

Abstract
Mutations in codons 12 and 13 of the KRAS oncogene are relatively common in colorectal and lung adenocarcinomas. Recent data indicate that these mutations result in resistance to anti-epidermal growth factor receptor therapy. Therefore, we assessed Sanger sequencing, pyrosequencing, and melting curve analysis for the detection of KRAS codon 12/13 mutations in formalin-fixed paraffin-embedded samples, including 58 primary and 42 metastatic colorectal adenocarcinomas, 63 primary and 17 metastatic lung adenocarcinomas, and 20 normal colon samples. Of 180 tumor samples, 62.2% were KRAS mutant positive, and 37.8% were negative. Melting curve analysis yielded no false positive or false negative results, but had 10% equivocal calls. Melting curve analysis also resulted in 4 cases with melting curves inconsistent with either wild-type or codon 12/13 mutations. These patterns were generated from samples with double mutants in codons 12/13 and with mutations outside of codons 12/13. Pyrosequencing yielded no false positive or false negative results as well. However, two samples from one patient yielded a pyrogram that was flagged as abnormal, but the mutation subtype could not be determined. Finally, using an electronic cutoff of 10%, Sanger sequencing showed 11.1% false positives and 6.1% false negatives. In our hands, the limit of detection for Sanger sequencing, pyrosequencing, and melting curve analysis was approximately 15 to 20%, 5%, and 10% mutant alleles, respectively.
The kirsten rat sarcoma viral oncogene homolog (K-ras), a GTPase binding protein encoded by the KRAS gene, serves as an intermediary signaling molecule in the epidermal growth factor receptor (EGFR) signaling pathway.1 KRAS mutations have been implicated in the pathogenesis of numerous tumors and are found in approximately 15 to 50% of lung adenocarcinomas, 30 to 60% of colorectal adenocarcinomas, and 90% of pancreatic adenocarcinomas.2,3 Mutation of the KRAS oncogene leads to its constitutive signaling and subsequent dysregulated cellular proliferation.4 KRAS mutations have been shown to occur relatively early in the development of colorectal adenocarcinoma and late in that of lung adenocarcinoma.5,6
Anti-EGFR monoclonal antibody therapies such as cetuximab and panitumumab are approved by the Food and Drug Administration for use in patients with metastatic colon cancer. Anti-EGFR tyrosine kinase inhibitors such as Erlotinib are approved for use in patients with locally advanced or metastatic non-small cell lung cancer, after failure of at least one prior chemotherapeutic regimen. Recent data indicate that the presence of a codon 12, 13, or 61 KRAS mutation results in resistance to these anti-EGFR therapies.7,8,9,10 Current guidelines from the National Comprehensive Cancer Network recommend the use of anti-EGFR therapies only in patients with wild-type KRAS lung and colorectal adenocarcinomas. (NCCN Clinical Practice Guidelines in Oncology. Non Small Cell Lung Cancer (Version 2.2009, available at: http://www.nccn.org/professionals/physician_gls/PDF/ncl.pdf) and Colon Cancer (Version 3.2009, available at http://www.nccn.org/professionals/physician_gls/PDF/colon.pdf). Both accessed October 9, 2009)
A variety of strategies can be used to assess KRAS mutation status in tumor samples including Sanger sequencing (SS), real-time PCR with or without melting curve analysis (MC), and pyrosequencing (PS). SS is considered by many to be the gold standard in mutation testing. SS can detect essentially all base substitutions, small insertions and deletions, but has a modest limit of detection, which can be highly variable depending on the exact sequence, and laboratory performing the test. Quantitative PCR with MC uses fluorescent probes to capitalize on differences in melting temperatures generated by genetic alterations. When a mutation is present in the target, the probe–target duplex is separated at a lower temperature as compared with a perfectly matching wild-type sequence. The differences in melting temperature (Tm) are detected by a loss of fluorescence as a function of increasing temperature. PS is a bioluminescence technique in which the pyrophosphate released during incorporation of a nucleotide into a growing DNA chain is converted to light through a series of enzymatic reactions. PS can identify individual bases or short stretches of nucleic acid sequence at predetermined positions. Here, we have compared these three platforms for the detection of KRAS codon 12 and 13 mutations in formalin-fixed paraffin-embedded (FFPE) lung and colorectal adenocarcinoma samples.
Materials and Methods
Samples
This study was covered under Institutional Review Board approval NA 00002948 for the use of de-identified clinical samples to evaluate new methods/technologies. Two hundred FFPE tissue samples from 191 patients were obtained from the tissue archives at the Johns Hopkins Hospital. These samples included 180 tumor samples, including 58 primary and 42 metastatic colorectal adenocarcinomas, 63 primary and 17 metastatic lung adenocarcinomas, from 171 patients (84 males and 87 females). Nine of these patients each contributed 2 tumor samples to the study, including 6 patients where 2 different blocks from the same tumor were tested and 3 patients who had both primary and metastatic tumors tested. The mean age of the patients analyzed was 78 years (range, 31 to 98 years). In addition, we collected 20 histologically normal colon samples from 20 patients (10 males and 10 females) with diverticulosis or diverticulitis and no past medical history of adenomatous polyps or adenocarcinoma. The mean age for patients with diverticulitis or diverticulosis was 61 years (range, 38 to 71 years).
