

Abstract
Background/Aims
Interleukin-6 stimulates liver regeneration and promotes hepatoprotection following experimental liver injury, but underlying mechanisms have not been fully characterized. Because studies suggest matrix metalloproteinase-2 (MMP-2) may promote liver injury, we examined whether IL-6 exerted its protective effects via regulation of MMP-2.
Methods
MMP-2 was analyzed in livers of IL-6−/− and IL-6+/+ mice following CCl4 administration. IL-6−/− mice were pretreated with IL-6 and liver histology and MMP-2 expression were examined after liver injury. IL-6−/− mice were treated with an MMP-2 inhibitor and assessment of injury (histology and serum ALT levels), apoptosis by TUNEL assay, and hepatocyte proliferation by BRDU-labeling was performed. These studies were complemented by analysis of cultured stellate cells.
Results
MMP-2 mRNA, protein, and activity was increased in IL-6−/− livers. Restoration of IL-6 signaling in IL-6−/− mice rescued injury and restored MMP-2 expression to wild-type levels. Furthermore, pharmacologic inhibition of MMP-2 decreased hepatocellular injury and apoptosis in IL-6−/− mice. In cultured stellate cells, recombinant IL-6 suppressed endogenous MMP-2 mRNA and protein expression.
Conclusions
IL-6 may be hepatoprotective in acute injury through down-regulation of MMP-2. These findings suggest a role for MMP-2 in amplifying liver injury in vivo.
Keywords: Matrix degradation, Stellate cells, Fibrosis, Liver injury, Interleukin-6, Matrix metalloproteinase-2
1. Introduction
Interleukin-6 (IL-6) is a cytokine that is elevated in patients with hepatitis [1] and alcoholic liver disease [2]. It is considered a hepatoprotective factor by stimulating hepatocyte proliferation through activation of Stat-3 and MAPK signaling pathways [3,4]. In addition, IL-6 attenuates hepatocyte apoptosis by maintaining adequate levels of several anti-apoptotic factors [5]. IL-6−/− mice develop increased liver injury in response to CCl4, a tumor necrosis factor-alpha (TNF-α) mediated model of liver injury [6], suggesting IL-6 may function downstream of TNF-α to ameliorate the injury response.
Liver injury elicits a wound healing response characterized by hepatocyte proliferation, infiltration of inflammatory cells, and transformation of perisinusoidal stellate cells into myofibroblasts which degrade sinusoidal ECM by releasing matrix degrading proteases, yielding a scar matrix containing type I collagen [7]. Sustained expression of matrix proteases may provoke the rapid influx of inflammatory cells [8], loss of the scaffolding that maintains the normal liver architecture, and cellular changes resulting from altered cell—matrix interactions.
Matrix metalloproteinases are a family of zinc-dependent proteases capable of degrading hepatic ECM, thereby playing a central role in tissue remodeling and repair after injury [9]; however, persistent overexpression of MMPs may contribute to the pathogenesis of liver diseases. Inhibition of MMP-2, a 72 kDa protease produced by activated stellate cells, blocks lethal hepatitis and apoptosis induced by TNF-α [8]. Furthermore, MMP-2-deficient mice demonstrate decreased hepatocyte apoptosis and necrosis, and enhanced survival in this model. The protective effects of decreased MMP-2 activity were ascribed to reduced sinusoidal ECM breakdown and decreased influx of inflammatory cells. Taken together, these findings suggest a role for MMP-2 in promoting liver injury [8], yet upstream signals regulating MMP-2 activity have not been explored.
No studies have examined the effects of IL-6 on MMP-2 in in vivo models of liver injury. Given the importance of IL-6 in protecting against toxin-induced liver injury and apoptosis, and the participation of MMP-2 in promoting TNF-α mediated liver injury, we examined whether IL-6’s protective effects could be attributable to altered expression of MMP-2.
2. Materials and methods
2.1. Toxin-induced injury models
Studies were performed on C57BL6/SV129 IL-6−/− and IL-6+/+ mice 12–16 weeks of age [10]. Mice were used with the approval of IACUC and under National Institutes of Health guidelines. In studies of acute injury, IL-6+/+ and IL-6−/− mice were injected intraperitoneally (i.p.) with a 50% solution of CCl4 (Sigma) at a dose of 2 μl/g animal weight. IL-6−/− and +/+ livers express equal levels of cytochrome CYP 2E1 which is responsible for the metabolism of CCl4 [11]. Mice were sacrificed at 0, 6, 12, 24, 36, 48, 72, 96, 120 h, 1 week, and 2 weeks post-CCl4. Cohorts of IL-6−/− mice were also pretreated with a subcutaneous injection of recombinant IL-6 at a dose of 1 mg/kg 20 min before CCl4 injection [11]. For chronic liver injury, IL-6+/+ and −/− mice (n = 3 per group) were given biweekly i.p. injections of 10% CCl4 at a dose of 5 μl/g for 5 weeks. Under isoflourane anesthesia animals were sacrificed and livers processed for RNA, protein, histology, and immunostaining.
2.2. Acute toxin-induced injury with concomitant pharmacologic inhibition of MMP-2/9
IL-6−/− and IL-6+/+ mice were treated with either the MMP-2/MMP-9-specific cyclic decapeptide CTTHWGFTLC [8] i.p. at a dose of 8 μl/g or vehicle concomitant with CCl4 and sacrificed at 24 and 48 h. The IC50 of this inhibitor for active MMP-2 and MMP-9 is 10 μM (up to 500 μM there is no inhibition of MMP-8, MMP-13, or MT1-MMP, according to the manufacturer). Two hours before sacrifice animals were injected i.p. with bromodeoxyuridine (BrdU, Sigma) at a dose of 50 mg/kg. Serum was analyzed for biochemistries. The dose of inhibitor was selected based on its use in murine models of cancer [12] and was well tolerated.
