

Abstract
Inactivation of MLH1 due to promoter hypermethylation strongly suggests a sporadic origin, providing exclusion criteria for Lynch syndrome. The aim of this study is to compare the utility of methylation analysis of MLH1 and BRAF V600E mutations for the selection of patients with MLH1 negative colorectal cancer for genetic testing. MLH1 methylation status was evaluated by MethyLight and methylation-specific MLPA (MS-MLPA) in tumor DNA from 73 colorectal cancer patients with loss of MLH1 protein expression. These tumors were analyzed for BRAF V600E mutations, and genetic testing for germline MLH1 mutations was performed in all corresponding patients. Ten patients had germline mutations in MLH1 and none of their tumors showed significant MLH1 methylation or BRAF V600E mutation. MLH1 genetic testing excluded patients by MethyLight in 47 patients (64%), by MS-MLPA in 49 (67%), and BRAF V600E mutation in only 25 patients (34%) (χ2P = 0.00001). Specificity was 75% for MethyLight, 78% for MS-MLPA and 40% for BRAF V600E mutation. The use of MethyLight or MS-MLPA instead of BRAF mutation resulted in a cost reduction of 41% and 45%, respectively, per every MLH1 mutation detected. Taken together, methylation analysis of MLH1 shows better performance characteristics than BRAF V600E mutation in the selection of patients for genetic testing of MLH1, especially when using MS-MLPA.
Lynch syndrome is the most common cause of hereditary colon cancer, accounting for 3–4% of all colorectal cancers (CRC),1,2 and it is due to germline mutations of DNA mismatch repair (MMR) genes MLH1, MSH2, MSH6, and PMS2.1 The hallmark of tumors with mismatch repair deficiency is high level microsatellite instability (MSI-H), and therefore almost all Lynch syndrome tumors show the MSI-H phenotype.1,3,4 However, only 15% of MSI-H tumors have a hereditary origin and the remaining 85% are largely sporadic. Exceptions include patients with hyperplastic polyposis in which MSI-H MLH1 deficient CRC can occur against a background of polyposis. In these tumors the MSI-H phenotype is due to epigenetic silencing of MLH1 caused by hypermethylation of its promoter region.5,6,7
According to the consensus criteria for the diagnostic strategy of Lynch syndrome, tumoral tissue from CRC patients fulfilling Amsterdam criteria or any of the revised Bethesda criteria should undergo MSI study or immunochemical staining for mismatch repair proteins.8 Patients whose tumors show MSI or lack of expression of any of these proteins must be submitted to genetic testing. However, only one third of tested patients following this strategy show germline mutations,9 and in the majority of these cases tumors develop as a result of epigenetically inactivated MLH1.
The study of MLH1 methylation status in MSI tumors has potential impact on the selection of CRC patients for genetic testing.10 A number of methods to determine DNA methylation in tumor tissues have been developed but most of them are technically complex, expensive and time consuming. For this reason methylation studies are not usually performed for diagnostic purposes. The considered gold-standard technique for the quantitative study of methylation, MethyLight,11,12 is technically difficult to perform and requires standardization arrangements that are not usually available in the majority of molecular biology laboratories. Recently, a novel and simpler quantitative method for the study of MLH1 methylation has become available: the methylation specific–multiplex probe amplification (MS-MLPA)13 (Salsa MS-MLPA kit ME011 mismatch repair genes (MMR), MRC–Holland, Amsterdam, The Netherlands). On the other hand, the BRAF gene mutation V600E has been identified in colorectal tumors showing MMR deficiency associated with the epigenetic silencing of the MLH1 gene and previous studies showed that tumors from patients with germline mutations in MMR genes do not show somatic mutations in BRAF.14 The aim of this study is to compare two methods of methylation analysis of MLH1 (MethyLight and MS-MLPA) with BRAF V600E mutation analysis for the selection of patients with MLH1 negative colorectal cancer for genetic testing of Lynch syndrome.
Materials and Methods
Subjects
Immunohistochemical analysis of MLH1 was performed in 2420 CRC surgical specimens. Tumor tissue was collected from a series of 2265 non-selected surgical CRC specimens from the EPICOLON study (n = 1.281)9,15 and from the Pathology Department of the Hospital General Universitario of Alicante, collected between the years 1999 to 2008 (n = 984). The remaining 155 tumors were collected from patients seen at the Genetic Counseling in Cancer Department of the Hospital General Universitario of Elche. Demographic, clinical, and tumor-related characteristics of probands, as well as a detailed family history, were obtained using a pre-established questionnaire, as described elsewhere. Loss of MLH1 expression was found in 149 tumors (6%). All these tumors showed normal expression of MSH2 and MSH6. Germline mutation analysis was completed in 92 patients that showed loss of MLH1 immunohistochemical expression. In 19 cases there was not enough tissue to perform molecular studies and they were excluded from this study. As a result, 73 patients with loss of MLH1 expression that had undergone germline mutation analysis were included in the study (Figure 1). The study was approved by the Ethics Committee of the participant hospitals and informed consent was obtained.