Sample Preparation and DNA Extraction
H&E-stained slides from FFPE tumor and normal samples were reviewed by a pathologist and tumor tissue was selected for analysis. Corresponding tissue from five unstained, 10-μm-thick tissue sections was removed using Pinpoint reagents according the manufacturer's protocol (ZymoResearch, Orange, CA). DNA was purified from the sample using QIAmp DNA kit (Qiagen, Valencia, CA) and quantified by OD 260 nm.
For our limit of detection study, we prepared dilutions of mutant KRAS cells from the LoVo cell line, which contains a heterozygous G13D mutation, into wild-type cells derived from a cell line of normal lymphoblasts. Total genomic DNA was isolated from cell pellets and purified with the Qiagen DNA-Mini kit. We analyzed each mixture for accuracy of the dilution using our clinical chimerism assay which employs the Identifiler reagents containing 15 short tandem repeat markers (Applied Biosystems, Foster City, CA).
Melting Curve Analysis
Reaction mixture consisted of LightCycler FastStart DNA Master Hybridization Probes Mix (Roche, Indianapolis Indiana), 500 nmol/L forward primer (5′-AAGGCCTGCTGAAAATGACTG-3′), 100 nmol/L reverse primer (5′-CCCTCCCCAGTCCTCATG-3′), 400 nmol/L sensor probe (5′-LC Red 640-TGCCTACGCCACCAGCTCCAA-phosphate-3′), and 200 nmol/L anchor probe (5′-CCACAAAATGATTCTGAATTAGCTGTATCGTCAAGGCACT-fluorescein-3′) in a final reaction volume of 20 μl. Reactions were thermal cycled in a LightCycler (Roche) as follows: 95°C for 10 minutes, followed by 40 cycles of 95°C for 10 seconds, 55°C for 10 seconds, and 72°C for 15 seconds. Melting curve analysis was performed through one cycle of 95°C for 20 seconds, 40°C for 30 seconds, and ramp to 85°C at 0.1°C/sec. Amplification and melting curves were generated using the LightCycler software. To validate the MC, we analyzed 29 pancreatic cancer samples that had previously been characterized for KRAS mutations (provided by Dr. Christine Iacobuzio-Donahue). Twenty-five of the 29 samples harbored one of four different KRAS mutations (G12D, G12V, G12R, G13D). The validation of lung and colorectal adenocarcinomas demonstrated three additional KRAS mutations (G12A, G12S, and G12C). We determined the Tm of wild-type alleles to be 71°C ± 2°C, while the seven most common KRAS mutations in lung and colorectal adenocarcinoma produced a Tm of 63°C ± 2°C.
Sanger Sequencing
PCR amplification products generated by the Lightcycler PCR/melting curve analysis were purified using QiaQuick reagents (Qiagen) and were cycle sequenced using Big Dye v3.1 reagents (Applied Biosystems) according to the manufacturer's protocol. Sequencing products were purified with CleanSEQ Sequencing Purification System (Agencourt Bioscience Corp., Beverly, MA) and automated sequencing performed by capillary electrophoresis on an ABI3700 (Applied Biosystems). Sequences were aligned and examined by two separate approaches: electronically with a set threshold of 10% and by visual inspection of the electropherogram, using Sequencher software (Gene Codes Corp., Inc.).
Pyrosequencing
Samples were PCR amplified using the KRAS v2.0 kit (Qiagen) according to the manufacturer's protocol. Each reaction contained 1× PCR buffer, 1.5 mmol/L MgCl2, 0.2 mmol/L of each dNTP, 5 pmol of forward primer, and 5 pmol of reverse primer (biotinylated), 0.8 Units of HotStar TaqDNA polymerase (Qiagen), 10 ng of template DNA, and dH2O to 25 μl final volume. Cycling conditions were as follows: 95°C 15 minutes, 38× (95°C 20 seconds, 53°C 30 seconds, 72°C 20 seconds), 72°C 5 minutes, 8°C hold. Following amplification, 10 μl of biotinylated PCR product was immobilized on streptavidin-coated sepharose beads (streptavidin sepharose high performance, GE Health care Bio-Sciences Corp., Piscataway, NJ) and washed in 70% EtOH. The purified biotinylated PCR product was released into the PyroMark Q24 (Biotage, SE) with PyroMark Gold reagents (Qiagen) containing 0.3 μmol/L sequencing primer and annealing buffer. The nucleotide dispensation order for codons 12/13 was: 5′-TACGACTCAGATCGTAG-3′.