2.3. Histology and immunohistochemistry
In all studies of acute injury, formalin-fixed, paraffin-embedded liver sections (5 μM) were stained with hematoxylin and eosin. Degree of centrilobular (perivenular) necrosis and inflammatory infiltrate were evaluated on a 4-point scale (Table 1) at prolonged time points after a single dose of CCl4 in 20 random fields at 10× magnification per animal (n = 3 per group) by a blinded pathologist (MIF). In addition, hepatocyte nuclear staining for BrdU was performed as described [4] in both IL-6+/+ and IL-6−/− receiving CCl4±MMP-2/9 inhibitor. Sirius Red staining for type I collagen quantitative histomorphometric analysis was performed on liver sections from chronically injured IL-6+/+ and IL-6−/− livers as described previously [13]. For MMP-2 immunostaining, the Vectastain ABC kit (Vector Laboratories; Burlinghame, CA) was used. MMP-2 antibody (Research Diagnostics; Flanders, NJ) and biotinylated horse anti-mouse secondary antibody (Vector Laboratories) in 1.5% horse serum in PBS was added to tissue at a 1:200 dilution and 1:1250 dilutions, respectively.
Table 1.
Histological assessment of IL-6−/− vs. IL-6+/+ livers following CCl4 administration
| Score | Centrilobular necrosis | Inflammation in centrilobular areas |
|---|---|---|
| 0 | None | None |
| 1 | Isolated necrotic hepatocytes or single row of hepatocyte drop-out in perivenular areas | Mild: inflammatory infiltrate affecting <50% centrilobular areas |
| 2 | >1 and up to 3 rows of perivenular necrotic hepatocytes | Moderate: inflammatory infiltrate affecting >50% and <75% centrilobular areas |
| 3 | >3 rows of perivenular necrotic hepatocytes with confluent and/or bridging necrosis | Severe: dense inflammatory infiltrate affecting >75% centrilobular areas |
Blinded pathologist (MIF) examined twenty 10× fields per animal (n = 3 per group) and scored centrilobular necrosis and inflammation independently on 4-point scale (0–3). Data analyzed using two-tailed independent Student’s t-test and represented graphically as means±SEM in Fig. 1(G and H).
2.4. Immunoblots
Preparation of whole liver extracts and western blot analysis was carried out as previously described [11]. Primary antibodies used were MMP-2 (Chemicon), MMP-9 (Chemicon), MT1-MMP (Chemicon), TIMP-2 (Chemicon), and β-actin (Sigma) at a dilution of 1:5000. β-Actin protein expression was used as a loading control. Protein expression was measured at 0, 6, 12, 24, 36, and 48 h to note any differences between IL-6−/− and IL-6+/+ livers. Significant time points were re-examined in duplicate or triplicate. Results were quantified by scanning densitometry using Bioquant software.
2.5. Gelatin zymography
Proteins (50 μg) were separated in a 10% polyacrylamide gel containing 1 mg/ml of bovine skin gelatin (Sigma). Purified MMP-2 and MMP-9 served as positive controls (Chemicon). Gels were washed twice for 30 min in 2.5% Triton X-100, then for 10 min in 0.1 M Tris (pH 7.4), and incubated for 18 h at 37°C in 0.1 M Tris (pH 7.4), 10 mM CaCl2, 5 mM ZnCl2. Staining with 0.5% Coomasie Blue and destaining performed as described [14].
2.6. TUNEL assay
The terminal deoxynucleotidyl transferase-mediated uDP nick-end labeling (TUNEL) assay was used to assess the degree of apoptosis (Cell Death Detection Kit; Boehringer-Mannheim). Quiescent and DNAse I pretreated IL-6−/− livers were used as negative and positive controls, respectively. Five (100×magnification) fields were randomly selected per slide and 100 hepatocytes counted per field. The mean percent of apoptotic hepatocytes was calculated and compared between different study groups.
2.7. Quantitative real time PCR analysis of MMP-2 mRNA expression (qRTPCR)
RNA was extracted using the Qiagen RNAeasy kit. 1 μg of RNA was reverse transcribed using first strand complementary DNA synthesis with random primers (Promega). The primers used were MMP-2 forward: 5′-GAT GTC GCC CCT AAA ACA AGA-3′ and reverse: 5′-GCC CAA AGA ACT TCT GCA TCA-3′. β2-Microglobulin forward: 5′-ATG CTG AAG AAC GGG AAA AA-3′ and reverse: 5′-CGG CCA TAC TGT CAT GCT TA-3′. Samples were analyzed in triplicate in an ABI PRISM 7900HT Sequence Detection System (Applied Biosystems) and normalized to β2-microglobulin.
