

Abstract
Self-renewing cancer stem cells (CSC) capable of spawning more differentiated tumor cell progeny are required for tumorigenesis and neoplastic progression of leukemias and several solid cancers. The mechanisms by which CSC cause tumor initiation and growth are currently unknown. Recent findings that suggest a negative correlation between degrees of host immunocompetence and rates of cancer development raise the possibility that only a restricted minority of malignant cells, namely CSC, may possess the phenotypic and functional characteristics to evade host antitumor immunity. In human malignant melanoma, a highly immunogenic cancer, we recently identified malignant melanoma initiating cells (MMIC), a novel type of CSC, based on selective expression of the chemoresistance mediator ABCB5. Here we present evidence of a relative immune privilege of ABCB5+ MMIC, suggesting refractoriness to current immunotherapeutic treatment strategies. We discuss our findings in the context of established immunomodulatory functions of physiologic stem cells and in relation to mechanisms responsible for the downregulation of immune responses against tumors. We propose that the MMIC subset might be responsible for melanoma immune evasion and that immunomodulation might represent one mechanism by which CSC advance tumorigenic growth and resistance to immunotherapy. Accordingly, the possibility of an MMIC-driven tumor escape from immune-mediated rejection has important implications for current melanoma immunotherapy.
Keywords: cancer stem cell, immunity, tolerance, melanoma, ABCB5, immunotherapy, regulatory T cells, anergy, MART-1, T cell activation
Introduction to the Cancer Stem Cell Theory of Tumor Initiation and Growth
Experimental evidence has been generated that tumors, like physiologic tissues, may be organized as a hierarchy of phenotypically heterogeneous cell populations with divergent self-renewal capacities, degrees of differentiation, and clonogenic potentials, and that cancer stem cells (CSC), which may comprise only a fraction of neoplastic cells but are nevertheless essential for its propagation, are found at the apex of this tumor cell hierarchy.1–13 CSC are operationally defined as a clinical tumor specimen–derived cancer minority population, prospectively identifiable by a molecular marker or marker combination, that, unlike cancer bulk populations negative for the particular marker or set of markers, can initiate tumor formation and growth in immunodeficient hosts in vivo.14 In addition, CSC populations should exhibit the selective ability to (1) self-renew, demonstrated by serial xenotransplantation and/or genetic lineage tracking experiments in vivo, and (2) to differentiate and give rise to non-tumorigenic cancer bulk components, thereby recapitulating the original patient tumor both morphologically and immunophenotypically in all serial xenografts.14 It is important to recognize that the CSC definition does not make any specific assumptions about the relative frequency of tumorigenic CSC populations or about any observed transdifferentiation plasticity of such cell subsets.14 Moreover, despite shared properties with physiologic stem cells (including self-renewal and differentiation), the term CSC does not refer to the putative cell-of-origin of a given tumor.14 Based on these defining criteria, the CSC hypothesis predicts (1) that the CSC pool is required for tumor maintenance and disease progression and (2) that targeted elimination of CSC could eradicate cancers currently resistant to systemic therapy. Consequently, researchers have embarked on the quest of prospectively identifying CSC populations in a multitude of tumors of diverse etiology1–13 and are beginning to elucidate the molecular pathways regulating their behavior.4,15–18 Importantly, critical links between CSC, tumor progression, and therapy resistance have emerged, and proof-of-principle has now been established that selective ablation of CSC can inhibit tumorigenic growth in experimental animal models.10,12,19 Despite these seminal advances, the processes through which CSC may sustain neoplastic growth and disease progression remain incompletely understood, and CSC-directed treatment strategies require further optimization to successfully translate such promising approaches to the clinic. For example, demonstration of a specific relationship of CSC to the evasion of host antitumor immunity would have important implications for cancer therapy.
Cancer Stem Cells in Human Malignant Melanoma and the Concept of Immune Surveillance
In human malignant melanoma, a highly aggressive and therapy-resistant cancer,20 studies from our group recently identified a novel type of CSC, malignant melanoma initiating cells (MMIC),10 based on their expression of the chemoresistance determinant ABCB5.21,22 ABCB5+ MMIC showed the exclusive capacity to establish melanoma xenografts in both primary and secondary nonobese diabetic/severe combined immunodeficiency disease (NOD/SCID) mouse recipients and gave rise to ABCB5+ and ABCB5− tumor cell progeny in serial xenotransplantation and in vivo genetic lineage tracking experiments, formally establishing a hierarchical organization among melanoma cells.10 Moreover, the frequency of ABCB5+ MMIC correlated significantly with disease progression in human patients, and specific targeting of MMIC for selective ablation via antibody-dependent cell-mediated cytotoxicity (ADCC) significantly inhibited tumor xenograft growth in experimental animal models.10 In addition, the ABCB5 gene was found preferentially expressed by in vitro self-renewing melanoma subpopulations,23 by melanomas of metastatic as opposed to primary tumor origin,24 and by melanoma cells with enhanced tumorigenic competence.25–27 However, the processes through which ABCB5+ MMIC and CSC in other cancers may sustain tumor growth and promote neoplastic progression remain incompletely understood.
Based on the established immunoprivilege and active immunoregulatory functions of physiologic stem cells,28–30 we previously hypothesized that CSC may foster tumor initiation and growth at least in part via attenuation of the antitumor immune response.20 A negative correlation between host immuno-competence and tumor initiation rates is also indicated by findings that immunocompromised patients, including solid organ transplant recipients on immunosuppressive medications and HIV-infected individuals following AIDS onset, show a markedly increased risk of developing malignant neoplasms of diverse etiologies.31 In addition, severely immunocompromised animals, such as SCID and Rag−/− mice, which bear deficiencies in both their innate and adaptive immune systems, display a significantly increased incidence of epithelial carcinoma development among other tumors late in life.32 Negative correlations between host immunocompetence and rates of tumor development may also apply to malignant melanoma.33 In this regard, recent findings by Quintana and colleagues showed that fewer human melanoma cells were required to cause tumor development in more severely compromised interleukin-2 receptor gamma chain null (IL-2Rγ−/−) NOD/SCID hosts compared with NOD/SCID recipients.34 Taken together with the findings of higher rates of cancer development in immunocompromised patients and animal models,31,32 these results lend support to the notion that an intact host immune system may be able to eliminate transformed cells to prevent tumorigenesis at early stages of disease.35 This “concept of immunosurveillance” may thus provide a potential explanation for the relatively low frequency of tumor development in healthy individuals.35 At the same time, the aforementioned findings indicate that only a restricted minority of tumor cells, that is, the CSC, may possess the phenotypic and functional properties to evade host immuno-surveillance and immune-mediated rejection in immunologically intact individuals.20 An immunoselection of CSC populations that would be expected to be more capable of surviving in an immunocompetent host compared to tumor bulk components20 might be especially relevant in a highly immunogenic cancer such as human malignant melanoma.36
Here we present initial evidence of a relative immune privilege of ABCB5+ MMIC and discuss our findings in the context of established mechanisms responsible for the downregulation of immune responses against tumors and in relation to immunomodulatory functions of physiologic stem cells.
Results and Discussion
Tumor Immune Evasion and Resistance to Immune-Mediated Rejection—Processes Regulated by the Cancer Stem Cell Pool?
