

Abstract
Herein we report a general synthesis of 1,3-diarylsubstituted indazoles utilizing a two-step Suzuki cross-coupling/deprotection/N-arylation sequence. This procedure proceeds in excellent overall yield starting from the 3-iodo-N-Boc indazole derivative allowing for rapid access to these compounds.
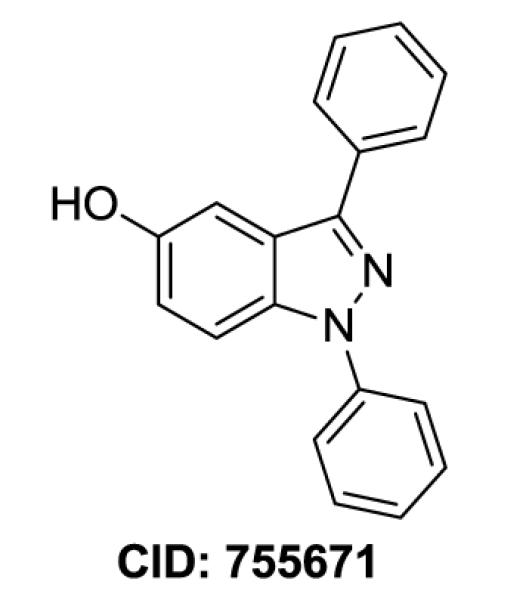
The Vanderbilt Specialized Chemistry Center for Accelerated Probe Development is a member of the Molecular Libraries Production Center Network (MLPCN) initiated and supported by the NIH Molecular Libraries Roadmap.1, 2 The MLPCN is a nationwide consortium of facilities that provide high-throughput small-molecule screening and medicinal chemistry expertise for the development of chemical probes for use as tools to explore biological targets or pathways for which small-molecule tools are unavailable.1 As part of this initiative a high-throughput screen identified a 1,3-diaryl substituted indazole compound as a potential lead (Figure 1, CID, Pubchem Compound ID). Benzannulated nitrogen heterocycles are ubiquitous in pharmaceutical research.3, 4 Over the past decade, the indazole structural variant (benzo[c]pyrazole) has received much attention due to a broad range of biological activity.5 The synthesis of the indazole core, as well as the functionalization of the indazole ring system has recently been reviewed.6, 7
Figure 1.
More important to our application, the metal catalyzed cross-coupling6 of both the C(3) position (with8-10 or without N-protection10-12), and the N(1) position has been reported both independently, and in a one-pot sequence.12 As part of a larger effort to discover novel heterocycles with interesting biological activities, we were in need of a flexible synthetic protocol for 1,3-diarylsubstituted indazoles to derivatize the N(1) and C(3) positions independently.
Recent reports from the Rault group11, 12 sparked our interest as this would provide us a method to synthesize differentially arylated N(1) and C(3) compounds. To this end, we sought to reproduce the findings in our labs (see Scheme 1). The first attempt was to obtain the C(3) iodinated compound followed by N-arylation and ultimately cross-couple the C(3) position (Scheme 1A). Although the initial iodination proceeded uneventfully (I2, KOH, quant.), the following N-arylation yielded the desired compound in a disappointingly low yield (20%).11 Next, the 3-iodo-5-methoxyindazole was reacted under Suzuki-Miyaura coupling conditions13, however, the reaction did not afford the desired product and only starting material was obtained (Scheme 1B).12 Lastly, we attempted the one-pot, diarylation procedure, which did yield product, but again in very low overall yield (Scheme 1C).12 Note the previously reported procedures employed unsubstituted indazole, whereas our substrate contains the 5-methoxy substrate, which was required for analog synthesis.
Scheme 1.
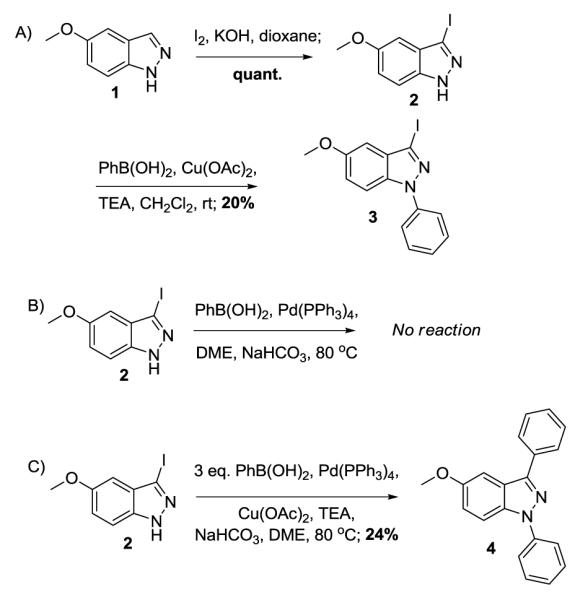
Selected attempts to arylate 5-methoxyindazole.
