosomal gene, presumably for the purpose of enhancing overexpression of heterologous genes. Correspondingly, the short multiple-cloning-site-derived sequence in pMG36e is preceded by a 75-bp coding sequence (14). To express pfKASIII without the fusion sequence, an in-frame stop codon followed by a ribosomal binding site (RBS) was appended to flanking regions of the gene encoding pfKASIII via PCR. The modified 1-kb DNA fragment was cloned into pMG36e and electroporated into the L. lactis ΔfabH strain. Notably, the ΔfabH strain was remarkably intransigent to electroporation, requiring modified electroporation methods and relatively high electroporation voltages (3.0 kV). We speculate that the high concentration of fatty acids in the growth medium may interfere with electroporation. Transformants were selected and inoculated into GM17 medium containing erythromycin. Transformants were capable of growth without supplementation by oleate, while transformants containing empty vector (pMG36e) were not, suggesting that pfKASIII functionally complemented the L. lactis ΔfabH strain. A strain containing the native fabH gene was also constructed from CL112. The recovered L. lactis fabH strain, designated YD038, was also capable of growth in GM17 medium not supplemented with fatty acid.
As these pMG36e-derived vectors were not present in sufficient copy numbers to detect by analytical gel electrophoresis, the presence of the gene encoding pfKASIII or L. lactis FabH was confirmed by amplifying and sequencing these genes from the aforementioned transformants. The sequenced PCR products confirmed the presence of plasmid and insert in these strains. Genomic DNA was separately purified from the pfKASIII strain YD037, and PCR confirmed that the 36-bp deletion was intact in the genomic copy of disrupted fabH.
Growth of YD037 and YD038.
The competence of the chimeric strains was evaluated on GM17 agar with or without bovine serum fatty acid supplementation (Fig. 2a). A similar pattern was observed for supplementation with pure oleate. The recombinant strains grew at rates and confluent densities similar to those of the wild-type strain IL1403 in the absence of exogenous fatty acids, whereas CL112 (ΔfabH) was capable of growing only with added fatty acids. Corresponding growth curves in liquid cultures (Fig. 2b) confirm this phenotype. Without fatty acid supplementation, the growth rate of pfKASIII-complemented strains was not distinguishable from that of L. lactis FabH-complemented strains but was somewhat lower than that of wild-type strain IL1403. With fatty acid supplementation, strains expressing pfKASIII grew at rates comparable to those of wild-type strain IL1403, which was higher than the supplemented ΔfabH strain (data not shown).
FIG. 2.
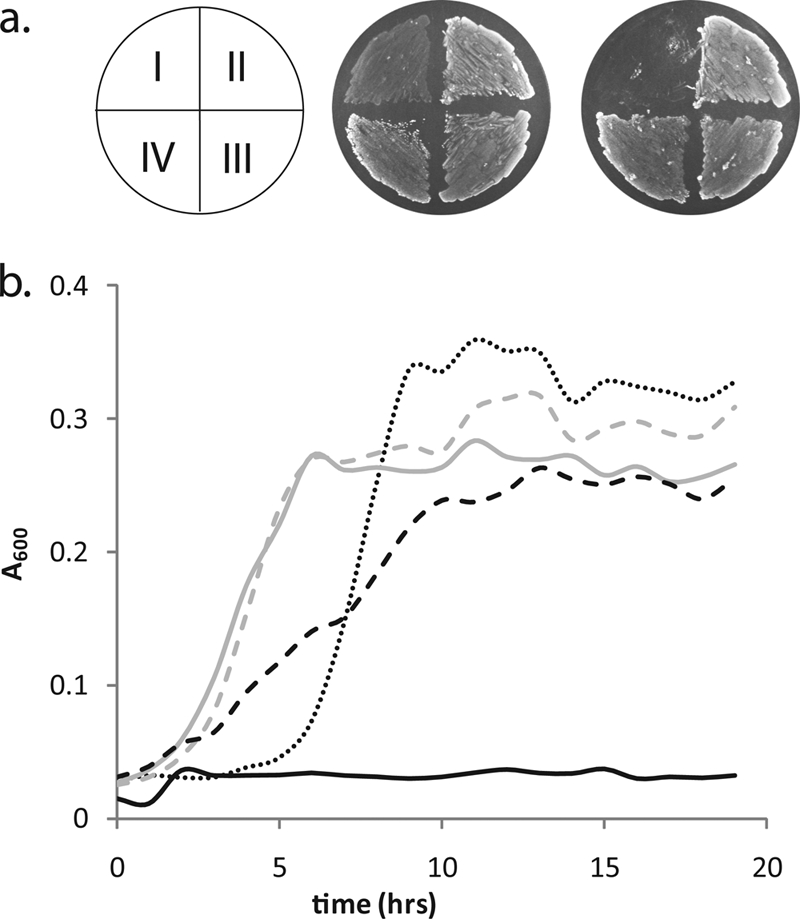
