

Abstract
The goal of this study was to define the anti-osteoclastogenic and/or anti-inflammatory role of IL-6 in inflammatory bone resorption using in vivo and in vitro methods. To this end, titanium particles were placed on murine calvaria, and bone resorption and osteoclast formation quantified in wild-type and IL-6-/- mice. In this model, calvarial bone loss and osteoclast formation were increased in titanium-treated IL-6-/- mice. Although basal numbers of splenic osteoclast precursors (OCP) were similar, IL-6-/- mice treated with particles in vivo had increased splenic OCP suggesting an enhanced systemic inflammatory response. In vitro osteoclastogenesis was measured using splenic (OCP) at various stages of maturation, including splenocytes from WT, IL-6-/- and TNFα transgenic mice. ELISA was used to measure TNFα production. IL-6 inhibited osteoclastogenesis in early OCP obtained from wild-type and IL-6-/- spleens. Pre-treatment of OCP with M-CSF for three days increased the CD11bhigh/c-Fms+ cell population, resulting in an intermediate staged OCP. Osteoclastogenesis was unaffected by IL-6 in M-CSF pre-treated and TNFα transgenic derived OCP. IL-6-/- splenocytes secreted greater concentrations of TNFα in response to titanium particles than WT; addition of exogenous IL-6 to these cultures decreased TNFα expression while anti-IL-6 antibody increased TNFα. While IL-6 lacks effects on intermediate staged precursors, the dominant in vivo effects of IL-6 appear to be related to strong suppression of early OCP differentiation and an anti-inflammatory effect targeting TNFα. Thus, the absence of IL-6 results in increased inflammatory bone loss.
Keywords: Osteoclastogenesis, IL-6, TNFα, inflammatory bone loss, macrophages
Introduction
Aseptic loosening is associated with the generation of particulate debris that is released into the joint space and becomes imbedded into the surrounding synovial tissues, where concentrations as high as 109 particles per gram are found [1]. These micron- and sub-micron sized particles are phagocytosed by macrophages and stimulate the release of pro-inflammatory cytokines [2]. TNFα, and interleukin-1 (IL-1) produced in this manner are considered mediators of the subsequent bone loss, since they directly and/or indirectly act as stimulators of osteoclast formation and bone resorption [3, 4].
Osteoclasts form by the fusion of mononuclear precursors from the macrophage/monocyte lineage. Cell surface markers characterize the maturation of osteoclast precursors (OCP). Important markers include c-Kit, a tyrosine kinase receptor that binds stem cell factor, c-Fms, the receptor for macrophage-colony-stimulating factor (M-CSF), CD11b, a beta2 integrin, and the receptor RANK [5, 6]. The earliest identifiable OCP are c-Kit+/c-fms-/CD11blow/RANK-. These cells progress to c-Kit+/c-Fms+/CD11blow/RANK-; the addition of M-CSF causes the progression to c-Kit-/c-Fms+/CD11bhigh/RANK+ cells, which are the first OCP responsive to receptor activator of NFκB ligand (RANKL) [5].
Prior work in our laboratory has established that CD11bhigh cells are more differentiated OCP. Over-expression of TNFα in vivo, through either administration of recombinant protein or in TNFα transgenic (TNFα-Tg) mice, results in a marked increase in the number of CD11bhigh cells in the spleen compared to untreated wild-type controls [7, 8]. In culture, these cells form osteoclasts several days earlier than controls following treatment with M-CSF and RANKL treatment. Thus, OCP from TNFα-Tg mice are at a more advanced stage of differentiation compared to OCP from wild-type mice.
c-Fms, the receptor for M-CSF, is an important marker for osteoclasts, and its expression commits cells to the osteoclast lineage. Osteoclast formation in the absence of M-CSF indicates that the precursors are of a more advanced state of maturation [5]. Thus, during osteoclastogenesis, precursors express different markers, and may have variable responses to exogenous factors.
Osteoclast formation is dependent upon two signals: M-CSF and RANKL. In the absence of these signals, osteoclastogenesis does not occur and osteopetrosis results, as is demonstrated in experiments in which osteoprotegerin, a secreted decoy receptor for RANKL, is over-expressed [9]. TNFα, while not necessary for osteoclast formation, greatly enhances the formation of osteoclasts by increasing both M-CSF and RANKL production [10, 11] in bone stromal cells, as well as by increasing the sensitivity of osteoclast precursor cells to RANKL [12]. Thus, in the presence of TNFα, lower concentrations of RANKL have increased potency; this may be one mechanism explaining the increased bone resorption observed in inflammatory conditions [4, 12]. Both RANKL and TNFα are produced by the inflammatory synovial-like membrane that forms during prosthetic loosening [3].
Recent work has suggested an alternate pathway of osteoclastogenesis involving TNFα but not RANKL. In vitro studies have shown that TNFα can stimulate osteoclast differentiation in the presence of M-CSF but not alone and that these osteoclasts also require IL-1 for full resorptive activity [13-15]. Inhibition of RANK signaling using a decoy receptor confirms that these osteoclasts can still develop in the absence of RANKL [13, 14]. However, in vivo, the overexpression of TNFα (in the presence of M-CSF) cannot induce osteoclastogenesis when RANK signaling is absent although it is sufficient to stimulate proliferation of osteoclast precursor cells [7].
While IL-6 is also highly expressed by macrophages and fibroblasts in the periprosthetic membrane [3, 16], its role in osteolysis is less clearly defined. Macrophages and fibroblasts stimulated by particles in vitro express high levels of IL-6 [2, 17]. Several studies suggest that IL-6 functions as a pro-osteoclastogenic and pro-inflammatory factor [18, 19] while others demonstrate anti-inflammatory [20] and anti-osteoclastogenic effects [21, 22]. Previously it has been shown that IL-6-/- mice have increased expression of both TNFα and IL-1 following systemic viral infection, a mechanism that may account for the anti-inflammatory effects of IL-6 [23]. Despite this somewhat controversial role for IL-6 in osteoclast development and inflammation, IL-6 deficient mice have no gross skeletal abnormalities [24, 25].
To define the role of IL-6 in particle-induced bone resorption, inflammation, and osteoclastogenesis, we utilized a mouse calvarial model of titanium-induced osteolysis [26], as well as in vitro splenocyte cultures to study the effects of IL-6 on osteoclast precursors of different maturational stages. In this paper we also attempt to clarify the role of IL-6 in conjunction with TNFα and M-CSF in driving the differentiation of osteoclasts.
Materials and Methods
Titanium particles
Pure titanium (Ti; 1-3 micron sized) particles were obtained from Johnson Matthey Chemicals (Ward Hill, MA) and prepared as previously described [17].