2.8. IL-6 regulation of MMP-2 in cultured stellate cells
To determine whether IL-6 suppresses the endogenous expression of MMP-2 protein and mRNA, we used the rat stellate cell line (HSC-T6), whose features closely resemble culture activated primary stellate cells [15]. Cells were serum starved for 24 h and treated with recombinant IL-6 (Peprotech, Inc.; NJ) at a dose of 100 ng/ml. Intact IL-6 signaling pathways were assessed in HSC-T6 cells by incubating ±IL-6 for 15 min, 30 min, and 1 h and assessment of STAT3 activation. IL-6 treated Hep G2 cells served as a positive control [16]. Cell extracts were harvested and 30 μg of protein were loaded, separated by polyacrylamide electorphoresis, transferred to PVDF membranes, and probed for phosphorylated STAT-3 (Cellular signaling; Beverly, MA) at a 1:2000 dilution. For RNA analysis, HSC-T6 cells were incubated ±IL-6 for 1, 2, 4, and 6 h, RNA extracted, and qRTPCR performed as before with the following primers: MMP-2 forward: 5′-ACC CAG ATG TGG CCA ACT AC-3′ and reverse: 5′-TAC TTT TAA GGC CCG AGC AA-3′ GAPDH forward: 5′-TGA TTC TAC CCA CGG CAA GT-3′ and reverse: 5′-AGC ATC ACC CCA TTT GAT GT-3′. All experiments performed in triplicate and normalized to GAPDH mRNA. For analysis of MMP-2 protein expression, HSC-T6 cells were plated at a density of 1 × 105 per well in 6-well format, serum-starved, and incubated ±IL-6 for 24 h. Culture supernatant collected and 30 μg of protein used for immunoblot analysis as described above.
2.9. Statistics
Comparisons between groups were performed using independent Student’s t-test and SPSS software. All data are represented as means±SEM.
3. Results
3.1. Increased injury, inflammation, and delayed recovery in IL-6−/− mice following CCl4
We reported that IL-6−/− mice have increased acute CCl4-mediated liver injury compared to IL-6+/+ mice [11], but we did not characterize their response at longer intervals and time to recovery. To do so, liver sections from IL-6−/− and IL-6+/+ mice at extended intervals after a single dose of CCl4 were examined by a blinded pathologist (MIF) and graded for degree of perivenular hepatocyte necrosis and accompanying inflammation as shown in Table 1. IL-6−/− livers demonstrated a 50% increase in inflammation (P<0.0001) and 24% increase in coagulative necrosis (P<0.024) compared to wild-type mice 120 h post-CCl4 which was recovered by 2 weeks (Fig. 1A–H).
Fig. 1.

Protracted injury and inflammation following CCl4 administration in IL-6−/− livers. Increased mononuclear and lymphoid inflammatory infiltrate, coagulative necrosis, and hemorrhage in IL-6−/− livers 120 h after a single intraperitoneal dose of CCl4 (B) compared to IL-6+/+ livers (A). Higher magnification demonstrates increased inflammatory cells in bridging areas of necrosis in IL-6−/− livers (D) compared to IL-6+/+ livers (C). Arrows indicating inflammation and coagulative necrosis. Complete restoration of liver architecture is noted by 2 weeks in both IL-6−/− (F) and IL-6+/+ livers (E) (original magnification, 50× A, B; 100× C, D; 10× E, F). Inflammation and perivenular necrosis was graded on a 4-point scale (Table 1) by a blinded pathologist in 20 random high power fields per animal (n = 3 in each group) and represented graphically in panels G and H. Data represents mean±SEM; **P<0.0001; ***P<0.024.
These results suggest an important role for IL-6 pathways in the early phase of injury and/or the wound healing/recovery process.
3.2. Increased fibrosis in IL-6−/− mice is associated with increased MMP-2 expression
We explored the possibility that IL-6 exerted its protective effects through preservation of the normal ECM. Because MMP-2 is capable of degrading normal ECM and is elevated in human models of chronic hepatitis and fibrosing liver injury [1], we examined whether increased fibrosis in IL-6−/− mice was associated with elevated MMP-2. As shown in Fig. 2, IL-6−/− mice developed 20% more fibrosis as determined by Sirius Red staining and quantitation using Bioquant software (Fig. 2A, B, and E; P<0.0001) which was associated with a 49% increase in sinusoidal staining for MMP-2 compared to IL-6+/+ livers (Fig. 2C, D, and F; P<0.001). These data led us to examine whether IL-6 signaling pathways are important in limiting MMP-2 expression in acute injury.
Fig. 2.

Increased MMP-2 is associated with fibrosis in IL-6−/− livers. Sirius Red staining shows increased fibrosis in IL-6−/− livers (B) compared to IL-6+/+ livers (A) after 5 weeks of chronic administration of CCl4 which is associated with increased MMP-2 expression as assessed by immunostaining (D vs. C). Arrows denote sinusoidal staining for MMP-2. Bioquant analysis performed on 36 images per animal (n = 3) demonstrates 20% increase in collagen I (E; P<0.0001) and 49% increase in MMP-2 expression (F; P<0.001) in IL-6−/− fibrotic livers. Original magnification 5× (A, B) and 200× (C, D).
3.3. MMP-2 expression and activity are increased in IL-6−/− livers during acute CCl4-induced injury
To explore the potential role of MMP-2 in mediating increased acute injury in IL-6−/− livers following CCl4, we examined its expression and activity. Twenty four hours after a single dose of CCl4 there was a ~4-fold increase in the expression of pro-MMP-2 (range 1.3–8.7) and a ~20-fold-increase (range 3.2–47; P<0.002) in active MMP-2 expression by immunoblot in IL-6−/− livers compared to wild-type animals (Fig. 3A and B). Increased protein expression correlated with enzyme activity using gelatin zymography (Fig. 3C). At 24 h, there was a >3-fold increase in MMP-2 activity in IL-6−/− livers. Since MMP-9 (Gelatinase B) also has type IV collagenase activity in the liver, we examined whether IL-6 deficiency also altered expression of this protease. There was no significant difference in the expression of active MMP-9, although latent MMP-9 was increased in IL-6−/− livers at 24 h (Fig. 4A). These results suggest that modulation of MMP-2 protein expression by IL-6 correlated with relative activity of the enzyme.
