

Abstract
Hemagglutination-based assays have several clinical shortcomings. To overcome this difficulty, we have developed a multiplex-PCR SNaPshot assay adapted to the Southern French population, which includes individuals from sub-Saharan Africa and the Comoros archipelago. Single nucleotide polymorphisms (SNPs) associated with clinically relevant blood antigens as well as with null phenotypes were profiled (i.e., K/k, Fya/Fyb/Fybw/Fynull, S/s/U–/U+var, Jka/Jkb, Doa/Dob, Yta/Ytb, and Coa/Cob). A single multiplex-PCR reaction was used to amplify nine gene regions encompassing 11 SNPs. Identification was obtained by incorporation of the complementary dye-conjugated single base at the 3′ end of each probe primer annealed proximal to the target SNP. After optimization, the SNaPshot assay was validated with 265 known allele or phenotype pairs. Results were found fully concordant with those of hemagglutination, allele-specific PCR, and/or sequencing. The assay was then evaluated on 227 blood samples in a clinical context. A total of 203 derived-phenotypes were generated, including 82 atypical phenotypes [i.e., Fy(b+w) (n = 32); K+ (n = 22); Co(b+) (n = 8); Yt(b+) (n = 18); S–s+U+var (n = 2), 105 null phenotypes, i.e., Fy(a–b–) (n = 97); S–s–U– (n = 6); S–s–U+var (n = 2)] and sixteen Fy-positive samples carried a FY*Fy allele. The findings show that this assay can provide a low-cost and fast genotyping tool well adapted to local ethnically mixed populations.
Hemagglutination is the traditional method for testing donor and patient blood group antigens and irregular antibodies. Although hemagglutination is a highly sensitive and specific tool that is inexpensive and easy to perform, it presents several clinical shortcomings that could benefit from newer technology.1 In this regard molecular analysis of genomic DNA now permits prediction of blood group phenotypes based on identification of single nucleotide polymorphisms (SNPs).2,3 This approach has great potential for resolving problems beyond the reach of conventional immunohematologic techniques (e.g., determination of blood group in patients who have undergone massive transfusion or have red cells covered with immunoglobulins and identification of fetal RhD status in pregnancies involving a risk for hemolytic disease of the new-born).4,5 Molecular analysis can also be useful for diagnosis in situations involving weakly reactive antibodies, weak or altered antigen expression, and genetic variability between populations requiring use of rare antibodies.
Determination of blood group antigens other than those of the ABO and RH systems depends mainly on the presence of one or more SNPs in the coding sequence of the relevant blood group gene. As a result blood group alleles can be predicted using DNA base assays such as allele-specific polymerase chain reaction (AS-PCR) and polymerase chain reaction restriction fragment length polymorphism (PCR-RFLP). However, these assays cannot be used routinely because throughput is too low. In the last few years, several large-scale genotyping assays (e.g., the BeadChip,6 Bloodchip,7 GenomeLab SNPstream,8,9 and other DNA microarray-based platforms) have been developed for identification of blood group SNPs.10,11,12 Because these assays are suitable for large-scale processing, they hold forth the possibility of routine SNP blood screening in hematological laboratories. The main obstacle to high-throughput genotyping platforms based on these technologies is that the necessary investment exceeds the resources and activity of most laboratories that require genetic support for a limited number of patients with unusual antibody combinations and/or phenotypes.
To overcome this limitation, we have developed and evaluated a rapid, sensitive, and low-cost three-step multiplex assay. The first step consists of a multiplex-PCR reaction to generate amplicons encompassing the target SNPs. The second step consists of a multiplex-PCR single-base extension assay of probe primers using the commercial (CE) SNaPshot Kit (Applied Biosystems, Foster City, CA).13 In this step, DNA polymerase incorporates the complementary dye-conjugated dideoxy nucleotide base at the 3′ end of each probe primer annealed proximal to the target SNP. In a third step, capillary electrophoresis is performed to determine the size of extended probe primers and fluorescence dye types.
The SNaPshot method has already been used for typing Y chromosome and mitochondrial SNPs in population analysis14,15 and for identifying mutations commonly associated in human gene expression and pathologies.16,17 In 2004, a Japanese team reported development of a 39-multiplex primer extension assay including 15 blood group loci.18 Trial data showed it to be a highly discriminating method allowing detection of SNP types even from short stretches of DNA, like in degraded DNA specimens. In July of the same year, the same team reported the simultaneous detection of six SNP sites in the ABO gene.19 More recently Chaudhuri's group at the New York Blood Center reported detection of 17 blood group SNPs using three independent multiplex SNaPshot reactions.20
The single PCR-multiplex SNaPshot reaction described herein was optimized to identify red cell SNPs determining well characterized clinically relevant blood group phenotypes (K/k, Fya/Fyb, Fybw, Fynull, S/s, U–, U+var, Jka/Jkb, Doa/Dob, Yta/Ytb and Coa/Cob).21 Selection of these phenotypes was based on i) the genetic variability of blood groups in different population groups living in the south of France that includes individuals from sub-Saharan Africa and the Comoros archipelago,22 and ii) the absence of specific antibodies directed against antigens of DO, YT, and CO systems and the description of hemolytic transfusion reactions caused by antibodies to these antigens.23 The goal was to develop a simple clinically useful genotyping tool for simultaneous detection of 11 SNPs defining 18 blood group alleles adapted to the local population. After optimization this genotyping assay was validated against data from serological, allele-specific, and sequencing investigations and tested clinically for one year.
Materials and Methods
Blood Samples
Ethylenediaminetetraacetate-anticoagulated (EDTA) peripheral blood samples were collected with informed consent by the Etablissement Français du Sang Alpes Méditerranée (Marseille, France). Detection of KEL2 (k, Cellano), FY1(Fya), FY2(Fyb), JK1(Jka), JK2(Jkb), MNS3(S), and MNS4(s) antigens was performed by tube tests or column agglutination (Scangel Biorad, France) using two different commercial sera for each antigen. Typing of KEL1(K) antigen was performed using Olympus microplate technology according to manufacturer's instructions (Diagast, France). In clinical context, patients (n = 227) including 50% from sub-Saharan Africa and Comoros archipelago were genotyped.