Results
Twenty non-tumor and 180 tumor samples were tested for mutations in codons 12 and 13 of KRAS by MC, SS, and PS. After reviewing our data, we decided to use PS as the gold standard for mutations in KRAS codons 12 and 13 because we felt that interpretation of the PS data were less subjective than for SS and MC. The 20 nontumor specimens all tested negative for KRAS mutations on all three platforms. Of the 180 tumor samples, 112 (62.2%) were KRAS mutant positive as determined. Our study deliberately included tumor samples known to harbor KRAS mutations and thus, our positive rate does not reflect the true incidence of KRAS mutations in our patient population. The mutations identified are summarized in Table 1. Consistent with the findings in the literature, mutations in codon 12 were far more common [98 of 112 (87.5%)] than in codon 13 [11 of 112 (9.8%)].11,12 The G12D mutation was the most common mutation found in primary [13 of 56, (23%)] and metastatic [(10 of 42, (24%)] colorectal carcinoma. The G12C mutation was the most common mutation found in primary lung adenocarcinoma [14 of 63 (23%)], while the G12V mutation was the most common mutation found in metastatic lung adenocarcinoma [5 of 17 (29%)]. An example of a wild-type and mutant (G12D) result from all three platforms is demonstrated in Figure 1.
Table 1.
KRAS Mutation Distribution by Body Site of Tested Tumor
| KRAS mutation | Colorectal adenocarcinoma (primary) | Colorectal adenocarcinoma (metastatic) | Lung adenocarcinoma (primary) | Lung adenocarcinoma (metastatic) | Total |
|---|---|---|---|---|---|
| G12V | 7 | 6 | 7 | 5 | 25 |
| G12A | 3 | 2 | 2 | 1 | 8 |
| G12C | 1 | 0 | 14 | 4 | 19 |
| G12D | 13 | 10 | 9 | 1 | 33 |
| G12R | 2 | 0 | 2 | 2 | 6 |
| G12S | 3 | 0 | 0 | 0 | 3 |
| G12F | 1 | 1 | 0 | 0 | 2 |
| G13D | 4 | 5 | 1 | 1 | 11 |
| Q22K | 0 | 0 | 1 | 0 | 1 |
| L19F | 0 | 1 | 0 | 0 | 1 |
| G12C/G13D | 2 | 0 | 0 | 0 | 2 |
| G12V/G13D | 1 | 0 | 0 | 0 | 1 |
| Total | 37 | 25 | 36 | 14 | 112 |
Bold type highlights the most frequent mutation at each tumor site.
Figure 1.
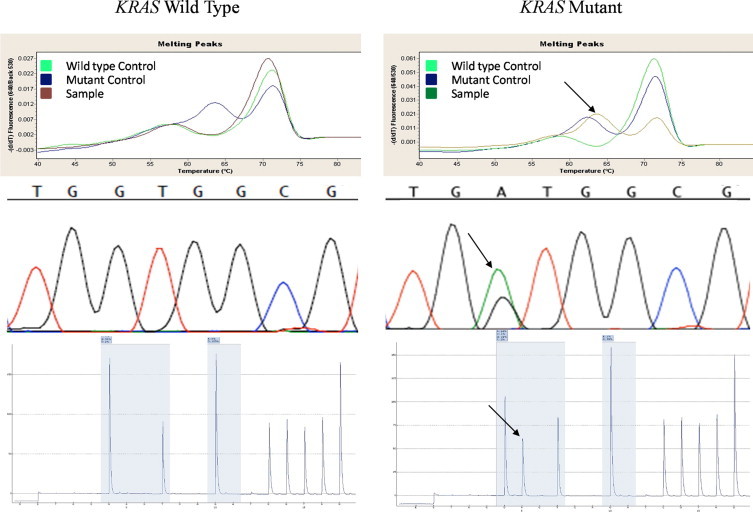
Melting curve, Sanger sequencing, and pyrosequencing data from a wild-type KRAS specimen and mutant KRAS specimen (G12D) are shown. Arrows indicate the mutation.
A comparison of the results from all three platforms is summarized in Table 2. MC yielded 94 KRAS mutants in the 180 tumor samples (52.2%) with a Tm consistent with a codon 12/13 mutation, 4 (2.2%) KRAS mutants with a Tm not consistent with a codon 12/13 mutation, 18 (10%) equivocal, and 64 (35.6%) negative. Out of the 18 equivocal samples, 13 were positive, and 4 negative by both SS and PS.
Table 2.
KRAS Mutation Testing Results by Platform
| Interpretation | Sanger sequencing (visual inspection) | Sanger sequencing (electronic 10% cutoff) | Melt curve analysis | Pyrosequencing |
|---|---|---|---|---|
| Positive | 111 | 120 | 98 | 110 |
| Negative | 69 | 60 | 64 | 70 |
| Equivocal | 0 | 0 | 18 | 0 |
SS yielded 100 of 180 (55.6%) KRAS mutants and 49 of 180 (27.2%) wild-type by both visual inspection and by automated interpretation with a 10% threshold. Eleven (6.1%) samples were interpreted as mutants by visual inspection but were not detected by the computer algorithm with a 10% threshold, and 20 cases (11.1%) were called positive using the automated interpretation, but were determined to be negative on visual inspection. Thus, the automated interpretation with a 10% threshold yielded both false positive and false negative mutation calls.
PS identified 110 of 180 (61.1%) KRAS mutants and 70 (38.9%) wild-type. Two samples from the same patient yielded a PS pyrogram that was flagged as abnormal, but the pattern could not be definitively interpreted (See below).
