

Abstract
Donor cell neoplasms are rare complications of treatment regimens that involve stem cell transplantation for hematological malignancies, myelodysplastic processes, or certain genetic or metabolic disorders. We report a case of donor cell leukemia in a pediatric patient with a history of acute myeloid leukemia that manifested as recurrent AML FAB type M5 fourteen months after umbilical cord blood transplantation. Although there was some immunophenotypic drift from the patient's original AML and their posttransplant presentation, the initial pathological impression was of recurrent disease. Bone marrow engraftment analysis by multiplex PCR of short tandem repeat markers performed on the patient's diagnostic specimen showed complete engraftment by donor cells, with a loss of heterozygosity in the donor alleles on chromosome 7. This led to the reinterpretation of this patient's disease as donor-derived leukemia. This interpretation was supported by a routine karyotype and fluorescence in situ hybridization analysis showing loss of chromosome 7 and a male (donor) chromosome complement in this female patient. Also noted was a loss of the patient's presenting chromosomal abnormality, t(11;19)(q23;p13). This case highlights the need for close coordination between all aspects of clinical testing for the transplant patient, including molecular engraftment studies, when distinguishing the very common complication of recurrent disease from the exceedingly rare complication of donor cell leukemia.
Unrelated donor umbilical cord blood transplantation (UCBT) is an increasingly used treatment in pediatric patients with a variety of different diseases, including neoplastic hematological diseases such as acute myeloid leukemia and myelodysplastic syndromes1,2 and inherited disorders of metabolism, including lysosomal and peroxisomal storage diseases.3 The growing adoption of UCBT stems both from its efficacy in treating these diseases and from its two distinct benefits over traditional bone marrow transplantation (BMT) and peripheral blood stem cell (progenitor cell) transplantation (PBPC): the availability of donor umbilical cord blood stem cells and the decrease in certain complications including acute and chronic graft versus host disease.4,5 In the context of neoplastic indications, an additional devastating complication that occurs in similar frequency in BMT, PBPC, and UCBT is recurrence of the patient's original disease. In pediatric patients receiving UCBT for acute lymphoblastic leukemia, overall three-year leukemia-free survival ranges from 20 to 35% depending on timing of transplantation (i.e., during the first or second complete remission, CR1 or CR2, or in patients with advanced disease). Similar rates of recurrence have been reported for pediatric patients with AML receiving UCBT (two-year leukemia-free survival of 22% in patients with advanced disease, 43% in patients treated during CR1 and 65% for patients treated during CR2; reviewed in 5).
Although very rare compared with recurrent disease, secondary neoplastic complications also occur in transplant patients. The best described are posttransplant lymphoproliferative disorders (PTLD). PTLD is an Epstein-Barr virus–driven neoplastic proliferation, usually of B-cells, in transplant patients that can progress from polymorphic PTLD, which has a good prognosis, to the more frankly malignant monomorphic PTLD, which includes diffuse large B-cell lymphoma, Burkitt lymphoma, plasmacytoma, and myeloma. PTLD is a rare complication in the setting of UCBT, occurring in fewer than 2% of patients.6,7 Although PTLD in solid organ transplants is usually of recipient origin, in patients who have had successful UCBT, PTLD is usually of donor origin.6,7,8,9
An even less common secondary neoplastic complication seen in BMT and UCBT patients is donor cell leukemia (DCL). Because of its rarity, the exact percentage of stem cell transplant patients that develop DCL is difficult to determine (reviewed in 10). In one large survey study, only 14 cases of donor cell leukemia were identified among approximately 10,000 transplant patients, making the incidence around 0.1%.11 However, some studies suggest the incidence may be much higher (2 of 40 patients in one institutional experience).12 Given that UCBT is a relatively new approach for stem cell transplantation, donor cell leukemia in UCBT patients is a very rarely reported event. A little more than 50 cases of donor cell leukemia have been reported in the literature in BMT, PBPC, and UCBT patients (reviewed in 10,13–18). Only ten cases of DCL have been previously described in UCBT patients.15,16,19,20,21
Here we report a new case of DCL after umbilical cord blood transplantation that manifested as a phenotypic recurrence of acute myeloid leukemia. This case was detected because of two incidental unusual findings on a PCR-based short tandem repeat (STR) analysis for bone marrow engraftment: maintained engraftment in the face of recurrent disease, and a loss of heterozygosity on chromosome 7 in cells of donor origin. The correct diagnosis of DCL in this patient by engraftment analysis was essentially an incidental finding and not the primary objective of the test. This case demonstrates how easily this could have been diagnosed as a simple recurrence and lends further weight to the assertion that donor cell leukemias are likely under-diagnosed. It also highlights the need for close coordination between all aspects of clinical testing for the transplant patient and the need for close attention to unusual or unexpected bone marrow engraftment patterns.