Genomic DNA Extraction
Genomic DNA was extracted from 200 μl of total blood sample using the QIAmp Blood DNA Mini kit (Qiagen, Courtaboeuf, France) according to the manufacturer instructions and quantified by optical density absorption measurement.
Blood Group Genotyping by Singleplex PCR
Allele-specific PCR genotyping was performed to identify SNPs determining the following alleles: CO*1, CO*2, KEL*1, KEL*2, YT*1, YT*2, JK*1, JK*2, FY*1, FY*2, FY*Fy, FY*X, DO*1, DO*2, MNS*3, and MNS*4 using previously described primer sets and methods.24,25,26,27,28,29,30
PCR and Probe Primer Design
All primers were designed using the Primer 3 program (http://frodo.wi.mit.edu/primer3, version: 0.4.0, 2000). Integrated DNA Technologies program (http://www.idtdna.com/analyzer/applications/oligoanalyzer, version: 3.0, 2008) was used to test for potential primer-dimer conflicts and second hairpin structures. Forward and reverse primers used in the nine-pair multiplex reaction for blood group gene amplifications are shown in Table 1. Primers used as probes were designed to anneal immediately adjacent to the SNP site on either the sense or anti-sense DNA strand (for JK838G>A and FY-33T>C). Their lengths differ from at least five bases by addition to the 5′ end of a polyT tail to distinguish them by capillary electrophoresis (Table 2). All probe Primers were synthesized by Eurogentec (Seraing Belgium) and purified by HPLC to remove incomplete synthesis products.
Table 1.
Primer Sequences Used for Blood Group Amplification in the 9-Pair Multiplex PCR
| System | GenBank accession number | Allele | Forward | Reverse | Amplicon size (bp) | Conc. (μM) |
|---|---|---|---|---|---|---|
| CO | NM_198098 | CO*1/CO*2 | 5′-gCTCTCAGAGGGAATTGAGC-3′ | 5′-GACACCTTCACGTTGTCCTG-3′† | 214 | 0.06 |
| KEL | NM_000420 | KEL*1/KEL*2 | 5′-gagggagatggagatggaaa-3′ | 5′-GACTGGTGTGTGTGGAGAGG-3′ | 350 | 0.12 |
| YT | NM_015831 | YT*1/YT*2 | 5′-ACTGGTGGGAATGACACAGA-3′ | 5′-ggagggatgcagagaaagag-3′ | 398 | 3.80 |
| JK | NM_015865 | JK*1/JK*2 | 5′-ctttcaatcccaccctcagt-3′ | 5′-ctcaataggctcctgccttc-3′ | 435 | 0.24 |
| FY | NM_002036 | FY*Fy | 5′-ATGGCCCTCATTAGTCCTTG-3′ | 5′-CATGGCACCGTTTGGTTCAG-3′† | 97 | 2.00 |
| FY*1/FY*2/FY*X | 5′-TCCCCCTCAACTGAGAACTC-3′† | 5′-AAGGCTGAGCCATACCAGAC-3′† | 392 | 0.23 | ||
| DO | NM_021071 | DO*1/DO*2 | 5′-GACAGCATCATGGAGAATGG-3′ | 5′-TGTGCTCAGGTTCCCAGTTG-3′ | 315 | 0.80 |
| MNS | NM_002100 | MNS*3/MNS*4 | 5′-cctccagaaaagaaaaacgt-3′ | 5′-ACAGTGAAACGATGGACAAGTT-3′ | 986 | 2.60 |
| GYPBS−230/GYPBS-Int5 | 5′-ctgtcttatttttctattgctatg-3′ | 5′-ctgtttctcttctgagtttaactg-3′ | 260 | 0.70 |
Multiplex PCR primers were selected according to following three criteria: (1) similar Tm value (60°C), (2) calculated ΔG between 0 and −10 kcal per mol, and (3) PCR products length less than 1000 bp and easily differentiable on agarose gel electrophoresis. Seven fragments encompassed one target SNP and two fragments encompassed two SNPs. Coamplification in the same fragment were performed to detect 230C>T and IVS5 + 5g>t changes in the GYPBS gene and 125G>A and 265C>T changes in the FY gene. Lower case letters refer to intron sequences.
Sequence primers already published by Reid team.6
Table 2.
Probe Primer Sequences Used for Multiplex Detection of 11 Blood Group SNPs
| System | Allele | Polymorphism | Position change | Location | Forward/ reverse | Extension primer | Size (b) | Conc. (μM) |
|---|---|---|---|---|---|---|---|---|
| FY | FY*1/FY*2 | Gly42Asp | 125G>A | Exon-2 | Forward | 5′-TGAATGATTCCTTCCCAGATGGAGACTATG-3′ | 30 | 0.04 |
| FY | C-wt/FY*X | Arg89Cys | 265C>T | Exon-2 | Forward | 5′-13T-GCTTTTCAGACCTCTCTTC-3′ | 32 | 1.20 |
| DO | DO*1/DO*2 | Asn265Asp | 793A>G | Exon-2 | Forward | 5′-TTGTTTAAAGTTATAAATATGAGCTACCACCCAAGAGGA-3′ | 39 | 0.25 |
| MNS | MNS*3/MNS*4 | Met29Thr | 143T>C | Exon-4 | Forward | 5′-cttatttggacttacattgaaattttgctttatagGAGAAA-3′ | 41 | 2.30 |
| JK | JK*1/JK*2 | Asp280Asn | 838G>A | Exon-9 | Reverse | 5′-16T-TGTTGAAACCCCAGAGTCCAAAGTAGATGT-3′ | 46 | 0.35 |
| CO | CO*1/CO*2 | Ala45Val | 134C>T | Exon-1 | Forward | 5′-20T-TTCAAATACCCGGTGGGGAACAACCAGACGG-3′ | 51 | 0.28 |
| MNS | g-wt/GYPBS-Int5 | IVS5, +5 | +5g>t | Intron-5 | Forward | 5′-24T-TTACASTATTCGCCGACTGATAAAGgtga-3′ | 53 | 0.016 |
| KEL | KEL*1/KEL*2 | Met193Thr | 698T>C | Exon-6 | Forward | 5′-24T-TGGTAAATGGACTTCCTTAAACTTTAACCGAA-3′ | 56 | 0.3 |
| YT | YT*1/YT*2 | His353Asn | 1057C>A | Exon-2 | Forward | 5′-40T-CTCATCAACGCGGGAGACTTC-3′ | 61 | 1.60 |
| MNS | C-wt/GYPBS−230 | Thr58Met | 230C>T | Exon-5 | Forward | 5′-43T-TGATGGCTGGTATTATTGGAA-3′ | 64 | 2.30 |
| FY | FY*wt/FY*Fy | Gata wt/mt | -33T>C | Promoter | Reverse | 5′-48T-AGCGCCTGTGCTTCCAAG-3′ | 66 | 0.02 |
Lower case letters refer to intron sequences. Bold characters refer to the 5′ poly-thymidine tail.
