

Cyanide (CN−) and carbon monoxide (CO) have long been of particular interest as classic inhibitors of respiration.[1] CO competes with oxygen, binding to reduced iron (Fe2+) to form the stable low-spin hemoprotein carbonyls, whereas CN− inhibits O2 reduction.[2] CN− is iso-electronic and isosteric with CO; both are linearly bound to heme centers and their complexes can often be found with the same symmetries.[3] Unlike CO and O2, CN− binds to both iron(II) and iron(III) hemoproteins, in most cases, but not all,[4] leading to low-spin states. The reaction of cyanide with ferrohemes has been relatively little studied due to the low stability of the complexes even at alkaline pH values (up to 9.4).[2] In most hemoproteins, only the kinetics of cyanide dissociation has been investigated as a transient species during reduction of the ferric cyanide complex.[5] The dissociation constants (Kdiss.) are of the order of ~1 M,[6, 7] compared to ~10−4–10−9 M in ferrihemes.[8]
We recently reported the characterization of the first cyano-ferroheme, five-coordinate [K(222)]-[Fe(TPP)(CN)].[4, 9] The cyanide ligand field is insufficient to yield a completely low-spin complex, rather the species is a S=0 ⇋ S=2 spin-crossover complex.[10] We now report a new polymorphic form of [K(222)][Fe(TPP(CN)], also a spin-crossover complex.[12] More importantly, we also present examples of new six-coordinate cyano-ferroheme species: bis(cyano), [K(222)]2[Fe(Por)(CN)2], and mixed ligand, [K(222)][Fe(TPP)(CN)(1-MeIm)], which are all low spin.[11] Although a number of [FeIII(Por)(CN)2] − and [FeIII(Por)(CN)(L)] complexes have been well-studied,[13] no isolated six-coordinate (cyano)iron(II) porphyrinates or structures had been reported.
In thermodynamic and kinetic studies of binding to hemoglobins, cyanide shows some note-worthy properties compared to CO and O2. For example, the geometric and electronic structure change induced by Fe–CN bonding may not trigger a cooperative process unlike that of Fe–CO bonding.[14, 15] The CN− ligand in the iron(II) hemoproteins can be replaced when exposed to CO.[2, 16] It appears that a direct comparison between cyanide and carbon monoxide ligation in iron(II) porphyrinates will be fruitful.[14]
The reaction of [Fe(TPP)] with CN− in chlorobenzene solution can be monitored by UV-vis spectra (shown in S.I.), and suggests the presence of two cyano species, [Fe(TPP)(CN)]− and [Fe(TPP)(CN)2]2−. Binding constants for the mono- and bis(cyano) species can be determined from a least-squares analysis of the spectral data.[17] Values are K1 = 4.3 × 105 M−1 and K2 = 3.1 × 103 M−1, comparable to the values found by Goff and Morgan[18] for heme c and also comparable to the binding constants found for the reaction of [Fe(OEP)] with CO [19] (K1 = 3.3 × 104 M−1 and K2 = 2.3 × 102 M−1). As in the CO species,[20] both five-coordinate [Fe(TPP)(CN)] − and six-coordinate [Fe(TPP)(CN)2]2− can be isolated.
X-ray structural characterization of bis(cyano)iron(II) porphyrinates reveals that cyanide can exhibit two different coordination modes. The cyanide ligands are either coordinated to the iron center without interaction with the K(222) cations or with interaction with the cations to form Fe–CN–K bridges (Figures 1a and 1b). Six-coordinate [K(222)][Fe(TPP)(CN)(1-MeIm)] (Figure 1c) can be prepared by addition of 1-methylimidazole to a solution of [K(222)][Fe(TPP)-(CN)]. Careful control of imidazole concentration is important to yield the desired mixed ligand complex. A number of structural features are illustrated in Figure 2 including iron atom displacements, ligand tilts, and equatorial Fe–Np bond distances. A key to information summarized is given in the box (upper left). Cyano complex structural parameters are consistent with low-spin states for all derivatives. The corresponding parameters for three analogous carbonyl complexes are given in the right hand column.
Figure 1.