Fig. 3.

Increased expression and activity of MMP-2 in IL-6−/− livers after single dose of CCl4. (A) Representative immunoblot for MMP-2 on whole liver extracts harvested from IL-6−/− and IL-6+/+ mice after administration of CCl4 demonstrates a 20-fold increase (range 3.2–47; ***P<0.002) in expression of active MMP-2 in IL-6−/− vs. IL-6+/+ animals. (B) Quantitation of increases of both active and latent MMP-2 from three independent experiments. β-Actin expression was used as a loading control. (C) Gelatin zymography performed on liver extracts demonstrates 3-fold increase in MMP-2 activity at 24 h in the IL-6−/− livers. [This figure appears in colour on the web.]
Fig. 4.

Expression of active MMP-9, TIMP-2, MT1-MMP, or uPA are not increased in IL-6−/− mice following administration of CCl4. Immunoblot of whole liver extracts prepared from IL-6+/+ and IL-6−/− livers following a single dose of CCl4 were probed with antibodies to MMP-9, TIMP-2, MT1-MMP, and uPA. (A) An increase in latent but not active MMP-9 is apparent in IL-6−/− livers. (B) Decreased TIMP-2 expression in IL-6−/− livers is apparent, concomitant with increased MMP-2. (C) A transient peak of MT1-MMP expression at 12 h precedes activation of MMP-2 then declines rapidly in IL-6−/− animals at 36 and 48 h. (D) uPA expression declines in concert with MMP-2 activation in IL-6−/− livers. β-Actin protein expression was used as a loading control on each blot. Experiments were performed in duplicate.
Since activation of MMP-2 requires both tissue inhibitor of metalloproteinase-2 (TIMP-2) and membrane-type 1 matrix metalloproteinase (MT1-MMP) [17] we examined the expression of TIMP-2 and MT1-MMP in this model. As demonstrated in a representative immunoblot (Fig. 4B), expression of TIMP-2 was reduced in IL-6−/− livers from 24 to 48 h by 32 and 50%, respectively. This reduction in TIMP-2 could further increase the net activity of MMP-2 in the IL-6−/− animals. Interestingly, there was a progressive decline in both the pro- (65 kDa) and active (63 kDa) forms of MT1-MMP (Fig. 4C). We also examined uPA expression in this model because of its reported role in MMP-2 activation [18]. Similar to the findings with MT1-MMP, we observed a decrease in uPA levels in IL-6−/− animals (Fig. 4D). Thus, net MMP-2 activity was increased in spite of reduced MT1-MMP and uPA expression.
3.4. Restoration of IL-6 signaling rescues CCl4-induced injury and restores active MMP-2 levels to wild-type levels
Reconstitution of IL-6 signaling by administering recombinant IL-6 20 min prior to CCl4 attenuated histologic injury (Fig. 5A–D) and reduced the expression of active MMP-2 in IL-6−/− mice (Fig. 5E).
Fig. 5.

Restoration of IL-6 signaling in IL-6−/− mice rescues CCl4-induced injury and restores MMP-2 expression to wild-type levels. Hematoxylin and eosin stained liver sections from IL-6−/− mice pretreated with either vehicle (A) or recombinant IL-6 (B) prior to CCl4 (original magnification 10×). Area outlined shown at higher magnification (100×; C, D). Those receiving recombinant IL-6 (B, D) had decreased injury at 24 h compared to control group (A, C). (E) Western blot of MMP-2 protein expression performed on extracts from IL-6−/− mice receiving vehicle (lane 2) compared with those receiving recombinant IL-6 (lanes 3 and 4). At 24 h, there is a significant increase in active MMP-2 expression in IL-6−/− animals, which is decreased in animals receiving recombinant IL-6. The blot probed for β-actin to confirm equal protein loading. Experiments were performed in triplicate.
3.5. Pharmacologic inhibition of MMP-2 attenuates injury and apoptosis in IL-6−/− mice without improvement in hepatocyte proliferation
In order to assess whether the improvements in histologic injury resulted directly from effects of IL-6 on MMP-2, IL-6−/− mice were administered a specific MMP-2/9 inhibitor i.p. concomitant with CCl4. Since there was no significant increase in active MMP-9 levels by immunoblot or zymography in the IL-6−/− livers, effects of the inhibitor were likely due to inhibition of MMP-2 alone. In the presence of the inhibitor, CCl4-induced injury was significantly reduced histologically (Fig. 6B vs. A) as well biochemically (Fig. 6C; ***P< 0.03). No significant improvement was noted in IL-6+/+ mice receiving the inhibitor. In addition, IL-6−/− livers receiving inhibitor demonstrated an 80% reduction in hepatocyte apoptosis as assessed by TUNEL-staining as compared to the control group (Fig. 6E vs. D, and G vs. F, H; ***P<0.001). Since IL-6 is known to be critical for the proliferative response of hepatocytes to CCl4-injury [11], we examined whether effects on hepatocyte proliferation were related to MMP-2 dysregulation. The peak proliferative response at 48 h was blunted in IL-6−/− animals receiving CCl4 compared to IL-6+/+ animals (Fig. 7A–F; **P<0.0001) and was not affected by the administration of the MMP-2 inhibitor. BrDU labeled hepatocytes in both treatment groups are shown in Fig. 7B–E and quantitated in panel F.
Fig. 6.