At this time we decided to explore other reaction conditions in an attempt to improve the overall yield of the sequence; and in addition, develop a reliable synthesis that allows for both arylation at the C(3) and N(1) in order to fully evaluate the SAR surrounding these compounds. Other reports in the literature utilize the N-Boc protected indazole under conventional thermal conditions.8, 10 However, and much to our surprise, a SciFinder search showing C(3) arylation followed by N(1) arylation of an indazole had only the aforementioned reference.12 With this sequence in mind, we set out to first arylate the C(3) position of 3-iodo-N-Boc-5-methoxyindazole (Scheme 2). Thus, N-Boc formation ((Boc)2O, TEA, DMAP; 95%) yielded the N-protected indazole in a straight forward manner.14 Next, Suzuki-Miyaura coupling under thermal conditions did not lead to product and only yielded the starting material (Scheme 2A). However, Suzuki-Miyaura cross-coupling utilizing microwave heating conditions not only affected the C-C bond formation, but also had concomitant Boc deprotection (Scheme 2B). There has been a report of N-deprotection of N-Boc indazole during the cross-coupling conditions9 in which the only product obtained was the 3-iodo-NH-indazole. This same report also demonstrates the use of a p-tosyl group which also undergoes N-deprotection; however, the overall yield of product was 45% (with equivalent amounts of unreacted de-tosylated starting material).9 The use of the N-Boc protecting group under microwave heating conditions has led to high yield of the desired cross-coupled, NH-indazole.
Scheme 2.
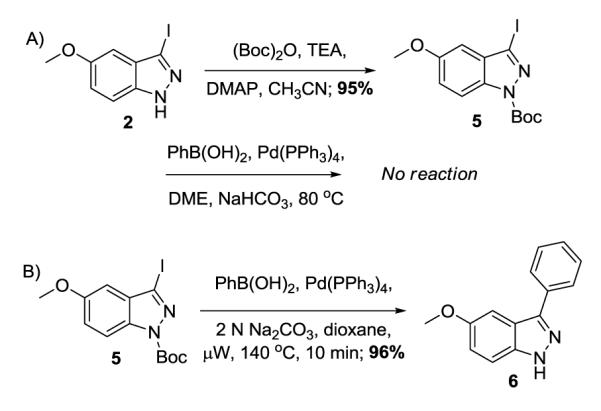
Suzuki cross-coupling of N-Boc-3-iodo-5-methoxyindazole.
Next we turned our attention to the generality of this cross-coupling, N-deprotection sequence (Table 1).15 The reactions were performed in commercially available heavy-walled Pyrex tubes. The indazole iodide 5 was reacted with 2 equiv of boronic acid in the presence of an aqueous base in dioxane with Pd(PPh3)4 as catalyst. The results and isolated yields are summarized in Table 1. The cross-coupling/N-deprotection is compatible with a variety of aryl and heteroaryl groups all proceeding in high yield (>80%). Both electron withdrawing groups and electron donating groups proceeded in high overall yield (6c, 6e, 6f, 6d). In addition to arylboronic acids, heteroarylboronic acids also performed well under these conditions (6b, 6i-6k). These reaction conditions allow for access to NH-3-(hetero)aryl substituted indazoles that have shown to be difficult to access previously.9
Table 1.
| Entry | Cmpd | Ar | Yield (%)a |
|---|---|---|---|
| 1 | 6a | phenyl | 96 |
| 2 | 6b | 3-pyridyl | 95 |
| 3 | 6c | 3-chlorophenyl | 98 |
| 4 | 6d | 3-methylphenyl | 94 |
| 5 | 6e | 3-trifluoromethylphenyl | 81 |
| 6 | 6f | 2,4-difluorophenyl | 81 |
| 7 | 6g | 4-trifluoromethylphenyl | 83 |
| 8 | 6h | 4-pyridyl | 87 |
| 9 | 6i | 1-methyl-1H-pyrazol-4-yl | 92 |
| 10 | 6j | 2-furyl | 96 |
| 11 | 6k | 2-thiophene | 97 |
| 12 | 6l | 4-methoxyphenyl | 87 |
Isolated yield
Following Buchwald’s procedure for the copper-diamine catalyzed N-arylation16, 17, we then proceeded to arylate the 3-substituted indazole substrates.18 The reaction was accomplished utilizing a general procedure which utilized the NH-indazole with 1.2 equiv of aryl iodide, 5 mol% CuI, 2 equiv of base (K3PO4),17 20 mol % ligand and toluene under thermal conditions.17 The results and isolated yields are summarized in Table 2. Table 2 shows that substitution around the aryl group (ortho, meta, para) are tolerated in this reaction – although the 2-methoxy and 2-fluoro-substiuted compounds reacted in lower yield (7k, 30%; 7l, 28%). Several functional groups were also tolerated in this reaction such as, amino, methoxy, halogen, and nitro. In addition, heteroaryl iodides were also well tolerated (7m, 98%).
