

Abstract
Ligation of two oligonucleotide probes hybridized adjacently to a DNA template has been widely used for detection of genome alterations. The multiplex ligation-dependent probe amplification (MLPA) technique allows simultaneous screening of multiple target sequences in a single reaction by using pairs of probes that carry tails for binding of common amplification primers. Resolution of the various targets is achieved by electrophoresis on the basis of predefined differences in amplicon length. In the conventional MLPA approach, one of the two target probes is generated by cloning in a single-stranded bacteriophage vector to introduce a sequence of defined length between the primer binding site and the specific target sequence. Here we demonstrate that differences in amplicon length can be achieved by using multiple short synthetic probes for each target sequence. When joined by a DNA ligase, these probes will form a single amplifiable template whose length is defined by the number and lengths of the individual probes. We have used this principle to establish a methylation-specific MLPA (MS-MLPA) assay that simultaneously determines the methylation status of five promoter CpG islands, and we have used this assay to analyze DNA from tumor tissue and corresponding urine samples from patients with bladder cancer. Our data show that the use of multiple short synthetic probes provides a simple means for custom-designed MS-MLPA analysis.
The principle of using a DNA ligase to covalently join two oligonucleotide probes hybridized adjacently on a DNA or RNA template has been widely used to study genome alterations. First described in 1984 as a means of reducing nonspecific probe binding,1 DNA ligase-based assays have been developed for detection of disease-causing mutations and single-nucleotide polymorphisms,2,3 sequence copy number variations,4 and aberrant DNA methylation.5,6 Ligation of two probes requires that the 3′ probe is phosphorylated at its 5′ end, and the reaction is efficient only when the two probes lie immediately adjacent to one another and there is a perfect match between template and probes at the ligation junction. DNA ligases are particularly sensitive to 3′ mismatches,7,8,9 which is used for allele discrimination in the oligonucleotide ligation assay2 and the ligase chain reaction10 and for detection of DNA methylation using bisulfite-treated DNA as a template.6
Multiplex ligation-dependent probe amplification (MLPA) was first developed as a technique for determining relative sequence copy numbers, which uses probe pairs for up to 40 individual loci in a single reaction.4 Each probe consists of a target-specific sequence and a nonhybridizing tail containing a universal primer binding site (Figure 1A). As a means to modulate the length of the ligation product, at least one of the probes in each pair contains a “stuffer sequence” of defined length between the primer binding site and the specific target sequence. After hybridization to the template, the probe pairs are joined by ligation, the ligated products are amplified with a common primer pair, and the probe-derived amplicons can then be separated by electrophoresis according to size. Quantitative analysis of the products provides a measure of the relative sequence copy number, which has been successfully applied to the detection of deletions in human disease-related genes as well as gene copy number variations in tumor samples.11 More recent developments of MLPA include assays to measure relative mRNA expression levels and assays to detect segmentally duplicated genes and changes in DNA methylation patterns.11 The principle of methylation-specific MLPA (MS-MLPA) is the same as for conventional MLPA, except that the binding site for each probe pair contains a site for a methylation-sensitive restriction enzyme, usually HhaI. When the probe-template hybrids are subjected to digestion with this enzyme, the generation of an amplifiable ligation product will depend on the methylation status of the target sequence.5 MS-MLPA assays have been used to study aberrations at imprinted loci in patients with inherited disorders and at tumor-suppressor loci in various cancers.11
Figure 1.
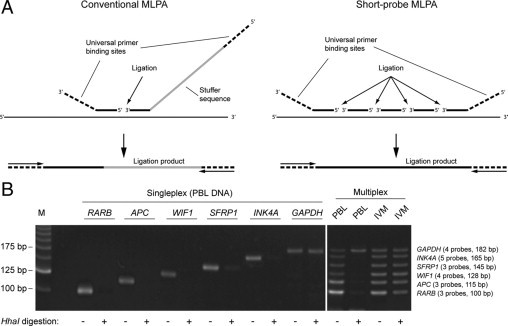
A: Principle of short-probe MLPA. In conventional MLPA, two oligonucleotide probes are generated for each target sequence, including a short probe generated by chemical synthesis and a long probe containing a stuffer sequence introduced by cloning in bacteriophage vectors. In short-probe MLPA, multiple short synthetic probes are included for each target, which hybridize adjacently and can be joined by DNA ligase into a single amplifiable product. The length of the final ligation product is determined by the number and lengths of the probes. The arrows in the lower portion of A indicate universal amplification primers. B: Short-probe MS-MLPA assay for methylation analysis of the RARB, APC, WIF1, SFRP1, and INK4A promoter regions. Sets of three to five probes for these targets and the GAPDH control sequence (lacking a HhaI recognition site) were hybridized to 50 ng of genomic DNA in singleplex and multiplex. After digestion with HhaI, in vitro-methylated DNA (IVM) gave positive signals from all six targets, whereas only a GAPDH signal was obtained when PBL DNA was used as template. M indicates 25-bp ladder.