(a) Growth of L. lactis strains with or without bovine serum fatty acid on GM17 agar. Left, with 60 μg/ml bovine serum fatty acid; right, without fatty acid. I, CL112 (ΔfabH); II, IL1403 (wild type); III, YD037 (ΔfabH:pffabH); IV, YD038 (ΔfabH:L. lactis fabH). The phenotype is identical for oleate-supplemented samples. (b) Growth trends of L. lactis strains. CL112 is the ΔfabH strain; IL1403 is the wild type. GM17 is the L. lactis growth medium containing oleate (O) and/or erythromycin (E). IL1403 in GM17 (dotted line); YD038 in GM17E (gray dashed line); YD037 in GM17E (gray solid line); CL112 in GM17O (black dashed line); CL112 in GM17 (black solid line).
RT-PCR of the L. lactis strains.
To evaluate the efficiency of transcription in strains expressing pfKASIII and L. lactis fabH, relative transcription levels of the encoding genes were determined by a quantitative RT-PCR. L. lactis fabH was transcribed in all four strains, but since the ΔfabH strain and the pfKASIII-complemented strain contain the 36-bp-deleted L. lactis fabH gene, the melting temperature of the amplified products in these strains was measurably lower (82.5°C) than that of the wild-type gene (84.5°C). In YD038 (ΔfabH:L. lactis fabH), the 36-bp-deleted products were suppressed by the presence of the highly transcribed wild-type gene. The relative transcription levels of the inactivated fabH in the ΔfabH strain and the pfKASIII-complemented strain were comparable, at 2.5 and 1.3 times the wild-type fabH expression, respectively. Conversely, the relative transcription level of the complete fabH in the recovered wild-type strain was substantially higher, up to 661.7-fold, indicating that the promoter in vector pMG36e is highly efficient in L. lactis CL112. As expected, no mRNA of pfKASIII was detected in IL1403, CL112, or YD038. However, in strain YD037, the gene encoding pfKASIII was transcribed 760-fold higher than was fabH in IL1403.
KASIII enzyme assays.
P. falciparum KASIII has two reported enzymatic activities: KAS and ACAT activities (10, 19). KAS activities of IL1403 (wild type), CL112 (ΔfabH mutant), and YD037 (pfKASIII-complemented strain) were measured in whole-cell lysates. Lysates generated by chemical disruption were incubated with a mixture of pfACP, pfMCAT, malonyl-CoA, and 1-14C-acetyl-CoA. As shown in Fig. 3, substantial KAS activity was detected in the whole-cell lysates of wild-type and pfKASIII-complemented strains but not detected in the parent ΔfabH strain. Negative-control reactions for KAS activity assays without pfACP displayed background levels of radioactivity (200 to 400 cpm).
FIG. 3.

Top panel, pathways for acetyl and malonyl metabolites in the context of pfKASIII; bottom panel, KAS activity (black) and ACAT activity (gray) of the L. lactis strains IL1403 (wild type), CL112 (ΔfabH), and YD037 (ΔfabH strain complemented by pfKASIII). Residual activity in the ΔfabH strain is consistent with previously measured data in L. lactis.