Murine Calvaria Model
Animal studies were conducted in accordance with principles and procedures approved by the University Committee on Animal Resources. Male and female C57/BL6 (n=10) or IL-6-/- (n=9) mice [24], (Jackson Laboratories, Bar Harbor, ME), six to eight weeks old, were used. Mice were anesthetized with 70 - 80mg/kg ketamine and five to seven mg/kg xylazine. The calvaria were exposed with a one-centimeter midline sagittal incision and thirty milligrams of autoclaved titanium was implanted. The incision was closed using 4-0 non-absorbable braided silk suture. Mice designated as “sham” received surgery, but no particles (n=5). Seven days post surgery, mice were euthanized and the calvaria removed for histological processing and micro-computed tomography (micro-CT) analysis. Three micron sections were stained with orange G or for tartrate-resistant acid phosphatase (TRAcP). Bone resorption and osteoclast number were measured as previously described [26]. Briefly, each section was digitally photographed and the image was oriented with the midline suture in the center of the field. The sagittal suture area in Orange G stained sections was determined by tracing the area of soft tissue within the midline suture and quantified using Osteometrics® software (Osteometrics, Decatur, GA). The number of osteoclasts in the midline suture area was determined by counting the number of TRAcP+ cells within the suture area of a 40x field.
For micro-CT analysis, the mice were euthanized, and the calvaria removed. Micro-computed tomography scans (VivaCT 40, ScanCo Medical, Basserdorf, Switzerland) were performed. A three-dimensional reconstruction of the mouse calvarium was generated using the integrated software.
In vitro Osteoclast Formation
Spleens from age and sex matched C57/BL6 wild-type, TNFα-Tg (obtained from Dr. G. Kollias, [27]), and IL-6-/- mice were harvested (some at the time of sacrifice post-titanium implantation, and some from naïve animals) and spleen cells isolated by passing the spleen through a 40μm wire mesh. Red blood cells were lysed with ammonium chloride, and after washing, spleen cells were plated in 96 well plates at a density of 1.75×105 cells/well. Cells were cultured in α-modified essential medium (Gibco BRL, Grand Island, NY) with 10% fetal calf serum (Hyclone, Logan, UT). Cultures were treated with 10 ng/ml M-CSF (R&D Systems, Minneapolis, MN) and 100 ng/ml RANKL (Amgen, Seattle, WA). Cultures were fixed and stained for TRAcP between days five and seven, and osteoclast area was measured as previously described [28]. All experiments were performed in quadruplicate (n=4).
Fluorescence-activated cell sorting (FACS) analysis and cell sorting
Surface protein staining was performed on freshly isolated spleen cells and bone marrow cells. After red blood cell lysis, a single-cell suspension was incubated with anti-murine CD16/32 to block Fc receptor-mediated antibody binding. Cells were then labeled with fluorescent antibodies, as described previously [29]. Data were acquired using a FACSCalibur instrument (Becton Dickinson, Bedford, MA) and analyzed with CellQuest software (Becton Dickinson). Pooled spleen cells from IL-6-/- or wild-type mice were labeled with anti-murine CD11b (eBioscience, San Diego, CA); bone marrow cultures were double stained with anti-murine CD11b and c-Fms antibodies (eBioscience) and then sorted. Unstained controls were used to calibrate instrument settings. Selected cells were collected, reanalyzed to assure their purity (>98%), and used for osteoclastogenesis assays as described above.
ELISA
WT and IL-6-/- splenocytes were harvested and cultured as described above. ANA-1 macrophages (a gift from Dr. G. Cox) are immortalized cells derived from the bone marrow of C57BL/6 mice that express markers of the differentiated macrophage, including Ly-5, Mac-1, and FcγR, but do not express markers of either B or T lymphocytes. ANA-1 macrophages are responsive to titanium particles and remain viable following particle treatment [30]. Cells were cultured in DMEM (Gibco BRL) with 10% fetal bovine serum (Gibco BRL) and 1% penicillin/streptomycin (Gibco BRL). All cells were plated in 24 well culture dishes at a concentration of 1×106 cells per well. Various concentrations of recombinant murine IL-6 or anti-IL-6 antibody (R&D Systems) were added in the presence or absence of titanium particles (1×107 particles/ml). Cells were pre-treated with anti-IL-6 antibody for one hour prior to addition of titanium particles to abolish any endogenous activity. Media was collected after 24 hours of titanium exposure. ELISA was performed per manufacturer’s guidelines, using a TNFα detection system with antibodies obtained from R&D systems as previously described [17]. Each sample was analyzed in quadruplicate; each treatment was replicated in triplicate, and each experiment was performed at least twice.
Statistical Analysis
Comparisons were made using ANOVA or ANCOVA with Tukey’s multiple range test. Significance was assigned for p values less than 0.05.
Results
IL-6 deficiency promotes titanium-induced osteoclastogenesis and bone resorption
The role of IL-6 in particulate-induced bone resorption was investigated using a murine calvaria model of wear debris-induced osteolysis [26]. Wild-type and IL-6-/- mice were evaluated seven days post-surgery, both with and without placement of titanium particles onto the calvarial surface. Titanium treatment resulted in abundant bone resorption and a marked increase in osteoclasts on the calvarial surface compared to sham operated controls (Figure 1). This effect was enhanced in titanium-treated IL-6-/- mice. Indeed, IL-6-/- but not wild-type animals showed evidence of full-thickness resorption of the parietal bones by histology and micro-CT (Figure 1). In wild-type mice, sagittal suture resorption area increased 4.7-fold compared to sham treated animals (p<0.001). In contrast, IL-6-/- mice had a 6.6-fold increase compared to control (p<0.00001) and a 42% increase compared to wild-type animals (p<0.05, Figure 2A). Titanium-treated wild-type and IL-6-/- mice also had increased numbers of osteoclasts along the calvarial surface compared to sham operated controls (2.4 and 3.7 fold, respectively). Although osteoclast number in IL-6-/- mice following titanium treatment was 58% greater in IL-6-/- mice compared to wild-type mice, this did not achieve statistical significance (Figure 2B). No differences were observed between the wild-type and IL-6-/- animals treated with sham surgery (Data not shown).
Figure 1. IL-6 deficiency increases particle induced osteolysis and osteoclast formation.
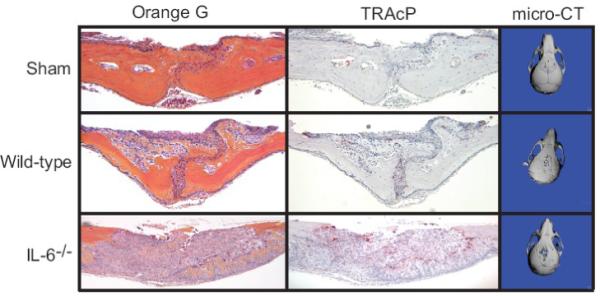
Wild-type (n=10) and IL-6-/- (n=9) mice underwent surgery, during which 30 mg of autoclaved titanium was implanted onto the calvaria, or surgery without titanium implantation (Sham; n=5). Seven days post surgery, mice were euthanized and calvaria processed for tartrate-resistant acid phosphatase (TRAcP) and Orange G staining. Representative histologic sections are shown at 100x original magnification. MicroCT scans were carried out on post-implantation day seven, and three-dimensional reconstructions generated. Representative images from post-implantation calvaria are shown, demonstrating increased osteolysis (full thickness) in IL-6-/- mice, consistent with the histologic findings.
Figure 2. IL-6 deficiency results in quantitatively more osteolysis and osteoclasts in response to titanium particles.