PCR Multiplex Amplification and Probe Primer Extension SNaPshot Reaction
The first step consisted of multiplex PCR using primers flanking each blood group SNP site that was developed to coamplify nine genomic fragments ranging from 97 to 986 bp (Figure 1). Multiplex PCR amplification was performed from 250 ng of genomic DNA in a final volume of 25 μl containing 1.8× PCR buffer, 3.6 mmol/L MgCl2, 0.2 mmol/L of each dNTP, 0.1 unit of TaqDNA-polymerase (Invitrogen, Cergy Pontoise, France), and a defined concentration of each primer (for final concentration see Table 1). Amplification was performed under stringent conditions (i.e., 95°C for 5 minutes for 1 cycle; 95°C for 30 seconds, 60°C for 45 seconds, 72°C for 120 seconds for 40 cycles; 72°C for 7 minutes for 1 cycle). Multiplex PCR amplicons were controlled on 3% (w/v) agarose gel before treatment with Exo/SAP master mix containing 10 U/μl of Exonuclease-I and 0.5 U/μl of Shrimp Alkaline Phosphatase (Euromedex, Souffelweyersheim, France) to remove unincorporated primers and dNTPs. PCR product (10 μl) was incubated with 3.3 μl of Exo/SAP master mixture for 1 hour at 37°C followed by 15 minutes at 80°C for enzyme inactivation.
Figure 1.
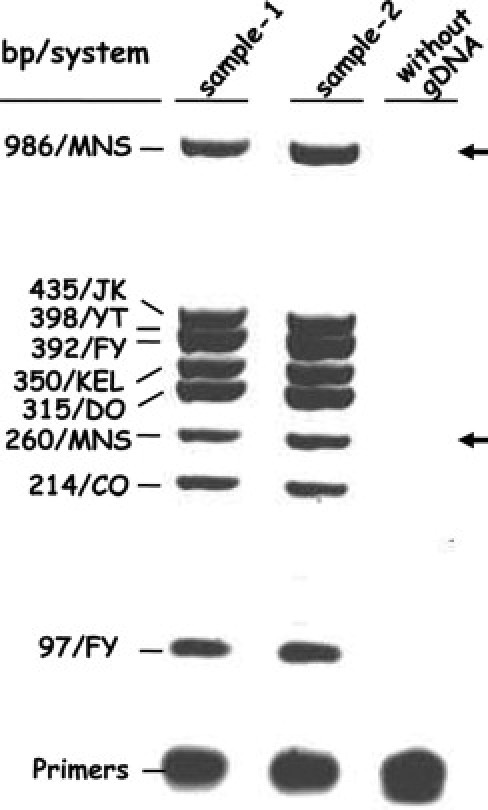
Multiplex PCR amplification of nine blood gene fragments. A 3% agarose gel of the PCR products from two common blood samples (lanes 1 and 2). Eight amplicon bands were observed on the agarose gel because YT and FY bands have similar molecular weights (398 and 392, respectively). Lane 3 shows control PCR without gDNA. Arrows indicate the two missing fragments (260 and 986 bp) when GYPB gene deletion occurred in blood sample phenotyped S–s–U–.
The second step consisted of a multiplex single-base primer extension reaction using the SNaPshot kit (Applied Biosystems, Courtaboeuf, France) according to the manufacturer's protocol. The final reaction volume was 10 μl containing 3 μl of the treated first-step PCR reaction, 5 μl of SNaPshot ready reaction premix containing fluorescent dideoxy nucleotides (A = dR6G, green; C = dTAMRA, black; G = dR110, blue; T = dROX, red), and probe primers (for final concentration see Table 2). The reaction was performed under stringent conditions (95°C for 10 seconds, 50°C for 10 seconds, 60°C for 30 seconds for 25 cycles). An aliquot of SNaPshot extension reaction (10 μl) was then treated with 1 unit of SAP for 1 hour at 37°C followed by 15 minutes at 80°C for enzyme inactivation, before the third step of capillary electrophoresis.
Capillary Electrophoresis Conditions
The fluorescence and size of extended products were determined by capillary electrophoresis on an ABI PRISM 3130 genetic analyser (Applied Biosystem) using POP-7 polymer and 36-cm length capillary. Before loading onto the genetic analyzer, an aliquot of treated SNaPshot multiplex extension reaction (0.5 μl) was mixed with 9.25 μl of deionized formamide (Applied Biosystems) and 0.25 μl of size standard (GeneScan-120 LIZ ladder, Applied Biosystem). Injection was performed during 16 seconds at 1.2 kV and electrophoresis for 20 minutes at 15 kV and 5 μA at 60°C. The laser was set at a constant power of 9.9 mW. Data were analyzed using GeneMapper v4.0 with specific detection parameters.
Sequence Analysis
Whenever discordance was found, PCR products were sequenced on both strands by BigDye Terminator v1.1 Sequencing Kit (Applied Biosystems) and analyzed on an automated fluorescence-based ABI PRISM 3130 Genetic analyser.
