

Abstract
Population screening has been proposed for Fragile X syndrome to identify premutation carrier females and affected newborns. We developed a PCR-based assay capable of quickly detecting the presence or absence of an expanded FMR1 allele with high sensitivity and specificity. This assay combines a triplet repeat primed PCR with high-throughput automated capillary electrophoresis. We evaluated assay performance using archived samples sent for Fragile X diagnostic testing representing a range of Fragile X CGG-repeat expansions. Two hundred five previously genotyped samples were tested with the new assay. Data were analyzed for the presence of a trinucleotide “ladder” extending beyond 55 repeats, which was set as a cut-off to identify expanded FMR1 alleles. We identified expanded FMR1 alleles in 132 samples (59 premutation, 71 full mutation, 2 mosaics) and normal FMR1 alleles in 73 samples. We found 100% concordance with previous results from PCR and Southern blot analyses. In addition, we show feasibility of using this assay with DNA extracted from dried-blood spots. Using a single PCR combined with high-throughput fragment analysis on the automated capillary electrophoresis instrument, we developed a rapid and reproducible PCR-based laboratory assay that meets many of the requirements for a first-tier test for population screening.
Expansions of polymorphic (CGG)n repeats beyond the normal range in the 5′ untranslated region of the Fragile X Mental Retardation (FMR1) gene on the X chromosome give rise to a variety of developmental and degenerative disorders, such as Fragile X syndrome (FXS), autism, Fragile X-associated tremor/ataxia syndrome (FXTAS) and primary ovarian insufficiency.1,2,3 FXS is the most common inherited form of mental retardation and related intellectual and developmental disabilities in affected individuals.1 In FXS patients, CGG repeats expanded beyond 200 (full mutation) become abnormally methylated.2,3 Hypermethylation of the CpG islands in the 5′-untranslated region of the FMR1 gene in the full-mutation individuals leads to FMR1 gene silencing and complete absence of the gene product FMRP protein.3 FMRP is an RNA-binding protein, which has been shown to play an important role in mRNA translation, dendritic transport of mRNAs, and protein-synthesis dependent synaptic plasticity.2,3,4
The frequency of full mutations in the general population is thought to be approximately 1 in 4000 males and 1 in 8000 females.1,5,6,7 Higher full-mutation allele frequency (1 in 2500 in females) has been documented in some countries.8,9,10,11,12,13
Alleles between 55 and 200 CGG repeats are classified as premutations. CpG islands in premutation FMR1 alleles are not methylated, and carriers of premutation alleles generally have normal levels of FMRP; however, CGG repeats in premutation alleles are unstable and may increase in size from generation to generation by maternal transmission, ultimately resulting in an affected newborn with a fully expanded allele. In fact, CGG expansion from 56 repeats in the mother to full mutation (538 repeats) in a child was reported.9 Recent studies have estimated a premutation allele frequency of 1 in 113–259 females and 1 in 260–800 males.5,10,11 It has been described that premutation allele carriers have higher prevalence of learning disabilities, ADHD, and autism than carriers of non-expanded alleles.12,13 Premutation females have higher rates of primary ovarian insufficiency: 20% in carriers versus 1% in normals.14 The size of the premutation may be a contributing factor to this condition.15 Furthermore, Fragile X-associated tremor/ataxia syndrome (FXTAS), a neurological disorder with symptoms including intention tremor, cerebellar ataxia, memory loss, dementia, and anxiety affects 20–40% premutation male and 8% of premutation female carriers who are over the age of 50.16,17
Molecular testing to diagnose Fragile X syndrome or identify premutation allele carriers has been available for over a decade. Methodologies for FXS diagnostic testing include Southern blot of the genomic DNA and PCR amplification of CGG repeat regions. Methylation-specific PCR can identify males with full mutations. Antibody-based detection of FMRP expression, and recently a mass spectrometry–based assay for FMRP18,19 are less common diagnostic methods. Each method has certain advantages and disadvantages. For example, Southern analysis can detect full mutation, methylation and large premutations, but it cannot detect small premutations. An antibody test for FMRP can be used for affected males but cannot identify female carriers or distinguish between normal and premutation carriers.20
PCR amplification of the CGG repeat region in FMR1 in combination with Southern blot are the most commonly used techniques for CGG repeat detection and sizing.21,22 PCR can easily detect normal alleles and most premutations; however, amplification of large premutations and full mutations is technically challenging. This is particularly difficult when working with specimens from females. An apparently normal homozygous result does not rule out the possibility of preferential amplification of the normal allele in the presence of full mutation that was not amplified by PCR.