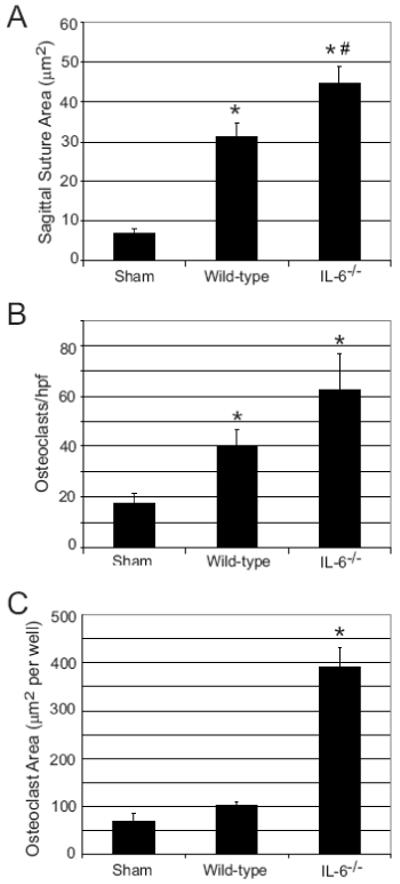
Wild-type (n= 10) and IL-6-/- (n=9) mice underwent titanium implantation surgery as detailed above. A) The area of osteolysis from Orange G stained sections was quantified using Osteometrics software (* indicates p<0.001 compared to Sham (n=5); # indicates p<0.05 compared to titanium-treated wild-type by ANOVA). B) The number of TRAcP positive stained cells per high-powered field was quantified (* indicates p<0.05 compared with Sham by ANOVA). C) Spleens were harvested from mice 7 days following implantation of titanium particles or after sham surgery. Cells were cultured for five days in the presence of M-CSF (10 ng/ml) and RANKL (100 ng/ml). The TRAcP+ multinucleated cell area was quantified. (* indicates p<1×10-8 compared to Sham by ANOVA).
Since we have shown that inflammatory conditions can increase splenic osteoclast precursor population [7, 8], osteoclastogenesis assays were performed on spleen cells harvested at the time of sacrifice. While splenocytes obtained from wild-type mice treated with titanium particles had a 48% increase in osteoclast formation compared to sham controls, this did not reach statistical significance (Figure 2C). In contrast, a 5.7-fold increase in osteoclast formation was observed in splenocyte cultures obtained from titanium-treated IL-6-/- mice (p<1×10-8, Figure 2C). Thus, treatment of IL-6-/- mice with particles in vivo increases calvarial bone resorption, and results in enhanced OCP in the spleen, consistent with an anti-osteoclastogenic role for IL-6.
IL-6 inhibits osteoclast formation in early splenic osteoclast precursor populations
A potential mechanism for the increased bone resorption observed following titanium implantation in IL-6-/- mice involves an increase in the baseline number of OCP. This mechanism accounts for the enhanced inflammatory bone resorption observed in transgenic mice over-expressing TNFα [7, 8]. Flow cytometry was performed on isolated spleen cells obtained from naïve wild-type and IL-6-/- mice in order to determine the proportion of CD11b+ cells. Wild-type and IL-6-/- mice had similar ratios of CD11b+ cells within the spleen (3-5%, Figure 3A).
Figure 3. IL-6 has differential effects on OCP differentiation from wild-type, IL-6-/-, and TNFα-Tg mice.
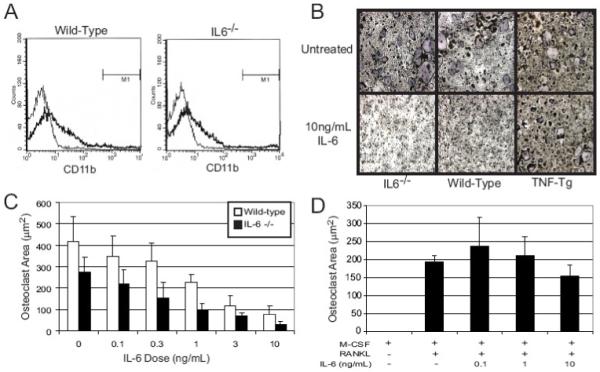
A) Spleens from WT and IL-6-/- mice were harvested from untreated mice. CD11b+ cells (bold line) present within the spleen were quantified using flow cytometry. B-D) Spleen cells were obtained from wild-type, IL-6-/-, and TNFα-Tg mice and placed in culture. B) Spleen cells were cultured for 5 days in the presence of M-CSF (10 ng/ml) and RANKL (100 ng/ml) with or without IL-6 (10 ng/ml) and representative photomicrographs of TRAcP stained cultures are shown. C) Wild-type and IL-6-/- spleen cell cultures were treated with M-CSF (10 ng/ml), RANKL (100 ng/ml), and various concentrations of recombinant IL-6 (n=4 per treatment group). Linear regression analysis shows dose dependent inhibition of osteoclast formation in both WT and IL-6-/- cells (p<1×10-4 and 0.0005 respectively). D) TNFα transgenic spleen cells were cultured with M-CSF (10 ng/ml) and RANKL (100 ng/ml) in the presence or absence of exogenous IL-6. The TRAcP+ multinucleated cell area per well was quantified (n=4 wells per group).
Recently we have shown TNFα-Tg mice have increased numbers of OCP within the spleen (four to seven fold greater) and that these cells are in a more advanced state of maturation (CD11bhigh) compared to cells derived from wild-type and IL-6-/- mice (CD11blow). Spleen cells from TNFα-Tg mice typically form osteoclasts approximately two days earlier than wild-type spleen cells [8]. To determine the effect of IL-6 on these intermediate OCP, wild-type, IL-6-/- and TNFα-Tg spleen cell cultures were treated with M-CSF and RANKL in the presence and absence of IL-6. IL-6 markedly inhibited osteoclast formation in wild-type and IL-6-/- spleen cell cultures but not TNFα-Tg mice (Figure 3B). Quantitatively, it can be seen that IL-6 has similar dose-dependent inhibitory effects on osteoclastogenesis in early OCP obtained from wild-type and IL-6-/- murine spleens following treatment with of M-CSF (10 ng/ml) and RANKL (100 ng/ml) for five days (Figure 3C). The effect was maximal at 10 ng/ml, where 82% and 89% reductions occurred in wild-type (p=7.5×10-5)and IL-6-/- (p<0.0005) derived OCP respectively (Figure 3C).
Since TNFα is an important regulator of inflammatory bone resorption [31], the interaction of IL-6 with TNFα was examined. Treatment of osteoclast precursors from TNFα-Tg mice with recombinant IL-6 did not significantly impair osteoclastogenesis, even at concentrations (10 ng/ml) that were markedly inhibitory in immature wild-type osteoclast precursors (Figure 3D). In order to confirm that these differences were not due to differences in IL-6 receptor expression by these various cell strains, flow cytometric analysis was performed. WT, IL-6-/- and TNFα-Tg splenocytes were all found to have equivalent cell surface expression of the IL-6 receptor (data not shown).