Measured ACAT activity of the reversible transfer of an acetyl group from acetyl-CoA to ACP to produce acetyl-ACP is reportedly ∼0.15% that of KAS activity. ACAT activity is monitored by incubating 1-14C-acetyl-CoA with ACP in the presence of pfKASIII and detecting the formation of [1-14C]acetyl-ACP by acid precipitation of ACP (6, 8). Extracts of strains were generated from resuspended total protein cell extracts and assayed for ACAT activity by using standard protocols. The resulting trends are shown in Fig. 4. ACAT activity was detectable in the pfKASIII-complemented strain extracts at significantly higher levels than that of the ΔfabH strain but at lower levels than that of the wild type.
FIG. 4.
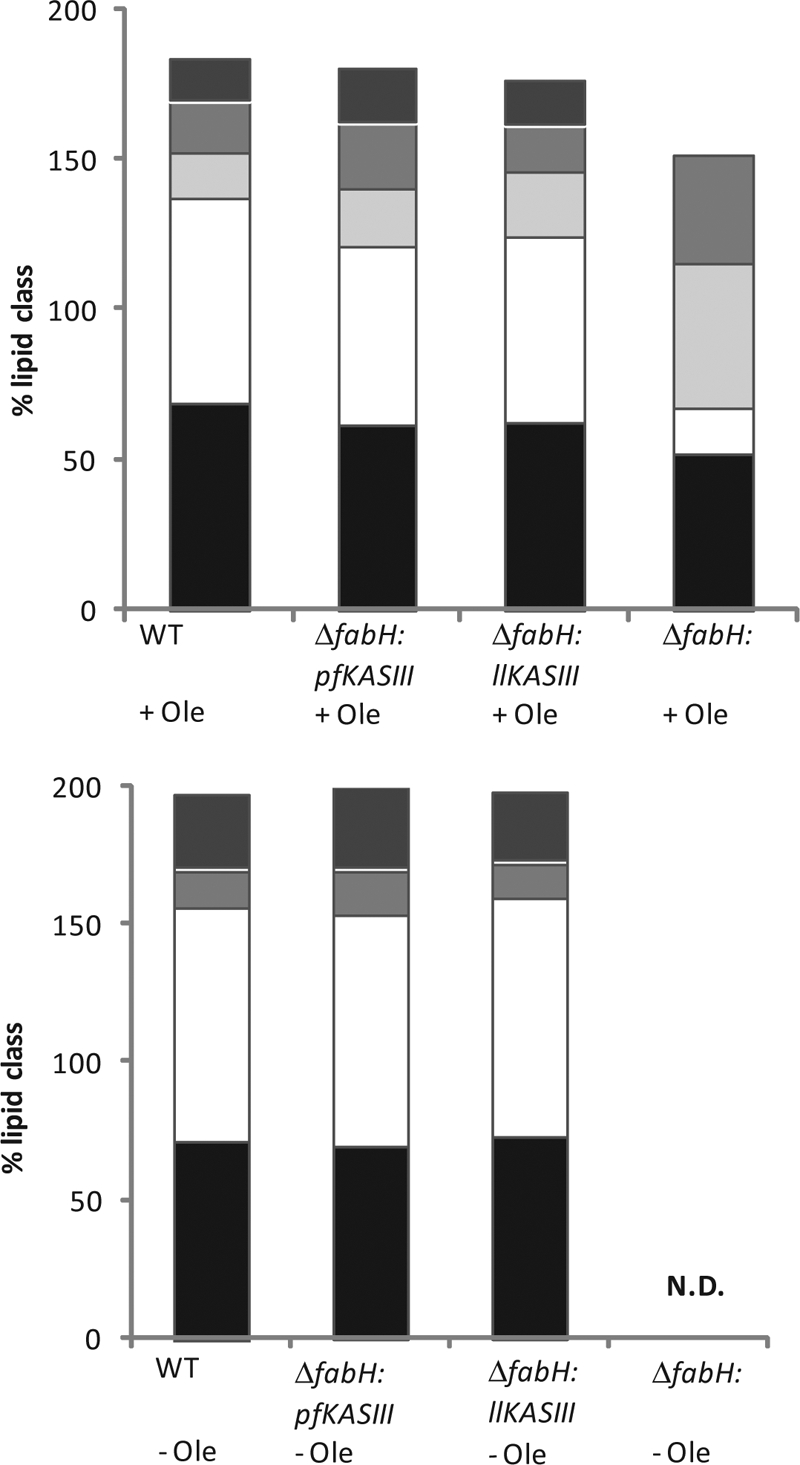
Fatty acid analysis of L. lactis strains grown on GM17 agar with and without oleate in wild-type (WT; IL1403), ΔfabH:pffabH (YD037), and ΔfabH:fabH (YD038) strains. Top panel, fatty acid groupings, from bottom to top as follows: black, straight-chain fatty acids; white, all saturated fatty acids; light gray, unsaturated fatty acids; medium-dark gray, unknown fatty acids; dark gray, cyclic fatty acids. Bottom panel, fatty acid groupings, from bottom to top as follows: black, straight-chain fatty acids; white, all saturated fatty acids; light gray, unsaturated fatty acids; dark gray, cyclic fatty acids.