Population screening has been proposed to identify premutation allele carrier females and affected newborns.7,23,24,25,26 To make screening viable, a simple, inexpensive test with high sensitivity and specificity is necessary. We previously reported a simple triplet repeat primed PCR (TRP PCR) based assay that amplifies CGG repeats in the FMR1 gene and allows rapid detection of the “stutters” formed by the chimeric primer which binds inside the CGG repeat region.27 Briefly, in TRP PCR, one primer is anchored completely outside of the CGG repeat region, while the other overlaps the CGG repeat and the adjacent unique sequence. Multiple amplicons are produced, each with a length difference of 3 bases. The “stutter” or ‘ladder’ produced on electrophoresis is the basis of this assay. Similar approaches have been reported by other groups using ethidium-stained agarose gels to detect “smears” formed by the chimeric primer.28 In an effort to develop a high-throughput research Fragile X assay capable of identifying the presence or absence of an expanded FMR1 allele in a sample, we combined a TRP PCR28,29,30 using a fluorescently labeled primer with fragment analysis on an Applied Biosystems PRISM 3100 genetic analyzer (3100). In this study we evaluated the assay using archived samples from routine clinical practice representing a range of FMR1 CGG repeats from normal through fully expanded alleles. We also show feasibility of running the TRP PCR assay with dried blood spot (DBS) samples.
Materials and Methods
Samples
Genomic DNAs from the Coriell Institute (http://www.coriell.org, last accessed Dec. 11, 2009) were used as templates in the development of the assay. Sample identification numbers and FMR1 genotypes are listed in Table 1. Genotypes were previously determined using materials and the common platform method as described elsewhere.31 Coriell DNA was diluted to 10 ng/μL before PCR amplification by the new TRP PCR assay.
Table 1.
FMR1 Genotypes for Coriell Samples Used in Proof of Concept Testing
| Coriell ID | Sex | FMR1 genotype |
|---|---|---|
| NA06891 | Male | 118 CGG repeats (premutation) |
| NA11880 | Female | 29/30 repeats (normal) |
| NA12548 | Female | 30/30 repeats (normal) |
| NA06910 | Female | 30/88 repeats (premutation) |
| NA20239 | Female | 20/193 repeats (premutation) |
| NA04025 | Male | 645 repeats (full mutation) |
To validate the new assay, 205 previously characterized blood samples were tested. DNA was extracted from 200 μl (PCR only) or 800 μl (Southern blot) of whole blood using the MagNA Pure LC 2.0 system (Roche Applied Science, Indianapolis, IN). Samples were previously genotyped concurrently using two methods: i) a laboratory-developed and validated PCR sizing test using ASR and GPR reagents obtained from Celera Corporation, Alameda, CA; and ii) a laboratory-developed and validated Southern blot analysis. For dried blood spot feasibility, 50 μl of whole blood was spotted onto 903-specimen collection paper (Whatman Inc., Florham Park, NJ). Circular punches (6 mm in diameter) were made and extracted by one of two methods. The first method follows a manual extraction procedure as described in detail elsewhere.32 The second method follows an automated extraction protocol using the BioSprint96 instrument (Qiagen, Valencia, CA) according to the manufacturer's protocol for dried blood spots. Of the resulting elution, 3 μl was used in the PCR reaction.
PCR
All PCR reagents used in this study were manufactured by Celera Corporation. A pair of PCR primers was designed to generate different sized amplicons depending on the size of the CGG-repeat region. The forward PCR primer is located upstream of the FMR1 CGG region. The fluorescently labeled reverse primer binds inside the CGG region and contains an eight-nucleotide-long tail. PCR reactions (20 μl) were set up as follows: 13 μl of high GC PCR buffer, 1.5 μl of TR PCR enzyme mix, 0.8 μl of FMR1 Primers-2, 1.7 μl of water, and 3 μl of 10 ng/μL DNA template. PCR reactions were set up on ice or on cold blocks. PCR was performed on an ABI GeneAmp PCR System 9700 thermal cycler (Applied Biosystems, Foster City, CA) with cycling conditions of 98.5°C for 30 seconds, 53°C for 30 seconds, and 75°C for 60 seconds for 50 cycles followed by a hold at 4°C. Fifty cycles of PCR allow a sufficient amount of PCR product for high fluorescent signals, thus increasing robustness for clinical testing from blood and dried blood spots.