Figure 1.
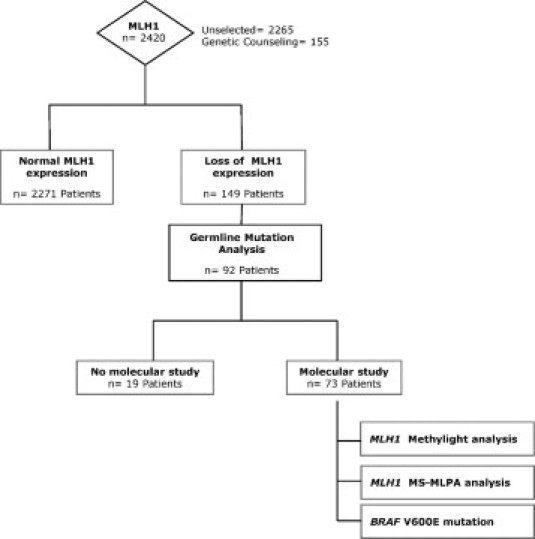
Flow diagram for the immunohistochemical and molecular analysis performed in patients and tumors.
Colorectal Cancer Tissue Samples and Immunohistochemistry
The representative tumor regions were identified by our pathologists (C.A., A.P.) in a multiview microscope, selecting the highest density of tumor cells with at least 70% of tumor cells, marked on the H&E-stained slides and subsequently identified on the corresponding tissue blocks. Tissue cylinders of a diameter of 1 mm were punched out from the marked areas of each block and incorporated into a recipient paraffin block using a tissue arrayer.
Genomic DNA was extracted from two tissue paraffin-embedded cylinders. Immunohistochemical analysis of MLH1, MSH2, MSH6, and PMS2 was performed in 5-μm sections of formalin-fixed, paraffin-embedded tumor tissue as previously described.16,17
BRAF V600E Mutation
The V600E BRAF mutation was detected using real-time chemistry TaqMan probes in an ABI Prism 7500 (Applied Biosystems, Foster City, CA) sequence detection system with allelic discrimination, all designed and previously described by Benlloch et al18 including primers and probes; the BRAF primer forward is 51F: 5′-CTACTGTTTTCCTTTACTTACTACACCTCAGA-3′, the BRAF primer reverse is 176R: 5′-ATCCAGACAACTGTTCAAACTGATG-3′, the BRAF probe for mutant allele is −5′-6FAM–5′-TAGCTACAGAGAAATC–MGB-3′ and the BRAF probe for wild-type allele is 5′-VIC-5′- CTAGCTACAGTGAAATC–MGB-3′ using 6-carboxyfluorescein (FAM). The reporter fluorophore was used for the mutant allele and VIC as a fluorophore for the wild-type allele. Fluorescence data were analyzed using the allelic discrimination software of the ABI Prism 7500 instrument.
Real-Time PCR (MethyLight) for Quantitative DNA Methylation Analysis
Real-time PCR(MethyLight) for quantitative DNA methylation analysis19 is based on bisulfite conversion of normal (unmethylated) cytosine nucleotide into uracil, leaving methylated cytosine intact. Bisulfite conversion of unmethylated cytosine was performed using the EZ DNA methylation-Gold kit according to the manufacturer (Zymo Research, Orange, CA). As the specific methylation sites in the promoter are known, a subsequent real-time polymerase chain reaction can be performed, The MLH1 forward primer is 5′-AGGAAGAGCGGATAGCGATTT-3′ the MLH1 reverse primer is 5′-TCTTCGTCCCTCCCTAAAACG-3′, and the MLH1 probe is 6FAM-5′-CCCGCTACCTAAAAAAATATACGCTTACGCG–TAMRA12 located in the hypermethylation region that specifically generates amplification products in methylated DNA. Primers and probe were chosen within the specific C-region of the reporter fluorophor for the mutant allele and VIC as reporter fluorophor for the wild-type allele. Fluorescence data were analyzed with the allelic discrimination software of the ABI Prism 7500 instrument.