M-CSF enhances OCP differentiation and establishes resistance to the anti-osteoclastogenic effect of IL-6
To determine if IL-6 treatment would have the same effect on more advanced OCP from wild-type mice, bone marrow cells or splenocytes were pre-treated with M-CSF for three days prior to addition of RANKL and IL-6. M-CSF was used based on prior findings showing that in vivo delivery of M-CSF increases CD11b expression and results in an increased proportion of CD11bhigh cells [32]. Bone marrow cells were treated for 72 hours with media containing no cytokines, M-CSF, TNFα, or both and then examined for CD11b and c-Fms expression by flow cytometry (Figure 4A). Treatment with M-CSF markedly increased the number of cells expressing high levels of CD11b and resulted in an increase in the proportion of cells expressing both CD11b and c-Fms, consistent with a more advanced stage osteoclast progenitor. Furthermore, while c-Fms expression was enhanced by TNFα alone, addition of both TNFα and M-CSF further increased the number of cells expressing both c-Fms and high levels of CD11b (Figure 4A).
Figure 4. Treatment of OCP with M-CSF enhances c-Fms and CD11b expression, increases cell proliferation, and prevents IL-6 mediated inhibition of differentiation.
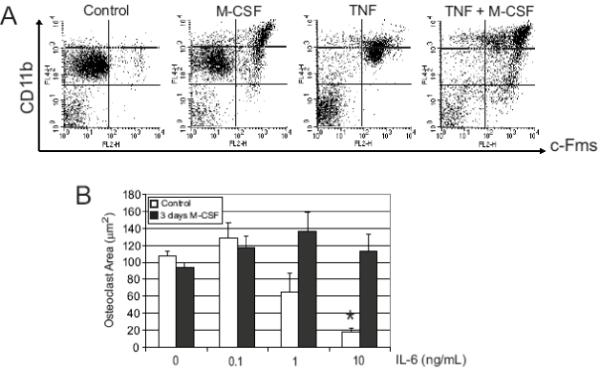
A) Bone marrow cultures were treated with exogenous M-CSF (10 ng/ml) and/or TNFα (10 ng/ml), and flow cytometry was performed using anti-c-Fms and anti-CD11b antibodies. CD11bhigh was defined by fluorescence levels greater than 103, as denoted by the dashed line in the upper boxes. Representative dot plots are shown. B) Wild-type spleen cells were pre-treated with control medium or medium containing M-CSF (10 ng/ml) for 3 days. All cultures were then treated with M-CSF (10 ng/ml) and RANKL (100 ng/ml) with or without IL-6 and TRAcP+ multinucleated cell area was quantified (n=4 per group). Control cells demonstrated marked inhibition of osteoclastogenesis (* indicates p<0.001 compared to 0 ng/mL IL-6 by ANOVA), whereas pretreated cells showed no inhibition of osteoclast formation.
M-CSF pre-treated spleen cultures were used as a model of intermediate OCP. Similar to spleen cells from TNFα-Tg mice, M-CSF pre-treated cultures consistently required a shorter duration of RANKL exposure (2 days less) for osteoclast formation to occur (data not shown). In wild-type spleen cultures pre-treated for 72 hours with M-CSF, IL-6 failed to inhibit RANKL induced osteoclast formation (p=0.36, Figure 4B); untreated cells again showed decreased osteoclast formation (p<0.001). These findings suggest that the inhibitory effect of IL-6 is limited to early OCP, as no inhibition of osteoclast formation is seen in cultures of more mature OCP such as those seen in TNFα-Tg and these M-CSF pre-treated splenocytes.
Inhibition of IL-6 increases expression of TNFα by macrophages
In addition to inhibiting early OCP, IL-6 may reduce inflammatory bone resorption in response to titanium particles through an anti-inflammatory effect, as described previously [20-22]. IL-6-/- splenocytes treated with titanium particles had significantly enhanced TNFα production compared to titanium-treated WT splenocytes (p=0.002, Figure 5A). Addition of exogenous IL-6 to these cultures significantly decreased TNFα formation by IL-6-/- splenocytes compared to untreated (p=0.02); the difference between IL-6-treated IL-6-/- splenocytes and WT splenocytes was not statistically different (p=0.339, Figure 5A).
Figure 5. IL-6 inhibits TNFα secretion by titanium treated splenocytes and macrophages.
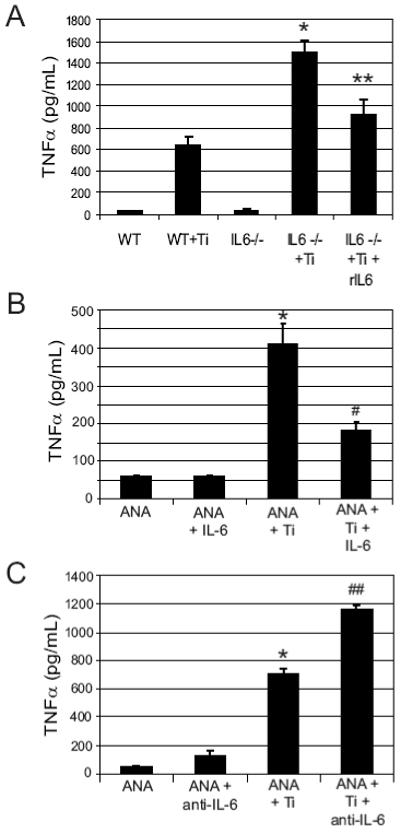
WT and IL-6-/- splenocytes (A), were treated with or without titanium particles (1×107particles/ml) in the presence or absence of 5 pg/mL of IL-6 for 24 hours (n=3 per group) and then media was collected for TNFα ELISA. Results of one experiment are shown. IL-6-/- cells treated with titanium showed significantly increased TNFα formation compared to titanium-treated WT (* indicates p=0.00005 by ANOVA); addition of exogenous IL-6 significantly reduced the TNFα production (** compared to titanium-treated IL-6-/-). ANA-1 macrophages were treated with titanium and/or 5 pg/ml IL-6 (B) or 5 μg/ml anti-IL-6 antibody (C) and TNFα concentration was measured by ELISA after 24 hours. Titanium stimulates TNFα production in these cells (* indicates p<0.0005 by ANOVA compared to untreated ANA cells). Recombinant IL-6 decreases TNFα production (# indicates p<4×10-11 compared to ANA + Ti by ANOVA). Addition of anti-IL-6 antibody significantly increased TNFα production compared to ANA + Ti, (## indicates p<0.02 by ANOVA).
ANA-1 macrophages treated with titanium particles (1×107 particles/ml) had an 8-fold increase in TNFα production (Figure 5B). Addition of IL-6 to the cultures markedly reduced TNFα production in response to titanium particles, with a 2.3-fold decrease observed following addition of 5 pg/ml of IL-6 (p<1×10-10). In contrast, TNFα production was increased when anti-IL-6 antibody was added to titanium-treated cultures (p<0.02, Figure 5C). The findings demonstrate that IL-6 suppresses the production of TNFα by macrophages in response to titanium particles, and that the absence of IL-6 is associated with an increased inflammatory response.