Capillary Electrophoresis Analysis
Hi-Di formamide, 20 μl (Applied Biosystems), and 2 μl of ROX 1000 size standard (Celera Corporation) were combined with 2 μl of PCR product. Samples were denatured at 95°C for 2 minutes before loading onto an ABI 3100 with POP-6 polymer on a 36-cm or 50-cm array. See Table 2 for injection conditions. Data from the run were visualized using either GeneMapper v.3.7 (Applied Biosystems) or GeneMarker (SoftGenetics, State College, PA) software. A threshold of 55 CGG repeats was set to distinguish between normal and expanded FMR1 alleles. The size (in base pairs) of the threshold was calculated using the following formula: No. of CGG repeats = (peak size − 134)/3, which is based on the primer binding sites and the number of base pairs in the amplicon excluding CGG repeats. The sample was considered normal if the CGG repeat ladder in the electropherogram did not pass the threshold. The sample was identified as having an expanded allele if the CGG repeat ladder crossed the threshold.
Table 2.
ABI 3100 Run Module Settings for Data Collection v.2.0 (36-cm Array) and v.1.1 (50-cm Array)
| Parameter name | v. 2.0 value (36 cm array) | v. 1.1 value (50 cm array) |
|---|---|---|
| Run temperature | 60°C | 60°C |
| Cap fill volume | n/a | 184 steps |
| Current tolerance | n/a | 100 μA |
| Run current | n/a | 100 μA |
| Voltage tolerance | n/a | 0.6 kV |
| Pre run voltage | 15 kV | 15 kV |
| Pre run time | 180 seconds | 180 seconds |
| Injection voltage | 15 kV | 15 kV |
| Injection time | 8 seconds | 8 seconds |
| Run voltage | 15 kV | 15 kV |
| Number of steps | 20 nk | 20 nk |
| Voltage step interval | 30 seconds | 30 seconds |
| Data delay time | 240 seconds | 240 seconds |
| Run time | 2000 seconds | 6500 seconds |
Results
Coriell Samples—Proof of Concept
Genomic Coriell DNA samples representing a range of FMR1 genotypes were used to optimize the conditions of the FMR1 TRP PCR and to establish run parameters for fragment analysis. PCR products were analyzed on an ABI 3100 with a 36-cm capillary array and POP-6 polymer. A “ladder” was observed for CGG repeats with each peak occurring 3 bp apart. Using standard analysis tools, an assay can be configured to provide a qualitative “yes” or “no” answer using a predefined cut-off value based on the termination of the “ladder” relative to the cut-off. In this study, we chose 55 repeats (300 bp) as the cut-off. A “no” is obtained when the ladder does not extend beyond 55 repeats (the sample does not contain an expanded allele and, therefore, does not require additional analysis). A “yes” is obtained when the ladder extends beyond 55 repeats (the sample has an expanded allele and requires further testing). Figure 1, A–G, shows representative electropherograms from the six Coriell DNA templates tested. Each sample was correctly classified as either normal or having an expanded allele present based on genotypes obtained from allele sizing PCR combined with fragment analysis (Table 1).
Figure 1.
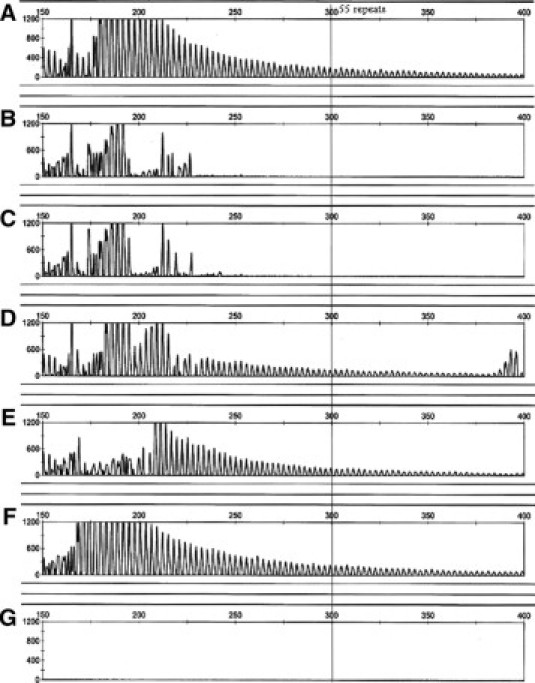
Representative screening electropherograms for previously genotyped Coriell samples with normal or expanded CGG alleles. Genotypes are noted below. The threshold cutoff for normal alleles of 55 repeats is shown. A sample with a ladder motif extending beyond the 55 repeat threshold was considered to have an expanded allele. CGG repeat sizes A: Coriell NA06891, premutation male with 118 CGG repeats. B: Coriell NA11880, normal female heterozygote with 29 and 30 repeats. C: Coriell NA12548, normal female homozygote with 30 repeats. D: Coriell NA06910, premutation female heterozygote with 30 and 88 repeats. E: Coriell NA20239, premutation female heterozygote with 20 and 193 repeats. F: Coriell NA04025, full-mutation male with ∼645 repeats. G: No template.