There are several mechanisms by which CSC might modulate immune responses.20 It is important to recognize, however, that the degree of natural antitumor immunity may be limited and often insufficient to destroy a rapidly growing neoplasm, including its CSC pool, which arises from the organism’s own tissue and therefore predominantly expresses self-antigens to which host immune cells have been tolerized.35 Immunogenic tolerance to a particular set of antigens is the absence of an immune response to those antigens, which can be achieved by processes that result in physical elimination (clonal deletion through apoptotic cell death) or functional inactivation (clonal anergy) of antigen-reactive cells.37 The induction of tolerance through positive and negative selection mechanisms equips the body with an immunocyte repertoire that recognizes and protects self major histocompatibility complex (MHC)/self peptide complex–bearing cells and is, at the same time, capable of launching a defense against cells expressing MHC/foreign peptide complexes.38–41 While self-tolerance under physiological conditions functions to protect the body against the emergence of autoimmunity, tolerance toward a tumor in the cancer context may permit neoplastic development and growth.35 This problem has been characterized for example by Theobald and colleagues for the tumor suppressor protein p53, which is expressed at elevated levels by approximately 50% of human cancers.42 However, because of its low-level expression in physiologic tissues, tolerance by clonal deletion of p53-reactive T cells could potentially impede antitumor immunity. Indeed, the avidity (relative intensity of reactivity) of p53-specific HLA-A2.1-restricted cytotoxic T lymphocytes (CTL) was 10-fold lower in p53+/+ compared to p53−/− HLA-A2.1/Kb transgenic mice.42
Nevertheless, antitumor immune responses can be detected in the tumor-bearing host and are particularly relevant to disease progression and anticancer therapeutic approaches in highly immunogenic tumors, including human malignant melanoma.36 Two distinct groups of tumor antigens can be distinguished: (1) tumor-specific antigens (TSAs), which are encoded by mutant or rearranged genes such as the leukemia-specific fusion protein BCR-ABL, and (2) tumor-associated antigens (TAAs), which are encoded by normal, cell lineage-specific genes.43 A number of TAAs have been characterized in melanoma, including the melanocyte differentiation antigens MART-1 (melanoma antigen recognized by T cells-1), tyrosinase, gp100, and the cancer testis antigen NY-ESO-1.44–47 Moreover, both circulating T cell clones and tumor-infiltrating lymphocytes (TILs) reactive against TAAs can be detected in melanoma patients.45,46,48 However, while MART-1 and tyrosinase-specific CD8+ immune effector cell populations are detectable in cancer patients, they are often incapable of rejecting the tumor.46 It is conceivable, that tumors may escape the anticancer immune response by downregulating TAAs, as indicated by a separate study by Khong and colleagues.48 In light of the CSC concept, it is further plausible that more undifferentiated tumor minority populations responsible for tumor initiation and growth, that is, CSC, may selectively not express TAAs associated with differentiation and may therefore be resistant to immune-mediated rejection. To experimentally address this possibility, we costained human melanomas for the MMIC-determinant ABCB510 and the TAA MART-1. While MART-1 can be expressed by both ABCB5− and ABCB5+ populations,21 certain melanomas, such as aggressive human A375 tumor cells, can exhibit relative negativity of the MMIC component for the differentiation antigen MART-1. Specifically, MART-1 was expressed by 14.1% of all melanoma cells. On the other hand, ABCB5+ MMIC detected among A375 melanoma cells did not express this TAA at significant levels (Fig. 1). Consistent with these findings, in the context of the association of pigmentation with the activity of tyrosinase,49 our previous results also demonstrated that ABCB5+ MMIC correlated with nonpigmented, undifferentiated regions in human patient biopsies.10 Our data indicate that MMIC might preferentially evade the antitumor immune response directed at tumor antigens associated with a more differentiated malignant phenotype (Fig. 2), providing for a potential explanation for the relative ineffectiveness of CD8+ tumor-reactive T cells in achieving major antitumor responses.46
Figure 1. Immune phenotype of ABCB5+ malignant melanoma initiating cells (MMIC).
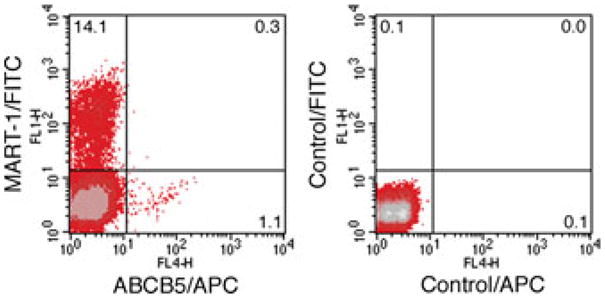
Representative dual-color flow cytometry analysis of human A375 melanoma cells costained for ABCB5 (APC, FL4 fluorescence) and the melanoma antigen recognized by T cells-1 (MART-1) (FITC, FL1 fluorescence). ABCB5+ cells co-expressing MART-1 are found in the top right quadrant of the left fluorescence plot. The right fluorescence plot depicts isotype control mAb-stained melanoma cells.
Figure 2.
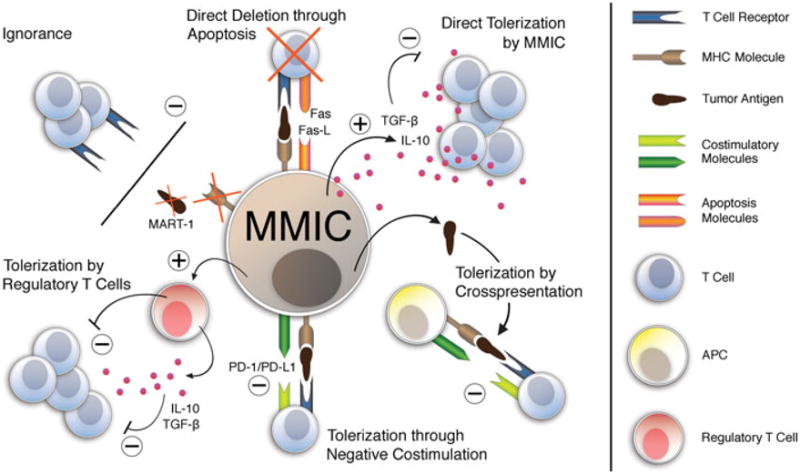
Schematic model of candidate mechanisms underlying malignant melanoma initiating cell (MMIC)-driven tumor immune evasion. Many of the processes underlying tolerance induction by physiologic stem cells parallel mechanisms of immune evasion relevant to neoplastic development, disease progression, and therapy-resistance of immunogenic cancers, including human malignant melanoma. It is possible that the CSC component within a tumor may possess a preferential capacity to mitigate the antitumor immune response, and that such a potential function of ABCB5+ MMIC might contribute to melanomagenesis. MMIC could evade rejection by tumor antigen–specific CD8+ immune effector populations through absent or reduced expression of tumor-associated antigens, such as MART-1, or decreased MHC class I molecule expression. Alternatively, T cell ignorance of MMIC may also result from spatial separation. Direct deletion of immune effector cells through induction of apoptotic cell death (e.g., through expression of apoptosis-inducing ligands, such as Fas-L) might represent an additional mechanism of MMIC-driven tumor escape from immune destruction. ABCB5+ MMIC could possibly also tolerize tumor-reactive T cells via direct secretion of immunosuppressive factors, including TGF-β or IL-10. Cross-presentation of tumor-associated or MMIC-specific antigens by APC incapable of providing adequate costimulation might likewise induce tumor tolerance by rendering melanoma-reactive T cells anergic. Furthermore, ABCB5+ MMIC could potentially also tolerize melanoma-specific T cells by delivering a negative costimulatory signal (e.g., via PD/PD-L1 or B7/CTLA-4 pathways) at the time of alloantigen recognition. Finally, MMIC might actively induce or recruit regulatory T cells to disrupt the antimelanoma immune response.
Another mechanism through which ABCB5+ MMIC might evade host immuno-surveillance may be through absence or downregulation of MHC class I molecules (Fig. 2). MHC class I antigens play a key role in the immune recognition of transformed cells50 and, accordingly, selective or total downregulation of class I molecules may render class I MHC-restricted CTLs unable to lyse melanoma target cells,48,51 including CSC. For instance, mesenchymal stem cells express decreased levels of MHC class I,52 further supporting the possibility of reduced class I expression by CSC populations. Moreover, it is known that class I MHC molecules are regulated not only on immune cells and physiologic stem cells but also on cancer cells,35 and that decreased MHC class I expression by human melanoma cells correlates positively with disease progression, therapeutic unresponsiveness, and adverse clinical outcome in human patients.53–55 In addition, given the fact that TAA-specific T cells are often unresponsive to melanoma cells expressing both the respective TAA and the appropriate MHC molecule,46,56 active tolerance induction by tumors may be operative. In support of this hypothesis, CD4+ T cells specific for a tumor antigen were unable to be primed and showed a diminished antigenic response upon adoptive transfer to tumor-bearing mice.57
One major mechanism of tolerance induction involves clonal deletion of antigen-reactive T cells through apoptotic cell death.37 An important mediator of T cell deletion is the Fas/Fas-L system.58 Neoplasms, including melanoma, may actively destroy T cells by expressing Fas-L, a function associated with enhanced experimental tumor formation.58–61 Indeed, Fas-L-expressing melanoma cells were found to trigger apoptosis of lymphoid cells,59 and autocrine secretion of soluble Fas-L shielded melanomas from Fas-mediated killing by CTLs.62 In addition, tumor cells can evade an immune attack by downregulation of the Fas receptor.58 In melanoma patients, loss of Fas-L expression in metastases was associated with better overall survival.63 Another tumor-expressed ligand, RCAS1 (receptor-binding cancer antigen expressed on SiSo cells), was also found to induce apoptotic cell death of T, B, and NK (natural killer) cells expressing its receptor.64 Importantly, human physiologic stem or progenitor cells can likewise regulate both the Fas/Fas-L system and RCAS1-mediated apoptosis.65–67 Together, these findings allow for the possibility that the ABCB5+ MMIC subset within melanomas might actively downregulate the antitumor immune response through apoptotic deletion of activated T cell clones (Fig. 2).