A major drawback of MLPA lies in the requirement for long probes, which cannot be made efficiently by chemical synthesis. In the original MLPA protocol,4 which also forms the basis for commercially available MLPA kits, a library of probes with different stuffer sequences was generated by cloning target-specific sequences into a panel of specially designed M13 vectors. This procedure is not feasible for custom design of MLPA assays containing a limited number of specific targets. Here we describe an alternative and simple approach to MLPA analysis, which involves the use of multiple short synthetic probes for each target site. The length of the probe-derived amplicons can be easily and accurately preset by the number and lengths of probes in the individual probe sets. We have applied this strategy to establish an MS-MPLA assay that simultaneously detects hypermethylation of the promoter regions of the genes encoding retinoic acid receptor β (RARB), adenomatous polyposis coli (APC), WNT inhibitory factor 1 (WIF1), secreted frizzled-related protein 1 (SFRP1), and the cyclin-dependent kinase inhibitor, p16 (INK4A). All five target sequences have previously been shown to be frequently hypermethylated in bladder cancer.12,13,14,15,16,17 Here we have used this assay to study methylation patterns in tumors and corresponding urine samples from patients with bladder cancer.
Materials and Methods
Cell Lines, Clinical Samples, and DNA Isolation
Human bladder cancer (T24) and melanoma (FM3, FM28, and FM79) cell lines were cultured in RPMI medium supplemented with 10% fetal bovine serum and 100 units each of penicillin and streptomycin (Invitrogen, Gaithersburg, MD). Tumor biopsies from 46 patients with bladder cancer (36 males and 10 females, median age = 67.5 years, range 45–90 years) and normal bladder mucosa samples from one bladder cancer patient (CB27) and six patients (3 males and 3 females, median age = 69.8 years, range 53–86 years) with noncancerous urinary lesions (three with benign prostate hyperplasia and three with painful bladder syndrome [interstitial cystitis]) were snap-frozen in liquid nitrogen and stored at −80°C. Naturally voided morning urine samples from 26 bladder cancer patients (19 males and 7 females, median age = 66.5 years, range 45–90 years) and seven healthy nonsmoking individuals (four males and three females, median age = 32 years, range 24–47 years) were centrifuged at 2000g for 10 minutes, and the sediment was washed once with phosphate-buffered saline and stored at −80°C. DNA was extracted using the QIAamp DNA Mini Kit (Qiagen GmbH, Hilden, Germany). Informed consent was obtained from all subjects, and the project was approved by the Ethical Committee of Copenhagen and Frederiksberg.
Oligonucleotide Probes
Oligonucleotide probes were designed for RARB, APC, WIF1, SFRP1, INK4A, and GAPDH (Table 1). Each of the target sequences except GAPDH contained a recognition site for the methylation-sensitive restriction enzyme HhaI (GCGC). In general, we adhered to previously described guidelines for probe design (MRC-Holland, Amsterdam, the Netherlands; date of accession 04/16/2008). Care was taken to position the ligation sites in regions devoid of single-nucleotide polymorphisms and other sequence variation that could potentially interfere with the ligation reaction. Gene sequences were retrieved from the UCSC Genome Browser (accession numbers listed in Table 1). The 5′ and 3′ probes in each set were synthesized with tails containing binding sites for universal primers (Figure 1A), and all probes except the 5′ probes were synthesized with a 5′ phosphate group to allow for ligation. All probes were synthesized at 0.2 μmol scale and purified by polyacrylamide gel electrophoresis (Sigma-Genosys, Haverhill, UK).
Table 1.
Oligonucleotide Probes for the MS-MLPA Assay
| Gene | Number of probes | GenBank accession no. | Probe sequences | Product length (bp) |
|---|---|---|---|---|
| RARB | 3 | NM_016152 | [X]−5′-GAGGCGAGCGGGCGCAGGCG-3′ | 100 |
| 5′-P-GAACACCGTTTTCCAAGCTA-3′ | ||||
| 5′-P-AGCCGCCGCAAATAAAAAGG-3′-[Y] | ||||
| APC | 3 | U02509 | 5′- [X]-GTCGGGAAGCGGAGAGAGAAGCAGC-3′ | 115 |
| 5′-P-TGTGTAATCCGCTGGATGCGGACCA-3′ | ||||
| 5′-P-GGGCGCTCCCCATTCCCGTCGGGAG-3′-[Y] | ||||
| WIF1 | 4 | AF122922 | [X]-CCCAAGTGGCGGCCGCCCAG-3′ | 128 |
| 5′-P-GCCTCGCGGGCCCCACTCCTCGC-3′ | ||||
| 5′-P-TCGCACCTCGCTCGCGCCAGCC-3′ | ||||
| 5′-P-CTTCCCGCTCTTCTGTTCTCGCT-3′-[Y] | ||||
| SFRP1 | 3 | NM_003012 | [X]-CTGGTTCTAGTAAACCGAACCCGCTCGCGAGGGAG-3′ | 145 |
| 5′-P-GCGATTGGCTCCCGCGCCGGTGACGGACGTGGTAA-3′ | ||||
| 5′-P-CGAGTGCGGCTCGCCCCGCCGGGAGCTGATTGGCT-−3′ [Y] | ||||
| INK4A | 5 | NM_000077 | [X]-TCACCAGAGGGTGGGGCGGACCGCG-3′ | 165 |
| 5′-P-TGCGCTCGGCGGCTGCGGAGAGGGG-3′ | ||||
| 5′-P-GAGAGCAGGCAGCGGGCGGCGGGGA-3′ | ||||
| 5′-P-GCAGCATGGAGCCGGCGGCGGGGAG-3′ | ||||
| 5′-P-CAGCATGGAGCCTTCGGCTGACTGG-3′-[Y] | ||||
| GAPDH | 4 | NM_002046 | [X]-CCAGCACCGATCACCTCCCATCGGGCCAATCTCAGTC-3′ | 182 |
| 5′-P-CCTTCCCCCCTACGTCGGGGCCCACACGCTC-3′ | ||||
| 5′-P-GGTGCGTGCCCAGTTGAACCAGGCGGCTGC-3′ | ||||
| 5′-P-GGAAAAAAAAAAGCGGGGAGAAAGTAGGGCCCGGCTACTAGCGG-3′-[Y] |
Recognition sites for the methylation-sensitive restriction enzyme, HhaI, are underscored.