Assay Validation
Two hundred five previously genotyped DNA samples were tested to validate the new assay in a clinical laboratory environment. Samples were amplified using FMR1 TRP PCR reagents and run on an ABI 3100 with a 50-cm array (laboratory preference) and POP-6 polymer. Equal result quality was seen with either 36-cm or 50-cm arrays. Table 3 summarizes the results. There were 132 samples with expanded alleles (59 premutations, 71 full mutations, 2 mosaics) and 73 normal/intermediate samples. All 205 samples showed 100% concordance with previous results obtained with sizing PCR and Southern blot analysis. Thirteen of the samples were assayed in triplicate for within-run variability and repeated over three separate runs for between-run variability. Each replicate yielded consistent results across all runs (data not shown).
Table 3.
Summary of Results for 205 Previously Genotyped Samples
| Number of samples | Genotype (sizing) classification | Screening classification |
|---|---|---|
| 73 | Normal/intermediate | Normal |
| 59 | Premutation | Expanded |
| 71 | Full mutation | Expanded |
| 2 | Mosaic | Expanded |
Assay Specificity
The specificity of the assay is its ability to identify true negatives within the test population. In this case, we defined negative as below the premutation range at 55 repeats. The validation runs included intermediate allele samples (45–54 CGG repeats). Figure 2, A–D, shows the electropherograms for four representative intermediate allele samples. Note that the ladder motif abruptly ends before the threshold mark of 55 repeats. The sudden increase in peak height just before the ladder ends is a result of the reverse primer design, which shares additional homology with sequence adjacent to the 3′ end of the CGG region of the FMR1 promoter. The added stability due to this homology of the reverse primer to template DNA allows for more efficient amplification of the entire CGG region, resulting in greater signal. The intensity of the signal (due to a full length amplification of the template) will decrease as the CGG region increases. A rescaled electropherogram (Figure 2D, inset) shows clearly the difference between the “stutter”' and a high allele with fluorescent levels returning to background.
Figure 2.
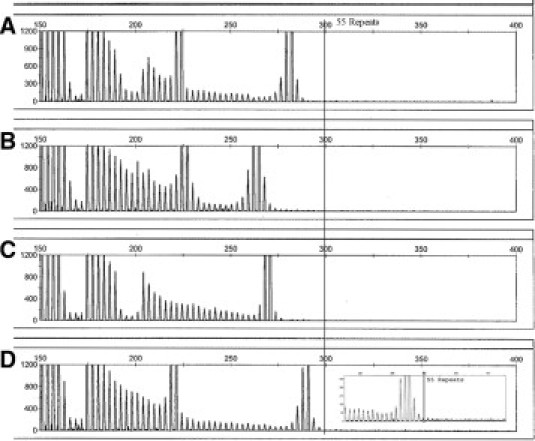
Screening electropherograms for intermediate allele (45–54) samples. Samples were previously genotyped by sizing and/or Southern blot. A: 30 and 50 repeats. B: 31 and 45 repeats. C: 47 repeats. D: 29 and 54 repeats (inset: rescaled y axis).
Assay Sensitivity
To test the ability of the new assay to detect mosaicism, mock mosaic samples were prepared by serial dilution of the full-mutation Coriell DNA (NA04025) with a normal Coriell DNA (NA12548). Each mock mosaic sample was tested by FMR1 TRP PCR assay followed by fragment analysis on the 3100 (Figure 3, A–E). The results show that expanded alleles can be detected at levels as low as 12.5% in the sample (3.75 ng of 30 ng total DNA in the reaction). We also detected expanded alleles in both mosaic clinical samples, although the level of mosaicism was not determined.
Figure 3.

Screening electropherograms for mock mosaic dilutions. Coriell NA04025 (full-mutation male) was serially diluted twofold with Coriell NA12548 (normal female) and then processed with the FX Screening assay. The electropherograms were rescaled to visualize the difference between a “stutter” and baseline. A: Undiluted NA12548 (normal) 30 ng/rxn. B: Undiluted NA04025 (full mutation) 30 ng/rxn. C: First dilution—each Coriell at 15 ng/rxn. D: Second dilution—Coriell NA04025 at 7.5 ng/rxn and NA12548 at 22.5 ng/rxn. E: Third dilution—Coriell NA04025 at 3.75 ng/rxn and NA12548 at 26.25 ng/rxn.