Immunogenic tolerance may also be achieved through nondeletional processes, such as through functional inactivation (clonal anergy) of antigen-reactive cells.39 One mechanism that has been implicated in the induction of anergy is direct T cell inhibition via immunosuppressive cytokines, including interleukin (IL)-10 and transforming growth factor-beta (TGF-β).68 For instance, TGF-β secretion may underlie human mesenchymal stem cell–mediated suppression of T cell proliferation, because antibodies against the soluble factor were found to partially restore T cell activation and proliferative responses in mesenchymal stem cell/T lymphocyte cocultures.66,68 TGF-β and IL-10 may also be produced by tumors, including melanomas, to avoid immune-mediated destruction.70–72 Indeed, T cell-specific blockade of TGF-β signaling was found to enhance the antitumor immune response in mice challenged with live tumor cells.71 Most notably, the TGF-β signaling pathway is specifically activated in the CSC fraction of breast cancers73 and secreted morphogen members of the TGF-β superfamily and their receptor transcripts are also preferentially expressed by CD133+ brain tumor CSC18 and by ABCB5+ MMIC.10 Taken together, these findings suggest that, in melanoma, it might be the MMIC subset that renders antigen-reactive lymphocytes anergic through secretion of immunosuppressive factors such as TGF-β or IL-10 to promote tumor immune evasion and ultimately tumorigenic growth (Fig. 2).
Induction of anergy may also result from impaired stimulation of antigen-specific lymphocytes.74 According to the “two signal” paradigm, full activation of T cells in response to antigens requires two separate but complementary signals: on antigen encounter, naïve T cells receive signal 1 through cognate interaction of their T cell receptor (TCR/CD3) with the MHC/antigenic peptide complex on an antigen presenting cell (APC) (Fig. 3). Signal 2 is an antigen-nonspecific signal triggered by the interaction of a pair of positive costimulatory molecules expressed on the T cell and APC, respectively, leading to full immune activation74–76 (Fig. 3A). Such positive costimulatory stimuli to the T cell induce IL-2 production, alloantigen-specific clonal expansion, and acquisition of a memory/effector phenotype77 (Fig. 3A). Negative costimulatory signals, on the other hand, function to downregulate T cell immune responses74 (Fig. 3B). Costimulatory molecules can be grouped broadly into two distinct superfamilies based on structural homology: The CD28:B7 superfamily, which includes CD28 and CTLA-4 (cytotoxic T lymphocyte antigen-4), and the tumor necrosis factor (TNF):TNF-R superfamily, in which the CD40-CD40L pathway is preeminent (Fig. 4). It is increasingly recognized, that costimulatory signaling events are not limited to T cell–APC interactions, but also comprise T cell–T cell, T cell–B cell, and T cell–nonlymphoid cell, including T cell–cancer cell interactions.78 Importantly, absence of effective costimulation may result in T cell anergy and/or apoptosis.79
Figure 3.
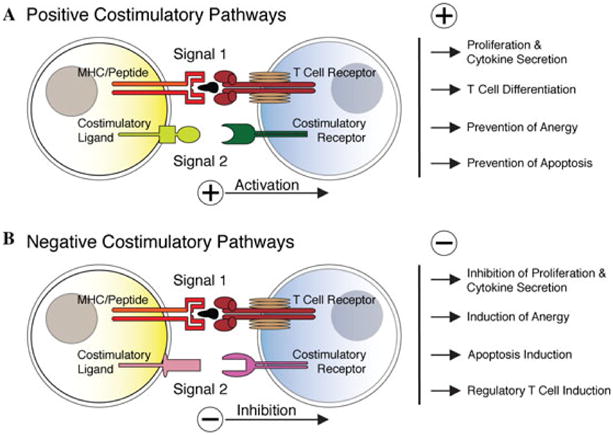
The “two-signal” paradigm of antigen-dependent T cell activation. According to the “two-signal” paradigm, antigen-dependent T cell activation requires two distinct but synergistic signals. (A) On antigen encounter, naïve T cells receive signal 1 through T cell receptor engagement with the MHC/antigenic peptide complex expressed on an APC, and signal 2 through ligation of a positive costimulatory receptor/ligand pair leading to full activation. Positive costimulation leads to T cell activation, proliferation, and cytokine production, drives naïve T helper cells into differentiation, and may inhibit T cell anergy and apoptosis. (B) By contrast, negative T cell costimulatory signals function to downregulate immune responses. T cell receptor engagement coincident with negative costimulation inhibits T cell proliferation and cytokine secretion, induces T cell anergy and apoptosis, and may control the suppressive function of regulatory T cells. Signal 1–associated molecules (MHC class I and II) and signal 2 pathway members (positve and negative costimulatory molecules) are regulated not only on T cells and APC; expression can also be observed on other lymphocyte compartments as well as in nonlymphoid cells, including cancer cells.
Figure 4.
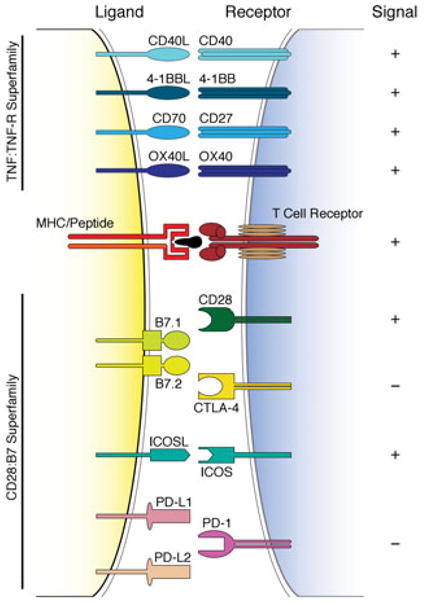
Schematic overview of costimulatory receptors and their ligands. Illustrated are costimulatory ligands and their respective receptors, which can deliver signals that function to either activate (+) or downregulate (−) immune responses in tandem with alloantigen-specific T cell receptor engagement with the MHC/peptide complex. Costimulatory molecules can be grouped broadly into two distinct superfamilies based on structural homology: the CD28:B7 superfamily, which includes CD28 and CTLA-4, and the TNF:TNF-R superfamily, in which the CD40-CD40L pathway is pre-eminent. Costimulatory molecules were originally described in immune cells. It is increasingly recognized, however, that costimulatory signaling can also occur in nonlymphoid cells, including cancer cells.
In this regard, it was recently demonstrated that physiologic bone marrow–derived stem cells could inhibit differentiation and function of APC leading to T cell unresponsiveness.80,81 In the cancer context, CSC might therefore reduce T cell responses to tumor antigens by actively modulating the activation state of APC (Fig. 2). Indeed, tolerance to immunogenic tumor epitopes may be achieved through cross-presentation of a particular TAA by APC incapable of providing adequate costimulation.82 In vivo activation of such APC by positive CD40/CD40L-mediated costimulation resulted in conversion of this T cell tolerance to T cell priming and subsequent regression of established neoplasms in experimental animal models,83 further emphasizing the importance of costimulatory signaling for a functional antitumor immune response.
It is also conceivable that tumor cells themselves may serve as functional APC.84 In fact, findings of class II MHC expression by human melanoma cells50,85 provide experimental support for this possibility. Importantly, maintenance of a nonresponsive state of anergic T cells requires their continuous exposure to the respective antigen.40 Direct presentation of tumor antigens by MHC class II–expressing melanoma cells in the absence of positive costimulation could therefore potentially perpetuate tolerogenic effects of tumor-specific T cells. Consistent with this hypothesis, MHC class II+ melanoma cells were able to induce clonal anergy in MHC class II–restricted T cell clones ex vivo.86 In vivo, T cell homing to lymph nodes is required for tolerance induction to alloantigens.87 Antigen presentation by MHC class II–expressing melanoma cells in metastases to lymph nodes could therefore play a particularly important role with regard to tolerance induction of tumor antigen–reactive T cells. In support of this possibility, expression of MHC class II antigen in melanoma cells was associated with disease progression and adverse clinical outcome in human patients.85,88 Strikingly, the frequency of ABCB5+ MMIC likewise correlates with disease progression in melanoma patients.10 Moreover, metastases to lymph nodes display an enhanced abundance of ABCB5+ MMIC as compared to visceral metastases or primary melanomas,10 indicating that MMIC may possess a preferential ability to evade host immunosurveillance. In the context of tolerance induction and costimulation, these findings could further support a model whereby ABCB5+ MMIC might actively present TAAs or CSC-specific antigens in the absence of positive costimulation to render tumor-reactive T cells anergic.