Table 2.
| Entry | Cmpd | R2 | Ar | Yield (%)a |
|---|---|---|---|---|
| 1 | 7a | H | phenyl | 86 |
| 2 | 7b | H | 4-fluorophenyl | 75 |
| 3 | 7c | H | 3-methoxyphenyl | 62 |
| 4 | 7d | H | 4-methylphenyl | 68 |
| 5 | 7e | H | 4-methoxyphenyl | 84 |
| 6 | 7f | H | 4-aminophenyl | 90 |
| 7 | 7g | H | 3-aminophenyl | 86 |
| 8 | 7h | H | 2-aminophenyl | 73 |
| 9 | 7i | H | 3-fluorophenyl | 65 |
| 10 | 7j | H | 3-nitrophenyl | 97 |
| 11 | 7k | H | 2-methoxyphenyl | 30 |
| 12 | 7l | H | 2-fluorophenyl | 28 |
| 13 | 7m | H | 2-pyridyl | 98 |
| 14 | 7n | 3-Cl | phenyl | 93 |
| 15 | 7o | 3-methyl | phenyl | 73 |
| 16 | 7p | 3-CF3 | phenyl | 80 |
| 17 | 7q | 2,4-difluoro | phenyl | 98 |
| 18 | 7r | 3-pyridyl | phenyl | 94 |
Isolated yield
Having established a three-step, two-pot Suzuki cross-coupling/deprotection/Buchwald coupling sequence to access the 5-methoxy indazoles we next were able to demethylate to access the initial screening hit in high yield. To this end, 7a was exposed to 48% HBr, AcOH under microwave conditions to yield 8 in 90% yield.19 Compound 8 was obtained in an overall yield of 71% from the commercially available 5-methoxyindazole allowing for library synthesis of a number of analogs for further testing in the MLPCN network of centers.
In conclusion, 3-aryl-NH-indazoles have been prepared in high yield from the corresponding 3-iodo-N-Boc indazoles under microwave irradiation. The reaction sequence has high functional group tolerance and excellent overall yield. The concomitant deprotection of the Boc group led to NH-indazoles that were directly coupled under Buchwald conditions to yield the desired 1,3-disubstituted indazole compounds. Further the 5-hydroxy compound was realized after demethylation under microwave conditions.
Supplementary Material
Scheme 3.
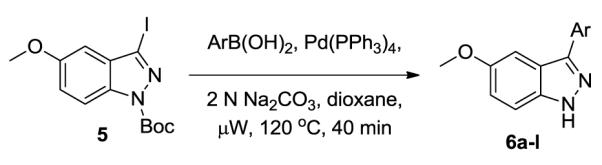
Demethylation of indazole.
Acknowledgments
The authors warmly thank the Vanderbilt Department of Pharmacology and the NIH/MLPCN (5U54MH084659-02) for support of this research. Vanderbilt is a member of the MLPCN and houses the Vanderbilt Specialized Chemistry Center for Accelerated Probe Development.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary data Supplementary data associated with this article can be found, in the online version, at doi:
References and Notes
- 1.For information on the Molecular Libraries Probe Production Centers Network (MLPCN) http://mli.nih.gov/mli/
- 2.For information on the Vanderbilt Specialized Chemistry Center http://www.mc.vanderbilt.edu/centers/mlpcn/index.htm l.