Five samples from three patients demonstrated two different mutations within codons 12/13. In one case, both a primary colon cancer and omental metastasis demonstrated mutations at each of the first two positions of codon 12. SS demonstrated G to T transversions at the first two positions of codon 12, indicating either a G12F mutation if the mutations occurred on the same allele, or G12C and G12V mutations if they occurred on different alleles (Figure 2A). MC demonstrated 1 mutant melt peak with a Tm of 55.9°C, which is significantly lower than that expected for a single mutation in either codon 12 or 13 (62 to 64.5°C), suggesting that the mutations were on the same allele (Figure 2B). PS yielded an error message in interpreting the data (Figure 2C). In light of the SS data, the PS data confirms that the two mutations were on the same allele, since the pyrogram would have yielded a different, distinct pattern had the mutations occurred on different alleles (Figure 2D).
Figure 2.
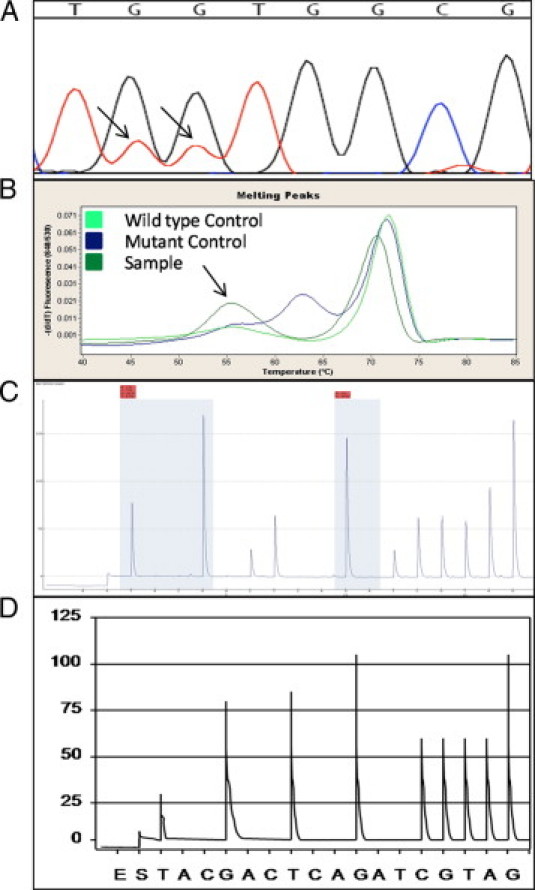
Results of SS, MC, and PS for a G12F mutation. A: SS demonstrated G to T transversions at positions 1 and 2 of codon 12. Arrows indicate the mutation. B: MC showed a Tm significantly lower than expected for a single mutation. Arrows indicate the mutation. C: PS generated a pyrogram that was flagged as abnormal, but the pattern could not be definitively interpreted with regard to the specific mutation subtype. D: A hypothetical PS pyrogram that would have resulted had the G to T transversions at positions 1 and 2 of codon 12 been on separate alleles.
In a second case, two separate blocks from a primary colon adenocarcinoma demonstrated mutations in both codons 12 and 13. For these samples, SS and PS demonstrated G12C and G13D mutations. The MC assay demonstrated a single mutant melt peak with a Tm consistent with a codon 12 or 13 mutation, suggesting that the mutations were likely to occur on separate alleles, either on different alleles in the same cells, or in separate cell populations. Interestingly, although histological review indicated that both samples contained 75 to 80% tumor cells, the PS data demonstrated 26% G12C mutant alleles and 49% G13D mutant alleles in one specimen, and 8% G12C and 46% G13D mutant alleles in the other specimen. These results highlight intratumoral mutant heterogeneity that can exist between different tissue blocks from the same specimen.
The third case demonstrating two KRAS mutations was from a primary colon cancer specimen. In this case, SS and PS demonstrated G12V and G13D mutations, and the MC assay demonstrated a single mutant melt peak with a Tm consistent with a codon 12 or 13 mutation, suggesting that the mutations were likely to occur on separate alleles. Pathology review of this specimen indicated that it contained 75% tumor cells. The PS data demonstrated 5% G12V mutant alleles and 17% G13D mutant alleles.
Two samples demonstrated mutations outside of codons 12/13. SS detected a Q22K mutation in one sample and L19F mutation in the other. Both yielded MC results with mutant peaks with Tms that were significantly different from that expected from a codon 12/13 mutation (68.7°C and 66.96°C respectively). PS was programmed to detect mutations at codons 12 and 13 only, and thus did not identify these mutations. Mutations outside of codons 12 and 13 might be detected on this platform if the instrument were programmed to interrogate additional codons.
To determine the reproducibility of each platform, eight samples were run at least three times on each platform. These samples included five wild-type, and one each of the G12C, G12V, and G13D mutants. Each platform yielded 100% reproducibility for qualitative interpretation of these samples. We also reviewed the quantitative reproducibility of the PS platform. Ten mutant positive samples, four G12C, four G12V, and two G13D, with percent mutant alleles ranging from 5% to 60%, were analyzed at least three times each. The data are summarized in Table 3. In addition, all 20 nontumor samples were run in duplicate. The average percent call at any base other than wild-type was 0.7, with a SD of 0.4 (data not shown).
Table 3.