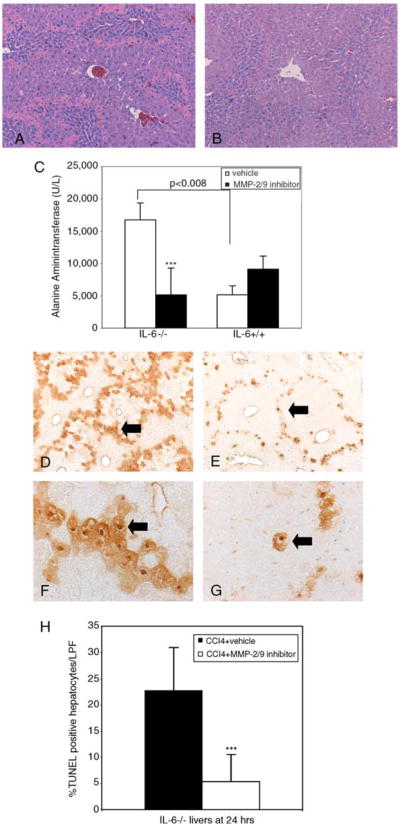
Pharmacologic inhibition of MMP-2 rescues CCl4-induced liver injury and apoptosis in IL-6−/− mice but has no significant impact in IL-6+/+ mice. Liver sections 24 h after co-administration of MMP-2/9 inhibitor at a dose of 8 μl/g or vehicle control and CCl4 were stained with hematoxylin and eosin (100×). IL-6−/− mice receiving the inhibitor developed less injury (B) compared to control animals receiving vehicle control (A). (C) ALT levels measured in IL-6−/− and IL-6+/+ with and without MMP-2/9 inhibitor. IL-6+/+ mice had significantly less injury as measured by ALT levels (P<0.008). IL-6−/− mice receiving inhibitor had a ~75% reduction in serum ALT (***P<0.03). TUNEL staining was performed on liver sections from IL-6−/− mice 24 h after the administration of CCl4 with or without MMP-2/9 inhibitor. IL-6−/− mice receiving inhibitor (E) demonstrated significantly less apoptosis than vehicle control group (D). High power magnification (400×) demonstrates fewer TUNEL-positive nuclei in animals receiving inhibitor (G) compared to those receiving vehicle (F). Arrows denote TUNEL-positive cells. (H) Five (100× magnification) fields were randomly selected per slide and 100 hepatocytes counted per field. Mean percent of apoptotic hepatocytes was calculated and compared between different study groups. IL-6−/− mice receiving inhibitor demonstrated a 17.4% absolute reduction and 80% relative reduction in hepatocyte apoptosis (***P<0.001). Bars represent means±SEM from three mice in each group.
Fig. 7.
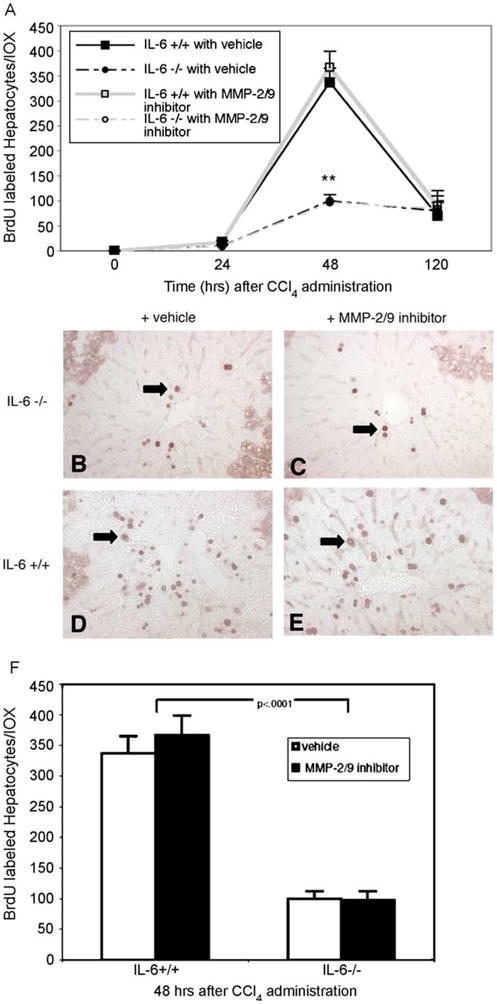
Administration of MMP-2 inhibitor does not restore hepatocyte proliferative response in IL-6−/− mice following CCl4 administration. (A) Peak proliferative defect in IL-6−/− livers notable at 48 h after single dose of CCl4. Data points represent means±SEM (**P<0.0001; IL-6−/− vehicle vs. IL-6+/+ vehicle). (B–E) Representative photomicrographs of BRDU immunohistochemistry at 48 h with or without MMP-2/9 inhibitor (original magnification, 100×). Arrows indicate large round positively stained hepatocyte nuclei. (F) Quantification of percent BRDU-labelled hepatocytes per 10× field at 48 h after CCl4 administration in IL-6+/+and IL-6−/− demonstrates no significant impact of inhibitor within either group. [This figure appears in colour on the web.]
3.6. IL-6 suppresses MMP-2 mRNA levels in vivo
Active MMP-2 protein was disproportionately increased compared to latent MMP-2 following CCl4-injury in IL-6−/− animals. To assess the potential site(s) of regulation of MMP-2 by IL-6, we analyzed MMP-2 mRNA expression by real-time PCR using RNA extracted from IL-6−/− and IL-6+/+ livers following a single dose of CCl4. As shown in Fig. 8, increased MMP-2 mRNA was observed at early time points in IL-6−/− livers, most notably at 24 h (14-fold). This finding is consistent with IL-6’s role in primarily stimulating early responses including gene expression, suggesting that early changes in MMP-2 expression after an acute injury are more likely to be IL-6 dependent than later changes.