- 3.Gribble GW. Katritzky AR, Rees CW, Scriven EFV, editors. Compr. Heterocycl. Chem. II. 1996;Vol. 2:207. [Google Scholar]
- 4.Gribble GW. J. Chem. Soc., Perkins Trans. 2000;1:1045. [Google Scholar]
- 5.Cerecetto H, Gerpe A, González M, Arán VJ, de Ocáriz Ochoa C. Mini-Rev. Med. Chem. 2005;5:869. doi: 10.2174/138955705774329564. [DOI] [PubMed] [Google Scholar]
- 6.Bräse S, Gil C, Knepper K. Bioorg. Med. Chem. 2002;10:2415. doi: 10.1016/s0968-0896(02)00025-1. [DOI] [PubMed] [Google Scholar]
- 7.Elguero J. Katritzky AR, Rees CW, Scriven EFV, editors. Comprehensive Heterocyclic Chemistry. 1996;Vol. 3:1. [Google Scholar]
- 8.Vazquez J, De SK, Chen L-H, Riel-Mehan M, Emdadi A, Cellitti J, Stebbins JL, Rega MF, Pellecchia M. J. Med. Chem. 2008;51:3460. doi: 10.1021/jm800068w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bouissane L, Kazzouli SE, Léonce S, Pfeiffer B, Rakib EM, Khouili M, Guillaumet G. Bioorg. Med. Chem. 2006;14:1078. doi: 10.1016/j.bmc.2005.09.037. [DOI] [PubMed] [Google Scholar]
- 10.Swahn B-M, Huerta F, Kallin E, Malmström J, Weigelt T, Viklund J, Womack P, Xue Y, öhberg L. Bioorg. Med. Chem. Lett. 2005;15:5095. doi: 10.1016/j.bmcl.2005.06.083. [DOI] [PubMed] [Google Scholar]
- 11.Collot V, Dallemagne P, Bovy PR, Rault S. Tetrahedron. 1999;55:6917. [Google Scholar]
- 12.Collot V, Bovy PR, Rault S. Tetrahedron Lett. 2000;41:9053. [Google Scholar]
- 13.Miyaura N, Suzuki A. Chem. Rev. 1995;95:2457. [Google Scholar]
- 14.3-Iodo-5-methoxy-1H-indazole (2). To a stirred solution of 5-methoxyindazole (0.50 g, 3.4 mmol) in 1,4-dioxane (34 mL) at 0 °C under argon was added KOH (1.10 g, 20.0 mmol), followed by iodine (1.71 g, 6.75 mmol). The reaction was stirred vigorously for 15 minutes, then warmed to room temperature and stirred overnight. The reaction’s color changed from black to yellow overnight. The reaction pH was adjusted to pH 5 by addition of 20% aqueous citric acid, aqueous Na2S2O3 (saturated, 50 mL) was added, and the mixture extracted with EtOAc. The combined organic layers were washed with water, brine, dried over MgSO4 and concentrated under vacuum to give 929 mg (100%) of the crude product as a yellow solid. This material was used in the next reaction without further purification. 1H NMR (400 MHz, DMSO-d6): δ7.44 (d, J = 9.0 Hz, 1 H), 7.05 (dd, J = 9.0, 2.1 Hz, 1 H), 6.74 (d, J = 1.7 Hz, 1 H), 3.81 (s, 3 H). LC/MS: RT = 1.27 min., m/z = 274.9 [M + 1]+.tert-Butyl 3-iodo-5-methoxy-1H-indazole-1-carboxylate (5). 3-Iodo-5-methoxy-1H-indazole, 2, (580 mg, 2.12 mmol), triethylamine (0.442 mL, 3.17 mmol) and DMAP (13 mg, 0.11 mmol) were stirred in acetonitrile (5 mL) for 10 minutes. Di-tert-butyl dicarbonate (508 mg, 2.33 mmol) was added and the reaction was stirred at room temperature for about 10 hours. The reaction mixture was concentrated under vacuum. The residue was dissolved in EtOAc and water. The aqueous layer was separated and extracted with EtOAc. The combined organic layers were washed with brine, dried over Na2SO4, concentrated and the residue chromatographed on silica gel (12 grams) eluting with a 0-to-15 % EtOAc/hexane gradient to give 749 mg (95%) of the pure product as a yellow solid. 1H NMR (400 MHz, DMSO-d6): δ 7.94 (d, J = 9.1 Hz, 1 H), 7.28 (dd, J9 = 9.1, 2.5 Hz, 1 H), 6.88 (d, J = 2.4 Hz, 1 H), 3.86 (s, 3 H), 1.63 (s, 9 H). LC/MS: RT = 1.65 min., m/z = 397.0 [M + Na]+.