Results
The Multiplex-Assay
An assay based on a multiplex PCR coupled with a single base extension assay using genomic DNA was designed for simultaneous detection of 11 SNPs defining 18 blood group alleles. These alleles belong to 7 common clinically relevant blood group systems (i.e., Kell, Duffy, MNS, Kidd, Dombrock, Cartwright, and Colton). This assay included primer extension reactions for the 230C>T and IVS5, +5g>t nucleotide changes reported to silence GYPBS alleles namely respectively GYPBS−230 and GYPBS-Int5,31 for the FY-33T>C change known to silence the FY*2 allele namely FY*Fy allele and for the FY265C>T change corresponding to the FY*X allele.32,33,34
Optimization
First, the specificity of each amplicon was evaluated in singleplex reaction and in multiplex reactions, the PCR conditions, DNA template, and primer concentrations were adjusted to obtain amplicon bands of same intensity (Figure 1). Then, the results of the entire assay including the multiplex-PCR, primer extension, electrophoresis, and fluorescent typing steps were evaluated, and the volume of the first multiplex-PCR used as well as the concentrations of probe primers were balanced so that peak height was at least 700 but not greater than 4000 relative fluorescence units (rfus). Probe primer concentrations for the balanced 11-multiplex detection are shown in Table 2. Because the mobility of the extension product and the signal intensity depend on the fluorescent dye labeled dideoxy nucleotide added, concomitant tests using samples exhibiting various known allele combinations were performed. Four different fluorescent dye combinations [i.e., black-red (6xC-T), blue-green (3xG-A), blue-red (1xG-T), and green-black (1xA-C)] were used to take into account the alleles present at the 11 SNPs and the reverse orientation of extension primers for JK838G>A and FY-33T>C. The signals from the different fluorescent nucleotide added were roughly balanced except for A–C SNP because of the difference in yield of the two dyes (1/3.5). Finally, the length of some extension primers was adjusted via thymidine number of the tail to obtain an electrophoretic peak at similar intervals to each locus.
Figure 2, A–G shows the typical GeneMapper electropherograms obtained by a series of 7 multiplex reactions after capillary electrophoresis in a POP7 polymer system, exhibiting allele variation for each polymorphic site tested in the multiplex SNaPshot assay. Alleles were identified according to the fluorescent dideoxy nucleotide added to the 3′-end of probe primer and the distance of migration. A single peak representing a single fluorescent dideoxy nucleotide added was observed in a homozygous position, whereas two peaks with somewhat different mobilities representing two different fluorescent dideoxy nucleotides added were generated in a heterozygous position. Homozygous and heterozygous signals were observed for all blood group SNPs tested except the homozygote genotypes FY*X/*X, CO*2/*2, KEL*1/*1, and YT*2/*2, which were not sufficiently available or prevalent.
Figure 2.
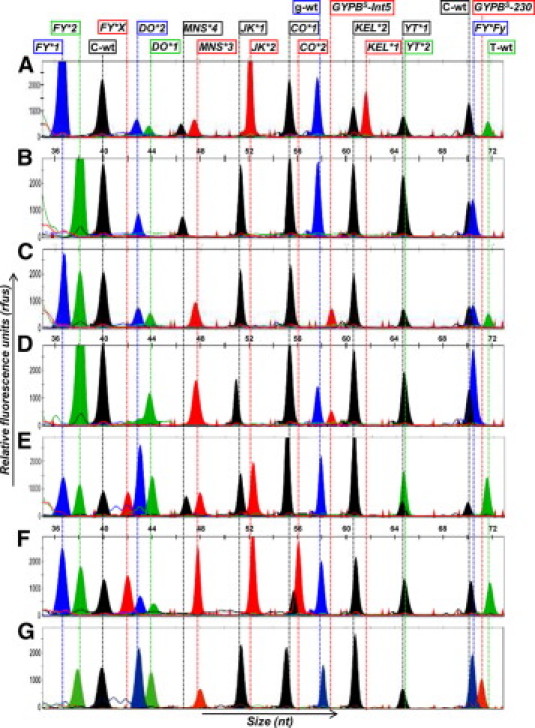
GeneMapper electropherograms of SNaPshot reactions. Plots of size (nt) versus relative fluorescence units (rfus) for 7 DNA samples exhibiting variations at the 11 SNP sites analyzed. The electropherograms correspond to extended genotype following: A:FY*1/*1, DO*1/*2, MNS*3/*4, JK*2/*2, CO*1/*1, KEL*1/*2, YT*1/*1; B:FY*Fy/*Fy, DO*2/*2, MNS*4/*4, JK*1/*1, CO*1/*1, KEL*2/*2, YT*1/*1; C:FY*1/*Fy, DO*1/*2, GYPBS-Int5/GYPBS-Int5, JK*1/*1, CO*1/*1, KEL*2/*2, YT*1/*1; D:FY*Fy/*Fy, DO*1/*1, MNS*3/GYPBS-Int5, JK*1/*1, CO*1/*1, KEL*2/*2, YT*1/*1; E:FY*1/*X, DO*1/*2, MNS*3/*4, JK*1/*2, CO*1/*1, KEL*2/*2, YT*1/*2; F:FY*1/*X, DO*1/*2, MNS*3/*3, JK*2/*2, CO*1/*2, KEL*2/*2, YT*1/*1. G shows the electropherogram for a rare patient in whom homozygous T230 and T243 for GYPB gene was identified, suggesting a GYPBS−230 allele either in double or single dose in trans with the GYPB gene deletion. Its extended genotype was as follows: FY*Fy/*Fy, DO*1/*2, GYPBS−230/GYPBS−230, JK*1/*1, CO*1/*1, KEL*2/*2, YT*1/*1. All plots were collected using POP-7 polymer under the described conditions. The top line indicates positions and blood allele names.