MLH1 promoter (−248∼−178) corresponding to a CpG island that when hypermethylated correlates with the absence of gene expression.20 COL2A1 (the collagen 2A1 gene) was used to normalize the amount of input bisulfite DNA, because of the lack of CpGs in its amplicon sequence, the COL2A forward primer is 5′-TCTAACAATTATAAACTCCAACCACCAA-3′, the COL2A reverse primer is 5′-GGGAAGATGGGATAGAAGGGAATAT-3′, and the COL2A probe is 6FAM-5′-CCTTCATTCTAACCCAATACCTATCCCACCTCTAAA–TAMRA.12 After sodium bisulfite conversion, genomic DNA was amplified by fluorescence-based, real-time quantitative PCR using ABI 7500. The percentage of methylated reference or degree of methylation at a specific locus was calculated with MLH1:COL2A1 ratio of a sample divided by the MLH1:COL2A1 ratio of the complete methylated DNA control. A percentage of methylated reference equal to 4 was chosen as the dichotomization threshold to establish a bimodal distribution: positive methylated samples (percentage of methylated reference >4) versus negative ones (percentage of methylated reference ≤4). The cutoff of 4 had been validated in a previous study because it highly correlates with loss of protein expression.12
Methylation-Specific Multiplex Ligation-Dependent Probe Amplification
The SALSA MS-MLPA kit ME011 mismatch repair genes (MMR) (MRC-Holland, Amsterdam, The Netherlands) is used to study aberrant CpG island methylation in the promoter of MMR genes, including MLH1, MSH2, MLH3, PMS2, MSH3, MSH6, and MGMT. The MS-MLPA method is based on probes that recognize specific sequences in DNA that contains a restriction site for a methylation-sensitive HhaI enzyme. This kit contains five MLPA probes specific to MLH1 regions: MLH1 1 (237 bp; −659 bp distance to ATG start); MLH1 2 (265 bp; −383 bp distance to ATG start); MLH1 3 (189 bp; −246 bp distance to ATG start); MLH1 4 (166 bp; −13 bp distance to ATG start); MLH1 5 (292 bp; +208 bp distance to ATG start). The target regions for the MLH1 gene silenced by promoter hypermethylation appear to be −248 to −178 nt, tested within probe MLH1 3, which corresponds to the specific C-region and −109 to +15 nt tested within probe MLH1 4.
MS-MLPA assays were performed as described by the manufacturer: in each reaction we used 200 ng of DNA (5 μl at 40 ng/μl). Similar to a conventional MLPA assay, genomic DNA is first denatured and subsequently cooled down to 25°C, followed by the addition of MS-MLPA probes and a 16-hour hybridization step. MS-MLPA assay is then split into two tubes; one tube is processed as a standard MLPA reaction: ligation of hybridized probe oligonucleotides followed by PCR amplification. The other tube of the MLPA hybridization reaction is incubated with the methylation-sensitive HhaI endonuclease. PCR was performed as described by the manufacturer, although we added double DNA–probe mix amount (10 μl) in each PCR reaction to improve results. Then, PCR fragments were separated and quantified by electrophoresis on an ABI 310 capillary analyzer (Applied Biosystems, Foster City, CA).
Methylation status for a tumor sample was calculated using GeneMapper v. 4.0 analysis software (Applied Biosystems). Peak height parameter is proportional to the amount of PCR product generated. To calculate the methylation ratio, each peak height from HhaI-digested tumor DNA was divided by its corresponding peak height from the undigested tumor DNA. To compensate for differences in PCR efficiency of the individual samples, each peak height (digested and undigested) was normalized dividing each probe amplification product by the average value of the 11 control probes without a HhaI enzyme site.21
The mean of MLH1 probe 3 and 4 corresponding to regions C and D, respectively, in MLH1 promoter were considered to calculate the methylation ratio. The dichotomization threshold to distinguish methylated versus non methylated samples was established at 15% based on a previous study associated with gene silencing.22
MLH1 Germline Genetic Testing
Germline mutation studies were performed on genomic DNA isolated from peripheral blood leukocytes or from non-tumor colon tissue as previously described.9 Point mutation analysis of MLH1 gene was done by PCR amplification and direct sequencing of the entire coding region and the exon-intron boundaries. PCR primers and conditions have been described elsewhere.23,24,25 Large genomic rearrangements (insertions and/or deletions) in MLH1 loci were screened by multiplex ligation-dependent probe amplification according to the manufacturer protocols (Salsa MLPA kit P003 and P008; MRC-Holland, Amsterdam, The Netherlands).
Data Management and Analysis
Data were collected and entered into the computer using Microsoft Access software for storage and initial analysis. Further analysis was done using SPSS software (SPSS 15.0, Chicago, IL). For continuous variables relevant measures of central tendency (means for normally distributed data, and medians and interquartile ranges for skewed data) were used to explore data. The χ2 test was used for comparison of qualitative variables. A Student's t-test was used for comparison of normally distributed continuous variables and a Mann-Whitney U-test was used for unpaired comparison of non-normally distributed continuous variables. The Pearson rank correlation test (for variables with normal distribution) was used to calculate correlation coefficient between variables. A P value of less than 0.05 was considered significant.
Results
Characteristics of patients regarding the presence of germline mutation and the methylation status can be seen in Table 1. Patients were placed in the methylation category if MLH1 methylation were found in tumor by either method.
Table 1.