Materials and Methods
Case Reports
The patient is a 3-year-old female who was first diagnosed with acute myeloid leukemia with CNS involvement at the age of 7 months. By flow cytometry, the leukemic cells were positive for CD4, CD14, CD64, and CD36, with partial expression of CD15, CD13, CD33, HLA-DR, and MPO. This is a typical cell surface antigen expression pattern for acute monoblastic leukemia (previously designated as AML FAB M5a). Cytogenetics showed t(11;19)(q23;p13). This translocation most likely represents the t(11;19)(q23;p13.3) MLL-ENL translocation associated with AML M4/M5 in children less than 1-year-old and not the t(11;19)(q23;p13.3) MLL-ELL translocation that is very difficult to detect with traditional G-banding. Histological and clinical laboratory features of this patient's leukemia are summarized in Table 1.
Table 1.
Histologic and Immunophenotypic Findings
| Study | Results |
|---|---|
| Original diagnostic findings | |
| Histology | 90% monoblasts on bone marrow biopsy |
| Histochemistry | Myeloperoxidase positive |
| Flow cytometry | Positive for CD4, CD14, CD64, CD36; partial positivity for CD13, CD15, CD33, HLA-DR, and MPO |
| Cytogenetics | t(11;19)(q23;p13) detected; negative for t(9;22), t(4;11), and inv16 |
| Diagnostic findings at the time of “recurrence” | |
| Histology | Blasts with large nuclei with 1–3 nucleoli; occasional cytoplasmic granules; no Auer rods |
| Flow cytometry | Positive for CD13, CD33, CD117, CD34, HLA-DR, CD4, CD7, CD38, and CD71 |
| Cytogenetics | Monosomy 7; negative for 11q23 translocation |
The patient was treated according to the Pediatric Oncology Group protocol 9421 with cytarabine (including an intrathecal dose), daunorubicin, and thioguanine.22 She achieved a complete remission after the first cycle of therapy and remained disease-free for approximately nine months, at which time she presented to her physician with leg pain. Peripheral blood analysis and cerebrospinal fluid cytology showed monoblasts (25% circulating blasts by flow cytometry). Cytogenetic analysis showed persistence of the t(11;19)(q23;p13) translocation consistent with recurrence of this patient's original neoplastic clone.
The patient was treated with 2-CDA, intrathecal methotrexate, hydrocortisone, and cytarabine. Although this course of chemotherapy was associated with a number of severe complications, including pancytopenia, Clostridium difficile colitis, neutropenic enterocolitis, and respiratory distress, the therapy was successful and the patient was free of CNS and bone marrow disease at the end of treatment. The original and relapse diagnosis and treatment occurred elsewhere, and she was referred to our facility for UCBT. At the time of transplantation (approximately 1.5 months after the completion of therapy), she remained free of disease.
The patient was conditioned for transplant using busulfan (14 mg IV every 6 hours for 16 doses) followed by melphalan (3 does of 26 mg) and anti-thymocyte globulin (411 mg IV daily for 3 doses). She then received an unrelated umbilical cord blood transplant that was from an ABO type O-positive male donor. Maternal granulocytes were also infused on days 1–29. The patient received low-dose heparin drip for veno-occlusive disease prophylaxis, IV hydration, and total parenteral nutrition starting at day 0 for anorexia. She also received CellCept, cyclosporine, and weekly IVIG for graft versus host disease prophylaxis. Prophylactic antimicrobials included voriconazole, acyclovir, and Bactrim pretransplant then pentamidine posttransplant for PCP prophylaxis. The success of the patient's UCBT was monitored by an STR based chimerism assay as described in the results section below.