Fatty acid analysis.
To ascertain the effects, if any, of pfKASIII substitution on bacterial membrane lipid composition, strains were cultivated on GM17 agar with or without oleate supplementation and analyzed by FAME analysis. As shown in Fig. 4, the lipid extracts of the pfKASIII-complemented strain membranes have clustered fatty acid profiles similar to those of L. lactis fabH complemented and wild-type strains. Only minor differences in composition were apparent (data not shown). All strains produced very low levels of branched-chain fatty acids, and YD037 produces somewhat smaller amounts of C14 fatty acids (∼10% versus ∼20%) and slightly more long-chain fatty acids, such as C18 (∼18% versus ∼11%) and C19:CYCLO (∼27% versus ∼24%), than do YD038 and IL1403. An unknown peak (retention time of 18.85 min) that existed only in oleate-supplemented cultures may be a fatty acid derived from oleate. Unsurprisingly, the ΔfabH strain produced a substantially different fatty acid profile, which contains an extremely high ratio of unsaturated fatty acid, especially C18:1. Most of this additional unsaturated fatty acid is probably oleate itself (C18:1 cis 9) which has been added to the medium for growth. These data demonstrate that pfKASIII fully restores fatty acid biosynthesis in L. lactis with minimal effects on overall lipid biosynthesis and composition. These data also suggest that membrane integrity of chimeric strains should be comparable to that of the wild type.
Antibiotic assays.
To determine whether the pfKASIII-complemented strain could be used as a prescreen for pfKASIII inhibitors, YD037 was grown in a 96-well plate format in medium with or without fatty acid supplementation with a dilution series of various known antibiotics. As shown in Fig. 5, growth inhibition of YD037 by vancomycin (targeting cell wall biosynthesis), rifampin (targeting RNA polymerase), and kanamycin (ribosomal targeting) was not reversed by added fatty acids. Cerulenin, an antibiotic targeting FAS II enzymes FabF and FabB, demonstrated potent cytotoxicity against YD037. However, added bovine-serum-derived fatty acids or oleic acid completely reversed all cerulenin-associated toxicity (data not shown).
FIG. 5.