Characteristics of Patients
| No germline mutation |
|||
|---|---|---|---|
| Characteristics | Germline mutation (n = 10) N (%) | Methylation (n = 51) N (%) | No methylation (n = 12) N (%) |
| Age at diagnosis | |||
| <50 years | 6 (60) | 7 (13.7)* | 2 (16.7)* |
| >50 years | 4 (40) | 44 (86.3) | 10 (83.3) |
| Sex | |||
| Male | 3 (30) | 21 (41.2) | 8 (66.6) |
| Female | 7 (70) | 30 (58.8) | 4 (33.4) |
| Revised Bethesda | |||
| Fulfilling | 10 (100) | 18 (35.3)* | 8 (66.6) |
| Not fulfilling | 0 (0) | 33 (64.7) | 4 (33.4) |
| Amsterdam II criteria | |||
| Fulfilling | 4 (40) | 1 (2)* | 4 (33.4) |
| Not fulfilling | 6 (60) | 50 (98) | 8 (66.6) |
| Tumor location | |||
| Right-sided | 4 (40) | 43 (84.3)* | 7 (58.3) |
| Left-sided | 6 (60) | 8 (15.7) | 5 (41.7) |
| Histological type | |||
| NOS | 6 (60) | 23 (45) | 7 (58.3) |
| Special | 4 (40) | 28 (55) | 5 (41.7) |
| Grade | |||
| High | 3 (30) | 25 (49) | 4 (33.3) |
| Low | 7 (70) | 26 (51) | 8 (66.6) |
| BRAF mutation | |||
| Mutated | 0 (0) | 26 (51)*† | 0 (0) |
| Not mutated | 10 (100) | 25 (49) | 12 (100) |
P < 0.05 compared with germline mutation patients.
P < 0.05 compared with non-mutated, non-methylated patients.
The correlation of the results of the two methylation techniques was 92%. This correlation is statistically significant (P < 0.001), with 47 of 73 cases found to be methylated by MethyLight and 49 of 73 methylated by MS-MLPA. Germline mutations in MLH1 were found in 10 patients (14%). None of these cases showed MLH1 promoter hypermethylation using any of the used methylation techniques. None of the patients with MLH1 germline mutation showed BRAF V600E mutations (Table 2).
Table 2.
Performance of Different Techniques for the Selection of Patients with MLH1-Negative Tumors for Genetic Testing of MLH1 Germline Mutation
| Germline mutation (n = 10) |
No germline mutation (n = 63) | |||||
|---|---|---|---|---|---|---|
| Techniques | N | % | N | % | Odds ratio | P |
| MethyLight | ||||||
| Not methylated | 10 | 100 | 16 | 25.4 | 1.6 (1.2–2.2) | <0.001 |
| Methylated | 0 | 0 | 47 | 74.6 | ||
| MS-MLPA | ||||||
| Not methylated | 10 | 100 | 14 | 22.2 | 1.7 (1.2–2.4) | <0.001 |
| Methylated | 0 | 0 | 49 | 77.8 | ||
| BRAF mutation | ||||||
| Not mutated | 10 | 100 | 38 | 60.3 | 1.3 (1.1–1.5) | <0.05 |
| Mutated | 0 | 0 | 25 | 39.7 | ||
Regarding patients with lack of MLH1 germline mutation, significant methylation was found in 47 of 63 tumors (75%) measured by MethyLight and 49 of 63 (78%) measured by MS-MLPA. However, only 25 of these tumors showed BRAF V600E mutation (40%) (Table 2). Using the different techniques to exclude patients for MLH1 genetic testing, MethyLight would exclude 47 out of 73 (64%), MS-MLPA 49 out of 73 (67%), and BRAF V600E mutation would only exclude 25 patients (34%) (χ2 P = 0.00001).
Table 3 shows sensitivity, specificity, positive predictive value, negative predictive value, positive likelihood ratio, and number needed to test for the different strategies to detect germline mutations. It also included the combination of molecular tools or the combination of both clinical and molecular tools. Strategies including methylation techniques have the highest specificity and positive predictive value clearly demonstrating their superiority over the study of BRAF V600E mutations. Among the MLH1 methylation tests studied, specificity and positive predictive value of MS-MLPA were slightly better than MethyLight.
Table 3.
Value of Different Strategies for the Detection of Germline Mutations in Patients with MLH1-Negative Tumors
| Strategies | Sensitivity | Specificity | PPV | NPV | +LR | NNT |
|---|---|---|---|---|---|---|
| MethyLight* | 100 | 74.6 | 38.5 | 100 | 3.94 | 1.34 |
| MS-MLPA† | 100 | 77.8 | 41.7 | 100 | 4.50 | 1.28 |
| BRAF‡ mutation | 100 | 40.3 | 21.3 | 100 | 1.67 | 2.48 |
| MethyLight + BRAF | 100 | 74.6 | 38.5 | 100 | 3.94 | 1.34 |
| MS-MLPA + BRAF | 100 | 77.8 | 41.7 | 100 | 4.50 | 1.28 |
| Bethesda§ | 100 | 58.1 | 27.8 | 100 | 2.38 | 1.72 |
| Bethesda + MethyLight | 100 | 84.1 | 50 | 100 | 6.30 | 1.19 |
| Bethesda + MS-MLPA | 100 | 87.3 | 55.6 | 100 | 7.87 | 1.14 |
| Bethesda + BRAF | 100 | 69.8 | 34.5 | 100 | 3.32 | 1.43 |
| Bethesda + MethyLight + BRAF | 100 | 84.1 | 50 | 100 | 6.30 | 1.19 |
| Bethesda + MS-MLPA + BRAF | 100 | 88.9 | 58.8 | 100 | 9.00 | 1.12 |
PPV, positive predictive value; NPV, negative predictive value; +LR, positive likelihood ratio; NNT, number needed to test for detecting one MLH1 germline mutation.