Pathologic Evaluation and Bone Marrow Engraftment Studies
Bone marrow aspirates were prepared and stained with a Wright stain using standard protocols. Flow cytometric analysis was performed on bone marrow samples using a BD FACSCalibur flow cytometer (BD BioSciences, San Jose, CA) with antibodies from BD BioSciences, San Jose, CA. Computer software used for gating was Paint-A-Gate (BD BioSciences, San Jose, CA) and CellQuest (BD BioSciences, San Jose, CA). Karyotyping was performed on bone marrow aspirates using standard protocols; approximate band resolution was 350. Interphase fluorescence in situ hybridization (FISH) analysis on bone marrow aspirates was performed using the dual color Vysis (Abbott Molecular, Abbott Park, IL) probes CEP7 and LSI D7S486, which identify the chromosome 7 centromere and a locus on 7q, respectively.
Bone marrow engraftment analysis was performed using PCR-mediated amplification and subsequent size analysis of STRs to determine the recipient or donor cell composition of the patient's peripheral blood and bone marrow. For fractionated samples, lymphocytes or granulocytes were first isolated from posttransplant samples using magnetically labeled anti-human CD3 or CD15 antibodies (isotype: mouse IgG1 and IgM kappa, respectively) and the RoboSep automated cell separator (StemCell Technologies, Vancouver, BC, Canada). Genomic DNA was then extracted from the purified fractionated cells and/or the whole unfractionated sample. This DNA was used in a multiplexed PCR-mediated amplification reaction targeting a total of eight autosomal STRs (D5S818, D13S317, D7S20, D16S539, vWA, TH01, TPOX, and CSF1P0) and one STR marker on the pseudo-autosomal region of the X and Y chromosomes, amelogenin (Promega Corporation, Madison, WI). After PCR amplification, fluorescently labeled PCR products were resolved by capillary electrophoresis on the ABI 3130xl Genetic Analyzer (Applied Biosystems, Carlsbad, California). GeneMapper software (Applied Biosystems, Carlsbad, California) was used to calculate the number of repeats and relative abundance of each repeat for each STR locus. These data were then used to calculate the percentage of donor and recipient cells in the original sample using donor and pretransplant recipient STR profiles. As little as a 2% population of donor or recipient cells can be detected with this assay.
Results
The success of the patient's UCBT was closely followed using a laboratory-developed bone marrow engraftment analysis assay that consists of PCR-mediated amplification and subsequent size analysis of eight polymorphic STRs: D5S818, D13S317, D7S20, D16S539, vWA, TH01, TPOX, and CSF1P0. Bone marrow and/or blood analysis at 1, 3, 6, and 9 months showed identical findings, eight informative markers with >98% male donor cells and no definite recipient or maternal cells. Figure 1 shows the results for one STR marker, D7S820 on chromosome 7, at nine months posttransplant for a peripheral blood specimen. For this marker, the patient (recipient) cells, UCBT donor cells, and maternal granulocyte donor cells are heterozygous for different repeat lengths. The patient is heterozygous for 9 repeats and 11 repeats, the UCBT donor is heterozygous for 11 repeats and 12 repeats, and the maternal cells are heterozygous for 9 repeats and 14 repeats. Thus, this is a highly informative marker with unique alleles in all three cell types. As seen in the posttransplant sample, the 9 repeat allele and 14 repeat allele are absent. Only the 11 repeat and 12 repeat alleles are present, making this a completely engrafted sample with only donor alleles present. A bone marrow biopsy performed at the same time showed cellular marrow with no evidence of recurrent leukemia.
Figure 1.
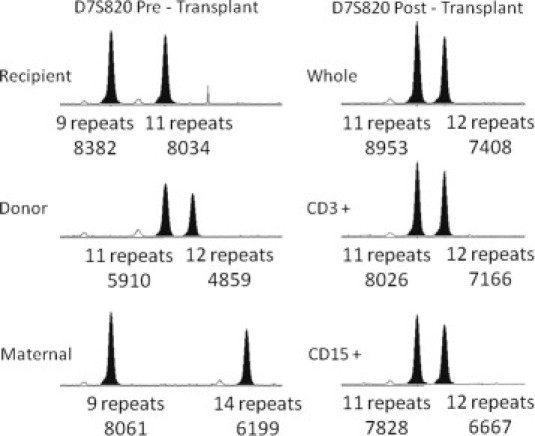
Post-UCBT Bone marrow engraftment analysis. Shown are capillary electropherograms of PCR amplification products for a single chromosome 7 short tandem repeat marker (D7S820). Pretransplant samples are shown on the left. A 9-month posttransplant peripheral blood sample is shown on the right. Whole, CD3+, and CD15+ denote an unfractionated sample, a CD3 selected whole blood fraction (T-cells), and a CD15 selected whole blood fraction (myeloid cells), respectively. Below each peak, the number of repeats and integrated fluorescence intensity are provided.