ORTEP diagrams of two [K(222)]2[Fe(TPP)(CN) 2] structures, showing two different coordination modes. [K(222)]2[Fe(TPP)(CN)2]·4PhCl is shown in (a) and one of two [K(222)]2[Fe(TPP)(CN)2]·PhCl units is shown in (b). The structure of [K(222)][Fe(TPP)(CN)(1-MeIm)] is illustrated in (c). All structures illustrated are at 100 K with thermal ellipsoids are contoured at the 50% probability level. Hydrogens omitted for clarity.
Figure 2.

Formal diagrams of the coordination groups of 100 K structures of (a) [K(222)][Fe(TPP)(CN)], (b) [K(222)]2[Fe(TPP)(CN)2], (c) [K(222)]2[Fe(TTP)(CN)2] (d) [K(222)]2[Fe(TPP)(CN)2], and (e) [K(222)][Fe(TPP)(CN)(1-MeIm)]. Comparable 100 K structures of (f) [Fe(OEP)(CO)],[20] (g) [Fe(OEP)(CO)2][20] and (h) [Fe(TPP)(CO)(1-MeIm)][24] are also given. As shown in the boxed key (upper left) a number of structural parameters are given for each structure. The dihedral angle between imidazole plane and closest Np–Fe–Nax (in degrees) are also shown for the imidazole derivatives.
The axial Fe–CN bond distance in the five-coordinate low-spin complex [Fe(TPP)(CN)]− at 1.8783 (10) Å (polymorph 1) and 1.869 (2) Å (polymorph 2) are the shortest observed in all of the iron(II) cyanide complexes. This is consistent with a sole cyano axial ligand. As reported previously for polymorph form 1,[4] the Fe–CN distance increases to 2.108 (3) Å as the complex undergoes a spin-state transition and becomes a high-spin species.[21] The addition of a second cyanide ligand to form the low-spin [Fe(TPP)(CN)2]2− complex leads to an ~0.1 Å increase in the Fe–CN distance (two equal axial bonds) compared to the five-coordinate low-spin form. The interaction of the CN− ligands with the [K(222)]+ cations leads to a further small increase in the Fe–CN distance to 1.988 (3) Å. It is to be presumed that the increases in the Fe–CN bond distances in the bis(cyano) complexes are the result of increased competition for bonding to the d6 iron(II) center. The effect of binding an imidazole ligand, a weaker π-accepting ligand than the cyanide ion, is to lead to an increase of only ~0.05 Å in the Fe–CN bond length, relative to the five-coordinate parent.
The Fe–CN bond distances show little variation with iron oxidation state change. The Fe–CN bond distance in bis(cyano) iron(III) derivatives range from 1.949 (4) Å to 1.990 (5) Å,[13, 22] bracketing the values observed for the iron(II) species. Similarly, the Fe–CN distances (values 1.918 (2)–1.929 (3) Å)[13, 23] in the iron(III) derivatives [Fe(Por)(CN)(1-MeIm)] are equivalent to that observed in the iron(II) complex.
The bond distance changes in this series of cyano derivatives are strikingly similar to those of an analogous series of carbonyl complexes (Figure 2). Since comparisons of CO and CN− as ligands in coordination chemistry have long been of interest,[25] this similarity has lead us to examine the correspondence of properties of the cyano and carbonyl series. It is to be noted that the longer Fe–C bond lengths in the cyano complexes is consistent with the idea that cyanide is a better σ donor but a poorer π-acceptor than the the carbonyl ligand.
Comparisons of changes in the stretching frequency of the two diatomic ligands and variation in the Mössbauer parameters should provide further enlightenment. Values of νCN and νCO are given in Table 1. There are large variations in the patterns of CN− and CO frequency changes across the series. The change in νCO between the five-coordinate complex (1944–1948 cm−1) and the bis(carbonyl) complex (2021 cm−1) is a difference of ~77 cm−1. This large difference strongly suggests a significant competition for π-donation by the two carbonyls from the d6 iron center in the bis complex. However the difference in the cyanide stretching frequency in the analogous pairs of cyanide complexes is virtually nil, with the differences between the five-coordinate species and the various bis(cyano) complexes ranging from 1 to 14 cm−1. The absence of significant variation in νCN between the mono and bis species suggests that change in the π bonding are much smaller in the cyano complexes. As shown in Table 1, the addition of a neutral nitrogen donor (imidazole) to the cyano complex has a modest effect; the same transformation in the carbonyl complexes leads to much larger changes with both increased and decreased shifts in νCO relative to the five-coordinate species.[24] The large variation in νCO in the six-coordinate mixed ligand species reflects the sensitivity of the vibration to its immediate environment.[24] We were thus surprised that the cyano derivative with ion pair interactions to the terminal cyanide nitrogen atoms had no effect on the CN frequency.