MethyLight: Absence of MLH1 promoter methylation by MethyLight.
MS-MLPA: Absence of MLH1 promoter methylation by MS-MLPA.
BRAF: Mutation V600E in BRAF gene.
Bethesda: Fulfillment of Bethesda criteria.
A cost-effectiveness analysis was made comparing different strategies (Figure 2). Performing germline mutation analysis in all patients with MLH1 negative tumors had a cost of 5840Є per detected mutation ($7893). The introduction of BRAF V600E mutation analysis in the algorithm achieved a reduction of this cost by 15%. However, the use of MethyLight or MS-MLPA reduced the cost by 41% and 45%, respectively. Moreover, the average technician time needed to do the work (including all of the previous process from DNA extraction) is 1.7 hours (102 minutes) per case for MethyLight and 0.8 hours (48 minutes) per case for MS-MLPA. This difference is due to the fact that MethyLight includes a bisulfite modification step and previous standardization arrangements that are avoided with MS-MLPA.
Figure 2.
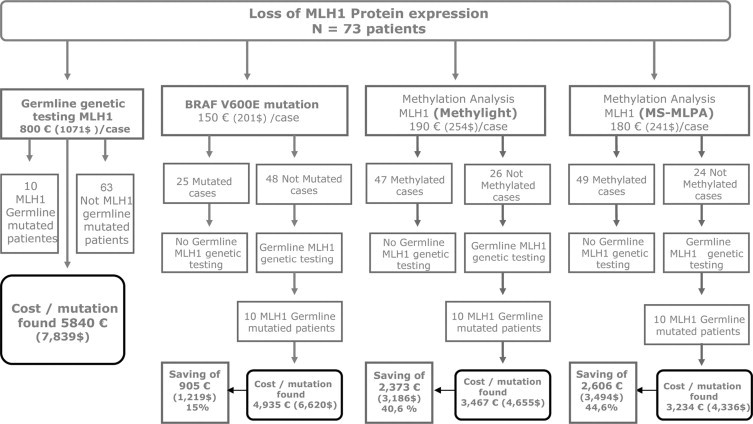
Analysis of cost per detected mutation using different molecular tools for patient selection.
Discussion
This study shows that MS-MLPA and MethyLight are equivalent techniques for MLH1 methylation analysis and both are of great value in the pre-selection of patients for genetic testing in Lynch syndrome, showing better performance than the more currently used strategy BRAF V600E mutation analysis. The main advantage of MS-MLPA technique is its simplicity, as it does not require previous experience in gene methylation techniques. The selection of patients with tumors showing lack of MLH1 expression for genetic testing can be improved using methylation analysis with a significant reduction of costs.
The selection of patients for genetic testing to diagnose Lynch syndrome is frequently difficult in clinical practice, as Amsterdam criteria detects Lynch syndrome with a high specificity but low sensitivity.26 On the other hand, the use of the revised Bethesda criteria improves sensitivity but lacks specificity.8 As a result, some Lynch syndrome patients are overlooked; on the other hand a high number of patients without mismatch repair gene mutations are sent for genetic testing leading to a significant increase in laboratory costs. Therefore, additional molecular methods are needed. Microsatellite instability and mismatch repair protein immunohistochemistry are widely used and have demonstrated its appropriateness for this purpose.9 Nevertheless, a number of patients with loss of MLH1 expression still undergo unnecessary genetic testing, as only one-third of patients tested after careful selection using current clinical and molecular tools have germline mutations.9 Previous preliminary studies have evaluated the usefulness of methylation analysis methods in the strategy for the identification of Lynch syndrome,10,27 showing excellent results. However, this technique is not usually used in clinical practice because of its complexity. Here we show for the first time results of a novel methylation analysis technique, the MS-MLPA, in terms of comparison with other techniques. MS-MLPA is easier to perform than other methylation analysis and it is feasible for routine molecular biology laboratories.