DBS Feasibility
DNA extracted from 14 DBS samples, covering the range of FMR1 genotypes, was tested to show feasibility for newborn screening. DNA was extracted with a manual procedure (see Materials and Methods) and with the Qiagen BioSprint96 instrument. Figure 4, A–F, shows the electropherograms obtained for six samples extracted using the BioSprint96 instrument. The results show robust signals for all FX genotypes tested. Results from the BioSprint96 extractions yielded higher signals than results from the manual extraction (data not shown). This observation may be a result of the amount of starting material used (a 6-mm disk for BioSprint, 3-mm disk for manual extraction). The use of an automated extraction protocol to process DBS samples demonstrates the feasibility of establishing the FMR1 TRP PCR assay in a high-throughput environment necessary for population screening.
Figure 4.

Feasibility of using dried blood spots as a sample source the FX Screening assay. One 6-mm punch from each blood spot was extracted on the Qiagen BioSprint96 following the standard protocol. A: Female intermediate. B: Female premutation. C: Male full mutation. D: Male intermediate. E: Male normal. F: Female full mutation.
Discussion
Diagnostic testing for Fragile X has traditionally included both Southern blot analysis to detect large expansions (higher premutation alleles through full-mutation alleles) and methylation status, and PCR to accurately size normal, intermediate, and smaller premutation alleles. PCR is routinely able to accurately size up to 100 repeats,31 although higher repeats may be detected. In fact, amplification into the full-mutation range is possible.31 The combination of Southern blots and PCR results in a highly sensitive and specific test for Fragile X premutations, full mutations, and mosaics (GeneReviews: FMR1-Related Disorders, http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=fragile, last accessed Dec. 8, 2009).
Since Southern blot analysis is costly and time consuming, methods have been sought to reduce the number of Southern blots. A testing algorithm may begin with a PCR sizing assay and reflex to Southern blot under the following scenarios: i) only one allele is detected in females (apparent homozygotes); ii) no alleles are detected in males; or iii) a premutation allele is detected (to identify premutation/full-mutation mosaics). In cases of apparent homozygosity in females, the normal allele amplifies more efficiently, and therefore, the expanded allele could fail to amplify sufficiently to be detected. Efforts have been made to quantitatively determine the presence of 1 or 2 copies of alleles in females by comparison with a reference (2 copies) sequence (E. Lyon and C.S. Richards, personal communications). Although results were promising, this method is not yet robust enough to use clinically. In rare cases, full-mutation/normal allele mosaic males could be missed completely.
Screening for FMR1 expanded alleles, either for carriers or for newborns, has long been discussed.5,7,8,23,24,25,26 Affected children may be born into families without a positive history, and the diagnosis can be delayed for several years. Screening for carriers would alert families to the possibility of having a child with Fragile X syndrome, allowing them options such as prenatal testing or family planning. An early diagnosis for newborns (or within the first year) could provide early behavioral intervention for the child and information for parents before a second at-risk child is born. However, screening requires an inexpensive, high-throughput assay, meaning that Southern blot analysis will not be able to be run on every sample. For newborn screening, scenarios have been described to identify methylated alleles in affected males, but this will not identify affected females.26,33 A methylation-sensitive assay would not be useful for premutation carrier testing in females. Chimeric primers and agarose gels have also been described, but since agarose gels are not conducive to high throughput, and “smears” may be difficult to score, it has been used as a second-tier test.28 In this example, the number of samples clearly identified as negative after the first-tier PCR is less than 50% for females.28 The majority of samples will need further testing.26,33 Such methods also depend on accurately identifying females and males, since algorithms may be different based on gender. As gender often is not included or is labeled incorrectly, additional follow-up from the laboratory would be necessary.
In contrast, combining a chimeric primer with single base resolution fragment analysis, the “smear” seen on a lower resolution agarose gel becomes “stutters” or “ladders” that are easily distinguished from background. A cut-off at 55 repeats (or whatever number a laboratory chooses to validate) can be set that will accurately distinguish premutation and full-mutation alleles from normal and intermediate alleles. Expanded alleles are detected in both males and females and therefore gender confirmation is no longer necessary for the FX screen. Our method resolves the challenges associated with apparent homozygous female and normal/mosaic males, since mosaicism may be detected down to 12.5%. By combining automated sample prep with capillary electrophoresis, this approach is conducive to high-throughput applications, with a possibility of 96 samples/24 hours. Higher throughput may be achieved by adapting the protocol for use on an Applied Biosystems 3730xl DNA analyzer. All samples with non-expanded alleles will be identified as such and require no further testing (Figure 5). Any allele that is identified as expanded by the screening approach can then be tested further by the “diagnostic” test, such as Southern blot and/or sizing by PCR. This single TRP-PCR/fragment analysis method can be used for both carrier and newborn screening as a first-tier test. By using this algorithm, the number of samples requiring Southern analysis is reduced by 98%. Of those samples reflexed to further testing, approximately 5 to 7% will be affected with full mutations, with the rest being premutations.