Table 1.
Multi-temperature Möossbauer data and C–N/O stretching frequencies of [K(222)][Fe(TPP)(CN)], two forms of [K(222)]2[Fe(TPP)(CN)2], [K(222)] 2[Fe(TTP)(CN) 2] and [K(222)][Fe(TPP)(CN)(1-MeIm)], and the comparable values for [Fe(OEP)(CO)],[20] [Fe(OEP)(CO) 2],[20] [Fe(OEP)(CO)(1-MeIm)][24] and [Fe(TPP)(CO)(1-MeIm)].[24] ΔEq and δ values in mm/s.
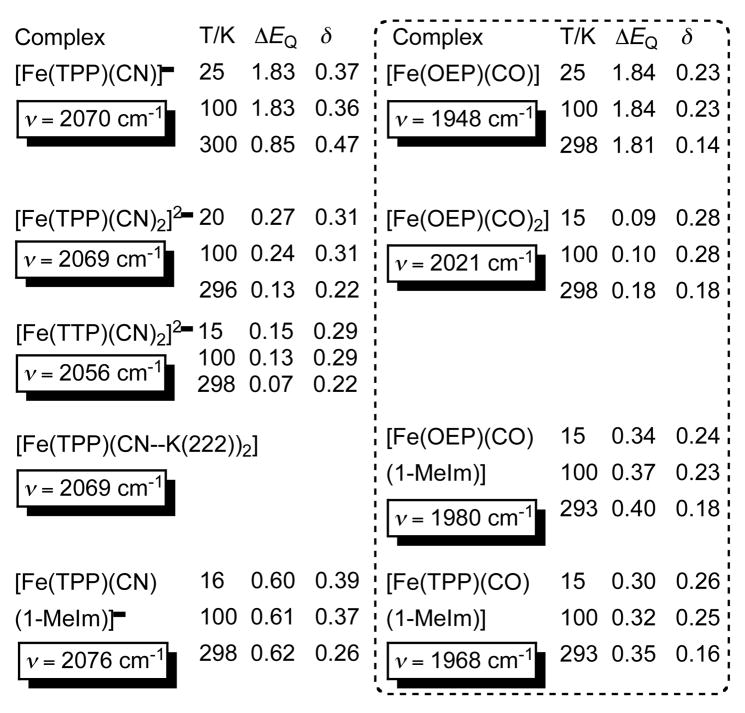 |
Temperature-dependent Mössbauer spectral values are shown in Table 1. We start by considering the mixed ligand species. Mössbauer spectra of many mixed ligand, six-coordinate carbonyl complexes have been measured. Typical features are 1) the relatively low value of the isomer shift, lower than typical for a formally iron(II) state and consistent with strong π back-donation from iron to CO,[26] and 2) the small value of the quadrupole splitting that is consistent with a nearly symmetric electron distribution at iron. The analogous cyano-imidazole derivative shows interesting differences. The larger value of the isomer shift (0.39 vs. 0.24 mm/s) is consistent with the expected lowered π acceptance by cyanide and the larger value of the quadrupole splitting (0.60 vs. 0.34 mm/s) is that expected for greater σ donation to iron. As expected, the five-coordinate species yield larger values of the quadrupole splitting. Again the differences in the isomer shift values are consistent with the differences in the π bonding characteristics of CN− and CO. The large change in the temperature-dependent quadrupole splitting values for the five-coordinate cyanide is the result of a LS ⇋ HS spin crossover. No evidence for a higher spin state in the five-coordinate CO complex is seen, consistent with its stronger ligand field character.
However, the two types of bis-ligated species show many (and unexpected) similarities. First, the isomer shift values are more similar; the bis(cyano) derivatives have isomer shifts that are smaller than the other groups of (cyano)iron(II) whereas the bis(carbonyl) derivative display a modest increase in the isomer shift relative to other carbonyl species. Clearly for the bis-ligated species, relative to all others species, the Fe s-electron density is at a minimum and in the bis(carbonyl) derivative, iron π-donation must be at a maximum. Second, the quadrupole splitting values are all near zero and are the smallest for any of the members of the cyano or the carbonyl systems, consistent with a near spherical distribution of d-electron density. Third, the ΔEq values show substantial temperature dependencies as seen in Table 1. Interestingly, the direction of the change is opposite in the cyano and carbonyl species. A large temperature dependence for ΔEq is usually considered to be the result of low-lying excited state(s), which in this case must be true for both classes. In view of the differing directions of change, it is most likely that the ΔEq signs are opposite. Obtaining the sign of ΔEq is difficult when the magnitude is small as in these cases[27] and the signs are currently not determined.