[X] = 5′-TATGTAAAACGACGGCCAGT-3′.
[Y] = 5′-ACCCAATTCGCCCTATAATA-3′.
Short-Probe MS-MLPA Assay
The MLPA reagent kit EK1 was obtained from MRC-Holland (Amsterdam, The Netherlands), and hybridization, ligation, and enzyme digestion were performed in a Peltier Thermal Cycler with heated lid (PTC-200; MJ Research), essentially as described.5 Briefly, DNA in 5 μl of TE buffer (10 mmol/L Tris–HCl and 1 mmol/L EDTA, pH 8.5) was denatured at 98°C for 10 minutes and incubated overnight at 60°C after the addition of 1.5 μl of MLPA Buffer (MRC-Holland) and 1.5 μl of a probe mix containing 20 fmol of each GAPDH probe, 12 fmol of each RARB and WIF1 probe, and 4.5 fmol of all other probes. After hybridization, 3 μl of Ligase buffer A (MRC-Holland) and 10 μl of H2O were added at room temperature, and the mixture was divided into two tubes and then subjected to ligation and combined ligation and digestion, respectively. For combined ligation and digestion, 0.25 μl of Ligase-65 (MRC-Holland), 0.5 μl of HhaI (Promega; 10 U/μl), 1.5 μl of Ligase buffer B (MRC-Holland), and H2O were added to a final volume of 20 μl. After initial incubation at 54°C for 15 minutes, the temperature was lowered to 37°C, and incubation was continued at 37°C for 30 minutes after the addition of 0.5 μl of HhaI. In the ligation reaction, the HhaI enzyme was replaced with an equal volume of H2O, and the mixtures were incubated at 54°C for 30 minutes. After inactivation of the enzymes at 98°C for 5 minutes, 3.7 μl of the reaction mixtures were subjected to PCR in a total volume of 12.5 μl containing 1 × Multiplex PCR Master Mix (QIAGEN, Valencia, CA), 1 × Q-Solution (QIAGEN), and 1 μmol/L each primer (5′-TATGTAAAACGACGGCCAGT-3′ and 5′-TATTATAGGGCGAATTGGGT-3′). PCR conditions were: 95°C for 15 minutes to activate the enzyme, followed by 40 cycles at 95°C for 30 seconds, 52°C for 30 seconds, and 72°C for 30 seconds. The PCR products were resolved by electrophoresis in 4% NuSieve GTG agarose gels (Cambrex, East Rutherford, NJ) and visualized by ethidium bromide staining. In vitro-methylated DNA (CpGenome Universal Methylated DNA; Millipore, Billerica, MA) was used as a positive control for methylation.
For PCR confirmation of INK4A deletions, the following primers were used: 5′-GGAAATTGGAAACTGGAAGC-3′ and 5′-TCTGAGCTTTGGAAGCTCT-3′ for INK4A, and 5′-CTTTCCTCTCGGGATTTCTTG-3′ and 5′-GGGTATACATGGGCTTGGATC-3′ for PAH.
Methylation-Specific Melting Curve Analysis
Two micrograms of genomic DNA were treated with sodium bisulfite according to standard procedures.18 The bisulfite-modified DNA was resuspended in 30 μl of TE buffer (10 mmol/L Tris–HCl and 1 mmol/L EDTA, pH 8.5) and stored at −80°C until use. Complete bisulfite conversion was confirmed by lack of amplification in a TaqMan assay with primers for a normal, nonconverted ACTB sequence.19 Methylation-specific melting curve analysis (MS-MCA)20 was performed using the LightCycler 1.2 instrument (Roche) and the FastStart DNA Master SYBR Green I Kit (Roche). Primer sequences and PCR conditions are listed in Table 2.