Figure 5.

Proposed testing strategy for the implementation of the TRP PCR assay for population screening. The model describes the algorithm which uses the TRP PCR assay as a first-tier test to decrease the number of samples that require diagnostic testing (sizing by PCR or Southern blot) from 50,000 to 660 samples based on the frequency of premutation and full-mutation carriers in the general population.
In this study we set the cut-off at 55 repeats, consistent with premutations. By not including the intermediate alleles, women at risk for affected children would be identified, while women at risk for affected grandchildren (an intermediate allele expanded to a premutation in a daughter) would not be identified. In this scenario the daughters would be identified in the next generation of screening. This would avoid a difficult counseling issue. To be conservative, the cut-off could be lowered to 50 repeats (284 bp) to ensure accurate sizing of the boundaries between intermediate and premutations. The cut-off could be further reduced to include the intermediate alleles, setting the cut-off at 45 repeats (∼269 bp), if this assay was to be used as a first-tier diagnostic test as well. Alternatively, when accurate sizing is desired for intermediate alleles, accurate PCR sizing tests are available.
A necessary component of any screening assay is low cost. Currently, the cost of an assay built using these reagents is comparable with the cost of cystic fibrosis carrier screening, removing this barrier to premutation carrier screening. For newborn screening to be broadly adopted, it is likely that costs will need to be reduced by several fold, which may be achievable at high volumes of testing.
We describe a simple research assay and a test algorithm that is suitable for population screening. This single PCR followed by capillary electrophoresis at a single base resolution has a high sensitivity and specificity for detecting expanded alleles. The simple yes/no answer for expansions is suitable for a first-tier screening test. If no expansion is detected, no further testing is necessary. When an expansion is detected, the sample is reflexed to a diagnostic test including Southern blot analysis and accurate sizing by PCR. This provides a possible solution for Fragile X screening either at a carrier or newborn screening level.
Footnotes
Supported in part by Associated Regional and University Pathologists (ARUP) and Celebra.
T.L., K.Y., M.Z., and N.M. are employed by Celera. E.L. discloses the receipt of two honoraria from Celera. Celera provided reagents to ARUP for assay validation purposes.
References
- 1.Turner G, Webb T, Wake S, Robinson H. Prevalence of fragile X syndrome. Am J Med Genet. 1996;64:196–197. doi: 10.1002/(SICI)1096-8628(19960712)64:1<196::AID-AJMG35>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 2.O'Donnell WT, Warren ST. A decade of molecular studies of fragile X syndrome. Annu Rev Neurosci. 2002;25:315–338. doi: 10.1146/annurev.neuro.25.112701.142909. [DOI] [PubMed] [Google Scholar]
- 3.Bechara EG, Didiot MC, Melko M, Davidovic L, Bensaid M, Martin P, Castets M, Pognonec P, Khandjian EW, Moine H, Bardoni B. A novel function for fragile X mental retardation protein in translational activation. PLoS Biol. 2009;7:e16. doi: 10.1371/journal.pbio.1000016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bourgeois JA, Coffey SM, Rivera SM, Hessl D, Gane LW, Tassone F, Greco C, Finucane B, Nelson L, Berry-Kravis E, Grigsby J, Hagerman PJ, Hagerman RJ. A review of fragile X premutation disorders: expanding the psychiatric perspective. J Clin Psychiat. 2009;70:852–862. doi: 10.4088/JCP.08m04476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Toledano-Alhadef H, Basel-Vanagaite L, Magal N, Davidov B, Ehrlich S, Drasinover V, Taub E, Halpern GJ, Ginott N, Shohat M. Fragile-X carrier screening and the prevalence of premutation and full-mutation carriers in Israel. Am J Hum Genet. 2001;69:351–360. doi: 10.1086/321974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crawford DC, Meadows KL, Newman JL, Taft LF, Scott E, Leslie M, Shubek L, Holmgreen P, Yeargin-Allsopp M, Boyle C, Sherman SL. Prevalence of the fragile X syndrome in African-Americans. Am J Med Genet. 2002;110:226–233. doi: 10.1002/ajmg.10427. [DOI] [PubMed] [Google Scholar]