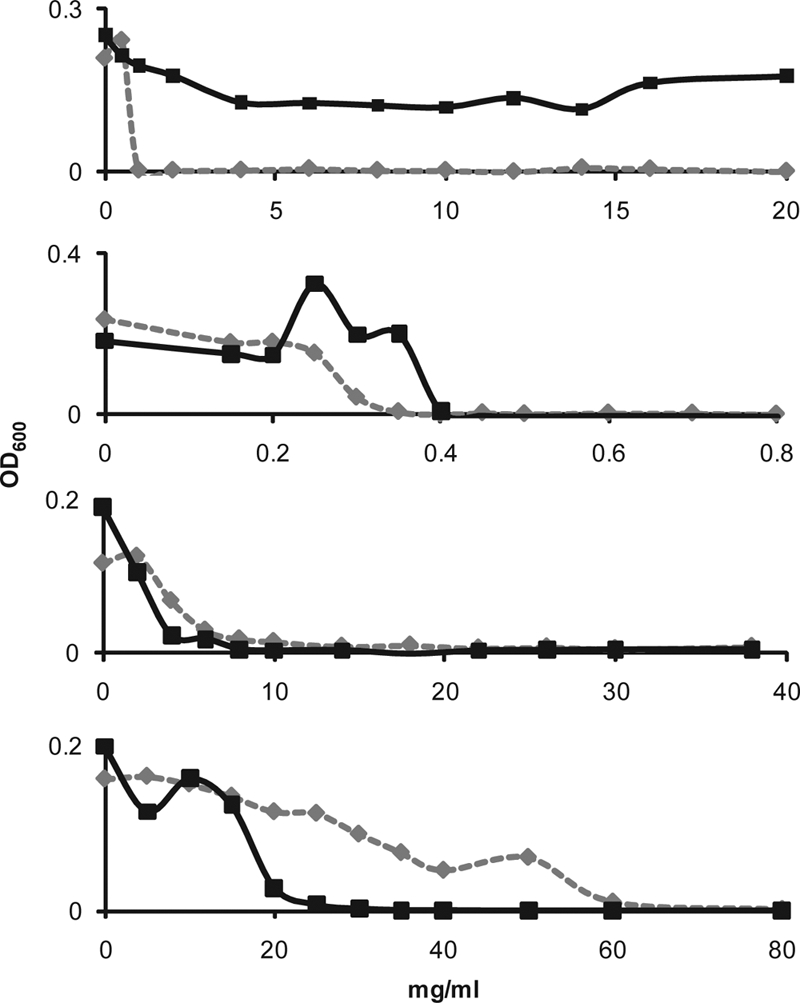
Inhibitor concentration response in the absence (dashed line) and presence (solid line) of added fatty acids. From top to bottom: cerulenin, vancomycin, rifampin, kanamycin.
DISCUSSION
Our first attempt at generating a pfKASIII-dependent bacterium involved generating the E. coli fabH knockout strain in the presence of a vector capable of expressing pfKASIII as a maltose binding protein (MBP) with a tobacco etch virus protease linker (4). In this system, the fusion protein is not catalytically competent whereas cleaved pfKASIII is active with E. coli ACP. Correspondingly, we observed that the E. coli ΔfabH:fabHpfKASIII-MBP strain was able to grow in medium lacking fatty acids when clones were transformed with a second vector containing TEV protease (Y. Du and B. Bachmann, unpublished data). Unfortunately, we also observed a high frequency of bypass formation in the knockout strains, which obfuscated characterization of pfKASIII in E. coli. Notably, a similar phenomenon of E. coli ΔfabH bypass mutation propensity was observed by Cronan and coworkers (personal communication), who have subsequently generated a stable fabH knockout in L. lactis (5). Correspondingly, we endeavored to use this strain as a host for expressing codon-optimized pfKASIII.
The plasmodial homolog of bacterial FabH, pfKASIII, possesses 36% amino acid identity with L. lactis FabH, and its cognate acyl carrier protein, pfACP, has only 32% identity with L. lactis ACP. Despite this moderately low homology, our data demonstrate that pfKASIII, expressed without the leader peptide, can functionally replace the bacterial FabH. The phenotype of permissive growth in fatty-acid-deficient medium is correlated to expression of pfKASIII by RT-PCR and biochemical activity through assay of KAS and ACAT activity in cell extracts. Together, these data suggest that L. lactis is functionally complemented by the P. falciparum FAS pathway initiating enzyme pfKASIII and that this enzyme can interact productively with the L. lactis ACP.
The growth properties of the L. lactis ΔfabH strains complemented with L. lactis fabH and the gene encoding pfKASIII are virtually indistinguishable. Both strains grow at comparable rates and possess highly similar fatty acid membrane profiles when grown in the absence of exogenously added fatty acids. Little or no variation in branched chain fatty acids was observed, suggesting, along with biochemical assay results, that pfKASIII and L. lactis FabH primarily accept acetyl-CoA as an initiating unit. This is in contrast to other Gram-positive KASIII enzymes, which have demonstrated a markedly increased flexibility for branched-chain starter units (7).