For most antithetical blood group markers, genotype-to-phenotype determination was straightforward based on electropherogram reading of one SNP. Conversely, more than one SNP was required to achieve genotype-to-phenotype determination for the Duffy and MNS systems. This raises the possibility that the FY*2 allele was either silent because of the −33T>C change in the GATA box corresponding to FY*Fy with a Fy(b−) phenotype or weakened because of C>T change at position 265 for a FY*X allele corresponding to a Fyx [Fy(b+weak)] phenotype.32,33,34 For the prediction of the S, s and U (+/−, +var) antigen status by GYPB gene analysis, it is necessary to take the two silencing nucleotide changes IV5, +5g>t and 230C>T into account in addition to the T>C nucleotide change at position 143 driving the MNS*3 and MNS*4 alleles.31
Validation
DNA blood samples partially phenotyped by hemagglutination and/or genotyped by allele-specific PCR reaction were used as controls to test the accuracy and reproducibility of our multiplex SNaPshot assay (Table 3). Results were found to be 100% in concordance with 63 hemagglutination phenotypings for KEL, JK, FY, and MNS. However, comparison of primer extension results with those of 202 allele pairs determined by allele specific PCR reactions showed five discordances (2.47%) involving the MNS and DO systems. Analysis of these discordances by sequencing reactions confirmed the accuracy of the multiplex SNaPshot assay. These discordances most probably result from visual misinterpretations on agarose electrophoresis. Thus, the multiplex SNaPshot assay was fully validated by results of allele specific PCR reaction, serology, and sequencing.
Table 3.
Comparison of Results Obtained by Primer Extension, Allele-Specific PCR, and Serology
| System | Genotypes | Primer extension results | Serology/primer extension | Allele specific/primer extension | Sequencing/primer extension |
|---|---|---|---|---|---|
| CO | CO*1/*1 | 27 | 27/27 | ||
| CO*1/*2 | 1 | 1/1 | |||
| CO*2/*2 | |||||
| KEL | KEL*1/*1 | 1 | 1/1 | ||
| KEL*1/*2 | 2 | 1/1 | 2/2 | ||
| KEL*2/*2 | 30 | 26/26 | 11/11 | ||
| YT | YT*1/*1 | 26 | 26/26 | ||
| YT*1/*2 | 3 | 3/3 | |||
| YT*2/*2 | 1 | 1/1 | |||
| JK | JK*1/*1 | 6 | 4/4 | 6/6 | |
| JK*1/*2 | 17 | 7/7 | 17/17 | ||
| JK*2/*2 | 8 | 2/2 | 8/8 | ||
| FY | FY*1/*1 | 4 | 3/3 | 2/2 | |
| FY*1/*2 | 10 | 4/4 | 6/6 | ||
| FY*2/*2 | 12 | 4/4 | 11/11 | ||
| FY*2/*Fy | 6 | 1/1 | 6/6 | ||
| FY*1/*Fy | 1 | 1/1 | |||
| FY*Fy/*Fy | 5 | 5/5 | |||
| FY*1/*X | 4 | 4/4 | |||
| DO | DO*1/*1 | 4 | 4/4 | ||
| DO*1/*2 | 19 | 16/17 | 1/1 | ||
| DO*2/*2 | 9 | 9/9 | |||
| MNS | MNS*3/*3# | 12 | 4/4 | 11/12 | 1/1 |
| MNS*3/*4 | 15 | 5/5 | 12/14 | 2/2 | |
| MNS*4/*4# | 5 | 2/2 | 4/5 | 1/1 | |
| GYPBS-Int5/MNS*4 | 2 | 2/2 | |||
| GYPBS-Int5/ GYPBS-Int5# | 1 | 1/1 | |||
| GYPBS−230/GYPSB-230# | |||||
| Total | 231 | 63/63 | 197/202 | 5/5 |
Primer extension reaction results are compared with serological and allele-specific PCR results when available; whenever a discordance appeared, a sequencing reaction was performed. # or single dose in trans with the GYPB gene deletion cannot be excluded.
One Year of Clinical Use
Since validation the multiplex SNaPshot assay has been evaluated for one year in the immunohematology laboratory of Marseille, France. Of the 406 patient blood samples addressed to the laboratory for molecular analysis, the multiplex tool has been used on 227 including 50% of patients from sub-Saharan African and Comore archipelago (Table 4). A total of 2497 SNP typings and 1589 DNA-derived phenotypes have been generated. Multiplex SNaPshot results were fully concordant with those partially provided by hemagglutination and/or sequencing. A number of atypical and null DNA-derived phenotypes have been identified including 32 samples genotyped FY*1/*X (n = 30) or FY*Fy/*X (n = 2) with, respectively, Fy(a+b+w) and Fy(a–b+w) derived phenotypes, 97 samples homozygous for the FY*Fy allele with Fy(a–b–) phenotype, and 22 samples genotyped KEL*1/*1 (n = 3) or KEL*1/*2 (n = 19) with a K-positive-derived phenotype.
Table 4.