- 7.Song FJ, Barton P, Sleightholme V, Yao GL, Fry-Smith A. Screening for fragile X syndrome: a literature review and modelling study. Health Technol Assess. 2003;7:1–106. doi: 10.3310/hta7160. [DOI] [PubMed] [Google Scholar]
- 8.Pesso R, Berkenstadt M, Cuckle H, Gak E, Peleg L, Frydman M, Barkai G. Screening for fragile X syndrome in women of reproductive age. Prenat Diagn. 2000;20:611–614. doi: 10.1002/1097-0223(200008)20:8<611::aid-pd881>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 9.Fernandez-Carvajal I, Lopez Posadas B, Pan R, Raske C, Hagerman PJ, Tassone F. Expansion of an FMR1 grey-zone allele to a full mutation in two generations. J Mol Diagn. 2009;11:306–310. doi: 10.2353/jmoldx.2009.080174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dombrowski C, Levesque S, Morel ML, Rouillard P, Morgan K, Rousseau F. Premutation and intermediate-size FMR1 alleles in 10572 males from the general population: loss of an AGG interruption is a late event in the generation of fragile X syndrome alleles. Hum Mol Genet. 2002;11:371–378. doi: 10.1093/hmg/11.4.371. [DOI] [PubMed] [Google Scholar]
- 11.Fernandez-Carvajal I, Walichiewicz P, Xiaosen X, Pan R, Hagerman PJ, Tassone F. Screening for expanded alleles of the FMR1 gene in blood spots from newborn males in a Spanish population. J Mol Diagn. 2009;11:324–329. doi: 10.2353/jmoldx.2009.080173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Farzin F, Perry H, Hessl D, Loesch D, Cohen J, Bacalman S, Gane L, Tassone F, Hagerman P, Hagerman R. Autism spectrum disorders and attention-deficit/hyperactivity disorder in boys with the fragile X premutation. J Dev Behav Pediatr. 2006;27:S137–S144. doi: 10.1097/00004703-200604002-00012. [DOI] [PubMed] [Google Scholar]
- 13.Goodlin-Jones BL, Tassone F, Gane LW, Hagerman RJ. Autistic spectrum disorder and the fragile X premutation. J Dev Behav Pediatr. 2004;25:392–398. doi: 10.1097/00004703-200412000-00002. [DOI] [PubMed] [Google Scholar]
- 14.Allingham-Hawkins DJ, Babul-Hirji R, Chitayat D, Holden JJ, Yang KT, Lee C, Hudson R, Gorwill H, Nolin SL, Glicksman A, Jenkins EC, Brown WT, Howard-Peebles PN, Becchi C, Cummings E, Fallon L, Seitz S, Black SH, Vianna-Morgante AM, Costa SS, Otto PA, Mingroni-Netto RC, Murray A, Webb J, MacSwinney F, Dennis N, Jacobs PA, Syrrou M, Georgiou I, Patsalis PC, Giovannucci Uzielli ML, Guarducci S, Lapi E, Cecconi A, Ricci U, Ricotti G, Biondi C, Scarselli B, Vieri F. Fragile X premutation is a significant risk factor for premature ovarian failure: the International Collaborative POF in Fragile X study–preliminary data. Am J Med Genet. 1999;83:322–325. [PMC free article] [PubMed] [Google Scholar]
- 15.Murray A. Premature ovarian failure and the FMR1 gene. Semin Reprod Med. 2000;18:59–66. doi: 10.1055/s-2000-13476. [DOI] [PubMed] [Google Scholar]
- 16.Hagerman PJ, Hagerman RJ. Fragile X-associated tremor/ataxia syndrome (FXTAS) Ment Retard Dev Disabil Res Rev. 2004;10:25–30. doi: 10.1002/mrdd.20005. [DOI] [PubMed] [Google Scholar]
- 17.Jacquemont S, Hagerman RJ, Leehey M, Grigsby J, Zhang L, Brunberg JA, Greco C, Des Portes V, Jardini T, Levine R, Berry-Kravis E, Brown WT, Schaeffer S, Kissel J, Tassone F, Hagerman PJ. Fragile X premutation tremor/ataxia syndrome: molecular, clinical, and neuroimaging correlates. Am J Hum Genet. 2003;72:869–878. doi: 10.1086/374321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iwahashi C, Tassone F, Hagerman RJ, Yasui D, Parrott G, Nguyen D, Mayeur G, Hagerman PJ. A quantitative ELISA assay for the fragile x mental retardation 1 protein. J Mol Diagn. 2009;11:281–289. doi: 10.2353/jmoldx.2009.080118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dodds ED, Tassone F, Hagerman PJ, Lebrilla CB. Polymerase chain reaction, nuclease digestion, and mass spectrometry based assay for the trinucleotide repeat status of the fragile X mental retardation 1 gene. Anal Chem. 2009;81:5533–5540. doi: 10.1021/ac9008918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pandey UB, Phadke SR, Mittal B. Molecular diagnosis and genetic counseling for fragile X mental retardation. Neurol India. 2004;52:36–42. [PubMed] [Google Scholar]