In summary, we have presented synthesis, molecular structures, and Mössbauer and vibrational spectroscopy of new six-coordinate cyano-ligated iron(II) porphyrinates. The invariant bond distances of the analogous iron(II) and -(III) porphyrinates are consistent with little π-back bonding between iron and cyanide. This conclusion is strengthened by the comparison with analogous carbonyl complexes. Clearly cyanide is a weaker 3eld ligand than CO in iron(II) porphyrinates.
Supplementary Material
Footnotes
Dedicated to Prof. Martin Gouterman
We thank the National Institutes of Health for support of this research under Grant GM-38401 to WRS and the NSF for X-ray instrumentation (Grant CHE-0443233)
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
Contributor Information
Dr. Jianfeng Li, Department of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, Indiana 46556 (USA), Fax (574) 631-6652
Dr. Bruce C. Noll, Department of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, Indiana 46556 (USA), Fax (574) 631-6652
Prof. Charles E. Schulz, Department of Physics, Knox College, Galesburg, Illinois 61401 (USA)
Prof. W. Robert Scheidt, Email: Scheidt.1@nd.edu, Department of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, Indiana 46556 (USA), Fax (574) 631-6652
References and Notes
- 1.Antonini E, Brunori M. Hemoglobin and Myoglobin in Their Reactions with Ligands. North-Holland; Amsterdam: 1971. [Google Scholar]
- 2.Yoshikawa S, O’Keeffe DH, Caughey WS. J Biol Chem. 1985;260:3518. [PubMed] [Google Scholar]
- 3.Sharpe AG. In: Comprehensive Coordination Chemistry. Wilkinson G, Gillard RD, McCleverty JA, editors. Vol. 2. Pergamon Press; Oxford, U. K: 1987. pp. 7–14. [Google Scholar]
- 4.Li J, Lord RL, Noll BC, Baik M-H, Schulz CE, Scheidt WR. Angew Chem Int Ed. 2008;47:10144. doi: 10.1002/anie.200804116. Ibid. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2008;120:10798. [Google Scholar]
- 5.Boffi A, Ilari A, Spagnuolo C, Chiancone E. Biochemistry. 1996;35:8068. doi: 10.1021/bi9601971. [DOI] [PubMed] [Google Scholar]
- 6.Brunori M, Antonini G, Castagnola M, Bellelli A. J Biol Chem. 1992;267:2258. [PubMed] [Google Scholar]
- 7.Mitchell R, Moddy AJ, Rich PR. Biochemistry. 1995;34:7576. doi: 10.1021/bi00023a003. [DOI] [PubMed] [Google Scholar]
- 8.Jafferji A, Allen JWA, Ferguson SJ, Fülöp V. J Biol Chem. 2000;275:25089. doi: 10.1074/jbc.M001377200. [DOI] [PubMed] [Google Scholar]
- 9.Abbreviations: Porph, generalized porphyrin dianion; Np, porphyrinato nitrogen; TPP, dianion of meso-tetraphenylporphyrin; TTP, meso-tetra-p-tolylporphyrin; OEP, dianion of 2,3,7,8,12,13,17,18-octaethylporphyrin; 1-MeIm, 1-methylimidazole; Kryptofix-222 or 222, 4,7,13,16,21,24-hexaoxo-1,10-diazabicyclo[8.8.8]hexacosane.
- 10.The unexpectedly lower ligand field strength of cyanide in this complex has been highlighted: Nakamura M. Angew Chem Int Ed. 2009;48:2.Ibid Angew Chem. 2009;121:2.
- 11.CCDC 723540, 723541, 723542, and 723543 ([K(222)][Fe(TPP(CN)] at 100, 296, 325, and 400 K), CCDC 723544 [K(222)]2[Fe(PTP)(CN)2]·PhCl, CCDC 723545 [K(222)]2-[Fe(TPP)(CN)2]·4PhCl, CCDC 723546 [K(222)]2[Fe(TTP)(CN)2], and CCDC 723547 [K(222)][Fe(TPP)(CN)(1-MeIm)] contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/datarequest/cif.