Table 2.
Primer Sequences and PCR Conditions for MS-MCA
| Gene | Forward primer | Reverse primer | Product length (bp) | Annealing temperature (°C) |
|---|---|---|---|---|
| RARB | 5′-GGTTTATTTTTTGTTAAAGGGG-3′ | 5′-AAAAATCCCAAATTCTCCTTC-3′ | 124 | 63 |
| APC | 5′-TGGGAGGGGTTTTGTGTTTTATT-3′ | 5′-CCATTCTATCTCCAATAACACCCTAA-3′ | 196 | 65 |
| WIF1 | 5′-AGGAGGTTTTGAGTAGTATGGTT-3′ | 5′-CCATAAATACAAACTCTCCTCCT-3′ | 134 | 65 |
| SFRP1 | 5′-AGTTTTGTAGTTTTAGGAGTTAGTGT-3′ | 5′-AAACAAAAAACTCAATCCCCAA-3′ | 128 | 65 |
| INK4A | 5′-GTTGTAGATTTTTTATTTATTTGG-3′ | 5′-AACCTTCCACTAACTAACTAACCAC-3′ | 142 | 65 |
GenBank accession numbers are shown in Table 1.
Results
Ligation of Multiple Short Synthetic Probes
The aim of our study was to establish a simple MS-MLPA assay for early detection of bladder cancer, which can be used in most laboratories with minimum equipment requirements. To circumvent the tedious and time-consuming generation of probes by cloning into phage vectors, we investigated the possibility of joining multiple short synthetic probes into an amplifiable template of defined length (Figure 1). A total of five sets of probes were designed for the promoter regions of RARB, APC, WIF1, SFRP1, and INK4A (Figure 1B; Table 1). One of the probes in each set contained a HhaI recognition site (GCGC) to allow methylation-dependent analysis. We also designed an additional probe set, which hybridizes to a GCGC-free region of GAPDH and will serve as a methylation-independent positive control for hybridization, ligation, and PCR. The lengths of the final products varied from 100 to 182 bp and were defined by the lengths (20–44 bp of target-specific sequence) and numbers (3–5) of probes to allow separation by agarose gel electrophoresis (Figure 1B).
To test whether multiple adjacent probes can be effectively joined by DNA ligase into an amplifiable product, we first hybridized the six probe sets individually to genomic DNA isolated from peripheral blood lymphocytes (PBL) from healthy donors, followed by ligation and amplification. All probe sets gave rise to a single band of the expected size (Figure 1B). Furthermore, a mix of the 22 probes representing all of the six targets gave rise to a ladder of six bands of equal intensity. These data show that ligation of multiple short synthetic probes provides a simple means for target detection in a multiplex format.
Detection of DNA Methylation by Short-Probe MS-MLPA
We next tested the ability of the short-probe MS-MLPA assay to distinguish between methylated and unmethylated targets. When the individual probe sets were hybridized to PBL DNA, digestion with HhaI abolished amplification from all targets except for GAPDH, which lacks a HhaI recognition site (Figure 1B). When all probe sets were combined in the multiplex format, ligation and digestion resulted in a single band (GAPDH) with PBL DNA as template and in all six bands with in vitro-methylated DNA (Figure 1B).
To further validate the multiplex assay, we analyzed DNA from 4 cancer cell lines (Figure 2A). These cell lines gave rise to positive signals for the control gene (GAPDH) and between 0 and 4 of the cancer-specific targets (see supplemental Table S1 at http://jmd.amjpathol.org). The methylation status of the RARB, APC, WIF1, SFRP1, and INK4A promoter regions was also examined by MS-MCA in these cells lines (Figure 2B). All fully and partially methylated sequences (as determined by MS-MCA) gave positive signals with the MS-MLPA assay; however MS-MLPA could not distinguish between the fully and partially methylated states (Figure 2B). Taken together, these results demonstrate that the short-probe MS-MLPA assay can reliably determine the methylation status at five genomic loci in the same reaction.
Figure 2.