ABCB5+ MMIC could also express negative costimulatory molecules to disrupt the antitumor immune response (Fig. 2). This possibility is supported by findings in physiologic stem cells, which may process antigens in a MHC class II–dependent fashion89 and inhibit lymphocyte activation at least in part through PD-1/PD-L1-mediated negative costimulatory signaling.90,91 Important insights have also been generated with regard to PD-1 negative costimulatory signaling in the context of melanoma development and immunotherapy.92–95 For example, monoclonal antibody–mediated blockade of PD-1 directed at thymocyte populations was found to enhance the expansion and function of human vaccine-induced T lymphocytes specific for melanoma-associated antigens95 and abrogated in vivo tumor growth in murine tumor models.94 Moreover, melanoma cell–expressed PD-L1 conferred resistance to CTL-mediated target cell lysis94 and induced T cell apoptosis in murine models of melanoma development.92
Another mechanism potentially responsible for downregulation of antitumor immune responses by MMIC might be the induction and/or active recruitment of regulatory T (Treg) cells (Fig. 2). Treg cells can potently suppress the activation, proliferation, and cytokine production of other T cells, and are essential for maintaining immunological self-tolerance and immune homeostasis.96–98 Phenotypically, Treg cells are generally defined by CD4+CD25+FoxP3+ marker expression,96 yet CD8+ Treg cell types have also been characterized.99 Signaling through negative costimulatory molecules expressed by Treg cells, such as CTLA-4 or PD-L1, may control their suppressive function.100,101 However, Treg cells may likewise require activation through positive costimulatory signaling events or proliferative cytokine stimulation to exert their tolerogenic effects.102,103 Notably, mesenchymal stem cells can induce tolerance to alloantigens via the generation and recruitment of Treg cells as demonstrated both in vitro and in experimental animal models in vivo.104–106 Moreover, in a murine melanoma model, Treg cell induction by mesenchymal stem cells fostered tumor growth, even in allogeneic mouse recipients.107 Recent evidence further suggests that Treg cells capable of inhibiting autologous T cell proliferation might also be involved in thwarting the antitumor immune response in oncology patients.108–112 Specifically, an increased prevalence of Treg cells both in peripheral blood and in tumor lesions was shown in breast and pancreatic cancer patients compared to healthy individuals.111 In additional malignancies, including melanoma, Treg cells are actively recruited from the blood and selectively accumulate in the tumor microenvironment, where they may form a dense infiltrate shielding the neoplasm from immune effector responses.108–110 Treg cell frequency was further associated with a high death hazard and reduced survival of tumor patients.110
In summary, many of the processes underlying tolerance induction by physiologic stem cells28,30 parallel mechanisms of immune evasion relevant to neoplastic development, disease progression, and therapy resistance of immunogenic cancers, including melanoma.35,84,113 It therefore seems plausible that the CSC component within a tumor may possess a preferential capacity to mitigate the antitumor immune response, and that such a potential function of ABCB5+ MMIC might contribute to melanomagenesis. Accordingly, the possibility of an MMIC-driven tumor escape from immune-mediated rejection has important implications for current immunotherapeutic strategies in human malignant melanoma.
Perspectives—Possible Relevance of Cancer Stem Cells to Melanoma Immunotherapy
In melanoma, immunotherapy has emerged as one of the most promising treatment options for patients with metastatic disease, with sometimes striking but nevertheless often limited success.36 A major challenge has been to develop strategies that efficiently overcome tumor evasion of host immune responses and immunotherapeutic resistance.35 Strategies for enhancing antitumor immunity can be broadly categorized into (1) nonspecific immunomodulation approaches aimed at activating the host’s immune response, (2) active immunization procedures directed at sensitizing the immune system against the autologous cancer (e.g., cancer vaccines with whole cells, peptides, or immunizing vectors), and (3) adoptive cell transfer of ex vivo expanded autologous or allogeneic lymphocytes with tumor target specificity.36
Nonspecific activators of tumor-reactive lymphocytes approved by the US Food and Drug Administration for the treatment of patients with metastatic melanoma include interferon (INF)-α and IL-2.36,114 However, most of the responses to monotherapies with INF-α and IL-2 have been partial, with complete response rates ranging from approximately 5–15%.115–119 Because of the established negative correlation of IL-2 cytokine levels with melanoma development in experimental animal models,120,121 it seems plausible that ABCB5+ MMIC might be refractory to IL-2-induced effects and/or might preferentially inhibit IL-2 production compared to melanoma bulk components, and that such a potential function of MMIC might contribute to melanomagenesis and disease progression. This possibility has implications for the design of experiments aimed at the identification and study of CSC subsets. If true, tumorigenicity experiments evaluated in the absence of an IL-2 effect due to lack of its functional receptor (such as in IL-2Rγ−/− NOD/SCID recipients34) would represent inadequate xenotrans-plantation assays for the detection of CSC. In the clinic, an obstacle of IL-2 immunotherapeutic regimens has been the simultaneous increase in Treg cells thought to contribute to poor treatment response in some melanoma patients.122 Depletion of the Treg cell pool may therefore represent a very promising approach to enhance immunotherapeutic efficacy, as indicated by findings in preclinical models.123 While Treg cell–depleting strategies have resulted in improved tumor control in some clinical studies,124 an inability of current regimens to mediate prolonged reduction of Treg cells and to induce efficient melanoma regression was observed by others.122,125 In light of the CSC hypothesis and its potential relevance to tumor immune evasion, specific targeting of MMIC-mediated mechanisms responsible for the induction or recruitment of Treg cells might circumvent such immune resistance phenomena.
More recently, additional immunotherapeutic agents have been developed that target critical regulatory elements of patient immune cells to enhance their antitumor cytotoxic efficacy.126 These include monoclonal antibodies that functionally block negative regulators of lymphocyte activation, including CTLA-4, PD-1, and TGF-β.36,93,126,127 Such regimens may potentially provide significant advantages over current immunotherapies because of their inhibitory effect on Treg cell function.93,126 Moreover, they might prove most efficient in attaining objective antitumor responses if potential MMIC-specific immune escape mechanisms are concurrently impaired (TGF-β secretion or negative costimulatory molecule expression by ABCB5+ MMIC, for example). In this regard, it might be interesting to analyze the efficacy of such novel agents for the treatment of metastatic melanoma not only with regard to the pattern and duration of immune responses128 but also in the context of their potential impact on the ABCB5+ MMIC pool.
Active immunization approaches (using whole cells, peptides, or antigen-pulsed APC) in human malignant melanoma are often directed at known targets of immune reactivity, which include markers of melanocyte differentiation such as tyrosinase, gp100, or MART-1.36 Despite the frequently observed induction of tumor antigen–specific T cells in response to such vaccination approaches,129 tumor progression can occur, and only rare and highly sporadic regressions have been achieved in melanoma patients.130 Our demonstration that the melanoma cell subset responsible for experimental tumor initiation and growth—ABCB5+ MMIC10—may not express melanoma-associated antigens could provide a novel explanation for the failure of such active immunization strategies. Epigenetic and targeted therapies capable of driving MMIC into differentiation, as described for other cancers that follow the CSC model of tumor development,18,131 could potentially improve the therapeutic efficacy of vaccination strategies directed at differentiation markers. In light of the CSC concept, it would appear, however, more promising to target markers specific to the MMIC compartment. For instance, ABCB5 might represent a promising target for MMIC-directed immunization strategies in human malignant melanoma, where the molecule is principally expressed.10,21,132 Our findings of significant experimental tumor growth inhibition via immune effector mechanisms induced by a monoclonal antibody specifically directed at ABCB5+ MMIC minority populations in the context of increased MMIC frequencies in metastatic compared to primary melanoma lesions in human patients10 would support such an approach. Selective eradication of tumorigenic minority populations was recently also demonstrated to halt experimental tumor development in other malignancies,12,19,133 further validating the potential therapeutic relevance of the CSC concept. Whether other antigens, including for example the cancer testis antigen NY-ESO-1 recently found expressed by more primitive melanoma cell subsets,134 might be present on ABCB5+ MMIC, represents an important future line of investigation.