Reproducibility Study
| Mutation | % Mutant per replicate | Average | SD |
|---|---|---|---|
| G12C | 11, 7, 10, 9, 9, 12 | 10 | 1.8 |
| G12C | 42, 43, 40 | 42 | 1.5 |
| G12C | 19, 17, 18 | 18 | 1.0 |
| G12C | 8, 8, 9, 10 | 9 | 1.0 |
| G12V | 7, 5, 6 | 6 | 1.0 |
| G12V | 59, 60, 59 | 59 | 0.60 |
| G12V | 11, 5, 12, 6, 3, 5 | 7 | 3.6 |
| G12V | 7, 12, 8 | 9 | 2.7 |
| G13D | 46, 46, 48, 47 | 47 | 1.0 |
| G13D | 53, 52, 51 | 52 | 1.0 |
SD, standard deviation.
Limit of detection studies were performed by mixing LoVo cells, which contain a heterozygous G13D mutation, with cells from a wild-type cell line. Short tandem repeat analysis of DNA isolated from the cell mixes indicated that the dilutions were accurate (Table 4). The undiluted mutant sample (100%) should have contained 50% mutant alleles if each cell contained one wild-type and one mutant allele. However, SS, PS, and MC all indicated that greater than 50% mutant alleles were present in the undiluted specimen (Table 4 and Figure 3). In fact, the percent mutant alleles identified by PS were consistent with LoVo cells carrying two mutant and one wild-type allele (Table 4). There are data in the literature indicating that some LoVo cell lines contain a gain of chromosome 12, which would be consistent with three copies of the KRAS gene.13,14 Cytogenetic analysis of our LoVo cell line demonstrated a gain of chromosome 12, which is consistent with this cell line having three copies of KRAS (data not shown). Thus, our predicted mutant alleles (Figure 3) are based on this cell line having one wild-type and two mutant copies of KRAS per cell (column 4 of Table 4).
Table 4.
Limit of Detection Study
| Dilution (% mutant DNA) | Identifiler (% mutant DNA) | Expected % mutated alleles if 1 WT, 1 MT allele per cell | Expected % mutated alleles if 1 WT, 2 MT allele per cell | Average % mutant alleles by PS | SS | MC |
|---|---|---|---|---|---|---|
| 100 | 100 | 50 | 66 | 66 | Positive | Positive |
| 50 | 48 | 25 | 40 | 39 | Positive | Positive |
| 20 | 21 | 10 | 18 | 18 | Positive | Positive |
| 10 | 11 | 5 | 10 | 9 | Not above baseline | Equivocal |
| 5 | 6 | 2.5 | 5 | 5 | Not above baseline | Equivocal |
WT, wild type; MT, mutant; PS, pyrosequencing; SS, Sanger sequencing; MC, melting curve analysis.
Figure 3.
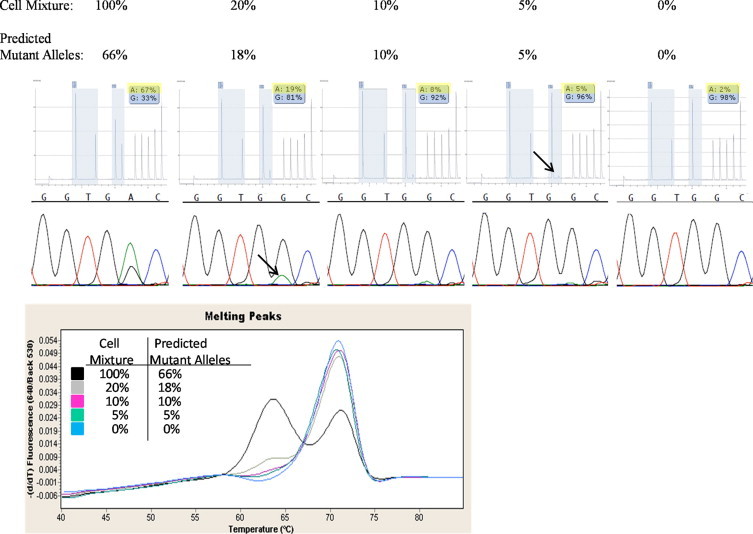
Dilution series data presented here include 100%, 20%, 10%, 5%, and 0% KRAS mutant cell mixtures and the corresponding predicted percentage of mutant alleles in each mixture. The dilutions representing the limit of detection for a KRAS mutation at codons 12/13 by SS, MC, and PS are designated by the arrow.
PS was able to clearly identify a mixture containing 5% mutant alleles. Using these data in conjunction with the reproducibility data of the normal samples (above), we concluded that the limit of detection for PS is 5% mutant alleles. SS was able to clearly identify a mixture containing 18% mutant alleles. Determining the limit of detection for SS can be somewhat subjective. We considered the small mutant peak seen in the cell mixture containing 10% predicted mutant alleles not significantly above baseline variability, and thus, the limit of detection for SS to be 15 to 20% mutant alleles. It is important to note that the limit of detection for SS can be mutation specific, in part due to the difference in fluorescence of the fluorophores. While reviewing the data from our clinical specimens, we noted that SS tended to be less sensitive at detecting mutations at the first position of codon 12 (data not shown). The limit of detection for the MC assay is even more subjective. Interestingly, the mutant peak dropped off significantly from the 100% to the 50% cell mixture. At a mixture of 10% predicted mutant alleles, the melting curve became difficult to distinguish from wild-type, and we considered it equivocal. Thus, we consider the limit of detection for MC to be approximately 10% mutant alleles, with the understanding that some of these would be interpreted as equivocal, but could be confirmed by another method.