Some studies have tested other screening strategies based on molecular or pathological analysis as a prior step before germline testing in MLH1 negative tumors. Somatic mutation in the oncogene BRAF (V600E) and p16 immunohistochemistry have been suggested as characteristic of sporadic colorectal tumors with MSI.14,28,29 In this study we show that methylation analysis techniques are clearly better than BRAF mutation in terms of specificity, positive predictive value, and cost. The methodology for detection of BRAF mutation needs similar equipment to MS-MLPA with a molecular biology core. The best performance of the latest technique makes it suitable for the selection of patients with absent MLH1 expression for MLH1 genetic testing. Recently we have shown the usefulness of p16 immunohistochemistry as a surrogate marker for p16 and MLH1 epigenetic silencing due to hypermethylation, showing that tumors with germline mutation of MLH1 do not show p16 hypermethylation and show normal staining of p16.29 p16 immunohistochemistry and BRAF have similar specificity for the detection of germline-mutated patients. However, according to our results, MLH1 methylation analysis doubles the specificity of these other molecular screening methods.
The main limitation of our study is the small number of patients with MLH1 germline mutations. However, the excellent correlation between MethyLight and MS-MLPA for the detection of hypermethylated tumors and the lack of MLH1 methylation among germline mutation carriers gives robustness to our data.
Another limitation is the diverse origin of our patients, with cases coming from the general population and a proportion of cases being selected from a genetic counseling unit. This approach hampers the evaluation of the appropriateness of the combination of clinical guidelines with tumor methylation analysis. A large population-based study would be necessary to confirm our data. Finally, germline epimutations in MLH1 have been recently described30 and, in this case, MLH1 methylation in tumor tissue can also be found as well as in blood and normal tissue. These MLH1 epimutations are very infrequent and must be specifically suspected in patients with multiple early-onset tumors and commonly without any significant family history. MS-MLPA for MLH1 and MSH2 has also been used for detection of germline epimutations in tumors and normal tissue.31
In summary, selection of patients for genetic testing in Lynch syndrome can be improved combining immunohistochemistry of MMR proteins with methylation analysis, especially using MS-MLPA, in cases with loss of MLH1 expression. This strategy is more cost-effective than the currently used of BRAF V600E mutation and requires similar technology, generally available in molecular biology facilities. The use of the combination of immunohistochemistry and methylation techniques for routine screening of Lynch syndrome should be tested in a population-based study to prove that this universal strategy can be more efficient than the ones used so far.
Footnotes
Supported in part by a grant from the Fundación de la CV para la investigación en el Hospital General Universitario de Alicante (Grupos consolidados 2008). L.P.-C. is the recipient of a grant from the Instituto de Salud Carlos III (FI07/00303). N.A. is supported by Schering-Plough.
References
- 1.Rustgi AK. The genetics of hereditary colon cancer. Genes Dev. 2007;21:2525–2538. doi: 10.1101/gad.1593107. [DOI] [PubMed] [Google Scholar]
- 2.Wheeler JM, Bodmer WF, Mortensen NJ. DNA mismatch repair genes and colorectal cancer. Gut. 2000;47:148–153. doi: 10.1136/gut.47.1.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Rodriguez-Bigas MA, Fodde R, Ranzani GN, Srivastava S. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58:5248–5257. [PubMed] [Google Scholar]
- 4.Ionov Y, Peinado MA, Malkhosyan S, Shibata D, Perucho M. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature. 1993;363:558–561. doi: 10.1038/363558a0. [DOI] [PubMed] [Google Scholar]
- 5.Cunningham JM, Christensen ER, Tester DJ, Kim CY, Roche PC, Burgart LJ, Thibodeau SN. Hypermethylation of the hMLH1 promoter in colon cancer with microsatellite instability. Cancer Res. 1998;58:3455–3460. [PubMed] [Google Scholar]