- 12.Comparisons of the structures of the two spin-crossover polymorphic forms at four temperatures between 100 and 400 K is given in the Supporting Information.
- 13.a) Li J, Noll BC, Schulz CE, Scheidt WR. Inorg Chem. 2007;46:2286. doi: 10.1021/ic061463u. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ikeue T, Ohgo Y, Saitoh T, Nakamura M, Fujii H, Yokoyama M. J Am Chem Soc. 2000;122:4068. [Google Scholar]; c) Ikezaki A, Nakamura M. Inorg Chem. 2002;41:2761. doi: 10.1021/ic0108383. [DOI] [PubMed] [Google Scholar]
- 14.Boffi A, Chiancone E, Takahashi S, Rousseau DL. Biochemistry. 1997;36:4505. doi: 10.1021/bi9618880. [DOI] [PubMed] [Google Scholar]
- 15.Boffi A, Chiancone E, Peterson ES, Wang J, Rousseau DL, Friedman JM. Biochemistry. 1997;36:4510. doi: 10.1021/bi961889s. [DOI] [PubMed] [Google Scholar]
- 16.a) Anson ML, Mirsky AE. J Gen Physiol. 1928;12:273. doi: 10.1085/jgp.12.2.273. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hill R. Proc Roy Soc B. 1929;105:112. [Google Scholar]; c) Keilin J. Biochem J. 1949;45:440. doi: 10.1042/bj0450440. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Keilin J. Biochem J. 1949;45:448. doi: 10.1042/bj0450448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leggett DJ. In: Computational Methods for the Determination of Formation Constants. Leggett DJ, editor. Chapter 6 Plenum; New York: 1985. [Google Scholar]
- 18.Goff H, Morgan LO. Inorg Chem. 1976;15:2069. [Google Scholar]
- 19.Strauss SH, Holm RH. Inorg Chem. 1982;21:863. [Google Scholar]
- 20.Silvernail NJ, Noll BC, Schulz CE, Scheidt WR. Inorg Chem. 2006;45:7050. doi: 10.1021/ic0613356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.This value is observed at 400 K; however, the spin-state transition is likely to not quite complete. In polymorph 2, this Fe–CN distance is observed to be 2.068 (4) at 400K (see Supporting Information).
- 22.a) Scheidt WR, Haller KJ, Hatano K. J Am Chem Soc. 1980;102:3017. [Google Scholar]; b) Schappacher M, Fischer J, Weiss R. Inorg Chem. 1989;28:390. [Google Scholar]; c) Balch AL, Noll BC, Safari N. Inorg Chem. 1993;32:2901. [Google Scholar]; d) Bartczak TJ, Wolowiee S, Latos-Grazynski L. Inorg Chim Acta. 1998;277:242. [Google Scholar]
- 23.Scott MJ, Lee SC, Holm RH. Inorg Chem. 1994;33:4651. [Google Scholar]
- 24.Silvernail NJ, Roth A, Schulz CE, Noll BC, Scheidt WR. J Am Chem Soc. 2005;127:14422. doi: 10.1021/ja053148x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.(a) Purcell KF. J Am Chem Soc. 1969;91:3487. [Google Scholar]; (b) DeKock RL, Sarapu AC, Fenske RF. Inorg Chem. 1971;10:38. [Google Scholar]; (c) Kettle SFA, Aschero GL, Diana E, Rossetti R, Stanghellini PL. Inorg Chem. 2006;45:4928. doi: 10.1021/ic0514041. [DOI] [PubMed] [Google Scholar]; (d) Ough EA, Stillman MJ. Inorg Chem. 1994;33:573. [Google Scholar]
- 26.(a) Calderazzo F, Frediani S, James BR, Pampaloni G, Reimer KJ, Sams JR, Serra AM, Vitali D. Inorg Chem. 1982;21:2302. [Google Scholar]; (b) Bancroft GM, Mays MJ, Prater BE. J Chem Soc A. 1970:956. [Google Scholar]
- 27.Debrunner P. In: Iron Porphyrins Part 3. Lever ABP, Gray HB, editors. Chapter 2 VCH Publishers Inc; New York: 1983. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.