Discussion
Here, we have compared three platforms for the detection of KRAS codon 12 and 13 mutations. Each platform has unique strengths and challenges (summarized in Table 5). In our hands, MC had the shortest turnaround time and required the least amount of hands-on tech time. MC analysis is a closed system, which should reduce the risk for contamination with amplicons carried over from previous PCR reactions. MC is a qualitative assay and does not identify the specific mutation present. Currently, there are no significant data indicating that the specific mutation type impacts prognosis or therapeutic choices. Therefore, reporting the specific mutation in KRAS 12/13 is not clinically necessary. However, this may change as more data are accumulated. Confirmatory testing is probably required for samples yielding mutant melt curves with a Tm outside of the expected range for wild-type and codon 12/13 mutations. As shown in this study, these patterns can be generated both by mutations outside of codons 12 and 13, and by double mutations within codons 12 and 13. The subjective nature of peak interpretation presents additional challenges in analyzing data from this platform.
Table 5.
Advantages and Disadvantages of Presented Platforms
| Platform | Advantages | Disadvantages |
|---|---|---|
| PS | Best LOD (5%) | Confirmation by another method occasionally required |
| Provides sequence information | ||
| Relatively fast run-time | ||
| SS | Interrogates the entire sequence | LOD 15−20% |
| Longest run time | ||
| Most labor-intensive | ||
| MC | Closed system | Does not define specific mutation |
| Fastest run-time | Confirmation frequently required | |
| Least labor-intensive |
PS, pyrosequencing; SS, Sanger sequencing; MC, melting curve analysis; LOD, limit of detection.
SS had the longest turnaround time and most hands-on time of the three platforms analyzed. SS identifies specific mutations and can detect mutations outside of codons 12/13. Using an automated interpretation algorithm with a 10% threshold, SS yielded 11.1% false positives and 6.1% false negatives, highlighting the need for manual review of all SS data. The limit of detection for mutation detection by SS is subjective, and may depend on the experience level of the person interpreting the data. In addition, SS's limit of detection can be variable depending on the specific mutation. Our findings did not replicate the 95.5% sensitivity of SS recently described in FFPE colorectal cancer samples.15 One benefit of this platform is its ability to evaluate a relatively long gene sequence for all possible mutations, although the clinical significance of KRAS mutations outside of codons 12 and 13 is unclear.
The most common mutations outside of 12/13 are missense mutations at codon 61, which occur in at least 1% and 2% of colon and lung adenocarcinomas respectively.16,17,18,19 Recent data indicate that these mutations also predict resistance to anti-EGFR therapies, and should therefore be considered for clinical testing.9 For each of the three platforms, detection of codon 61 mutations requires an additional PCR amplification and analysis due to the genomic distance between codons 61 and 12/13. For mutations outside of 12, 13, and 61, the data indicating their relevance to patient management are limited, and therefore, clinical testing is not recommended at this time.
PS detects specific mutations, but primarily at codons that it is programmed to analyze, typically codons 12/13. Our study confirmed a 5% limit of detection for mutant alleles as previously shown.20,21 The limit of detection for different KRAS mutations appears to be more uniform than for SS, due to the fact that the detection at each position is the same (light emission), rather than different fluorophores. PS provided specific mutation data with an improved limit of detection over both SS and MC.
PS and SS characterized three cases with two mutations within KRAS codons 12 and 13, which were identified as mutant by MC, but not as double mutants. The clinical significance of double mutations, if any, is unclear. These cases do, however, highlight the potential for tumor heterogeneity. Heterogeneity within the tumor cells, in conjunction with the fact that all specimens will contain some percentage of nontumor cells, may result in a relatively low percentage of mutated alleles within some specimens. In fact, we found a relatively poor correlation between the percentage of tumor cells in the population as estimated by a pathologist, and the percentage of mutant alleles by pyrosequencing. In our series, there were five samples with the percentage of tumor estimated to be between 40 and 80% by two separate pathologist (20% to 40% predicted mutant alleles), but with <10% KRAS mutant alleles identified by pyrosequencing. Thus, the discrepancy observed between the platforms due to differences in each platform's limit of detection could not solely be attributed to the tumor content of the samples. The clinical significance of low level mutants in relation to prognosis and therapeutic benefit has yet to be fully studied. Although limited data exist on the significance of KRAS mutations within a small subpopulation of a tumor, it is assumed that their identification will be clinically important. Interestingly, the percentage of KRAS mutant was not universally lower, but sometimes greater than that predicted based on the percent tumor in the specimen. We identified two samples in our series that had tumor percentages estimated to be ≤20% (≤10% predicted mutant alleles), but with >45% KRAS mutant alleles identified by pyrosequencing. This may be due to an underestimation of tumor by the pathologists, or individual tumor cells harboring extra copies of mutant KRAS alleles. In these cases, tumor percentage estimates, as determined independently by two pathologists, were concordant.