Fig. 8.
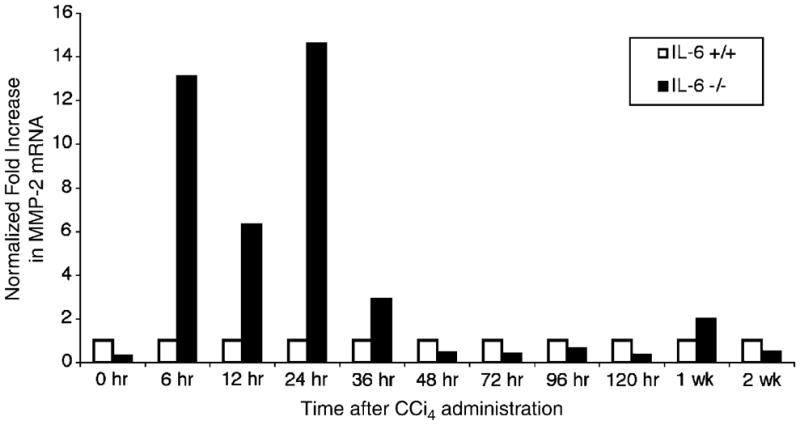
IL-6 suppresses MMP-2 mRNA levels in vivo. Real time PCR using RNA extracted from IL-6−/− and IL-6+/+ livers following a single dose of CCl4 was analyzed using primers specific for MMP-2 mRNA and normalized to β2-microglobulin. There was a significant increase in MMP-2 mRNA in IL-6−/− livers at 6, 12, 24, and 36 h after a single dose of CCl4. This increase was most notable at 24 h where a 13.6-fold increase in MMP-2 mRNA was noted in the IL-6−/− livers.
3.7. IL-6 suppresses endogenous MMP-2 mRNA and protein expression in cultured stellate cells
Because activated stellate cells are the predominant source of MMP-2 in liver injury, we examined whether IL-6 suppresses endogenous MMP-2 in a rat stellate cell line, HSC-T6 [15]. IL-6 signaling pathways are intact in these cells as demonstrated by phosphorylation of STAT-3 in the presence of IL-6 (Fig. 9A). In HSC-T6 cells there was early suppression of MMP-2 mRNA at 2 h following IL-6 incubation (Fig. 9B; ***P<0.0012) compared with control, which correlated with an 86% reduction in MMP-2 protein in IL-6 treated cells (Fig. 9C; P<.0015).
Fig. 9.
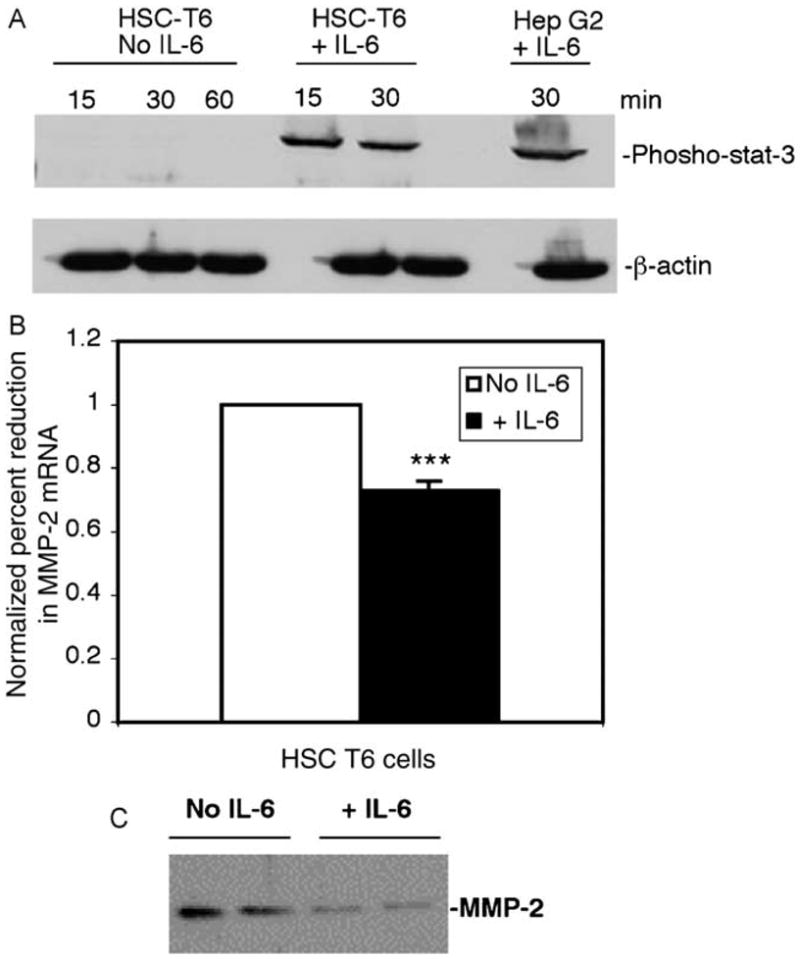
IL-6 suppresses endogenous MMP-2 mRNA and protein in a stellate cell line. (A) HSC-T6, an immortalized rat stellate cell line, demonstrates phoshphorylated STAT-3 after incubation with recombinant IL-6 for 15 and 30 min compared to vehicle control. Hep G2 cells incubated with IL-6 for 30 min served as positive control. (B) RNA extracted from HSC-T6 cells incubated with IL-6 for 2 h demonstrated a mean 27% reduction in MMP-2 mRNA (range 18–58%, ***P<0.0012) in MMP-2 mRNA compared with control as assessed by real time PCR. Data from representative experiment shown here. (C) Immunoblot analysis of cell culture supernatant of HSC-T6 cells after 24 h incubation with IL-6 demonstrates an average 86% reduction (range 60–93%, P<0.0015) in MMP-2 protein expression compared to control. Quantification performed using Bioquant image analysis software.