- 21.Haddad LA, Mingroni-Netto RC, Vianna-Morgante AM, Pena SD. A PCR-based test suitable for screening for fragile X syndrome among mentally retarded males. Hum Genet. 1996;97:808–812. doi: 10.1007/BF02346194. [DOI] [PubMed] [Google Scholar]
- 22.Brown WT, Nolin S, Houck G, Jr, Ding X, Glicksman A, Li SY, Stark-Houck S, Brophy P, Duncan C, Dobkin C, Jenkins E. Prenatal diagnosis and carrier screening for fragile X by PCR. Am J Med Genet. 1996;64:191–195. doi: 10.1002/(SICI)1096-8628(19960712)64:1<191::AID-AJMG34>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 23.Fanos JH, Spangner KA, Musci TJ. Attitudes toward prenatal screening and testing for Fragile X. Genet Med. 2006;8:129–133. doi: 10.1097/01.gim.0000200158.66554.7f. [DOI] [PubMed] [Google Scholar]
- 24.Musci TJ, Caughey AB. Cost-effectiveness analysis of prenatal population-based fragile X carrier screening. Am J Obstet Gynecol. 2005;192:1905–1912. doi: 10.1016/j.ajog.2005.02.052. discussion 1912–1915. [DOI] [PubMed] [Google Scholar]
- 25.Saul RA, Friez M, Eaves K, Stapleton GA, Collins JS, Schwartz CE, Stevenson RE. Fragile X syndrome detection in newborns-pilot study. Genet Med. 2008;10:714–719. doi: 10.1097/GIM.0b013e3181862a76. [DOI] [PubMed] [Google Scholar]
- 26.Coffee B, Keith K, Albizua I, Malone T, Mowrey J, Sherman SL, Warren ST. Incidence of fragile X syndrome by newborn screening for methylated FMR1 DNA. Am J Hum Genet. 2009;85:503–514. doi: 10.1016/j.ajhg.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu P, Sinitsyna I, Lyon E. A precise sizing method for fragile X (CGG)n repeats (Abstract) J Mol Diagn. 2006;8:627. [Google Scholar]
- 28.Tassone F, Pan R, Amiri K, Taylor AK, Hagerman PJ. A rapid polymerase chain reaction-based screening method for identification of all expanded alleles of the fragile X (FMR1) gene in newborn and high-risk populations. J Mol Diagn. 2008;10:43–49. doi: 10.2353/jmoldx.2008.070073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Warner JP, Barron LH, Goudie D, Kelly K, Dow D, Fitzpatrick DR, Brock DJ. A general method for the detection of large CAG repeat expansions by fluorescent PCR. J Med Genet. 1996;33:1022–1026. doi: 10.1136/jmg.33.12.1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ciotti P, Di Maria E, Bellone E, Ajmar F, Mandich P. Triplet repeat primed PCR (TP PCR) in molecular diagnostic testing for Friedreich ataxia. J Mol Diagn. 2004;6:285–289. doi: 10.1016/S1525-1578(10)60523-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Amos Wilson J, Pratt VM, Phansalkar A, Muralidharan K, Highsmith WE, Jr, Beck JC, Bridgeman S, Courtney EM, Epp L, Ferreira-Gonzalez A, Hjelm NL, Holtegaard LM, Jama MA, Jakupciak JP, Johnson MA, Labrousse P, Lyon E, Prior TW, Richards CS, Richie KL, Roa BB, Rohlfs EM, Sellers T, Sherman SL, Siegrist KA, Silverman LM, Wiszniewska J, Kalman LV. Consensus characterization of 16 FMR1 reference materials: a consortium study. J Mol Diagn. 2008;10:2–12. doi: 10.2353/jmoldx.2008.070105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Larsen LA, Gronskov K, Norgaard-Pedersen B, Brondum-Nielsen K, Hasholt L, Vuust J. High-throughput analysis of fragile X (CGG)n alleles in the normal and premutation range by PCR amplification and automated capillary electrophoresis. Hum Genet. 1997;100:564–568. doi: 10.1007/s004390050552. [DOI] [PubMed] [Google Scholar]
- 33.Tzeng CC, Liou CP, Li CF, Lai MC, Tsai LP, Cho WC, Chang HT. Methyl-CpG-binding PCR of bloodspots for confirmation of fragile X syndrome in males. J Biomed Biotechnol. 2009;2009:643692. doi: 10.1155/2009/643692. [DOI] [PMC free article] [PubMed] [Google Scholar]