- 15.Representative Suzuki Coupling Example: 5-methoxy-3-phenyl-1H-indazole (6a). To a microwave reaction vial was added tert-butyl 3-iodo-5-methoxy-1H-indazole-1-carboxylate, 5, (130 mg, 0.35 mmol), Pd(PPh3)4 (20 mg, 0.02 mmol), phenylboronic acid (85 mg, 0.69 mmol), 1,4-dioxane (3 mL) and aqueous 2N Na2CO3 (0.77 mL, 1.54 mmol). The vial was sealed and heated under microwave irradiation at 120 °C for 40 minutes. Upon cooling to room temperature, the reaction mixture was diluted with EtOAc and poured through Celite, washing with EtOAc. The solute was concentrated under vacuum and the residue purified by reverse-phase HPLC to afford 75 mg (96%) of the product as a white solid. 1H NMR (400 MHz, DMSO-d6): δ 7.95 (d, J = 7.4 Hz, 2 H), 7.53-7.48 (m, 3 H), 7.39-7.36 (m, 2 H), 7.04 (dd, J = 9.0, 2.1 Hz, 1 H), 3.83 (s, 3 H). 13C NMR (150 MHz, DMSO-d6) δ 154.9, 143.0, 137.8, 134.4, 129.2, 127.7, 127.0, 120.6, 118.5, 112.0, 100.2, 55.8. LC/MS: RT = 1.76 min., m/z = 225.0 [M + H]+.
- 16.Klapars A, Antilla JC, Huang X, Buchwald SL. J. Am. Chem. Soc. 2001;123:7727. doi: 10.1021/ja016226z. [DOI] [PubMed] [Google Scholar]
- 17.Antilla JC, Baskin JM, Barder TE, Buchwald SL. J. Org. Chem. 2004;69:5578. doi: 10.1021/jo049658b. [DOI] [PubMed] [Google Scholar]
- 18.Representative Buchwald coupling procedure: 5-methoxy-3-phenyl-1-(pyridin-2-yl)-1H-indazole (7m). To a microwave reaction vial was added 5-methoxy-3-phenyl-1H-indazole (70 mg, 0.31 mmol), CuI (3 mg, 0.02 mmol) and K3PO4 (139 mg, 0.66 mmol). The vial was sealed with a septum cap. The vial was evacuated and back-filled with argon twice. In a separate vial 2-iodopyridine (0.04 mL, 0.37 mmol), trans-N,N-diamine-1,2-cyclohexanediamine (0.010 mL, 0.06 mmol) and toluene (0.42 mL) were mixed, then syringed into the microwave vial. The vial was placed in a 110 °C oil bath and stirred overnight (18 hrs). Upon cooling to room temperature the reaction mixture was diluted with EtOAc and poured through Celite, washing with EtOAc. The solute was concentrated under vacuum and the residue purified by reverse-phase HPLC to give 92 mg (98%) of the desired product as a white solid. 1H NMR (400 MHz, DMSO-d6): δ 8.73 (d, J = 9.2 Hz, 1 H), 8.55 (d, J = 4.0 Hz, 1 H), 8.09-7.97 (m, 4 H), 7.60-7.57 (m, 2 H), 7.51-7.45 (m, 2 H), 7.31-7.24 (m, 2 H), 3.88 (s, 3 H). 13C NMR (150 MHz, DMSO-d6) δ 156.3, 153.7, 148.3, 146.0, 139.5, 135.6, 132.6, 129.5, 129.1, 127.9, 124.1, 120.8, 119.7, 116.6, 113.3, 101.3, 55.9. LC/MS: RT = 1.74 min., m/z = 302.2 [M + H]+.
- 19.1,3-diphenyl-1H-indazol-5-ol (8). 5-methoxy-1,3-diphenyl-1H-indazole, 7a, (51 mg, 0.17 mmol), was added to a microwave reaction vial followed by 2.0 mL of (1:1) 48% HBr/acetic acid. The vial was sealed and the reaction heated at 180 °C for 10 minutes. Upon cooling to room temperature the reaction mixture was diluted with EtOAc and aqueous (saturated) NaHCO3 was slowly added until the reaction pH was basic. The aqueous layer was extracted with EtOAc (3x). The combined organic layers were dried over Na2SO4, concentrated under vacuum and the residue purified by reverse-phase HPLC to give 44 mg (90%) of the product as light tan solid. 1H NMR (400 MHz, DMSO-d6): δ9.51 (s, 1 H), 7.95 (d, J = 7.2 Hz, 2 H), 7.80 (d, J = 7.6 Hz, 2 H), 7.74 (d, J = 9.0 Hz, 1 H), 7.61-7.54 (m, 4 H), 7.46-7.37 (m, 3 H), 7.07 (dd, J = 9.0, 2.2 Hz, 1 H). LC/MS: RT = 1.54 min., m/z = 287.0 [M + H]+.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.