- 6.Herman JG, Umar A, Polyak K, Graff JR, Ahuja N, Issa JP, Markowitz S, Willson JK, Hamilton SR, Kinzler KW, Kane MF, Kolodner RD, Vogelstein B, Kunkel TA, Baylin SB. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci USA. 1998;95:6870–6875. doi: 10.1073/pnas.95.12.6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miyakura Y, Sugano K, Konishi F, Ichikawa A, Maekawa M, Shitoh K, Igarashi S, Kotake K, Koyama Y, Nagai H. Extensive methylation of hMLH1 promoter region predominates in proximal colon cancer with microsatellite instability. Gastroenterology. 2001;121:1300–1309. doi: 10.1053/gast.2001.29616. [DOI] [PubMed] [Google Scholar]
- 8.Umar A, Boland CR, Terdiman JP, Syngal S, de la CA, Ruschoff J, Fishel R, Lindor NM, Burgart LJ, Hamelin R, Hamilton SR, Hiatt RA, Jass J, Lindblom A, Lynch HT, Peltomaki P, Ramsey SD, Rodriguez-Bigas MA, Vasen HF, Hawk ET, Barrett JC, Freedman AN, Srivastava S. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004;96:261–268. doi: 10.1093/jnci/djh034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pinol V, Castells A, Andreu M, Castellvi-Bel S, Alenda C, Llor X, Xicola RM, Rodriguez-Moranta F, Paya A, Jover R, Bessa X. Accuracy of revised Bethesda guidelines, microsatellite instability, and immunohistochemistry for the identification of patients with hereditary nonpolyposis colorectal cancer. JAMA. 2005;293:1986–1994. doi: 10.1001/jama.293.16.1986. [DOI] [PubMed] [Google Scholar]
- 10.Bettstetter M, Dechant S, Ruemmele P, Grabowski M, Keller G, Holinski-Feder E, Hartmann A, Hofstaedter F, Dietmaier W. Distinction of hereditary nonpolyposis colorectal cancer and sporadic microsatellite-unstable colorectal cancer through quantification of MLH1 methylation by real-time PCR. Clin Cancer Res. 2007;13:3221–3228. doi: 10.1158/1078-0432.CCR-06-3064. [DOI] [PubMed] [Google Scholar]
- 11.Ogino S, Cantor M, Kawasaki T, Brahmandam M, Kirkner GJ, Weisenberger DJ, Campan M, Laird PW, Loda M, Fuchs CS. CpG island methylator phenotype (CIMP) of colorectal cancer is best characterised by quantitative DNA methylation analysis and prospective cohort studies. Gut. 2006;55:1000–1006. doi: 10.1136/gut.2005.082933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ogino S, Kawasaki T, Brahmandam M, Cantor M, Kirkner GJ, Spiegelman D, Makrigiorgos GM, Weisenberger DJ, Laird PW, Loda M, Fuchs CS. Precision and performance characteristics of bisulfite conversion and real-time PCR (MethyLight) for quantitative DNA methylation analysis. J Mol Diagn. 2006;8:209–217. doi: 10.2353/jmoldx.2006.050135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nygren AO, Ameziane N, Duarte HM, Vijzelaar RN, Waisfisz Q, Hess CJ, Schouten JP, Errami A. Methylation-specific MLPA (MS-MLPA): simultaneous detection of CpG methylation and copy number changes of up to 40 sequences. Nucleic Acids Res. 2005;33:e128. doi: 10.1093/nar/gni127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bessa X, Balleste B, Andreu M, Castells A, Bellosillo B, Balaguer F, Castellvi-Bel S, Paya A, Jover R, Alenda C, Tito L, Martinez-Villacampa M, Vilella A, Xicola RM, Pons E, Llor X. A prospective, multicenter, population-based study of BRAF mutational analysis for Lynch syndrome screening. Clin Gastroenterol Hepatol. 2008;6:206–214. doi: 10.1016/j.cgh.2007.10.011. [DOI] [PubMed] [Google Scholar]
- 15.Pinol V, Andreu M, Castells A, Paya A, Bessa X, Rodrigo J. Frequency of hereditary non-polyposis colorectal cancer and other colorectal cancer familial forms in Spain: a multicentre, prospective, nationwide study. Eur J Gastroenterol Hepatol. 2004;16:39–45. doi: 10.1097/00042737-200401000-00007. [DOI] [PubMed] [Google Scholar]
- 16.Jover R, Paya A, Alenda C, Poveda MJ, Peiro G, Aranda FI, Perez-Mateo M. Defective mismatch-repair colorectal cancer: clinicopathologic characteristics and usefulness of immunohistochemical analysis for diagnosis. Am J Clin Pathol. 2004;122:389–394. doi: 10.1309/V9PG-K2Y2-60VF-VULR. [DOI] [PubMed] [Google Scholar]
- 17.Xicola RM, Llor X, Pons E, Castells A, Alenda C, Pinol V, Andreu M, Castellvi-Bel S, Paya A, Jover R, Bessa X, Giros A, Duque JM, Nicolas-Perez D, Garcia AM, Rigau J, Gassull MA. Performance of different microsatellite marker panels for detection of mismatch repair-deficient colorectal tumors. J Natl Cancer Inst. 2007;99:244–252. doi: 10.1093/jnci/djk033. [DOI] [PubMed] [Google Scholar]