Discussion
IL-6 has been shown to promote osteoclast formation and bone resorption in some studies, while others have shown an inhibitory effect. Mice administered IL-6 develop hypercalcemia, leukocytosis, and cachexia [33]. Engrafting of human bone cells infected with an IL-6-expressing retrovirus into nude mice resulted in an increase in osteoclast-lined mineralized trabecular bone surfaces [34]. Addition of IL-6 and its soluble receptor increased bone resorption in neonatal mouse calvaria organ cultures [35]. In contrast, transgenic mice over-expressing human IL-6 have a reduction in both osteoclast number and bone resorption [36]. In two studies using mouse neonatal calvaria and parietal bone organ cultures, IL-6 failed to induce bone resorption [37, 38]. Furthermore, blockade of IL-6 with antibodies did not prevent PTH, PGE2, or 1,25-dihydroxyvitamin D3 mediated osteoclastogenesis or bone resorption [38].
In this model we conclusively show by histomorphometry and micro-CT that osteolytic lesions are greater in IL-6 knockout mice indicating that IL-6 is anti-osteoclastogenic. Furthermore, the absence of IL-6 increased the number of OCP present in the spleens of titanium treated mice. Thus, IL-6 deficiency results in a systemic response to localized inflammation that increases the number of splenic OCP. We believe that IL-6 is part of a larger negative feedback loop targeting TNFα and osteoclastogenesis. Two possible downstream effects of IL-6 may be to inhibit the production of TNFα from particle-stimulated cells as well as inhibit early osteoclastogenesis (possibly by preventing the TNFα-mediated upregulation of early osteoclast differentiation genes). Our data (Figures 3 and 5) support both of these possible mechanisms by demonstrating the IL-6 treatment early in culture blocks osteoclastogenesis and TNFα production from splenocytes. Recently two other groups have also demonstrated an anti-osteoclastogenic effect of IL-6 on osteoclast precursors, one group showing that MAP kinase activation downstream of IL-6 inhibits RANK and JNK-mediated signals [22]. Duplomb et al. have also shown an anti-osteoclastogenic effect of IL-6 that involves re-direction of the precursor pool toward a monocyte/macrophage phenotype [39].
Interestingly, a new biologic therapy targeting IL-6 has emerged for rheumatoid arthritis (RA), called tocilizumab. This humanized IL-6 receptor monoclonal antibody has shown great promise at reducing swelling and radiographic evidence of joint damage in clinical trials for RA [40, 41]. Pre-clinical work with tocilizumab in collagen-induced arthrtitis in monkeys has demonstrated that IL-6 signaling blockade reduces RANKL expression and osteoclast numbers [42]. Although the success of tocilizumab would appear to contradict the anti-osteoclastogenic effect of IL-6 that we have observed, both pro- and anti-osteoclastogenic effects may result from IL-6 signal transduction through the “signal orchestration model” as proposed by Kamimura et al. which postulates that individual cytokines acting on a single cell may use complex intracellular signaling cascades with opposing effects [43]. IL-6 is also known to act synergistically with other cytokines such that the net effect on osteoclastogenesis may vary depending on the exact cytokine milieu that is present [44]. In this way it might be possible for non-particle related diseases to experience a pro-osteoclastogenic effect of IL-6 that is mechanistically different from the anti-osteoclastogenic effect in particle-mediated osteolysis. We would therefore predict then, that the precise cytokine milieu present in various pathologies would be different, as would the response to tocilizumab therapy.
Our goal was to better define the role of IL-6 during particle-induced osteolysis. Using a well-established and highly quantitative murine model, as well as qualitative micro-CT scans, IL-6 deficient mice developed increased osteoclasts and had enhanced bone resorption following implantation of titanium particles onto the calvaria. Increased bone resorption, osteoclast numbers, and IL-1 expression were also observed in an osteomyelitis model using IL-6-/- mice as well as in wild-type mice treated with anti-IL-6 antibody [21]. Finally, IL-6-/- mice treated with intra-articular zymosan had increased cartilage destruction compared with wild-type controls [45]. Thus, these other models of inflammatory bone or cartilage destruction using a genetic approach also show an IL-6 anti-resorptive effect.
A series of in vitro experiments were performed in order to define mechanisms through which IL-6 inhibits inflammatory bone resorption, focusing on OCP as key target cells. Initial experiments confirmed that the difference in osteoclast number was not due to an increased pool of precursor cells in IL-6 deficient mice. Both wild-type and IL-6-/- mice spleens contained similar percentages of osteoclast precursor cells (CD11b+) by flow cytometry. Furthermore, wild-type and IL-6-/- spleen cells responded similarly to M-CSF and RANKL. Thus, differences in bone resorption are not related to differences in basal numbers or responsiveness of OCP.
However, it is interesting to note that our in vitro results were much more dramatic in terms of the relative increase in osteoclasts (3.8 fold increase in osteoclast area compared to wild-type in Figure 2C vs. only a 58% increase in osteoclast number in vivo, Figure 2B). One explanation is that osteoclastogenesis in vivo is a temporally more variable process and that osteoclast numbers may peak at different times. We chose 7 days as our endpoint as this is the point of maximal osteolysis. It is also worth noting that the in vitro experiment was measured as osteoclast area and the in vivo data as the number of osteoclasts; one explanation for the discrepancy in magnitude of the response is that the dramatic increase in osteoclast area in vitro is due to a greater proportion of very large multinucleated cells. This would explain why we see a large osteoclast area but no significant difference in osteoclast number in vivo. Indeed there are several TRAcP+ osteoclasts visible in the histologic section from the IL-6 knockout that are much larger than any of the TRAcP+ cells in the wild-type or sham sections. Examination of mice deficient in gp130, a critical element in the active IL-6 receptor complex, finds that osteoclasts in these animals too are substantially larger than normal [46].
A unique aspect of our approach was the use of OCP in different stages of differentiation. Prior work by our laboratory has defined early osteoclast precursor cells to be CD11b+/c-Fmslow/RANKlow by FACS [7, 8]. Treatment of this early precursor population with IL-6 inhibited stimulation of osteoclastogenesis by RANKL and M-CSF in a dose-dependent manner. Effects were similar in wild-type and IL-6-/- mice and inhibition persisted following co-treatment with TNFα. Thus, IL-6 is a potent inhibitor of early osteoclast maturation.
Previous work from our lab has shown TNFα-Tg mice have an increased proportion of OCP in the spleen expressing high levels of CD11b [6, 8] and that these OCP form mature osteoclasts more rapidly in culture. Using CD11b as a marker of osteoclast maturation, here we show that in vitro treatment with M-CSF for three days enhanced OCP differentiation, consistent with previous reports showing that administration of M-CSF treatment increased the population of CD11bhigh cells in vivo [6, 32]. M-CSF increased the population of cells expressing CD11b and c-Fms; the effect was further enhanced by TNFα. M-CSF is also known to potently stimulate proliferation with the daughter cell population having a particularly high percentage of CD11bhigh cells; this effect is also enhanced by TNFα [6]. This synergistic enhancement of proliferation and differentiation by M-CSF and TNFα may be one mechanism responsible for the presence of more mature OCP in TNFα-Tg mice [6-8]. In the more mature OCP cultures obtained from either TNFα-Tg mice or following pre-treatment with M-CSF, IL-6 failed to inhibit osteoclastogenesis. These findings show that IL-6 has differential effects on osteoclastogenesis, dependent on the stage of maturation of the OCP, and the presence of other cytokines.