4. Discussion
These data identify an important new link between the hepatoprotective effects of IL-6 and MMP-2 suppression as both MMP-2 inhibition and IL-6 reconstitution, with resultant decreases in MMP-2 levels, ameliorated injury in IL-6 deficient animals. Interestingly, MMP-2 inhibition did not restore the hepatocyte proliferative defect observed in IL-6−/− livers, suggesting that this defect is independent of effects on MMP-2 expression in a CCl4-model of injury. Moreover, MMP-2 inhibition did not significantly impact injury in IL-6+/+ livers, indicating that low levels of MMP-2 are needed for normal healing. In the case of IL-6−/− animals in which there is an excessive amount of MMP-2, the inhibitor was clearly beneficial. Our findings reinforce recent observations in which MMP-2 deficient mice and mice treated with an MMP-2/9 inhibitor were protected from TNF-αinduced hepatitis and apoptosis [8]. Taken together, MMP-2 is not simply up regulated once injury is established but rather may amplify injury and inflammation, perhaps leading to increased hepatic fibrosis [11]. Physiologic IL-6 may therefore play an important role in limiting injury and possibly eventual fibrosis via its inhibitory effects on MMP-2 expression.
The precise mechanism by which IL-6 regulates MMP-2 in vivo is unclear. IL-6−/− injured livers express significantly increased MMP-2 mRNA levels. Since the cellular source of MMP-2 in the liver is thought to be the hepatic stellate cell [19], we confirmed that recombinant IL-6 suppressed the expression of endogenous MMP-2 protein and mRNA expression in an immortalized rat stellate cell line. The observed effects on mRNA suggests that IL-6 regulates MMP-2 to some degree at the transcriptional level, but alone they are unlikely to explain the profound differences in MMP-2 activation observed in vivo. Because significant differences in MMP-2 activation were noted in vivo, we measured the expression of MT1-MMP and uPA, the known activators of MMP-2 in the liver, and TIMP-2, an endogenous inhibitor of MMP-2. While protein expression of MT1-MMP and uPA declined in parallel with MMP-2 activation, TIMP-2 was also reduced and may have contributed to increased MMP-2 activity. In addition, others have reported that hepatocytes induce MMP-2 activation through a plasma membrane-dependent mechanism(s), suggesting that cell–cell interactions are involved in this process in vivo and may also be important in our model [19]. However, there may also be a novel mechanism of MMP-2 activation in this model, which will be explored in future studies.
It is unclear if decreased MMP-2 expression attenuates hepatocyte injury and apoptosis solely by limiting matrix degradation and/or whether important alternative mechanisms independent of matrix breakdown are involved. Increased MMP-2-induced ECM breakdown may: (1) promote the influx of inflammatory cells and consequently further hepatocellular damage [8]; (2) alter cell–matrix and cell–cell interactions and thereby enhance hepatocyte susceptibility to necrosis and apoptosis; (3) release matrix-bound cytokines, chemokines, adhesion molecules, growth factors, and/or apoptotic mediators important for regulating cellular behavior in response to injury [20]. Matrix-independent mechanisms for the role of MMP-2 in acute injury may include direct cleavage of cell-surface receptors that mediate intracellular pathways predisposing hepatocytes to injury. In summary, our findings demonstrate that IL-6 is hepatoprotective in part through its regulation of MMP-2 and suggest an important role for MMP-2 in amplifying liver injury in vivo.
Acknowledgments
This work was supported by University of Pennsylvania Digestive and Liver Center Grant P30 DK50306, National Institutes of Health Grants T32-DK07066 and KO8 DK60474 (to MBB), DK56621 (to SLF) and 1 F32 DK09732-01 (to KK), The Mount Sinai Feld Fibrosis Program and AASLD/Schering Advanced Hepatology Fellowship Grant (to MBB), and DK49629 and DK58315 (to RT). The authors gratefully acknowledge the helpful comments of Professor MJ Arthur in reviewing this manuscript and Dr Gennaro Cilberto for originally providing IL-6−/− mice.