- 18.Benlloch S, Paya A, Alenda C, Bessa X, Andreu M, Jover R, Castells A, Llor X, Aranda FI, Massuti B. Detection of BRAF V600E mutation in colorectal cancer: comparison of automatic sequencing and real-time chemistry methodology. J Mol Diagn. 2006;8:540–543. doi: 10.2353/jmoldx.2006.060070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eads CA, Danenberg KD, Kawakami K, Saltz LB, Blake C, Shibata D, Danenberg PV, Laird PW. MethyLight: a high-throughput assay to measure DNA methylation. Nucleic Acids Res. 2000;28:E32. doi: 10.1093/nar/28.8.e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deng G, Chen A, Hong J, Chae HS, Kim YS. Methylation of CpG in a small region of the hMLH1 promoter invariably correlates with the absence of gene expression. Cancer Res. 1999;59:2029–2033. [PubMed] [Google Scholar]
- 21.Jeuken JW, Cornelissen SJ, Vriezen M, Dekkers MM, Errami A, Sijben A, Boots-Sprenger SH, Wesseling P. MS-MLPA: an attractive alternative laboratory assay for robust, reliable, and semiquantitative detection of MGMT promoter hypermethylation in gliomas. Lab Invest. 2007;87:1055–1065. doi: 10.1038/labinvest.3700664. [DOI] [PubMed] [Google Scholar]
- 22.Joensuu EI, Abdel-Rahman WM, Ollikainen M, Ruosaari S, Knuutila S, Peltomaki P. Epigenetic signatures of familial cancer are characteristic of tumor type and family category. Cancer Res. 2008;68:4597–4605. doi: 10.1158/0008-5472.CAN-07-6645. [DOI] [PubMed] [Google Scholar]
- 23.Hampel H, Frankel W, Panescu J, Lockman J, Sotamaa K, Fix D, Comeras I, La Jeunesse J, Nakagawa H, Westman JA, Prior TW, Clendenning M, Penzone P, Lombardi J, Dunn P, Cohn DE, Copeland L, Eaton L, Fowler J, Lewandowski G, Vaccarello L, Bell J, Reid G, de la Chapelle A. Screening for Lynch syndrome (hereditary nonpolyposis colorectal cancer) among endometrial cancer patients. Cancer Res. 2006;66:7810–7817. doi: 10.1158/0008-5472.CAN-06-1114. [DOI] [PubMed] [Google Scholar]
- 24.Kolodner RD, Tytell JD, Schmeits JL, Kane MF, Gupta RD, Weger J, Wahlberg S, Fox EA, Peel D, Ziogas A, Garber JE, Syngal S, Anton-Culver H, Li FP. Germ-line msh6 mutations in colorectal cancer families. Cancer Res. 1999;59:5068–5074. [PubMed] [Google Scholar]
- 25.Wahlberg SS, Schmeits J, Thomas G, Loda M, Garber J, Syngal S, Kolodner RD, Fox E. Evaluation of microsatellite instability and immunohistochemistry for the prediction of germ-line MSH2 and MLH1 mutations in hereditary nonpolyposis colon cancer families. Cancer Res. 2002;62:3485–3492. [PubMed] [Google Scholar]
- 26.Vasen HF, Watson P, Mecklin JP, Lynch HT. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC. Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology. 1999;116:1453–1456. doi: 10.1016/s0016-5085(99)70510-x. [DOI] [PubMed] [Google Scholar]
- 27.Nakagawa H, Nagasaka T, Cullings HM, Notohara K, Hoshijima N, Young J, Lynch HT, Tanaka N, Matsubara N. Efficient molecular screening of Lynch syndrome by specific 3′ promoter methylation of the MLH1 or BRAF mutation in colorectal cancer with high-frequency microsatellite instability. Oncol Rep. 2009;21:1577–1583. doi: 10.3892/or_00000390. [DOI] [PubMed] [Google Scholar]
- 28.Domingo E, Laiho P, Ollikainen M, Pinto M, Wang L, French AJ, Westra J, Frebourg T, Espin E, Armengol M, Hamelin R, Yamamoto H, Hofstra RM, Seruca R, Lindblom A, Peltomaki P, Thibodeau SN, Aaltonen LA, Schwartz S., Jr BRAF screening as a low-cost effective strategy for simplifying HNPCC genetic testing. J Med Genet. 2004;41:664–668. doi: 10.1136/jmg.2004.020651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paya A, Alenda C, Pérez-Carbonell L, Rojas E, Soto JL, Guillen C, Castillejo A, Barberá VM, Carrato A, Castells A, Llor X, Andreu M, Koh J, Enders GH, Benlloch S, Jover R. Utility of p16 immunohistochemistry for the identification of Lynch syndrome. Clin Cancer Res. 2009;15:3156–3162. doi: 10.1158/1078-0432.CCR-08-3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hitchins MP, Wong JJ, Suthers G, Suter CM, Martin DI, Hawkins NJ, Ward RL. Inheritance of a cancer-associated MLH1 germ-line epimutation. N Engl J Med. 2007;356:697–705. doi: 10.1056/NEJMoa064522. [DOI] [PubMed] [Google Scholar]
- 31.Gylling A, Ridanpaa M, Vierimaa O, Aittomaki K, Avela K, Kaariainen H, Laivuori H, Poyhonen M, Sallinen SL, Wallgren-Pettersson C, Jarvinen HJ, Mecklin JP, Peltomaki P. Large genomic rearrangements and germline epimutations in Lynch syndrome. Int J Cancer. 2009;124:2333–2340. doi: 10.1002/ijc.24230. [DOI] [PubMed] [Google Scholar]