Bacterial models have previously been developed as bioassay strains for the discovery of novel FAS II-targeting inhibitors. Notably, an antisense differential sensitivity strategy has been developed for S. aureus in which antisense attenuation of bacterial fabF generated a strain hypersensitive to FAS II inhibitors (21). Potential FAS II-targeting agents were deconvoluted from off-target antibacterial compounds by replica testing with relatively insensitive (non-antisense-expressing) parent strain, and hits were confirmed by secondary in vitro biochemical assays. This two-stage method was subsequently used in a large-scale campaign for discovery of new natural product inhibitors of FAS II pathway biosynthetic enzymes, culminating in the discovery of several new FabB/H-targeting agents, including platensimycin and platencin (17, 18).
While the ultimate utility of FAS II-targeting agents as antibacterial agents remains somewhat controversial (1), the observation that FAS II deletions in P. yoelii are unable to transition from the liver to blood stage infection suggests that FAS II inhibitors may be useful for malaria prophylaxis. Correspondingly, we propose that, based on the biochemical and phenotypic data described herein, growth of L. lactis ΔfabH strains complemented with the gene encoding pfKASIII could be used as a component of a pfKASIII high-throughput screening assay system. In this two-stage process, a two-plate assay of YD037 antibacterial activity, with and without fatty acid complementation, can uncover potential FAS II-targeting compounds. In the second stage, the subset of pfKASIII-inhibiting compounds can be identified within hits by in vitro biochemical KAS assay, as we have described in this work. The advantage of this approach would be that it generates a target-based phenotypic screen for use as a primary assay.
This study comprises a new example of the successful complementation of an essential gene in a bacterium with a functionally equivalent gene from P. falciparum. To the best of our knowledge the only other previous example is that of P. falciparum ACP complementing an E. coli acpP knockout (2), a result that helped form the precedent for the present work. Moreover, attempts at bacterial KASIII interspecies complementation have historically proven to be challenging. For instance, the Gram-positive bacterium Streptomyces coelicolor was complemented with the E. coli homolog (ΔfabH:ecfabH). In contrast to this study, the resulting chimeras were slow growing and displayed substantial changes in membrane fatty acid composition (7).
Future studies will evaluate the ability of YD037 to serve as a proxy strain for the discovery pfKASIII-targeting antibiotics.
Acknowledgments
This work funded by ARMY STTR W81XWH-07-C-0092 and the Vanderbilt Institute of Chemical Biology (B.O.B.). Additional support was provided by NIH R01-AI065853, the Johns Hopkins Malaria Research Institute, and the Bloomberg Family Foundation (S.T.P.).
Plasmid pMG36e was a generous gift from Jan Kok at Groningen University. L. lactis IL1403 (wild type) and L. lactis CL112 (L. lactis ΔfabH) were kindly provided by John E. Cronan at University of Illinois at Urbana-Champaign.
Footnotes
Published ahead of print on 23 April 2010.
REFERENCES
- 1.Brinster, S., G. Lamberet, B. Staels, P. Trieu-Cuot, A. Gruss, and C. Poyart. 2009. Type II fatty acid synthesis is not a suitable antibiotic target for Gram-positive pathogens. Nature 458:83-86. [DOI] [PubMed] [Google Scholar]
- 2.De Lay, N. R., and J. E. Cronan. 2007. In vivo functional analyses of the type II acyl carrier proteins of fatty acid biosynthesis. J. Biol. Chem. 282:20319-20328. [DOI] [PubMed] [Google Scholar]