For research purposes, the platforms evaluated here provide unique and complementary data. For example, to identify mutations outside of codons 12/13, detect low level (<10%) mutant alleles, and to determine whether double mutants reside on a single or multiple alleles, may require the use of all three platforms. When choosing a clinical testing platform, factors to consider include sensitivity (false negatives), specificity (false positives), reproducibility, limit of detection, turnaround time, ease of interpretation, and cost (including instrument, reagent, and tech time). In terms of cost, all three assays require an investment in instrumentation; SS, capillary electrophoresis, PS, pyrosequencer, and MC, real-time PCR instrument. In our experience, the reagent costs were higher, but comparable, for PS and SS, and lowest for MC. Not surprisingly, we found tech time to be the biggest factor for determining assay cost. SS was the most labor intensive, and MC was the least, which in our hands, made SS the most expensive, and MC the least expensive to perform. In our experience, the difference in turnaround time for the three platforms was insignificant when considering the time to process the sample (ie, fix, embedded, cut, review to identify tumor, and extract DNA), and that runs were batched and run twice weekly in our lab. We found PS to have the best limit of detection, which we found important given sample heterogeneity. Although PS can require confirmatory testing in rare instances, we found the interpretation of PS data overall to be the most straightforward.
References
- 1.Ellis RW, Defeo D, Shih TY, Gonda MA, Young HA, Tsuchida N, Lowy DR, Scolnick EM. The p21 src genes of Harvey and Kirsten sarcoma viruses originate from divergent members of a family of normal vertebrate genes. Nature. 1981;292:506–511. doi: 10.1038/292506a0. [DOI] [PubMed] [Google Scholar]
- 2.Bos JL, Fearon ER, Hamilton SR, Verlaan-de Vries M, van Boom JH, van der Eb AJ, Vogelstein B. Prevalence of ras gene mutations in human colorectal cancers. Nature. 1987;327:293–297. doi: 10.1038/327293a0. [DOI] [PubMed] [Google Scholar]
- 3.Minamoto T, Mai M, Ronai Z. K-ras mutation: early detection in molecular diagnosis and risk assessment of colorectal, pancreas, and lung cancers–a review. Cancer Detect Prev. 2000;24:1–12. [PubMed] [Google Scholar]
- 4.Der CJ, Cooper GM. Altered gene products are associated with activation of cellular rasK genes in human lung and colon carcinomas. Cell. 1983;32:201–208. doi: 10.1016/0092-8674(83)90510-x. [DOI] [PubMed] [Google Scholar]
- 5.Sugio K, Kishimoto Y, Virmani AK, Hung JY, Gazdar AF. K-ras mutations are a relatively late event in the pathogenesis of lung carcinomas. Cancer Res. 1994;54:5811–5815. [PubMed] [Google Scholar]
- 6.Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AM, Bos JL. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;319:525–532. doi: 10.1056/NEJM198809013190901. [DOI] [PubMed] [Google Scholar]
- 7.Linardou H, Dahabreh IJ, Kanaloupiti D, Siannis F, Bafaloukos D, Kosmidis P, Papadimitriou CA, Murray S. Assessment of somatic k-RAS mutations as a mechanism associated with resistance to EGFR-targeted agents: a systematic review and meta-analysis of studies in advanced non-small-cell lung cancer and metastatic colorectal cancer. Lancet Oncol. 2008;9:962–972. doi: 10.1016/S1470-2045(08)70206-7. [DOI] [PubMed] [Google Scholar]
- 8.Massarelli E, Varella-Garcia M, Tang X, Xavier AC, Ozburn NC, Liu DD, Bekele BN, Herbst RS, Wistuba II. KRAS mutation is an important predictor of resistance to therapy with epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. Clin Cancer Res. 2007;13:2890–2896. doi: 10.1158/1078-0432.CCR-06-3043. [DOI] [PubMed] [Google Scholar]
- 9.Loupakis F, Ruzzo A, Cremolini C, Vincenzi B, Salvatore L, Santini D, Masi G, Stasi I, Canestrari E, Rulli E, Floriani I, Bencardino K, Galluccio N, Catalano V, Tonini G, Magnani M, Fontanini G, Basolo F, Falcone A, Graziano F. KRAS codon 61, 146 and BRAF mutations predict resistance to cetuximab plus irinotecan in KRAS codon 12 and 13 wild-type metastatic colorectal cancer. Br J Cancer. 2009;101:715–721. doi: 10.1038/sj.bjc.6605177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karapetis CS, Khambata-Ford S, Jonker DJ, O'Callaghan CJ, Tu D, Tebbutt NC, Simes RJ, Chalchal H, Shapiro JD, Robitaille S, Price TJ, Shepherd L, Au HJ, Langer C, Moore MJ, Zalcberg JR. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359:1757–1765. doi: 10.1056/NEJMoa0804385. [DOI] [PubMed] [Google Scholar]