Rare and Null Blood Samples Genotyped
| One year | |
|---|---|
| Number of blood samples | |
| Addressed | 406 |
| Analyzed by Multiplex SNaPshot (%) | 227 (54) |
| Total number of SNPs tested | 2.497 |
| Genotype | (n) | Derived phenotype |
|---|---|---|
| Atypical or null blood types | ||
| FY*1/*X | 30 | Fy(a+b+w) |
| FY*Fy/*X | 2 | Fy(a–b+w) |
| FY*Fy/*Fy | 97 | Fy(a–b–) |
| FY*1/*Fy or | 16 | Fy(a+b–) or |
| FY*2/*Fy | Fy(a–b+) | |
| GYPB deleted | 6 | S–s–U– |
| GYPBS-Int5/GYPBS-Int5# | 1 | S–s–U+var |
| GYPBS-Int5/MNS*4 | 2 | S–s+ U+var |
| GYPBS−230/GYPBS−230# | 1 | S–s–U+var |
| KEL*1/*1 | 3 | K+k– |
| KEL*1/*2 | 19 | K+k+ |
| CO*1/*2 | 8 | Co(a+b+) |
| YT*1/*2 | 15 | Yt(a+b+) |
| YT*2/*2 | 3 | Yt(a–b+) |
| Total | 203 |
# or single dose in trans with the GYPB gene deletion cannot be excluded.
In the MNS system, GYPB gene deletion was identified in six individuals of African descent who exhibited the S–s– phenotype associated with the absence of the high prevalence U antigen. Four of the six samples showed both S–s–U– and Fy(a–b–) predicted phenotypes resulting from a FY*Fy/*Fy genotype. One sample appeared to be homozygous for GYPBS-Int5/GYPBS-Int5 or GYPBS-Int5 in trans with the GYPB deletion with a S–s–U+var derived phenotype. Two other serologically phenotyped S_s+ samples were identified as heterozygous for GYPBS-Int5/MNS*4 with a S–s+U+var derived phenotype. Altogether these findings are consistent with a correlation between IVS5, +5g>T and the GYPBS allele. Similarly, the 230C>T silencing change detected at homozygote state in one sample correlated with the GYPBS allele and a S–s–U+var derived phenotype (see Figure 2G). As described above, the GYPBS−230 allele can be in either double or single dose in trans with the GYPB deletion. In addition, 681 genotype-to-phenotype determinations not obtained serologically were defined for DO, YT, and CO systems including identification of 26 atypical genotypes CO*1/*2 (n = 8), YT*1/*2 (n = 15), and YT*2*/2 (n = 3).
In this clinical setting (i.e., a pretransfusional immunology laboratory), our multiplex assay has proven to be a reliable tool that generated reproducible results in the hands of different operators. In addition, laboratory experience showed that this tool was time- and cost-efficient because results could be obtained within 24 hours after collection of blood samples and reagent cost per SNP detected was low in comparison with other genotyping technologies. Unit cost for one DNA extraction and one SNaPshot assay including technician and machine times, and kit cost is around of $2.9 per SNP analyzed.
Discussion
Because of the various limitations associated with agglutination-based assays and the need to manage patients to avoid alloimmunization with future transfusion-related complications, the challenge of this work has been to develop a multiplex assay to reliably predict common clinically relevant blood group antigens. Two strategies can be used to improve the accuracy of antigen typing. The first is to perform both phenotyping and genotyping in the same laboratory. The second consists in maintaining a database of blood group SNPs as a mining source for alleles and their blood gene products to accelerate the quest for matched bloods. The multiplex assay presented here is aligned with both these strategies.
Our assay is based on the well-known SNaPshot method and uses quality-controlled reagents from a commercial company. We selected primer sets to simultaneously amplify nine genomic regions of seven blood group genes [KEL, DARC(FY), GYPB, SLC14A1(JK), ART4(DO), ACHE(YT), and AQP1(CO)] in a single PCR reaction and then 11 probe primers for multiplex extension reaction at SNP sites defining 18 blood group alleles.21 Comparatively, Chaudhuri's team used three independent multiplex SNaPshot reactions to determine 17 SNPs.20 Detection of single-base extended probe primers is based on extended length and fluorescence detected by capillary electrophoresis on a sequencing machine. It is a direct determination of the nucleotide type present at SNP site, which is visualized by one or two different color peaks on the electropherogram. This eliminates the problem of the negative extension reaction because all probe primers must incorporate one or two nucleotide types at the SNP site except in the case of gene deletion. This is an advantage over large-scale genotyping technologies based on microarrays or microarrayed fluorescent beadships probing blood gene amplicons can lead to negative hybridization reactions with false-negative results. Another advantage of the SNaPshot approach described here is not to require expensive development of new siliconchips or microbeads for typing of new SNP sites.
The 18 blood group alleles corresponding to the 11 SNPs detected by our multiplex assay were chosen in function of clinical significance and frequency in population groups living in the south of France. Because persons from sub-Saharan Africa and the Comoros archipelago account for local population, there is a high risk of patients with a known predisposition to alloimmunization, such as those with sickle cell or thalassemic disorders that require multiple blood transfusions to ensure survival. For this reason the multiplex assay included probe primes against nucleotide changes known to impair the GYPB and DARC genes reported in individuals of African descent.31,32 Detection of SNPs defining the alleles of the DO, YT, and CO systems was also included because there are hemolytic transfusion reactions caused by antibodies to DO, YT, and CO system antigens and no available commercial antibodies to determine these antigens.23
This multiplex SNaPshot assay was initially validated by comparison with available allele-specific PCR, serological, and/or sequencing results. Clinical experience with this multiplex assay in our regional pretransfusion laboratory demonstrated that it was a powerful and highly accurate (100%) molecular tool that saves both time and money. Data analysis and interpretation are simple, thus making this assay relatively easy to use for technicians having a period of specialist training. During this year trial period, genotyping of 227 patient samples including characterization of 203 atypical or null blood types was performed. Genotyping in addition to the classical phenotyping by serology facilitated matching of blood components to the recipient's blood type and probably contributed to lowering alloimmunization of patients receiving multiple transfusions.
Nevertheless, providing compatible donors with specific antigen-negative blood remains problematic, particularly in cases involving chronically transfused patients with unusual antibody combinations and phenotypes. To overcome this problem, we will soon test the feasibility of automating our multiplex tool in a 96-well microplate format to allow genotyping of donor cohorts and identification of rare phenotypes at lower cost. This technique would provide an alternative to serological screening, reduce the need for rare antisera, and save time.
This study demonstrates that multiplex SNaPshot assay can provide a simple robust tool for DNA genotyping in clinical contexts not easily amenable to serological analysis. In our regional pretransfusional laboratory typing of 18 blood alleles using this approach proved to be a reliable complementary tool with a turnaround time of 24 hours. This molecular tool also provides much more information regarding donor/recipient compatibility than is possible by using serological methods alone. However, because genotype does not always reflect phenotype because novel mutations arise continually, serological phenotyping still remains an essential test to confirm donor/recipient compatibility when specific antibodies are available. In conclusion, we believe that our multiplex SNaPshot assay in tandem with serological methods is an effective RBC genotyping tool to reduce transfusion-related immunizations and to avoid hemolytic complications of transfusions in ethnically mixed populations.