The relationship between the 11 repeat and 12 repeat alleles is also informative. These alleles are present at near equal intensity, as is expected because they are present in a heterozygous state. The longer repeat length (12) is somewhat less efficiently amplified, showing smaller integrated peak heights. For the pretransplant donor sample and the three posttransplant samples (unfractionated, CD3-fraction, and CD15-fraction) shown in Figure 1, the ratios between the 12 repeat allele and 11 repeat allele are 0.82, 0.83, 0.89, and 0.85, respectively. Less efficient amplification of longer repeat lengths is a common finding in this and other STRs included in this assay. For repeat lengths separated by a single repeat unit, the integrated peak height of the smaller amplification product may also be increased by ‘downward stutter’ (the very small intensity peaks preceding high intensity peaks). An analysis of 100 independent PCR reactions performed in our laboratory for the D7S820 STR marker reveals that when repeat lengths are one repeat unit apart, the average ratio of the integrated peak height of the longer allele to the shorter allele is 0.89 (median 0.89; range 0.76–1.15; SD 0.073). The average ratio for alleles that differ by two repeat units is 0.91 (median 0.91; range 0.74–1.11; SD 0.066). A similar phenomenon is observed for the D5S818 allele on chromosome 5 (one repeat: average 0.91; median 0.91; range 0.74–1.12; SD 0.074; two repeats: average 0.93; median 0.94; range 0.69–1.12; SD 0.07). These two alleles are presented in some detail because of their particular relevance in the evaluation of loss of heterozygosity (LOH) in the context of myeloid leukemia and myelodysplastic syndromes as demonstrated below.
Approximately 14.5 months after UCBT, blasts were noted on a peripheral blood smear, and a bone marrow biopsy was performed. Numerous blasts were seen in the bone marrow biopsy and accompanying aspirate smear. The immunophenotype of these blasts was not identical to that previously documented, but expression of myeloid and monocytic markers was evident. This is not an unusual finding; cytogenetic evolution and immunophenotypic change is not uncommon in the context of recurrent leukemia, and change in immunophenotype or karyotype is not a reliable indicator of a de novo process.23,24 Thus, these histological and immunophenotypic findings were interpreted as recurrent acute monoblastic leukemia (Figure 2 and Table 1). These findings, however, are also consistent with a therapy-related de novo process (t-AML). As such, additional studies, including a conventional karyotype to evaluate for therapy-related chromosomal gains and losses, were initiated as detailed below.
Figure 2.
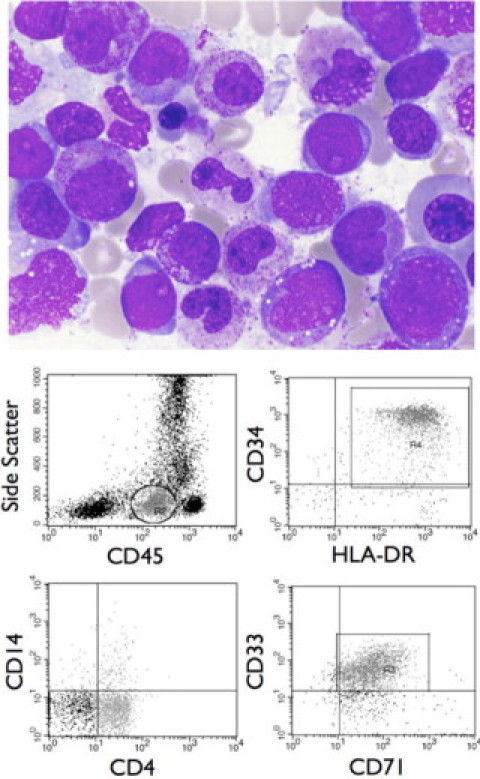
Post-UCBT diagnosis of AML. A Wright-stained bone marrow aspirate smear (×1000) showing blasts with histological characteristics consistent with acute monoblastic leukemia, including a moderate amount of finely granular cytoplasm with the absence of Auer rods, lacy chromatin with one to several prominent nucleoli, and occasional nuclear indentations or grooves (top). Flow cytometric analysis reveals a blast population with a CD45 expression level below normal lymphocytes and a low side scatter (circled/gated cells, top left). Analysis of these cells reveals strong expression of the early hematopoietic associated antigens CD34 and HLA-DR (top right), partial dim expression of the monocytic marker CD4 and lack of expression of the monocytic marker CD14 (bottom left), and dim expression of the myeloid marker CD33 and moderate expression of CD71, which is present on proliferating cells (bottom right). This immunophenotypic profile is consistent with acute monoblastic leukemia.