- 3.Gardner, M. J., N. Hall, E. Fung, O. White, M. Berriman, R. W. Hyman, J. M. Carlton, A. Pain, K. E. Nelson, S. Bowman, I. T. Paulsen, K. James, J. A. Eisen, K. Rutherford, S. L. Salzberg, A. Craig, S. Kyes, M. S. Chan, V. Nene, S. J. Shallom, B. Suh, J. Peterson, S. Angiuoli, M. Pertea, J. Allen, J. Selengut, D. Haft, M. W. Mather, A. B. Vaidya, D. M. Martin, A. H. Fairlamb, M. J. Fraunholz, D. S. Roos, S. A. Ralph, G. I. McFadden, L. M. Cummings, G. M. Subramanian, C. Mungall, J. C. Venter, D. J. Carucci, S. L. Hoffman, C. Newbold, R. W. Davis, C. M. Fraser, and B. Barrell. 2002. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 419:498-511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kapust, R. B., and D. S. Waugh. 2000. Controlled intracellular processing of fusion proteins by TEV protease. Protein Expr. Purif. 19:312-318. [DOI] [PubMed] [Google Scholar]
- 5.Lai, C. Y., and J. E. Cronan. 2003. Beta-ketoacyl-acyl carrier protein synthase III (FabH) is essential for bacterial fatty acid synthesis. J. Biol. Chem. 278:51494-51503. [DOI] [PubMed] [Google Scholar]
- 6.Lee, P. J., J. B. Bhonsle, H. W. Gaona, D. P. Huddler, T. N. Heady, M. Kreishman-Deitrick, A. Bhattacharjee, W. F. McCalmont, L. Gerena, M. Lopez-Sanchez, N. E. Roncal, T. H. Hudson, J. D. Johnson, S. T. Prigge, and N. C. Waters. 2009. Targeting the fatty acid biosynthesis enzyme, beta-ketoacyl-acyl carrier protein synthase III (PfKASIII), in the identification of novel antimalarial agents. J. Med. Chem. 52:952-963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li, Y., G. Florova, and K. A. Reynolds. 2005. Alteration of the fatty acid profile of Streptomyces coelicolor by replacement of the initiation enzyme 3-ketoacyl acyl carrier protein synthase III (FabH). J. Bacteriol. 187:3795-3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu, J. Z., P. J. Lee, N. C. Waters, and S. T. Prigge. 2005. Fatty acid synthesis as a target for antimalarial drug discovery. Comb. Chem. High Throughput Screen. 8:15-26. [DOI] [PubMed] [Google Scholar]
- 9.Muench, S. P., J. B. Rafferty, R. McLeod, D. W. Rice, and S. T. Prigge. 2003. Expression, purification and crystallization of the Plasmodium falciparum enoyl reductase. Acta Crystallogr. D Biol. Crystallogr. 59:1246-1248. [DOI] [PubMed] [Google Scholar]
- 10.Prigge, S. T., X. He, L. Gerena, N. C. Waters, and K. A. Reynolds. 2003. The initiating steps of a type II fatty acid synthase in Plasmodium falciparum are catalyzed by pfACP, pfMCAT, and pfKASIII. Biochemistry 42:1160-1169. [DOI] [PubMed] [Google Scholar]
- 11.Rademaker, J. L., H. Herbet, M. J. Starrenburg, S. M. Naser, D. Gevers, W. J. Kelly, J. Hugenholtz, J. Swings, and J. E. van Hylckama Vlieg. 2007. Diversity analysis of dairy and nondairy Lactococcus lactis isolates, using a novel multilocus sequence analysis scheme and (GTG)5-PCR fingerprinting. Appl. Environ. Microbiol. 73:7128-7137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ralph, S. A., G. G. van Dooren, R. F. Waller, M. J. Crawford, M. J. Fraunholz, B. J. Foth, C. J. Tonkin, D. S. Roos, and G. I. McFadden. 2004. Tropical infectious diseases: metabolic maps and functions of the Plasmodium falciparum apicoplast. Nat. Rev. Microbiol. 2:203-216. [DOI] [PubMed] [Google Scholar]
- 13.Schmittgen, T. D., and K. J. Livak. 2008. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 3:1101-1108. [DOI] [PubMed] [Google Scholar]
- 14.van de Guchte, M., J. M. van der Vossen, J. Kok, and G. Venema. 1989. Construction of a lactococcal expression vector: expression of hen egg white lysozyme in Lactococcus lactis subsp. lactis. Appl. Environ. Microbiol. 55:224-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vaughan, A. M., M. T. O'Neill, A. S. Tarun, N. Camargo, T. M. Phuong, A. S. Aly, A. F. Cowman, and S. H. Kappe. 2009. Type II fatty acid synthesis is essential only for malaria parasite late liver stage development. Cell. Microbiol. 11:506-520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vial, H. J., and M. L. Ancelin. 1998. Malarial lipids, p. 159-175. In I. W. Sherman (ed.), Malaria: parasite biology, pathogenesis, and protection. ASM Press, Washington, DC.