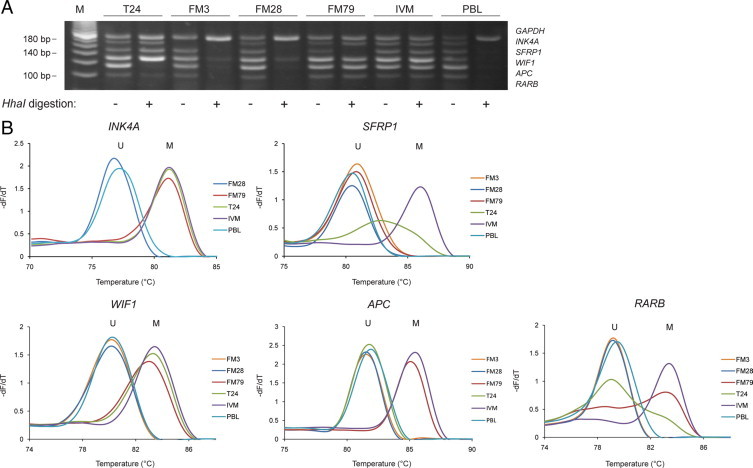
Short-probe MS-MLPA analysis (A) and MS-MCA (B) of cancer cell lines. MS-MCA distinguishes methylated (M) from unmethylated (U) sequences on the basis of differences in melting temperature after bisulfite treatment. Fluorescence data were converted into melting peaks by plotting the negative derivative of fluorescence over temperature (−dF/dT) versus temperature. Partially methylated sequences result in broadened melting peaks located between the melting peaks of the unmethylated and fully methylated sequences (RARB in the FM79 and T24 cell lines and SFRP1 in the T24 cell line). MS-MCA data on INK4A methylation are not shown for FM3, which harbors a homozygous INK4A deletion. IVM indicates in vitro–methylated DNA; M, 20-bp ladder.
Limit of Detection and Assay Sensitivity
To assess the minimum level of input DNA required for the short-probe MS-MLPA assay to work reliably, we analyzed serial dilutions of human genomic DNA. Using PBL DNA as template, all bands appeared at equal intensities and complete digestion was obtained with as little as 12.5 ng of input DNA (Figure 3A). With respect to the maximum amount of input DNA, we occasionally experienced unequal band intensities and incomplete digestion when using 200 ng or more of DNA (Figure 3A). On the basis of these experiments, the use of 20–100 ng of template DNA appears to be optimal for ensuring robust amplification and restriction enzyme digestion.
Figure 3.
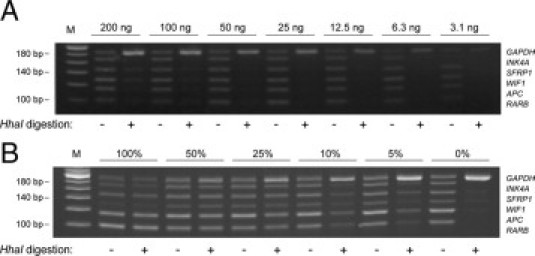
Limit of detection and assay sensitivity. A: Serial dilutions of genomic DNA isolated from PBL were subjected to short-probe MS-MPLA analysis. B:In vitro-methylated DNA (IVM) was diluted into PBL DNA and subjected to short-probe MS-MLPA using a total of 50 ng of these mixtures as template. M indicates 20-bp ladder.
To test the sensitivity of the assay, we serially diluted in vitro–methylated DNA into PBL DNA. With an input of 50 ng of DNA, the assay could simultaneously detect hypermethylated copies of all five targets down to the 5% level in a background of wild-type DNA (Figure 3B). As the total amount of DNA was kept constant in the mixtures of in vitro–methylated DNA and PBL DNA, the number of GAPDH targets also remained constant in these samples. Nevertheless, as amplification of the different targets with the primer set is competitive, the signal intensity of the GAPDH signal increased when the number of methylated targets is decreased (Figure 3B).
Multiplex Methylation Detection in Clinical Samples
To test the ability of the short-probe MS-MLPA assay to detect methylation patterns in heterogeneous clinical samples containing a mixture of tumor cells and normal cells, we first analyzed DNA isolated from 46 bladder tumor biopsies representing various tumor stages (Figure 4A; see supplemental Table S1 at http://jmd.amjpathol.org). All samples gave positive signals for the GAPDH control sequence but showed highly variable signal profiles for the cancer-specific targets. The frequencies of promoter hypermethylation were 17% for RARB, 35% for APC, 33% for WIF1, 39% for SFRP1, and 2% for INK4A (see supplemental Table S1 at http://jmd.amjpathol.org). The low frequency of INK4A hypermethylation in this series of bladder tumors is consistent with some previous studies17,21 and was confirmed by MS-MCA (see supplemental Figure S1 at http://jmd.amjpathol.org). For many of the samples, we observed that the methylation-specific signals were weaker than the GAPDH signal, which would be expected because of tumor heterogeneity and contamination of the samples with normal cells. However, in the current setting of the assay, it is not possible to obtain quantitative information. A total of four tumors (9%) showed loss of signal from the INK4A probe set in both the ligation and ligation-digestion reactions (see Figure 4A for examples). The lack of probe binding may be explained by homozygous deletions at this locus, which are common in bladder cancer.22,23 Indeed, the presence of INK4A deletions in these samples was confirmed by PCR analysis (see supplemental Figure S2 at http://jmd.amjpathol.org).
Figure 4.

Short-probe MS-MLPA analysis of bladder tumors, urine specimens, and normal bladder mucosa. A: Tumor samples. B: Paired tumor and urine samples. The origin of the band between the APC and RARB bands in samples T54 and U59 is unknown. C: Urine from healthy subjects. D: Bladder mucosa from patients with noncancerous urinary lesions. IVM indicates in vitro–methylated DNA; M, 20-bp ladder.