Abbreviations
- IL-6
interleukin-6
- ECM
extracellular matrix
- CCl4
carbon tetrachloride
- HSC
hepatic stellate cell
- TNF-α
tumor necrosis factor-alpha
- IACUC
Institutional Animal Care and Use Committee
- STAT
signal transducer and activator of transcription
- IC
inhibitory concentration
- BrdU
bromodeoxyuridine
- MT1-MMP
membrane-type 1 matrix metalloproteinase
- TIMP-2
tissue inhibitor of metalloproteinase-2
- TUNEL
terminal deoxynucleltidyl transferase-mediated uDP nick-end labeling assay
- qRTPCR
quantitative real-time polymerase chain reaction
- MMP
matrix metalloproteinase
References
- 1.Sun Y, Tokushige K, Isano E, Yamauchi K, Ohata H. Elevated serum interleukin-6 levels in patients with acute hepatitis. J Clin Immunol. 1992;12:192–200. doi: 10.1007/BF00918089. [DOI] [PubMed] [Google Scholar]
- 2.Hill D, Marsano L, Cohen D, Allen J, Shedlofsky S, McClain C. Increased plasma interleukin-6 concentrations in alcoholic hepatitis. J Clin Immunol. 1992;119:547–552. [PubMed] [Google Scholar]
- 3.Taub R. Hepatoprotection via the IL-6/stat 3 pathway. J Clin Investig. 2003;112:978–980. doi: 10.1172/JCI19974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cressman D, Greenbaum L, DeAngelis R, Cilberto G, Furth E, Poli V, et al. Liver failure and defective hepatocyte regeneration in interleukin-6 deficient mice. Science. 1996;274:1379–1383. doi: 10.1126/science.274.5291.1379. [DOI] [PubMed] [Google Scholar]
- 5.Kovalovich K, Li W, DeAngelis R, Greenbaum L, Cilberto G, Taub R. Interleukin-6 protects against Fas-mediated death by establishing a critical level of anti-apoptotic hepatic proteins FLIP, Bcl-2, and Bcl-XL. J Biol Chem. 2001;276:26605–26613. doi: 10.1074/jbc.M100740200. [DOI] [PubMed] [Google Scholar]
- 6.Czaja M, Xu J, Alt E. Prevention of carbon tetrachloride-induced rat liver injury by soluble tumor necrosis factor receptor. Gastroenterology. 1995;108:1849–1854. doi: 10.1016/0016-5085(95)90149-3. [DOI] [PubMed] [Google Scholar]
- 7.Friedman S. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem. 2000;275:2247–2250. doi: 10.1074/jbc.275.4.2247. [DOI] [PubMed] [Google Scholar]
- 8.Wielockx B, Lannoy K, Shapiro S, Itoh T, Itohara S, Vanderkerckhove J, et al. Inhibition of matrix metalloproteinases blocks lethal hepatitis and apoptosis induced by tumor necrosis factor and allows safe antitumor therapy. Nat Med. 2001;7:1202–1208. doi: 10.1038/nm1101-1202. [DOI] [PubMed] [Google Scholar]
- 9.Nagase H, Woessner J. Matrix metalloproteinases. J Biol Chem. 1999;1999:21491–21494. doi: 10.1074/jbc.274.31.21491. [DOI] [PubMed] [Google Scholar]
- 10.Poli V, Balena R, Fattori E, Markatos A, Yamamoto M, Tanaka H, et al. Interleukin-6 deficient mice are protected from bone loss caused by estrogen depletion. Eur Mol Biol Org J. 1994;13:1189–1196. doi: 10.1002/j.1460-2075.1994.tb06368.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kovalovich K, Angelis RD, Li W, Furth E, Cilberto G, Taub R. Increased toxin-induced liver injury and fibrosi in IL-6 deficient mice. Hepatology. 2000;31:149–159. doi: 10.1002/hep.510310123. [DOI] [PubMed] [Google Scholar]
- 12.Koivunen E, Arap W, Valtanen H, Rainisalo A, Medina O, Heikkila P, Kantor C, et al. Tumor targeting with a selective gelatinase inhibitor. Nat Biotech. 1999;17:768–774. doi: 10.1038/11703. [DOI] [PubMed] [Google Scholar]
- 13.Safadi R, Ohta M, Alvarez C, Fiel MI, Bansal M, Mehal W, et al. Immune stimulation of hepatic fibrogenesis by CD8 Cells and attenuation by transgenic interleukin-10 from hepatocytes. Gastroenterology. 2004;127:870–882. doi: 10.1053/j.gastro.2004.04.062. [DOI] [PubMed] [Google Scholar]
- 14.Le J, Dauchot P, Perrot J, Cambazard F, Frey J, Chamson A. Quantitative zymography of matrix metalloproteinases by measuring hydroxyproline: application to gelatinases A and B. Electrophoresis. 1999;20:2824–2829. doi: 10.1002/(SICI)1522-2683(19991001)20:14<2824::AID-ELPS2824>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 15.Vogel S, Piantedosi R, Frank J, Lalazar A, Rockey D, Friedman S, et al. An immortalized rat liver stellate cell line (HSC-T6): a new cell model for the study of retinoid metabolism in vitro. J Lipid Res. 2000;41:882–893. [PubMed] [Google Scholar]
- 16.Leu J, Crissey M, Leu J, Ciliberto G, Taub R. Interleukin-6 induced stat-3 and AP-1 amplify hepatocyte nuclear factor-1 mediated transactivation of hepatic genes, an adaptive response to liver injury. Mol Cell Biol. 2001;21:414–424. doi: 10.1128/MCB.21.2.414-424.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Overall C, Tam E, McQuibban G, Morrison C, Wallon U, Bigg H, et al. Domain interactions in the gelatinase A/TIMP-2/MT1-MMP activation complex. J Biol Chem. 2000;2000:39497–39506. doi: 10.1074/jbc.M005932200. [DOI] [PubMed] [Google Scholar]
- 18.Keski-Oja J, Lohi J, Tuuttila K, Tryggvaso K, Vartio T. Proteolytic processing of the 72,000-Da type IV collagenase by urokinase plasminogen activator. Exp Cell Res. 1992;202:471–476. doi: 10.1016/0014-4827(92)90101-d. [DOI] [PubMed] [Google Scholar]
- 19.Theret N, Musso O, L’Helgoualch A, Clement B. Activation of matrix metalloproteinase-2 from hepatic stellate cells requires interactions with hepatocytes. Am J Pathol. 1997;150:51–58. [PMC free article] [PubMed] [Google Scholar]
- 20.Schuppan D, Ruehl M, Somasundarum R, Hahn E. Matrix as a modulator of hepatic fibrogenesis. Semin Liver Dis. 2001;21:351–372. doi: 10.1055/s-2001-17556. [DOI] [PubMed] [Google Scholar]