Another strategy to enhance immune-mediated tumor destruction is to infuse melanoma patients with ex vivo–generated autologous or allogeneic T cell clones specifically reactive against melanoma antigens.36 While this so-called adoptive cell transfer (ACT) approach has yielded significant antitumor responses, the in vivo homing properties and lifespan of such tumor-reactive T cell clones may be limited.135 Opportunities for improving ACT treatment efficacy might involve transduction or stimulation of ex vivo expanded T cells with appropriate growth factors or positive costimulatory ligands,136 concurrent blockade of inhibitory signals on tumor-reactive lymphocytes,137 or prior depletion of host Treg cells.138 Because the CSC concept applies to tumor development and neoplastic progression in human patients,10 ex vivo expansion of T lymphocytes reactive against MMIC-specific epitopes might represent an additional promising avenue to further enhancing successful tumor eradication through ACT. In view of the potential ability of ABCB5+ MMIC to preferentially evade immune-mediated rejection, current immunotherapeutic regimens might generate more durable responses if they also block the mechanisms responsible for MMIC-driven tolerance induction.
Materials and Methods
Melanoma Cell Culture
The human A375 melanoma cell line was purchased from ATCC (Manassas, VA) and cultured in RPMI 1640 medium (Lonza Bio-Whittaker, Walkersville, MD) supplemented with 10% (v/v) fetal bovine serum (Invitrogen GIBCO, Carlsbad, CA) and 2% (v/v) penicillin/streptomycin (Lonza Bio-Whittaker) as described previously.10,21
Antibodies
The specific IgG1γ anti-ABCB5 mono-clonal antibody (mAb) 3C2–1D12141 was used in the herein reported studies. Unconjugated MOPC-31C mouse isotype control mAb and fluorescein isothiocyanate (FITC)-conjugated goat antimouse secondary Ab were purchased from BD PharMingen (San Diego, CA). Unconjugated antihuman MART-1 mAb was purchased from Abcam (Cambridge, MA), and allophycocyanin (APC)-conjugated donkey antimouse secondary Ab from eBioscience (San Diego, CA).
Flow Cytometric Analysis
Analysis of co-expression of ABCB5 with the MART-1 intracellular marker in A375 melanoma cells was performed as described previously.10,76 Briefly, cells were incubated with anti-ABCB5 or isotype control mAbs followed by counterstaining with APC-conjugated donkey antimouse IgG. Cells were then fixed in phosphate buffered saline containing 2% paraformaldehyde (30 min at 4°C) and subsequently incubated with unconjugated MART-1 or isotype control mAbs followed by counterstaining with FITC-conjugated anti immunoglobulin secondary antibody. Washing steps with staining buffer or 1% saponin permeabilization buffer were performed between incubations. Subsequently, dual-color flow cytometry was performed with acquisition of fluorescence at the FL1 (FITC) and FL4 (APC) emission spectra on a Becton Dickinson FACSCalibur™ (Becton Dickinson, San Jose, CA) and analysis was carried out using the CellQuest™ software package (Becton Dickinson) as described.10,21,76
Acknowledgments
Our work is supported by funds provided by the NCI/NIH (grants 1RO1CA113796-01A1 and 1R01CA138231-01 to M.H.F.), the NCI/NIH Specialized Program of Research Excellence (SPORE) in Skin Cancer (grant 2P50CA093683-05S2 to Thomas S. Kupper), and a Postdoctoral Fellowship Award from the American Heart Association Founders Affiliate (to T.S.).
Footnotes
Conflicts of Interest
The authors declare no conflicts of interest.
References
- 1.Al-Hajj M, et al. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–737. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 3.Dalerba P, et al. Phenotypic characterization of human colorectal cancer stem cells. Proc Natl Acad Sci USA. 2007;104:10158–10163. doi: 10.1073/pnas.0703478104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hermann PC, et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. 2007;1:313–323. doi: 10.1016/j.stem.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 5.Lapidot T, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–648. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 6.Li C, et al. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67:1030–1037. doi: 10.1158/0008-5472.CAN-06-2030. [DOI] [PubMed] [Google Scholar]
- 7.O’Brien CA, et al. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–110. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- 8.Prince ME, et al. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc Natl Acad Sci USA. 2007;104:973–978. doi: 10.1073/pnas.0610117104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ricci-Vitiani L, et al. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445:111–115. doi: 10.1038/nature05384. [DOI] [PubMed] [Google Scholar]
- 10.Schatton T, et al. Identification of cells initiating human melanomas. Nature. 2008;451:345–349. doi: 10.1038/nature06489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Singh SK, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–5828. [PubMed] [Google Scholar]
- 12.Yang ZF, et al. Significance of CD90(+) cancer stem cells in human liver cancer. Cancer Cell. 2008;13:153–166. doi: 10.1016/j.ccr.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 13.Zhang S, et al. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 2008;68:4311–4320. doi: 10.1158/0008-5472.CAN-08-0364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reya T, et al. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 15.Bao S, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 16.Bao S, et al. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006;66:7843–7848. doi: 10.1158/0008-5472.CAN-06-1010. [DOI] [PubMed] [Google Scholar]
- 17.Li X, et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst. 2008;100:672–679. doi: 10.1093/jnci/djn123. [DOI] [PubMed] [Google Scholar]
- 18.Piccirillo SG, et al. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature. 2006;444:761–765. doi: 10.1038/nature05349. [DOI] [PubMed] [Google Scholar]
- 19.Bao S, et al. Targeting cancer stem cells through L1CAM suppresses glioma growth. Cancer Res. 2008;68:6043–6048. doi: 10.1158/0008-5472.CAN-08-1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schatton T, Frank MH. Cancer stem cells and human malignant melanoma. Pigment Cell Melanoma Res. 2008;21:39–55. doi: 10.1111/j.1755-148X.2007.00427.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frank NY, et al. ABCB5-mediated doxorubicin transport and chemoresistance in human malignant melanoma. Cancer Res. 2005;65:4320–4333. doi: 10.1158/0008-5472.CAN-04-3327. [DOI] [PubMed] [Google Scholar]
- 22.Huang Y, et al. Membrane transporters and channels: role of the transportome in cancer chemosensitivity and chemoresistance. Cancer Res. 2004;64:4294–4301. doi: 10.1158/0008-5472.CAN-03-3884. [DOI] [PubMed] [Google Scholar]
- 23.Keshet GI, et al. MDR1 expression identifies human melanoma stem cells. Biochem Biophys Res Commun. 2008;368:930–936. doi: 10.1016/j.bbrc.2008.02.022. [DOI] [PubMed] [Google Scholar]
- 24.Sousa JF, Espreafico EM. Suppression subtractive hybridization profiles of radial growth phase and metastatic melanoma cell lines reveal novel potential targets. BMC Cancer. 2008;8:19. doi: 10.1186/1471-2407-8-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoek KS, et al. In vivo switching of human melanoma cells between proliferative and invasive states. Cancer Res. 2008;68:650–656. doi: 10.1158/0008-5472.CAN-07-2491. [DOI] [PubMed] [Google Scholar]
- 26.Hoek KS, et al. Stemming the flood. Pigment Cell Melanoma Res. 2009;22:6–7. doi: 10.1111/j.1755-148X.2008.00539.x. [DOI] [PubMed] [Google Scholar]
- 27.Suryo Rahmanto Y, Dunn LL, Richardson DR. Identification of distinct changes in gene expression after modulation of melanoma tumor antigen p97 (melanotransferrin) in multiple models in vitro and in vivo. Carcinogenesis. 2007;28:2172–2183. doi: 10.1093/carcin/bgm096. [DOI] [PubMed] [Google Scholar]
- 28.Frank MH, Sayegh MH. Immunomodulatory functions of mesenchymal stem cells. Lancet. 2004;363:1411–1412. doi: 10.1016/S0140-6736(04)16134-5. [DOI] [PubMed] [Google Scholar]