We next used the MS-MLPA assay to the analysis of DNA isolated from urine samples from 26 of the bladder cancer patients from whom tumor DNA was also available. Representative results for paired tumor and urine samples are shown in Figure 4B. Of 37 independent methylation events found in the tumor samples, 33 could also be detected in the corresponding urine samples (89%; see supplemental Table S1 at http://jmd.amjpathol.org).
Finally, we used the assay to analyze DNA from seven urine samples from healthy controls and seven normal bladder mucosa samples (Figure 4C and 4D). Although the majority of the urine samples were negative for all markers, with the exception of one being positive for WIF1 (HCU5) and one showing weak WIF1, APC, and RARB bands (HCU6), the majority of bladder tissue samples gave positive signals for WIF1 and SFRP1.
Discussion
The development of the MLPA technology has significantly advanced the study of genomic changes in inherited disorders and cancer.11 Commercially available MLPA kits usually contain up to 40 probe pairs for selected targets with amplicon lengths of up to 500 bp. However, as many MLPA applications require a special design of probes for a limited number of specific targets, which may not be included in currently available kits, there is an urgent need for new developments that allow simple alternatives to vector-based probe generation. Synthetic probes of up to 100 bp in length have been used for some MLPA applications,24,25 but chemical synthesis of longer oligonucleotides is still problematic. Here we have introduced a general approach to custom-designed MLPA, which is based on the use of multiple short synthetic probes for each target sequence. The probes hybridize adjacently to the DNA template and can be joined by a DNA ligase to form a single amplifiable template. The use of three or more probes results in highly specific target recognition as the formation of full-length ligation products requires at least two independent ligation events. This approach provides a simple, cost-effective, and flexible means of tailoring MLPA assays with respect to both the number and lengths of individual targets and may be adapted to any set of targets in a user-defined configuration.
We used the short-probe MLPA approach to analyze promoter methylation at five loci that have been reported relevant for bladder cancer. The MS-MLPA assay was designed to require only minimal instrumentation (a thermal cycler), and the product lengths were adjusted to allow resolution by agarose gel electrophoresis (i.e., with 10- to 15-bp-size increments). However, the number and lengths of the ligation products can be easily adjusted to suit other applications or separation techniques, such as capillary electrophoresis, which allows resolution of 3- to 4-bp differences.25 Short-probe MS-MLPA analysis of 46 cases of bladder cancer showed a great intertumor variation in the profiles of hypermethylation events, whereas paired tumor and urine samples in most cases showed identical patterns. Thus, in cases of bladder cancer where tumor cells are released into the urine, this assay may be used for noninvasive detection of bladder tumors and for disease monitoring. Urine samples from healthy donors were negative in this assay; however, this result should be interpreted with caution as these samples were not age matched to urine samples from bladder cancer patients.
The short-probe MS-MLPA approach described here has many of the same advantages and limitations as conventional MS-MLPA based on vector-generated probes.5 The advantages include the use of genomic DNA instead of a bisulfite-treated template, the ability to analyze multiple targets using as little as 50 ng of DNA as starting template, and the ability to detect homozygous deletions. One limitation of the assay is that it does not distinguish between fully and partially methylated sequences. Another important limitation is related to the use of a methylation-sensitive restriction enzyme to discriminate between methylated and unmethylated sequences. Incomplete digestion of unmethylated targets may lead to false-positive results and is a caveat inherent in most restriction enzyme-based methylation assays.26 We occasionally observed weak methylation signals in samples from healthy controls, which may be the result of incomplete digestion. However, most of these signals were from WIF1 and SFRP1, which have previously been reported to be frequently hypermethylated in bladder tissue from normal bladder mucosa.13,14,27 This suggests that WIF1 and SFRP1 may have limited use as markers for noninvasive detection of bladder tumors compared with the other loci examined (INK4A, APC and RARB), which showed consistent hypomethylation in urine from healthy subjects and bladder mucosa from patients with noncancerous urinary lesions.
A potential limitation specific for the short-probe MLPA approach is the need for a longer stretch of template DNA to assist in the generation of a full-length, amplifiable ligation product (cf. Figure 1A). As the signal intensity from a probe set is directly related to the number of intact template molecules, a reduced signal will be expected from the longer probe sets if the template DNA is degraded. As a control for DNA integrity, the longest probe set in our assay was designed for a target sequence with no HhaI recognition site, whose amplification will depend solely on presence of an intact template. Using DNA from cancer cell lines, urine samples, and fresh-frozen tissues, we did not observe a decrease in PCR performance with increasing target length. However, analysis of long ligation products may be problematic in samples characterized by a high degree of DNA degradation, such as formalin-fixed paraffin-embedded tissue. For analysis of low-quality DNA, we therefore recommend the use of only two or three short probes for each target sequence.