IL-6 also has anti-inflammatory properties, and has been shown to reduce TNFα expression in numerous models. LPS stimulates production of TNFα in mice, which in turn induces IL-6 secretion. IL-6 then acts in a negative feedback loop to decreases further production of TNFα [20]. Similarly, IL-6 deficient mice have a three-fold increase in TNFα production in response to LPS compared to wild-type mice [47]. Expression of both TNFα and IL-1 was increased in IL-6-/- mice with systemic viral infection [23]. Administration of recombinant IL-6 reduced inflammatory responses, including TNFα and IL-1, and mortality in mice treated with LPS [20, 48]. Similar effects were observed in mice with exposure to aerosolized endotoxin. IL-6 production was induced by the endotoxin, and higher concentrations of TNFα and MIP-2 were noted in mice deficient in IL-6 [49].
TNFα is a key mediator of inflammatory bone resorption. TNFα increases inflammation, induces pre-osteoclast differentiation, expands the osteoclast precursor pool, and makes OCP more sensitive to the effects of RANKL [8, 12]. TNFα also stimulates RANKL expression by stromal cells [10, 11]. Titanium-induced TNFα production by ANA-1 macrophages was markedly reduced in cultures treated with IL-6. In contrast, addition of anti-IL-6 antibody resulted in increased TNFα production. Spleen cell cultures from WT and IL-6-/- mice demonstrated enhanced TNFα secretion by titanium treated IL-6 deficient splenocytes; this enhancement was decreased by addition of exogenous IL-6. Thus, TNFα inhibition may be one of the targets through which IL-6 inhibits osteolysis in vivo.
The observed effect of IL-6 deficiency in vivo is to increase inflammatory bone resorption. Further experiments suggest this occurs both through the suppression of early maturation of osteoclasts by IL-6, as well as IL-6 decreasing production of TNFα by macrophages. IFN-γ and IL-4 inhibit osteoclast formation through the STAT signaling pathway [50]. IL-6 signals through STAT1 and STAT3, suggesting that perhaps IL-6 shares similar signaling pathways with other inhibitors of OCP differentiation [51]. Recent studies implicate the MAPK ERK1/2 pathway in the inhibition of osteoclastogenesis by IL-6 [39]. Further, there may be an interaction between the particles themselves and other pathways, including SOCS3, an inhibitor of IL-6 signaling [52]. Elucidation of the complex, stage-specific downstream signals that control the differential responses to IL-6 is an important area for future investigation.
These experiments show that IL-6 has diverse effects on osteoclast progenitor cells at various stages of maturation. IL-6 inhibits the differentiation of early osteoclast progenitors (CD11blow, c-Fms-) treated with M-CSF, RANKL, and TNFα. In OCP at an intermediate stage of differentiation (CD11bhigh, c-Fms+), IL-6 neither inhibits nor enhances osteoclastogenesis.
Acknowledgements
This work was supported by NIH grant R01-ARO48681 (RJO) and OREF grant O49956-001 (MD). The authors would like to thank Xing Qiu, Ph.D. for his assistance with the statistical analysis of these data.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Margevicius KJ, Bauer TW, McMahon JT, Brown SA, Merritt K. Isolation and characterization of debris in membranes around total joint prostheses. J Bone Joint Surg Am. 1994;76:1664–75. doi: 10.2106/00004623-199411000-00010. [DOI] [PubMed] [Google Scholar]
- [2].Chiba J, Schwendeman LJ, Booth RE, Jr., Crossett LS, Rubash HE. A biochemical, histologic, and immunohistologic analysis of membranes obtained from failed cemented and cementless total knee arthroplasty. Clin Orthop Relat Res. 1994:114–24. [PubMed] [Google Scholar]
- [3].Haynes DR, Crotti TN, Potter AE, Loric M, Atkins GJ, Howie DW, Findlay DM. The osteoclastogenic molecules RANKL and RANK are associated with periprosthetic osteolysis. J Bone Joint Surg Br. 2001;83:902–11. doi: 10.1302/0301-620x.83b6.10905. [DOI] [PubMed] [Google Scholar]
- [4].O’ Gradaigh D, Ireland D, Bord S, Compston JE. Joint erosion in rheumatoid arthritis: interactions between tumour necrosis factor alpha, interleukin 1, and receptor activator of nuclear factor kappaB ligand (RANKL) regulate osteoclasts. Ann Rheum Dis. 2004;63:354–9. doi: 10.1136/ard.2003.008458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Arai F, Miyamoto T, Ohneda O, Inada T, Sudo T, Brasel K, Miyata T, Anderson DM, Suda T. Commitment and differentiation of osteoclast precursor cells by the sequential expression of c-Fms and receptor activator of nuclear factor kappaB (RANK) receptors. J Exp Med. 1999;190:1741–54. doi: 10.1084/jem.190.12.1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Yao Z, Li P, Zhang Q, Schwarz EM, Keng P, Arbini A, Boyce BF, Xing L. Tumor necrosis factor-alpha increases circulating osteoclast precursor numbers by promoting their proliferation and differentiation in the bone marrow through up-regulation of c-Fms expression. J Biol Chem. 2006;281:11846–55. doi: 10.1074/jbc.M512624200. [DOI] [PubMed] [Google Scholar]
- [7].Li P, Schwarz EM, O’Keefe RJ, Ma L, Boyce BF, Xing L. RANK signaling is not required for TNFalpha-mediated increase in CD11(hi) osteoclast precursors but is essential for mature osteoclast formation in TNFalpha-mediated inflammatory arthritis. J Bone Miner Res. 2004;19:207–13. doi: 10.1359/JBMR.0301233. [DOI] [PubMed] [Google Scholar]
- [8].Li P, Schwarz EM, O’Keefe RJ, Ma L, Looney RJ, Ritchlin CT, Boyce BF, Xing L. Systemic tumor necrosis factor alpha mediates an increase in peripheral CD11b(high) osteoclast precursors in tumor necrosis factor alpha-transgenic mice. Arthritis Rheum. 2004;50:265–76. doi: 10.1002/art.11419. [DOI] [PubMed] [Google Scholar]
- [9].Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, Capparelli C, Morony S, Oliveira-dos-Santos AJ, Van G, Itie A, Khoo W, Wakeham A, Dunstan CR, Lacey DL, Mak TW, Boyle WJ, Penninger JM. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 1999;397:315–23. doi: 10.1038/16852. [DOI] [PubMed] [Google Scholar]