- 29.Le Blanc K, et al. Treatment of severe acute graft-versus-host disease with third party haploidentical mesenchymal stem cells. Lancet. 2004;363:1439–1441. doi: 10.1016/S0140-6736(04)16104-7. [DOI] [PubMed] [Google Scholar]
- 30.Le Blanc K, Ringden O. Immunomodulation by mesenchymal stem cells and clinical experience. J Intern Med. 2007;262:509–525. doi: 10.1111/j.1365-2796.2007.01844.x. [DOI] [PubMed] [Google Scholar]
- 31.Grulich AE, et al. Incidence of cancers in people with HIV/AIDS compared with immuno-suppressed transplant recipients: a meta-analysis. Lancet. 2007;370:59–67. doi: 10.1016/S0140-6736(07)61050-2. [DOI] [PubMed] [Google Scholar]
- 32.Shankaran V, et al. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410:1107–1111. doi: 10.1038/35074122. [DOI] [PubMed] [Google Scholar]
- 33.Haanen JB, et al. Melanoma-specific tumor-infiltrating lymphocytes but not circulating melanoma-specific T cells may predict survival in resected advanced-stage melanoma patients. Cancer Immunol Immunother. 2006;55:451–458. doi: 10.1007/s00262-005-0018-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Quintana E, et al. Efficient tumour formation by single human melanoma cells. Nature. 2008;456:593–598. doi: 10.1038/nature07567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mapara MY, Sykes M. Tolerance and cancer: mechanisms of tumor evasion and strategies for breaking tolerance. J Clin Oncol. 2004;22:1136–1151. doi: 10.1200/JCO.2004.10.041. [DOI] [PubMed] [Google Scholar]
- 36.Rosenberg SA, et al. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. 2008;8:299–308. doi: 10.1038/nrc2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ramsdell F, Fowlkes BJ. Clonal deletion versus clonal anergy: the role of the thymus in inducing self tolerance. Science. 1990;248:1342–1348. doi: 10.1126/science.1972593. [DOI] [PubMed] [Google Scholar]
- 38.Kappler JW, Roehm N, Marrack P. T cell tolerance by clonal elimination in the thymus. Cell. 1987;49:273–280. doi: 10.1016/0092-8674(87)90568-x. [DOI] [PubMed] [Google Scholar]
- 39.Ramsdell F, Lantz T, Fowlkes BJ. A non-deletional mechanism of thymic self tolerance. Science. 1989;246:1038–1041. doi: 10.1126/science.2511629. [DOI] [PubMed] [Google Scholar]
- 40.Ramsdell F, Fowlkes BJ. Maintenance of in vivo tolerance by persistence of antigen. Science. 1992;257:1130–1134. doi: 10.1126/science.257.5073.1130. [DOI] [PubMed] [Google Scholar]
- 41.Surh CD, Sprent J. T-cell apoptosis detected in situ during positive and negative selection in the thymus. Nature. 1994;372:100–103. doi: 10.1038/372100a0. [DOI] [PubMed] [Google Scholar]
- 42.Theobald M, et al. Tolerance to p53 by A2.1-restricted cytotoxic T lymphocytes. J Exp Med. 1997;185:833–841. doi: 10.1084/jem.185.5.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rosenberg SA. A new era for cancer immunotherapy based on the genes that encode cancer antigens. Immunity. 1999;10:281–287. doi: 10.1016/s1074-7613(00)80028-x. [DOI] [PubMed] [Google Scholar]
- 44.Houghton AN, Gold JS, Blachere NE. Immunity against cancer: lessons learned from melanoma. Curr Opin Immunol. 2001;13:134–140. doi: 10.1016/s0952-7915(00)00195-3. [DOI] [PubMed] [Google Scholar]
- 45.Jager E, et al. Induction of primary NY-ESO-1 immunity: CD8+ T lymphocyte and antibody responses in peptide-vaccinated patients with NY-ESO-1+ cancers. Proc Natl Acad Sci USA. 2000;97:12198–12203. doi: 10.1073/pnas.220413497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee PP, et al. Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nat Med. 1999;5:677–685. doi: 10.1038/9525. [DOI] [PubMed] [Google Scholar]
- 47.Stockert E, et al. A survey of the humoral immune response of cancer patients to a panel of human tumor antigens. J Exp Med. 1998;187:1349–1354. doi: 10.1084/jem.187.8.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Khong HT, Wang QJ, Rosenberg SA. Identification of multiple antigens recognized by tumor-infiltrating lymphocytes from a single patient: tumor escape by antigen loss and loss of MHC expression. J Immunother. 2004;27:184–190. doi: 10.1097/00002371-200405000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Davidson RL. Regulation of malanin synthesis in mammalian cells: effect of gene dosage on the expression of differentiation. Proc Natl Acad Sci USA. 1972;69:951–955. doi: 10.1073/pnas.69.4.951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guerry DT, et al. HLA-DR histocompatibility leukocyte antigens permit cultured human melanoma cells from early but not advanced disease to stimulate autologous lymphocytes. J Clin Invest. 1984;73:267–271. doi: 10.1172/JCI111201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aptsiauri N, et al. Role of altered expression of HLA class I molecules in cancer progression. Adv Exp Med Biol. 2007;601:123–131. doi: 10.1007/978-0-387-72005-0_13. [DOI] [PubMed] [Google Scholar]
- 52.Le Blanc K, et al. HLA expression and immunologic properties of differentiated and undifferentiated mesenchymal stem cells. Exp Hematol. 2003;31:890–896. doi: 10.1016/s0301-472x(03)00110-3. [DOI] [PubMed] [Google Scholar]
- 53.Cabrera T, et al. HLA class I expression in metastatic melanoma correlates with tumor development during autologous vaccination. Cancer Immunol Immunother. 2007;56:709–717. doi: 10.1007/s00262-006-0226-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Carretero R, et al. Analysis of HLA class I expression in progressing and regressing metastatic melanoma lesions after immunotherapy. Immunogenetics. 2008;60:439–447. doi: 10.1007/s00251-008-0303-5. [DOI] [PubMed] [Google Scholar]
- 55.van Houdt IS, et al. Favorable outcome in clinically stage II melanoma patients is associated with the presence of activated tumor infiltrating T-lymphocytes and preserved MHC class I antigen expression. Int J Cancer. 2008;123:609–615. doi: 10.1002/ijc.23543. [DOI] [PubMed] [Google Scholar]
- 56.Kawakami Y, et al. Recognition of shared melanoma antigens in association with major HLA-A alleles by tumor infiltrating T lymphocytes from 123 patients with melanoma. J Immunother. 2000;23:17–27. doi: 10.1097/00002371-200001000-00004. [DOI] [PubMed] [Google Scholar]
- 57.Staveley-O’Carroll K, et al. Induction of antigen-specific T cell anergy: An early event in the course of tumor progression. Proc Natl Acad Sci USA. 1998;95:1178–1183. doi: 10.1073/pnas.95.3.1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Strand S, et al. Lymphocyte apoptosis induced by CD95 (APO-1/Fas) ligand-expressing tumor cells—a mechanism of immune evasion? Nat Med. 1996;2:1361–1366. doi: 10.1038/nm1296-1361. [DOI] [PubMed] [Google Scholar]
- 59.Andreola G, et al. Induction of lymphocyte apoptosis by tumor cell secretion of FasL-bearing microvesicles. J Exp Med. 2002;195:1303–1316. doi: 10.1084/jem.20011624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gastman BR, et al. Fas ligand is expressed on human squamous cell carcinomas of the head and neck, and it promotes apoptosis of T lymphocytes. Cancer Res. 1999;59:5356–5364. [PubMed] [Google Scholar]
- 61.Hahne M, et al. Melanoma cell expression of Fas(Apo-1/CD95) ligand: implications for tumor immune escape. Science. 1996;274:1363–1366. doi: 10.1126/science.274.5291.1363. [DOI] [PubMed] [Google Scholar]
- 62.Hallermalm K, et al. Autocrine secretion of Fas ligand shields tumor cells from Fas-mediated killing by cytotoxic lymphocytes. Cancer Res. 2004;64:6775–6782. doi: 10.1158/0008-5472.CAN-04-0508. [DOI] [PubMed] [Google Scholar]
- 63.Neuber K, Eidam B. Expression of Fas ligand (CD95L) in primary malignant melanoma and melanoma metastases is associated with overall survival. Onkologie. 2006;29:361–365. doi: 10.1159/000094355. [DOI] [PubMed] [Google Scholar]
- 64.Nakashima M, Sonoda K, Watanabe T. Inhibition of cell growth and induction of apoptotic cell death by the human tumor-associated antigen RCAS1. Nat Med. 1999;5:938–942. doi: 10.1038/11383. [DOI] [PubMed] [Google Scholar]