Acknowledgements
We thank Thomas Granier for his expert technical assistance, Mhammed Touinssi for the available allele-specific PCR results to validate the SNaPshot assay, and the staff of hematology laboratories of EFS Alpes-Méditerranée for providing blood samples used in this study. We thank Andrew Corsini for proofreading the manuscript.
Footnotes
Supported by Établissement Français du Sang, Paris, France (all coauthors).
References
- 1.Reid ME. Overview of molecular methods in immunohematology. Transfusion. 2007;47(Suppl):10S–16S. doi: 10.1111/j.1537-2995.2007.01304.x. [DOI] [PubMed] [Google Scholar]
- 2.Daniels G. The molecular genetics of blood group polymorphism. Transpl Immunol. 2005;14:143–153. doi: 10.1016/j.trim.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 3.Storry JR, Olsson ML. Genetic basis of blood group diversity. Br J Haematol. 2004;126:759–771. doi: 10.1111/j.1365-2141.2004.05065.x. [DOI] [PubMed] [Google Scholar]
- 4.Daniels G, Finning K, Martin P, Soothill P. Fetal blood group genotyping from DNA from maternal plasma: an important advance in the management and prevention of haemolytic disease of the fetus and newborn. Vox Sang. 2004;87:225–232. doi: 10.1111/j.1423-0410.2004.00569.x. [DOI] [PubMed] [Google Scholar]
- 5.Rouillac-Le Sciellour C, Puillandre P, Gillot R, Baulard C, Metral S, Le Van Kim C, Cartron JP, Colin Y, Brossard Y. Large-scale pre-diagnosis study of fetal RHD genotyping by PCR on plasma DNA from RhD-negative pregnant women. Mol Diagn. 2004;8:23–31. doi: 10.1007/BF03260044. [DOI] [PubMed] [Google Scholar]
- 6.Hashmi G, Shariff T, Seul M, Vissavajjhala P, Hue-Roye K, Charles-Pierre D, Lomas-Francis C, Chaudhuri A, Reid ME. A flexible array format for large-scale, rapid blood group DNA typing. Transfusion. 2005;45:680–688. doi: 10.1111/j.1537-2995.2005.04362.x. [DOI] [PubMed] [Google Scholar]
- 7.Avent ND, Martinez A, Flegel WA, Olsson ML, Scott ML, Nogues N, Pisacka M, Daniels G, van der Schoot E, Muniz-Diaz E, Madgett TE, Storry JR, Beiboer SH, Maaskant-van Wijk PA, von Zabern I, Jimenez E, Tejedor D, Lopez M, Camacho E, Cheroutre G, Hacker A, Jinoch P, Svobodova I, de Haas M. The BloodGen project: toward mass-scale comprehensive genotyping of blood donors in the European Union and beyond. Transfusion. 2007;47(1 Suppl):40S–46S.. doi: 10.1111/j.1537-2995.2007.01309.x. [DOI] [PubMed] [Google Scholar]
- 8.Denomme GA, Van Oene M. High-throughput multiplex single-nucleotide polymorphism analysis for red cell and platelet antigen genotypes. Transfusion. 2005;45:660–666. doi: 10.1111/j.1537-2995.2005.04365.x. [DOI] [PubMed] [Google Scholar]
- 9.Montpetit A, Phillips MS, Mongrain I, Lemieux R, St-Louis M. High-throughput molecular profiling of blood donors for minor red blood cell and platelet antigens. Transfusion. 2006;46:841–848. doi: 10.1111/j.1537-2995.2006.00805.x. [DOI] [PubMed] [Google Scholar]
- 10.Beiboer SH, Wieringa-Jelsma T, Maaskant-Van Wijk PA, van der Schoot CE, van Zwieten R, Roos D, den Dunnen JT, de Haas M. Rapid genotyping of blood group antigens by multiplex polymerase chain reaction and DNA microarray hybridization. Transfusion. 2005;45:667–679. doi: 10.1111/j.1537-2995.2005.04319.x. [DOI] [PubMed] [Google Scholar]
- 11.Bugert P, McBride S, Smith G, Dugrillon A, Kluter H, Ouwehand WH, Metcalfe P. Microarray-based genotyping for blood groups: comparison of gene array and 5′-nuclease assay techniques with human platelet antigen as a model. Transfusion. 2005;45:654–659. doi: 10.1111/j.1537-2995.2005.04318.x. [DOI] [PubMed] [Google Scholar]
- 12.Karpasitou K, Drago F, Crespiatico L, Paccapelo C, Truglio F, Frison S, Scalamogna M, Poli F. Blood group genotyping for Jk(a)/Jk(b). Fy(a)/Fy(b), S/s, K/k, Kp(a)/Kp(b), Js(a)/Js(b), Co(a)/Co(b), and Lu(a)/Lu(b) with microarray beads. Transfusion. 2008;48:505–512. doi: 10.1111/j.1537-2995.2007.01555.x. [DOI] [PubMed] [Google Scholar]
- 13.Bell PA, Chaturvedi S, Gelfand CA, Huang CY, Kochersperger M, Kopla R, Modica F, Pohl M, Varde S, Zhao R, Zhao X, Boyce-Jacino MT, Yassen A, SNPstream UHT. ultra-high throughput SNP genotyping for pharmacogenomics and drug discovery. Biotechniques. 2002;Suppl:70–72. 74, 76–77. [PubMed] [Google Scholar]
- 14.Brion M. Y chromosome SNP analysis using the single-base extension: a hierarchical multiplex design. Methods Mol Biol. 2005;297:229–242. doi: 10.1385/1-59259-867-6:229. [DOI] [PubMed] [Google Scholar]
- 15.Vallone PM, Just RS, Coble MD, Butler JM, Parsons TJ. A multiplex allele-specific primer extension assay for forensically informative SNPs distributed throughout the mitochondrial genome. Int J Legal Med. 2004;118:147–157. doi: 10.1007/s00414-004-0428-5. [DOI] [PubMed] [Google Scholar]