In conclusion, we have shown that the use of multiple short synthetic probes may provide a simple and cost-effective approach to custom-designed MLPA. The use of entirely synthetic probes allows a flexible assay design, and the addition of a probe set or replacement of one probe set requires only few adjustments of the assay. The external probes of each set may be short and contain only the sequence required for primer binding, as in the assay described here, or they may be long synthetic probes containing stuffer sequences. We have specifically used the short-probe approach to MS-MLPA, but the same approach may be used for most other MLPA applications and thus provide the basis for a more widespread usage of the MLPA technology.
Footnotes
Supported by the Danish Cancer Society, the Neye Foundation, Henny Sophie Clausen and furniture architect Axel Clausen's Foundation, Lily Benthine Lund's Foundation, Dagmar Marshall's Foundation, the A. P. Møller Foundation for the Advancement of Medical Science, the Toyota Foundation, and the Copenhagen County University Hospital Foundation.
CME Disclosure: None of the authors disclosed any relevant financial relationships.
Supplemental material for this article can be found on http://jmd.amjpathol.org.
Web Extra Material
{kind=link}
{kind=link}
Results of MS-MLPA analysis of human cancer cell lines and tumor and corresponding urine samples from patients with bladder cancer.
References
- 1.Whiteley NM, Hunkapiller MW, and Glazer AN, inventors: Applied Biosystems, Inc. assignee 1989. Detection of specific sequences in nucleic acids. United States Patent US 4883750
- 2.Landegren U, Kaiser R, Sanders J, Hood L. A ligase-mediated gene detection technique. Science. 1988;241:1077–1080. doi: 10.1126/science.3413476. [DOI] [PubMed] [Google Scholar]
- 3.Nickerson DA, Kaiser R, Lappin S, Stewart J, Hood L, Landegren U. Automated DNA diagnostics using an ELISA-based oligonucleotide ligation assay. Proc Natl Acad Sci USA. 1990;87:8923–8927. doi: 10.1073/pnas.87.22.8923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002;30:e57. doi: 10.1093/nar/gnf056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nygren AO, Ameziane N, Duarte HM, Vijzelaar RN, Waisfisz Q, Hess CJ, Schouten JP, Errami A. Methylation-specific MLPA (MS-MLPA): simultaneous detection of CpG methylation and copy number changes of up to 40 sequences. Nucleic Acids Res. 2005;33:e128. doi: 10.1093/nar/gni127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dahl C, Guldberg P. A ligation assay for multiplex analysis of CpG methylation using bisulfite-treated DNA. Nucleic Acids Res. 2007;35:e144. doi: 10.1093/nar/gkm984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nilsson M, Barbany G, Antson DO, Gertow K, Landegren U. Enhanced detection and distinction of RNA by enzymatic probe ligation. Nature Biotechnol. 2000;18:791–793. doi: 10.1038/77367. [DOI] [PubMed] [Google Scholar]
- 8.Bhagwat AS, Sanderson RJ, Lindahl T. Delayed DNA joining at 3′ mismatches by human DNA ligases. Nucleic Acids Res. 1999;27:4028–4033. doi: 10.1093/nar/27.20.4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu J, Tong J, Feng H, Huang J, Afonso CL, Rock DL, Barany F, Cao W. Unique ligation properties of eukaryotic NAD+-dependent DNA ligase from Melanoplus sanguinipes entomopoxvirus. Biochim Biophys Acta. 2004;1701:37–48. doi: 10.1016/j.bbapap.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 10.Barany F. Genetic disease detection and DNA amplification using cloned thermostable ligase. Proc Natl Acad Sci USA. 1991;88:189–193. doi: 10.1073/pnas.88.1.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kozlowski P, Jasinska AJ, Kwiatkowski DJ. New applications and developments in the use of multiplex ligation-dependent probe amplification. Electrophoresis. 2008;29:4627–4636. doi: 10.1002/elps.200800126. [DOI] [PubMed] [Google Scholar]
- 12.Hoque MO, Begum S, Topaloglu O, Chatterjee A, Rosenbaum E, Van CW, Westra WH, Schoenberg M, Zahurak M, Goodman SN, Sidransky D. Quantitation of promoter methylation of multiple genes in urine DNA and bladder cancer detection. J Natl Cancer Inst. 2006;98:996–1004. doi: 10.1093/jnci/djj265. [DOI] [PubMed] [Google Scholar]