- 65.Benvenuto F, et al. Human mesenchymal stem cells promote survival of T cells in a quiescent state. Stem Cells. 2007;25:1753–1760. doi: 10.1634/stemcells.2007-0068. [DOI] [PubMed] [Google Scholar]
- 66.Liu J, et al. Suppression of human peripheral blood lymphocyte proliferation by immortalized mesenchymal stem cells derived from bone marrow of Banna Minipig inbred-line. Transplant Proc. 2004;36:3272–3275. doi: 10.1016/j.transproceed.2004.11.090. [DOI] [PubMed] [Google Scholar]
- 67.Matsushima T, et al. Receptor binding cancer antigen expressed on SiSo cells, a novel regulator of apoptosis of erythroid progenitor cells. Blood. 2001;98:313–321. doi: 10.1182/blood.v98.2.313. [DOI] [PubMed] [Google Scholar]
- 68.Taylor A, et al. Mechanisms of immune suppression by interleukin-10 and transforming growth factor-beta: the role of T regulatory cells. Immunology. 2006;117:433–442. doi: 10.1111/j.1365-2567.2006.02321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Di Nicola M, et al. Human bone marrow stromal cells suppress T-lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood. 2002;99:3838–3843. doi: 10.1182/blood.v99.10.3838. [DOI] [PubMed] [Google Scholar]
- 70.Chen Q, et al. Production of IL-10 by melanoma cells: examination of its role in immuno-suppression mediated by melanoma. Int J Cancer. 1994;56:755–760. doi: 10.1002/ijc.2910560524. [DOI] [PubMed] [Google Scholar]
- 71.Gorelik L, Flavell RA. Immune-mediated eradication of tumors through the blockade of transforming growth factor-beta signaling in T cells. Nat Med. 2001;7:1118–1122. doi: 10.1038/nm1001-1118. [DOI] [PubMed] [Google Scholar]
- 72.Inge TH, et al. Inhibition of tumor-specific cytotoxic T-lymphocyte responses by transforming growth factor beta 1. Cancer Res. 1992;52:1386–1392. [PubMed] [Google Scholar]
- 73.Shipitsin M, et al. Molecular definition of breast tumor heterogeneity. Cancer Cell. 2007;11:259–273. doi: 10.1016/j.ccr.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 74.Rothstein DM, Sayegh MH. T-cell costimulatory pathways in allograft rejection and tolerance. Immunol Rev. 2003;196:85–108. doi: 10.1046/j.1600-065x.2003.00088.x. [DOI] [PubMed] [Google Scholar]
- 75.Frank MH, et al. Specific MDR1 P-glycoprotein blockade inhibits human alloimmune T cell activation in vitro. J Immunol. 2001;166:2451–2459. doi: 10.4049/jimmunol.166.4.2451. [DOI] [PubMed] [Google Scholar]
- 76.Pendse SS, et al. P-glycoprotein functions as a differentiation switch in antigen presenting cell maturation. Am J Transplant. 2006;6:2884–2893. doi: 10.1111/j.1600-6143.2006.01561.x. [DOI] [PubMed] [Google Scholar]
- 77.Jenkins MK, et al. CD28 delivers a costimulatory signal involved in antigen-specific IL-2 production by human T cells. J Immunol. 1991;147:2461–2466. [PubMed] [Google Scholar]
- 78.Kroczek RA, Mages HW, Hutloff A. Emerging paradigms of T-cell costimulation. Curr Opin Immunol. 2004;16:321–327. doi: 10.1016/j.coi.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 79.Schwartz RH. A cell culture model for T lymphocyte clonal anergy. Science. 1990;248:1349–1356. doi: 10.1126/science.2113314. [DOI] [PubMed] [Google Scholar]
- 80.Beyth S, et al. Human mesenchymal stem cells alter antigen-presenting cell maturation and induce T-cell unresponsiveness. Blood. 2005;105:2214–2219. doi: 10.1182/blood-2004-07-2921. [DOI] [PubMed] [Google Scholar]
- 81.Jiang XX, et al. Human mesenchymal stem cells inhibit differentiation and function of monocyte-derived dendritic cells. Blood. 2005;105:4120–4126. doi: 10.1182/blood-2004-02-0586. [DOI] [PubMed] [Google Scholar]
- 82.Sotomayor EM, et al. Cross-presentation of tumor antigens by bone marrow-derived antigen-presenting cells is the dominant mechanism in the induction of T-cell tolerance during B-cell lymphoma progression. Blood. 2001;98:1070–1077. doi: 10.1182/blood.v98.4.1070. [DOI] [PubMed] [Google Scholar]
- 83.Sotomayor EM, et al. Conversion of tumor-specific CD4+ T-cell tolerance to T-cell priming through in vivo ligation of CD40. Nat Med. 1999;5:780–787. doi: 10.1038/10503. [DOI] [PubMed] [Google Scholar]
- 84.Dunn GP, et al. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991–998. doi: 10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- 85.Brocker EB, Suter L, Sorg C. HLA-DR antigen expression in primary melanomas of the skin. J Invest Dermatol. 1984;82:244–247. doi: 10.1111/1523-1747.ep12260181. [DOI] [PubMed] [Google Scholar]
- 86.Becker JC, et al. Tumor escape mechanisms from immunosurveillance: induction of unresponsiveness in a specific MHC-restricted CD4+ human T cell clone by the autologous MHC class II + melanoma. Int Immunol. 1993;5:1501–1508. doi: 10.1093/intimm/5.12.1501. [DOI] [PubMed] [Google Scholar]
- 87.Bai Y, et al. L-selectin-dependent lymphoid occupancy is required to induce alloantigen-specific tolerance. J Immunol. 2002;168:1579–1589. doi: 10.4049/jimmunol.168.4.1579. [DOI] [PubMed] [Google Scholar]
- 88.Ruiter DJ, et al. MHC antigens in human melanomas. Semin Cancer Biol. 1991;2:35–45. [PubMed] [Google Scholar]
- 89.Romieu-Mourez R, et al. Regulation of MHC class II expression and antigen processing in murine and human mesenchymal stromal cells by IFN-gamma, TGF-beta, and cell density. J Immunol. 2007;179:1549–1558. doi: 10.4049/jimmunol.179.3.1549. [DOI] [PubMed] [Google Scholar]
- 90.Augello A, et al. Bone marrow mesenchymal progenitor cells inhibit lymphocyte proliferation by activation of the programmed death 1 pathway. Eur J Immunol. 2005;35:1482–1490. doi: 10.1002/eji.200425405. [DOI] [PubMed] [Google Scholar]
- 91.Chang CJ, et al. Placenta-derived multipotent cells exhibit immunosuppressive properties that are enhanced in the presence of interferon-gamma. Stem Cells. 2006;24:2466–2477. doi: 10.1634/stemcells.2006-0071. [DOI] [PubMed] [Google Scholar]
- 92.Dong H, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 93.Fong L, Small EJ. Anti-cytotoxic T-lymphocyte antigen-4 antibody: the first in an emerging class of immunomodulatory antibodies for cancer treatment. J Clin Oncol. 2008;26:5275–5283. doi: 10.1200/JCO.2008.17.8954. [DOI] [PubMed] [Google Scholar]
- 94.Hirano F, et al. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res. 2005;65:1089–1096. [PubMed] [Google Scholar]
- 95.Wong RM, et al. Programmed death-1 blockade enhances expansion and functional capacity of human melanoma antigen-specific CTLs. Int Immunol. 2007;19:1223–1234. doi: 10.1093/intimm/dxm091. [DOI] [PubMed] [Google Scholar]
- 96.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–1061. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- 97.Nishizuka Y, Sakakura T. Thymus and reproduction: sex-linked dysgenesia of the gonad after neonatal thymectomy in mice. Science. 1969;166:753–755. doi: 10.1126/science.166.3906.753. [DOI] [PubMed] [Google Scholar]
- 98.Sakaguchi S, et al. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155:1151–1164. [PubMed] [Google Scholar]
- 99.Smith TR, Kumar V. Revival of CD8+ Treg-mediated suppression. Trends Immunol. 2008;29:337–342. doi: 10.1016/j.it.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 100.Wang L, et al. Programmed death 1 ligand signaling regulates the generation of adaptive Foxp3+CD4+ regulatory T cells. Proc Natl Acad Sci USA. 2008;105:9331–9336. doi: 10.1073/pnas.0710441105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wing K, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008;322:271–275. doi: 10.1126/science.1160062. [DOI] [PubMed] [Google Scholar]
- 102.Elpek KG, et al. Ex vivo expansion of CD4+CD25+FoxP3+ T regulatory cells based on synergy between IL-2 and 4-1BB signaling. J Immunol. 2007;179:7295–7304. doi: 10.4049/jimmunol.179.11.7295. [DOI] [PubMed] [Google Scholar]
- 103.Hombach AA, et al. Effective proliferation of human regulatory T cells requires a strong costimulatory CD28 signal that cannot be substituted by IL-2. J Immunol. 2007;179:7924–7931. doi: 10.4049/jimmunol.179.11.7924. [DOI] [PubMed] [Google Scholar]
- 104.Aggarwal S, Pittenger MF. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood. 2005;105:1815–1822. doi: 10.1182/blood-2004-04-1559. [DOI] [PubMed] [Google Scholar]