- [10].Cenci S, Weitzmann MN, Roggia C, Namba N, Novack D, Woodring J, Pacifici R. Estrogen deficiency induces bone loss by enhancing T-cell production of TNF-alpha. J Clin Invest. 2000;106:1229–37. doi: 10.1172/JCI11066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Collin-Osdoby P, Rothe L, Anderson F, Nelson M, Maloney W, Osdoby P. Receptor activator of NF-kappa B and osteoprotegerin expression by human microvascular endothelial cells, regulation by inflammatory cytokines, and role in human osteoclastogenesis. J Biol Chem. 2001;276:20659–72. doi: 10.1074/jbc.M010153200. [DOI] [PubMed] [Google Scholar]
- [12].Lam J, Takeshita S, Barker JE, Kanagawa O, Ross FP, Teitelbaum SL. TNF-alpha induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J Clin Invest. 2000;106:1481–8. doi: 10.1172/JCI11176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Azuma Y, Kaji K, Katogi R, Takeshita S, Kudo A. Tumor necrosis factor-alpha induces differentiation of and bone resorption by osteoclasts. J Biol Chem. 2000;275:4858–64. doi: 10.1074/jbc.275.7.4858. [DOI] [PubMed] [Google Scholar]
- [14].Kobayashi K, Takahashi N, Jimi E, Udagawa N, Takami M, Kotake S, Nakagawa N, Kinosaki M, Yamaguchi K, Shima N, Yasuda H, Morinaga T, Higashio K, Martin TJ, Suda T. Tumor necrosis factor alpha stimulates osteoclast differentiation by a mechanism independent of the ODF/RANKL-RANK interaction. J Exp Med. 2000;191:275–86. doi: 10.1084/jem.191.2.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sabokbar A, Kudo O, Athanasou NA. Two distinct cellular mechanisms of osteoclast formation and bone resorption in periprosthetic osteolysis. J Orthop Res. 2003;21:73–80. doi: 10.1016/S0736-0266(02)00106-7. [DOI] [PubMed] [Google Scholar]
- [16].Konttinen YT, Xu JW, Waris E, Li TF, Gomez-Barrena E, Nordsletten L, Santavirta S. Interleukin-6 in aseptic loosening of total hip replacement prostheses. Clin Exp Rheumatol. 2002;20:485–90. [PubMed] [Google Scholar]
- [17].Bukata SV, Gelinas J, Wei X, Rosier RN, Puzas JE, Zhang X, Schwarz EM, Song XY, Griswold DE, O’Keefe RJ. PGE2 and IL-6 production by fibroblasts in response to titanium wear debris particles is mediated through a Cox-2 dependent pathway. J Orthop Res. 2004;22:6–12. doi: 10.1016/S0736-0266(03)00153-0. [DOI] [PubMed] [Google Scholar]
- [18].Kim KJ, Hijikata H, Itoh T, Kumegawa M. Joint fluid from patients with failed total hip arthroplasty stimulates pit formation by mouse osteoclasts on dentin slices. J Biomed Mater Res. 1998;43:234–40. doi: 10.1002/(sici)1097-4636(199823)43:3<234::aid-jbm3>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- [19].Kotake S, Sato K, Kim KJ, Takahashi N, Udagawa N, Nakamura I, Yamaguchi A, Kishimoto T, Suda T, Kashiwazaki S. Interleukin-6 and soluble interleukin-6 receptors in the synovial fluids from rheumatoid arthritis patients are responsible for osteoclast-like cell formation. J Bone Miner Res. 1996;11:88–95. doi: 10.1002/jbmr.5650110113. [DOI] [PubMed] [Google Scholar]
- [20].Aderka D, Le JM, Vilcek J. IL-6 inhibits lipopolysaccharide-induced tumor necrosis factor production in cultured human monocytes, U937 cells, and in mice. J Immunol. 1989;143:3517–23. [PubMed] [Google Scholar]
- [21].Balto K, Sasaki H, Stashenko P. Interleukin-6 deficiency increases inflammatory bone destruction. Infect Immun. 2001;69:744–50. doi: 10.1128/IAI.69.2.744-750.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Yoshitake F, Itoh S, Narita H, Ishihara K, Ebisu S. Interleukin-6 directly inhibits osteoclast differentiation by suppressing receptor activator of NF-kappaB signaling pathways. J Biol Chem. 2008;283:11535–40. doi: 10.1074/jbc.M607999200. [DOI] [PubMed] [Google Scholar]
- [23].Benihoud K, Salone B, Esselin S, Opolon P, Poli V, Di Giovine M, Perricaudet M, Saggio I. The role of IL-6 in the inflammatory and humoral response to adenoviral vectors. J Gene Med. 2000;2:194–203. doi: 10.1002/(SICI)1521-2254(200005/06)2:3<194::AID-JGM102>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- [24].Kopf M, Baumann H, Freer G, Freudenberg M, Lamers M, Kishimoto T, Zinkernagel R, Bluethmann H, Kohler G. Impaired immune and acute-phase responses in interleukin-6-deficient mice. Nature. 1994;368:339–42. doi: 10.1038/368339a0. [DOI] [PubMed] [Google Scholar]
- [25].Poli V, Balena R, Fattori E, Markatos A, Yamamoto M, Tanaka H, Ciliberto G, Rodan GA, Costantini F. Interleukin-6 deficient mice are protected from bone loss caused by estrogen depletion. EMBO J. 1994;13:1189–96. doi: 10.1002/j.1460-2075.1994.tb06368.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Schwarz EM, Benz EB, Lu AP, Goater JJ, Mollano AV, Rosier RN, Puzas JE, Okeefe RJ. Quantitative small-animal surrogate to evaluate drug efficacy in preventing wear debris-induced osteolysis. J Orthop Res. 2000;18:849–55. doi: 10.1002/jor.1100180602. [DOI] [PubMed] [Google Scholar]
- [27].Keffer J, Probert L, Cazlaris H, Georgopoulos S, Kaslaris E, Kioussis D, Kollias G. Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. EMBO J. 1991;10:4025–31. doi: 10.1002/j.1460-2075.1991.tb04978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Huang W, O’Keefe RJ, Schwarz EM. Exposure to receptor-activator of NFkappaB ligand renders pre-osteoclasts resistant to IFN-gamma by inducing terminal differentiation. Arthritis Res Ther. 2003;5:R49–59. doi: 10.1186/ar612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Li P, Sanz I, O’Keefe RJ, Schwarz EM. NF-kappa B regulates VCAM-1 expression on fibroblast-like synoviocytes. J Immunol. 2000;164:5990–7. doi: 10.4049/jimmunol.164.11.5990. [DOI] [PubMed] [Google Scholar]
- [30].Soloviev A, Schwarz EM, Kuprash DV, Nedospasov SA, Puzas JE, Rosier RN, O’Keefe RJ. The role of p105 protein in NFkappaB activation in ANA-1 murine macrophages following stimulation with titanium particles. J Orthop Res. 2002;20:714–22. doi: 10.1016/S0736-0266(01)00180-2. [DOI] [PubMed] [Google Scholar]
- [31].Lam J, Abu-Amer Y, Nelson CA, Fremont DH, Ross FP, Teitelbaum SL. Tumour necrosis factor superfamily cytokines and the pathogenesis of inflammatory osteolysis. Ann Rheum Dis. 2002;61(Suppl 2):ii82–3. doi: 10.1136/ard.61.suppl_2.ii82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Misawa E, Sakurai T, Yamada M, Tamura Y, Motoyoshi K. Administration of macrophage colony-stimulating factor mobilized both CD11b+CD11c+ cells and NK1.1+ cells into peripheral blood. Int Immunopharmacol. 2004;4:791–803. doi: 10.1016/j.intimp.2004.03.004. [DOI] [PubMed] [Google Scholar]