- 13.Urakami S, Shiina H, Enokida H, Kawakami T, Kawamoto K, Hirata H, Tanaka Y, Kikuno N, Nakagawa M, Igawa M, Dahiya R. Combination analysis of hypermethylated Wnt-antagonist family genes as a novel epigenetic biomarker panel for bladder cancer detection. Clin Cancer Res. 2006;12:2109–2116. doi: 10.1158/1078-0432.CCR-05-2468. [DOI] [PubMed] [Google Scholar]
- 14.Urakami S, Shiina H, Enokida H, Kawakami T, Tokizane T, Ogishima T, Tanaka Y, Li LC, Ribeiro-Filho LA, Terashima M, Kikuno N, Adachi H, Yoneda T, Kishi H, Shigeno K, Konety BR, Igawa M, Dahiya R. Epigenetic inactivation of Wnt inhibitory factor-1 plays an important role in bladder cancer through aberrant canonical Wnt/beta-catenin signaling pathway. Clin Cancer Res. 2006;12:383–391. doi: 10.1158/1078-0432.CCR-05-1344. [DOI] [PubMed] [Google Scholar]
- 15.Catto JW, Azzouzi AR, Rehman I, Feeley KM, Cross SS, Amira N, Fromont G, Sibony M, Cussenot O, Meuth M, Hamdy FC. Promoter hypermethylation is associated with tumor location, stage, and subsequent progression in transitional cell carcinoma. J Clin Oncol. 2005;23:2903–2910. doi: 10.1200/JCO.2005.03.163. [DOI] [PubMed] [Google Scholar]
- 16.Friedrich MG, Weisenberger DJ, Cheng JC, Chandrasoma S, Siegmund KD, Gonzalgo ML, Toma MI, Huland H, Yoo C, Tsai YC, Nichols PW, Bochner BH, Jones PA, Liang G. Detection of methylated apoptosis-associated genes in urine sediments of bladder cancer patients. Clin Cancer Res. 2004;10:7457–7465. doi: 10.1158/1078-0432.CCR-04-0930. [DOI] [PubMed] [Google Scholar]
- 17.Dulaimi E, Uzzo RG, Greenberg RE, Al-Saleem T, Cairns P. Detection of bladder cancer in urine by a tumor suppressor gene hypermethylation panel. Clin Cancer Res. 2004;10:1887–1893. doi: 10.1158/1078-0432.ccr-03-0127. [DOI] [PubMed] [Google Scholar]
- 18.Clark SJ, Harrison J, Paul CL, Frommer M. High sensitivity mapping of methylated cytosines. Nucleic Acids Res. 1994;22:2990–2997. doi: 10.1093/nar/22.15.2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eads CA, Danenberg KD, Kawakami K, Saltz LB, Blake C, Shibata D, Danenberg PV, Laird PW. MethyLight: a high-throughput assay to measure DNA methylation. Nucleic Acids Res. 2000;28:E32. doi: 10.1093/nar/28.8.e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Worm J, Aggerholm A, Guldberg P. In-tube DNA methylation profiling by fluorescence melting curve analysis. Clin Chem. 2001;47:1183–1189. [PubMed] [Google Scholar]
- 21.Maruyama R, Toyooka S, Toyooka KO, Harada K, Virmani AK, Zochbauer-Muller S, Farinas AJ, Vakar-Lopez F, Minna JD, Sagalowsky A, Czerniak B, Gazdar AF. Aberrant promoter methylation profile of bladder cancer and its relationship to clinicopathological features. Cancer Res. 2001;61:8659–8663. [PubMed] [Google Scholar]
- 22.Chapman EJ, Harnden P, Chambers P, Johnston C, Knowles MA. Comprehensive analysis of CDKN2A status in microdissected urothelial cell carcinoma reveals potential haploinsufficiency, a high frequency of homozygous co-deletion and associations with clinical phenotype. Clin Cancer Res. 2005;11:5740–5747. doi: 10.1158/1078-0432.CCR-05-0411. [DOI] [PubMed] [Google Scholar]
- 23.Orlow I, Lacombe L, Hannon GJ, Serrano M, Pellicer I, Dalbagni G, Reuter VE, Zhang ZF, Beach D, Cordon-Cardo C. Deletion of the p16 and p15 genes in human bladder tumors. J Natl Cancer Inst. 1995;87:1524–1529. doi: 10.1093/jnci/87.20.1524. [DOI] [PubMed] [Google Scholar]
- 24.Stern RF, Roberts RG, Mann K, Yau SC, Berg J, Ogilvie CM. Multiplex ligation-dependent probe amplification using a completely synthetic probe set. Biotechniques. 2004;37:399–405. doi: 10.2144/04373ST04. [DOI] [PubMed] [Google Scholar]
- 25.Kozlowski P, Roberts P, Dabora S, Franz D, Bissler J, Northrup H, Au KS, Lazarus R, Domanska-Pakiela D, Kotulska K, Jozwiak S, Kwiatkowski DJ. Identification of 54 large deletions/duplications in TSC1 and TSC2 using MLPA, and genotype-phenotype correlations. Hum Genet. 2007;121:389–400. doi: 10.1007/s00439-006-0308-9. [DOI] [PubMed] [Google Scholar]
- 26.Dahl C, Guldberg P. DNA methylation analysis techniques. Biogerontology. 2003;4:233–250. doi: 10.1023/a:1025103319328. [DOI] [PubMed] [Google Scholar]
- 27.Yates DR, Rehman I, Abbod MF, Meuth M, Cross SS, Linkens DA, Hamdy FC, Catto JW. Promoter hypermethylation identifies progression risk in bladder cancer. Clin Cancer Res. 2007;13:2046–2053. doi: 10.1158/1078-0432.CCR-06-2476. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Results of MS-MLPA analysis of human cancer cell lines and tumor and corresponding urine samples from patients with bladder cancer.