- 105.Casiraghi F, et al. Pretransplant infusion of mesenchymal stem cells prolongs the survival of a semiallogeneic heart transplant through the generation of regulatory T cells. J Immunol. 2008;181:3933–3946. doi: 10.4049/jimmunol.181.6.3933. [DOI] [PubMed] [Google Scholar]
- 106.Di Ianni M, et al. Mesenchymal cells recruit and regulate T regulatory cells. Exp Hematol. 2008;36:309–318. doi: 10.1016/j.exphem.2007.11.007. [DOI] [PubMed] [Google Scholar]
- 107.Djouad F, et al. Immunosuppressive effect of mesenchymal stem cells favors tumor growth in allogeneic animals. Blood. 2003;102:3837–3844. doi: 10.1182/blood-2003-04-1193. [DOI] [PubMed] [Google Scholar]
- 108.Ahmadzadeh M, et al. FOXP3 expression accurately defines the population of intratumoral regulatory T cells that selectively accumulate in metastatic melanoma lesions. Blood. 2008;112:4953–4960. doi: 10.1182/blood-2008-06-163048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Clark RA, et al. Human squamous cell carcinomas evade the immune response by down-regulation of vascular E-selectin and recruitment of regulatory T cells. J Exp Med. 2008;205:2221–2234. doi: 10.1084/jem.20071190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Curiel TJ, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 111.Liyanage UK, et al. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J Immunol. 2002;169:2756–2761. doi: 10.4049/jimmunol.169.5.2756. [DOI] [PubMed] [Google Scholar]
- 112.Woo EY, et al. Cutting edge: Regulatory T cells from lung cancer patients directly inhibit autologous T cell proliferation. J Immunol. 2002;168:4272–4276. doi: 10.4049/jimmunol.168.9.4272. [DOI] [PubMed] [Google Scholar]
- 113.Ahmadzadeh M, Rosenberg SA. IL-2 administration increases CD4+CD25(hi) Foxp3+ regulatory T cells in cancer patients. Blood. 2006;107:2409–2414. doi: 10.1182/blood-2005-06-2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Schuchter LM. Adjuvant interferon therapy for melanoma: high-dose, low-dose, no dose, which dose? J Clin Oncol. 2004;22:7–10. doi: 10.1200/JCO.2004.10.907. [DOI] [PubMed] [Google Scholar]
- 115.Atkins MB, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 1999;17:2105–2116. doi: 10.1200/JCO.1999.17.7.2105. [DOI] [PubMed] [Google Scholar]
- 116.Flaherty LE, et al. Outpatient biochemotherapy with interleukin-2 and interferon alfa-2b in patients with metastatic malignant melanoma: results of two phase II cytokine working group trials. J Clin Oncol. 2001;19:3194–3202. doi: 10.1200/JCO.2001.19.13.3194. [DOI] [PubMed] [Google Scholar]
- 117.Rosenberg SA, et al. Observations on the systemic administration of autologous lymphokine-activated killer cells and recombinant interleukin-2 to patients with metastatic cancer. N Engl J Med. 1985;313:1485–1492. doi: 10.1056/NEJM198512053132327. [DOI] [PubMed] [Google Scholar]
- 118.Smith FO, et al. Treatment of metastatic melanoma using interleukin-2 alone or in conjunction with vaccines. Clin Cancer Res. 2008;14:5610–5618. doi: 10.1158/1078-0432.CCR-08-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tarhini AA, et al. Durable complete responses with high-dose bolus interleukin-2 in patients with metastatic melanoma who have experienced progression after biochemotherapy. J Clin Oncol. 2007;25:3802–3807. doi: 10.1200/JCO.2006.10.2822. [DOI] [PubMed] [Google Scholar]
- 120.Abdel-Wahab Z, et al. Transduction of human melanoma cells with interleukin-2 gene reduces tumorigenicity and enhances host antitumor immunity: a nude mouse model. Cell Immunol. 1994;159:26–39. doi: 10.1006/cimm.1994.1292. [DOI] [PubMed] [Google Scholar]
- 121.Zatloukal K, et al. Elicitation of a systemic and protective anti-melanoma immune response by an IL-2-based vaccine. Assessment of critical cellular and molecular parameters. J Immunol. 1995;154:3406–3419. [PubMed] [Google Scholar]
- 122.Powell DJ, Jr, et al. Inability to mediate prolonged reduction of regulatory T Cells after transfer of autologous CD25-depleted PBMC and interleukin-2 after lymphodepleting chemotherapy. J Immunother. 2007;30:438–447. doi: 10.1097/CJI.0b013e3180600ff9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Jones E, et al. Depletion of CD25+ regulatory cells results in suppression of melanoma growth and induction of autoreactivity in mice. Cancer Immun. 2002;2:1. [PubMed] [Google Scholar]
- 124.Dannull J, et al. Enhancement of vaccine-mediated antitumor immunity in cancer patients after depletion of regulatory T cells. J Clin Invest. 2005;115:3623–3633. doi: 10.1172/JCI25947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Attia P, et al. Inability of a fusion protein of IL-2 and diphtheria toxin (Denileukin Diftitox, DAB389IL-2, ONTAK) to eliminate regulatory T lymphocytes in patients with melanoma. J Immunother. 2005;28:582–592. doi: 10.1097/01.cji.0000175468.19742.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kirkwood JM, et al. Next generation of immunotherapy for melanoma. J Clin Oncol. 2008;26:3445–3455. doi: 10.1200/JCO.2007.14.6423. [DOI] [PubMed] [Google Scholar]
- 127.Maker AV, Attia P, Rosenberg SA. Analysis of the cellular mechanism of antitumor responses and autoimmunity in patients treated with CTLA-4 blockade. J Immunol. 2005;175:7746–7754. doi: 10.4049/jimmunol.175.11.7746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Reuben JM, et al. Biologic and immunomodulatory events after CTLA-4 blockade with ticilimumab in patients with advanced malignant melanoma. Cancer. 2006;106:2437–2444. doi: 10.1002/cncr.21854. [DOI] [PubMed] [Google Scholar]
- 129.Van Der Bruggen P, et al. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science. 1991;254:1643–1647. doi: 10.1126/science.1840703. [DOI] [PubMed] [Google Scholar]
- 130.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10:909–915. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Arce C, et al. A proof-of-principle study of epigenetic therapy added to neoadjuvant Doxorubicin cyclophosphamide for locally advanced breast cancer. PLoS ONE. 2006;1:e98. doi: 10.1371/journal.pone.0000098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Chen KG, et al. Principal expression of two mRNA isoforms (ABCB 5alpha and ABCB 5beta) of the ATP-binding cassette transporter gene ABCB 5 in melanoma cells and melanocytes. Pigment Cell Res. 2005;18:102–112. doi: 10.1111/j.1600-0749.2005.00214.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Jin L, et al. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med. 2006;12:1167–1174. doi: 10.1038/nm1483. [DOI] [PubMed] [Google Scholar]
- 134.Sigalotti L, et al. Cancer testis antigens in human melanoma stem cells: expression, distribution, and methylation status. J Cell Physiol. 2008;215:287–291. doi: 10.1002/jcp.21380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Dudley ME, et al. Adoptive transfer of cloned melanoma-reactive T lymphocytes for the treatment of patients with metastatic melanoma. J Immunother. 2001;24:363–373. doi: 10.1097/00002371-200107000-00012. [DOI] [PubMed] [Google Scholar]
- 136.Liu K, Rosenberg SA. Transduction of an IL-2 gene into human melanoma-reactive lymphocytes results in their continued growth in the absence of exogenous IL-2 and maintenance of specific antitumor activity. J Immunol. 2001;167:6356–6365. doi: 10.4049/jimmunol.167.11.6356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Phan GQ, et al. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc Natl Acad Sci USA. 2003;100:8372–8377. doi: 10.1073/pnas.1533209100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Antony PA, et al. CD8+ T cell immunity against a tumor/self-antigen is augmented by CD4+ T helper cells and hindered by naturally occurring T regulatory cells. J Immunol. 2005;174:2591–2601. doi: 10.4049/jimmunol.174.5.2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Frank NY, et al. Regulation of progenitor cell fusion by ABCB5 P-glycoprotein, a novel human ATP-binding cassette transporter. J Biol Chem. 2003;278:47156–47165. doi: 10.1074/jbc.M308700200. [DOI] [PubMed] [Google Scholar]