- 16.Filippini S, Blanco A, Fernandez-Marmiesse A, Alvarez-Iglesias V, Ruiz-Ponte C, Carracedo A, Vega A. Multiplex SNaPshot for detection of BRCA1/2 common mutations in Spanish and Spanish related breast/ovarian cancer families. BMC Med Genet. 2007;8:40. doi: 10.1186/1471-2350-8-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yan H. Snapshot of the allele-specific variation in human gene expression. Methods Mol Biol. 2005;311:31–38. doi: 10.1385/1-59259-957-5:031. [DOI] [PubMed] [Google Scholar]
- 18.Inagaki S, Yamamoto Y, Doi Y, Takata T, Ishikawa T, Imabayashi K, Yoshitome K, Miyaishi S, Ishizu H. A new 39-plex analysis method for SNPs including 15 blood group loci. Forensic Sci Int. 2004;144:45–57. doi: 10.1016/j.forsciint.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 19.Doi Y, Yamamoto Y, Inagaki S, Shigeta Y, Miyaishi S, Ishizu H. A new method for ABO genotyping using a multiplex single-base primer extension reaction and its application to forensic casework samples. Leg Med (Tokyo) 2004;6:213–223. doi: 10.1016/j.legalmed.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 20.Palacajornsuk P, Halter C, Isakova V, Tarnawski M, Farmar J, Reid ME, Chaudhuri A. Detection of blood group genes using multiplex SNaPshot method. Transfusion. 2009;49:740–749. doi: 10.1111/j.1537-2995.2008.02053.x. [DOI] [PubMed] [Google Scholar]
- 21.Reid ME, Lomas-Francis C. Blood group antigen factsbook. 2nd ed. Academic Press; San Diego: 2003. [Google Scholar]
- 22.Chiaroni J, Touinssi M, Frassati C, Degioanni A, Gibert M, Reviron D, Mercier P, Boetsch G. Genetic characterization of the population of Grande Comore Island (Njazidja) according to major blood groups. Hum Biol. 2004;76:527–541. doi: 10.1353/hub.2004.0053. [DOI] [PubMed] [Google Scholar]
- 23.Klein HG, Anstee DJ. Mollison's Blood Transfusion in Clinical Medicine. Blackwell; Oxford: 2005. [Google Scholar]
- 24.Eshleman JR, Shakin-Eshleman SH, Church A, Kant JA, Spitalnik SL. DNA typing of the human MN and Ss blood group antigens in amniotic fluid and following massive transfusion. Am J Clin Pathol. 1995;103:353–357. doi: 10.1093/ajcp/103.3.353. [DOI] [PubMed] [Google Scholar]
- 25.Hessner MJ, McFarland JG, Endean DJ. Genotyping of KEL1 and KEL2 of the human Kell blood group system by the polymerase chain reaction with sequence-specific primers. Transfusion. 1996;36:495–499. doi: 10.1046/j.1537-2995.1996.36696269506.x. [DOI] [PubMed] [Google Scholar]
- 26.Irshaid NM, Thuresson B, Olsson ML. Genomic typing of the Kidd blood group locus by a single-tube allele-specific primer PCR technique. Br J Haematol. 1998;102:1010–1014. doi: 10.1046/j.1365-2141.1998.00874.x. [DOI] [PubMed] [Google Scholar]
- 27.Joshi SR, Wagner FF, Vasantha K, Panjwani SR, Flegel WA. An AQP1 null allele in an Indian woman with Co(a-b-) phenotype and high-titer anti-Co3 associated with mild HDN. Transfusion. 2001;41:1273–1278. doi: 10.1046/j.1537-2995.2001.41101273.x. [DOI] [PubMed] [Google Scholar]
- 28.Touinssi M, Chiaroni J, Degioanni A, Granier T, Dutour O, Bailly P, Bauduer F. DNA-based typing of Kell. Kidd MNS, Dombrock, Colton, and Yt blood group systems in the French Basques. Am J Hum Biol. 2008;20:308–311. doi: 10.1002/ajhb.20720. [DOI] [PubMed] [Google Scholar]
- 29.Wu GG, Jin SZ, Deng ZH, Zhao TM. Polymerase chain reaction with sequence-specific primers-based genotyping of the human Dombrock blood group DO1 and DO2 alleles and the DO gene frequencies in Chinese blood donors. Vox Sang. 2001;81:49–51. doi: 10.1046/j.1423-0410.2001.00052.x. [DOI] [PubMed] [Google Scholar]
- 30.Yan L, Zhu F, Fu Q, He J. c17 ABO, Rh, MNS, Duffy, Kidd,Yt, Scianna, and Colton blood group systems in indigenous Chinese. Immunohematology. 2005;21:10–14. [PubMed] [Google Scholar]
- 31.Storry JR, Reid ME, Fetics S, Huang CH. Mutations in GYPB exon 5 drive the S-s-U+(var) phenotype in persons of African descent: implications for transfusion. Transfusion. 2003;43:1738–1747. doi: 10.1046/j.0041-1132.2003.00585.x. [DOI] [PubMed] [Google Scholar]
- 32.Tournamille C, Colin Y, Cartron JP, Le Van Kim C. Disruption of a GATA motif in the Duffy gene promoter abolishes erythroid gene expression in Duffy-negative individuals. Nat Genet. 1995;10:224–228. doi: 10.1038/ng0695-224. [DOI] [PubMed] [Google Scholar]
- 33.Tournamille C, Le Van Kim C, Gane P, Le Pennec PY, Roubinet F, Babinet J, Cartron JP, Colin Y. Arg89Cys substitution results in very low membrane expression of the Duffy antigen/receptor for chemokines in Fy(x) individuals. Blood. 1998;92:2147–2156. [PubMed] [Google Scholar]
- 34.Castilho L. The value of DNA analysis for antigens in the Duffy blood group system. Transfusion. 2007;47(Suppl):28S–31S. doi: 10.1111/j.1537-2995.2007.01307.x. [DOI] [PubMed] [Google Scholar]