- 16a.Waller, R. F., M. B. Reed, A. F. Cowman, and G. I. McFadden. 2000. Protein trafficking to the plastid of Plasmodium falciparum is via the secretory pathway. EMBO J. 19:1794-1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang, J., S. Kodali, S. H. Lee, A. Galgoci, R. Painter, K. Dorso, F. Racine, M. Motyl, L. Hernandez, E. Tinney, S. L. Colletti, K. Herath, R. Cummings, O. Salazar, I. Gonzalez, A. Basilio, F. Vicente, O. Genilloud, F. Pelaez, H. Jayasuriya, K. Young, D. F. Cully, and S. B. Singh. 2007. Discovery of platencin, a dual FabF and FabH inhibitor with in vivo antibiotic properties. Proc. Natl. Acad. Sci. U. S. A. 104:7612-7616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang, J., S. M. Soisson, K. Young, W. Shoop, S. Kodali, A. Galgoci, R. Painter, G. Parthasarathy, Y. S. Tang, R. Cummings, S. Ha, K. Dorso, M. Motyl, H. Jayasuriya, J. Ondeyka, K. Herath, C. Zhang, L. Hernandez, J. Allocco, A. Basilio, J. R. Tormo, O. Genilloud, F. Vicente, F. Pelaez, L. Colwell, S. H. Lee, B. Michael, T. Felcetto, C. Gill, L. L. Silver, J. D. Hermes, K. Bartizal, J. Barrett, D. Schmatz, J. W. Becker, D. Cully, and S. B. Singh. 2006. Platensimycin is a selective FabF inhibitor with potent antibiotic properties. Nature 441:358-361. [DOI] [PubMed] [Google Scholar]
- 19.Waters, N. C., K. M. Kopydlowski, T. Guszczynski, L. Wei, P. Sellers, J. T. Ferlan, P. J. Lee, Z. Li, C. L. Woodard, S. Shallom, M. J. Gardner, and S. T. Prigge. 2002. Functional characterization of the acyl carrier protein (PfACP) and beta-ketoacyl ACP synthase III (PfKASIII) from Plasmodium falciparum. Mol. Biochem. Parasitol. 123:85-94. [DOI] [PubMed] [Google Scholar]
- 20.Yin, H., W. Liu, K. Goleniewska, N. A. Porter, J. D. Morrow, and R. S. Peebles, Jr. 2009. Dietary supplementation of omega-3 fatty acid-containing fish oil suppresses F2-isoprostanes but enhances inflammatory cytokine response in a mouse model of ovalbumin-induced allergic lung inflammation. Free Radic. Biol. Med. 47:622-628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Young, K., H. Jayasuriya, J. G. Ondeyka, K. Herath, C. Zhang, S. Kodali, A. Galgoci, R. Painter, V. Brown-Driver, R. Yamamoto, L. L. Silver, Y. Zheng, J. I. Ventura, J. Sigmund, S. Ha, A. Basilio, F. Vicente, J. R. Tormo, F. Pelaez, P. Youngman, D. Cully, J. F. Barrett, D. Schmatz, S. B. Singh, and J. Wang. 2006. Discovery of FabH/FabF inhibitors from natural products. Antimicrob. Agents Chemother. 50:519-526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu, M., T. R. Kumar, L. J. Nkrumah, A. Coppi, S. Retzlaff, C. D. Li, B. J. Kelly, P. A. Moura, V. Lakshmanan, J. S. Freundlich, J. C. Valderramos, C. Vilcheze, M. Siedner, J. H. Tsai, B. Falkard, A. B. Sidhu, L. A. Purcell, P. Gratraud, L. Kremer, A. P. Waters, G. Schiehser, D. P. Jacobus, C. J. Janse, A. Ager, W. R. Jacobs, Jr., J. C. Sacchettini, V. Heussler, P. Sinnis, and D. A. Fidock. 2008. The fatty acid biosynthesis enzyme FabI plays a key role in the development of liver-stage malarial parasites. Cell Host Microbe 4:567-578. [DOI] [PMC free article] [PubMed] [Google Scholar]