- 11.Andreyev HJ, Norman AR, Cunningham D, Oates J, Dix BR, Iacopetta BJ, Young J, Walsh T, Ward R, Hawkins N, Beranek M, Jandik P, Benamouzig R, Jullian E, Laurent-Puig P, Olschwang S, Muller O, Hoffmann I, Rabes HM, Zietz C, Troungos C, Valavanis C, Yuen ST, Ho JW, Croke CT, O'Donoghue DP, Giaretti W, Rapallo A, Russo A, Bazan V, Tanaka M, Omura K, Azuma T, Ohkusa T, Fujimori T, Ono Y, Pauly M, Faber C, Glaesener R, de Goeij AF, Arends JW, Andersen SN, Lövig T, Breivik J, Gaudernack G, Clausen OP, De Angelis PD, Meling GI, Rognum TO, Smith R, Goh HS, Font A, Rosell R, Sun XF, Zhang H, Benhattar J, Losi L, Lee JQ, Wang ST, Clarke PA, Bell S, Quirke P, Bubb VJ, Piris J, Cruickshank NR, Morton D, Fox JC, Al-Mulla F, Lees N, Hall CN, Snary D, Wilkinson K, Dillon D, Costa J, Pricolo VE, Finkelstein SD, Thebo JS, Senagore AJ, Halter SA, Wadler S, Malik S, Krtolica K, Urosevic N. Kirsten ras mutations in patients with colorectal cancer: the ‘RASCAL II’ study. Br J Cancer. 2001;85:692–696. doi: 10.1054/bjoc.2001.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keohavong P, DeMichele MA, Melacrinos AC, Landreneau RJ, Weyant RJ, Siegfried JM. Detection of K-ras mutations in lung carcinomas: relationship to prognosis. Clin Cancer Res. 1996;2:411–418. [PubMed] [Google Scholar]
- 13.Soulie P, Poupon MF, Remvikos Y, Dutrillaux B, Muleris M. Distinct chromosomal alterations associated with TP53 status of LoVo cells under PALA selective pressure: a parallel with cytogenetic pathways of colorectal cancers. Oncogene. 1999;18:775–781. doi: 10.1038/sj.onc.1202336. [DOI] [PubMed] [Google Scholar]
- 14.Kleivi K, Teixeira MR, Eknaes M, Diep CB, Jakobsen KS, Hamelin R, Lothe RA. Genome signatures of colon carcinoma cell lines. Cancer Genet Cytogenet. 2004;155:119–131. doi: 10.1016/j.cancergencyto.2004.03.014. [DOI] [PubMed] [Google Scholar]
- 15.Tol J, Dijkstra JR, Vink-Börger ME, Nagtegaal ID, Punt CJ, van Krieken JH, Ligtenberg MJ. High sensitivity of both sequencing and real-time PCR analysis of KRAS mutations in colorectal cancer tissue. J Cell Mol Med. 2009 doi: 10.1111/j.1582-4934.2009.00788.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosell R, Monzó M, Pifarré A, Ariza A, Sánchez JJ, Moreno I, Maurel J, López MP, Abad A, de Anta JM. Molecular staging of non-small cell lung cancer according to K-ras genotypes. Clin Cancer Res. 1996;2:1083–1086. [PubMed] [Google Scholar]
- 17.Rosell R, Li S, Skacel Z, Mate JL, Maestre J, Canela M, Tolosa E, Armengol P, Barnadas A, Ariza A. Prognostic impact of mutated K-ras gene in surgically resected non-small cell lung cancer patients. Oncogene. 1993;8:2407–2412. [PubMed] [Google Scholar]
- 18.Winder T, Mündlein A, Rhomberg S, Dirschmid K, Hartmann BL, Knauer M, Drexel H, Wenzl E, De Vries A, Lang A. Different types of K-Ras mutations are conversely associated with overall survival in patients with colorectal cancer. Oncol Rep. 2009;21:1283–1287. doi: 10.3892/or_00000352. [DOI] [PubMed] [Google Scholar]
- 19.Oliveira C, Westra JL, Arango D, Ollikainen M, Domingo E, Ferreira A, Velho S, Niessen R, Lagerstedt K, Alhopuro P, Laiho P, Veiga I, Teixeira MR, Ligtenberg M, Kleibeuker JH, Sijmons RH, Plukker JT, Imai K, Lage P, Hamelin R, Albuquerque C, Schwartz S, Jr, Lindblom A, Peltomaki P, Yamamoto H, Aaltonen LA, Seruca R, Hofstra RM. Distinct patterns of KRAS mutations in colorectal carcinomas according to germline mismatch repair defects and hMLH1 methylation status. Hum Mol Genet. 2004;13:2303–2311. doi: 10.1093/hmg/ddh238. [DOI] [PubMed] [Google Scholar]
- 20.Ogino S, Kawasaki T, Brahmandam M, Yan L, Cantor M, Namgyal C, Mino-Kenudson M, Lauwers GY, Loda M, Fuchs CS. Sensitive sequencing method for KRAS mutation detection by Pyrosequencing. J Mol Diagn. 2005;7:413–421. doi: 10.1016/S1525-1578(10)60571-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dufort S, Richard MJ, de Fraipont F. Pyrosequencing method to detect KRAS mutation in formalin-fixed and paraffin-embedded tumor tissues. Anal Biochem. 2009;391:166–168. doi: 10.1016/j.ab.2009.05.027. [DOI] [PubMed] [Google Scholar]