- [33].Black K, Garrett IR, Mundy GR. Chinese hamster ovarian cells transfected with the murine interleukin-6 gene cause hypercalcemia as well as cachexia, leukocytosis and thrombocytosis in tumor-bearing nude mice. Endocrinology. 1991;128:2657–9. doi: 10.1210/endo-128-5-2657. [DOI] [PubMed] [Google Scholar]
- [34].Sandhu JS, Gorczynski RM, Waddell J, Nguyen H, Squires J, Boynton EL, Hozumi N. Effect of interleukin-6 secreted by engineered human stromal cells on osteoclasts in human bone. Bone. 1999;24:217–27. doi: 10.1016/s8756-3282(98)00172-0. [DOI] [PubMed] [Google Scholar]
- [35].Palmqvist P, Persson E, Conaway HH, Lerner UH. IL-6, leukemia inhibitory factor, and oncostatin M stimulate bone resorption and regulate the expression of receptor activator of NF-kappa B ligand, osteoprotegerin, and receptor activator of NF-kappa B in mouse calvariae. J Immunol. 2002;169:3353–62. doi: 10.4049/jimmunol.169.6.3353. [DOI] [PubMed] [Google Scholar]
- [36].Kitamura H, Kawata H, Takahashi F, Higuchi Y, Furuichi T, Ohkawa H. Bone marrow neutrophilia and suppressed bone turnover in human interleukin-6 transgenic mice. A cellular relationship among hematopoietic cells, osteoblasts, and osteoclasts mediated by stromal cells in bone marrow. Am J Pathol. 1995;147:1682–92. [PMC free article] [PubMed] [Google Scholar]
- [37].Barton BE, Mayer R. IL 3 and IL 6 do not induce bone resorption in vitro. Cytokine. 1990;2:217–20. doi: 10.1016/1043-4666(90)90019-p. [DOI] [PubMed] [Google Scholar]
- [38].Holt I, Davie MW, Braidman IP, Marshall MJ. Interleukin-6 does not mediate the stimulation by prostaglandin E2, parathyroid hormone, or 1,25 dihydroxyvitamin D3 of osteoclast differentiation and bone resorption in neonatal mouse parietal bones. Calcif Tissue Int. 1994;55:114–9. doi: 10.1007/BF00297186. [DOI] [PubMed] [Google Scholar]
- [39].Duplomb L, Baud’huin M, Charrier C, Berreur M, Trichet V, Blanchard F, Heymann D. Interleukin-6 inhibits receptor activator of nuclear factor kappaB ligand-induced osteoclastogenesis by diverting cells into the macrophage lineage: key role of Serine727 phosphorylation of signal transducer and activator of transcription 3. Endocrinology. 2008;149:3688–97. doi: 10.1210/en.2007-1719. [DOI] [PubMed] [Google Scholar]
- [40].Nishimoto N, Hashimoto J, Miyasaka N, Yamamoto K, Kawai S, Takeuchi T, Murata N, van der Heijde D, Kishimoto T. Study of active controlled monotherapy used for rheumatoid arthritis, an IL-6 inhibitor (SAMURAI): evidence of clinical and radiographic benefit from an x ray reader-blinded randomised controlled trial of tocilizumab. Ann Rheum Dis. 2007;66:1162–7. doi: 10.1136/ard.2006.068064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Smolen JS, Beaulieu A, Rubbert-Roth A, Ramos-Remus C, Rovensky J, Alecock E, Woodworth T, Alten R. Effect of interleukin-6 receptor inhibition with tocilizumab in patients with rheumatoid arthritis (OPTION study): a double-blind, placebo-controlled, randomised trial. Lancet. 2008;371:987–97. doi: 10.1016/S0140-6736(08)60453-5. [DOI] [PubMed] [Google Scholar]
- [42].Kato A, Matsuo S, Takai H, Uchiyama Y, Mihara M, Suzuki M. Early effects of tocilizumab on bone and bone marrow lesions in a collagen-induced arthritis monkey model. Exp Mol Pathol. 2008;84:262–70. doi: 10.1016/j.yexmp.2008.03.003. [DOI] [PubMed] [Google Scholar]
- [43].Kamimura D, Ishihara K, Hirano T. IL-6 signal transduction and its physiological roles: the signal orchestration model. Rev Physiol Biochem Pharmacol. 2003;149:1–38. doi: 10.1007/s10254-003-0012-2. [DOI] [PubMed] [Google Scholar]
- [44].Huang W, Drissi MH, O’Keefe RJ, Schwarz EM. A rapid multiparameter approach to study factors that regulate osteoclastogenesis: demonstration of the combinatorial dominant effects of TNF-alpha and TGF-ss in RANKL-mediated osteoclastogenesis. Calcif Tissue Int. 2003;73:584–93. doi: 10.1007/s00223-003-0059-8. [DOI] [PubMed] [Google Scholar]
- [45].van de Loo FA, Kuiper S, van Enckevort FH, Arntz OJ, van den Berg WB. Interleukin-6 reduces cartilage destruction during experimental arthritis. A study in interleukin-6-deficient mice. Am J Pathol. 1997;151:177–91. [PMC free article] [PubMed] [Google Scholar]
- [46].Shin HI, Divieti P, Sims NA, Kobayashi T, Miao D, Karaplis AC, Baron R, Bringhurst R, Kronenberg HM. Gp130-mediated signaling is necessary for normal osteoblastic function in vivo and in vitro. Endocrinology. 2004;145:1376–85. doi: 10.1210/en.2003-0839. [DOI] [PubMed] [Google Scholar]
- [47].Fattori E, Cappelletti M, Costa P, Sellitto C, Cantoni L, Carelli M, Faggioni R, Fantuzzi G, Ghezzi P, Poli V. Defective inflammatory response in interleukin 6-deficient mice. J Exp Med. 1994;180:1243–50. doi: 10.1084/jem.180.4.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Ulich TR, Guo KZ, Remick D, del Castillo J, Yin SM. Endotoxin-induced cytokine gene expression in vivo. III. IL-6 mRNA and serum protein expression and the in vivo hematologic effects of IL-6. J Immunol. 1991;146:2316–23. [PubMed] [Google Scholar]
- [49].Xing Z, Gauldie J, Cox G, Baumann H, Jordana M, Lei XF, Achong MK. IL-6 is an antiinflammatory cytokine required for controlling local or systemic acute inflammatory responses. J Clin Invest. 1998;101:311–20. doi: 10.1172/JCI1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Moreno JL, Kaczmarek M, Keegan AD, Tondravi M. IL-4 suppresses osteoclast development and mature osteoclast function by a STAT6-dependent mechanism: irreversible inhibition of the differentiation program activated by RANKL. Blood. 2003;102:1078–86. doi: 10.1182/blood-2002-11-3437. [DOI] [PubMed] [Google Scholar]
- [51].Heinrich PC, Behrmann I, Haan S, Hermanns HM, Muller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. 2003;374:1–20. doi: 10.1042/BJ20030407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Rakshit DS, Ly K, Sengupta TK, Nestor BJ, Sculco TP, Ivashkiv LB, Purdue PE. Wear debris inhibition of anti-osteoclastogenic signaling by interleukin-6 and interferon-gamma. Mechanistic insights and implications for periprosthetic osteolysis. J Bone Joint Surg Am. 2006;88:788–99. doi: 10.2106/JBJS.E.00711. [DOI] [PubMed] [Google Scholar]