An engraftment analysis was also performed at this time on both peripheral blood and bone marrow samples. This analysis showed eight informative markers with >98% male donor cells and no definite recipient or maternal cells, consistent with complete engraftment. Shown in Figure 3 are the results for the D7S820 marker. There is an absence of 9 repeat (recipient) and 14 repeat D7S820 alleles (maternal granulocyte donor) (Figure 3). For the peripheral blood specimen, the CD3+ (T-cell) fraction showed a normal ratio between the 11 repeat and 12 repeat (donor) alleles, with the 12 repeat allele PCR product having a slightly smaller integrated fluorescence intensity than the 11 repeat allele PCR product. This is in contrast to the unfractionated and CD15+ fraction, which showed a clear allelic imbalance between the 11 and 12 repeat alleles, with significant attenuation of the 11 repeat allele integrated peak height. This finding is consistent with a LOH for this marker on chromosome 7. Taken together, persistent complete ‘engraftment’ in the context of a histological recurrence and LOH for D7S820 on chromosome 7 is diagnostic of donor cell leukemia.
Figure 3.
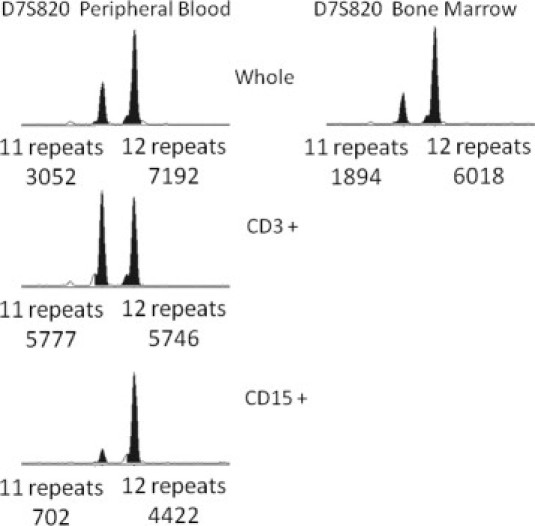
Bone marrow engraftment analysis after clinical recurrence. Shown are capillary electropherograms of PCR amplification products for a single chromosome 7 short tandem repeat marker (D7S820). A fractionated peripheral blood sample is shown on the left. A bone marrow sample is shown on the right. Whole, CD3+, and CD15+ denote an unfractionated sample, a CD3 selected whole blood fraction (T-cells), and a CD15 selected whole blood fraction (myeloid cells), respectively. Below each peak, the number of repeats and integrated fluorescence intensity are provided.
A conventional karyotype and FISH analysis confirmed monosomy 7. The karyotype also showed the presence of a Y chromosome, further confirming the donor origin of this patient's leukemia. Finally, FISH for 11q23 showed an absence of 11q23 abnormalities in these blasts, lending additional support to the hypothesis that this was a de novo process rather than a recurrence. This case of donor cell leukemia was refractory to two courses of clofarabine, etoposide, and cyclophosphamide, and to one course of fludarabine, idarubicin, Ara-C, and G-CSF. The patient ultimately succumbed to her disease.
Subsequent testing of the original umbilical cord blood was performed by FISH for chromosome 7. Two hundred nuclei were scored, and a normal signal pattern was seen in 97.5% of cells. This is within the range of normal for this probe set. The donor is also without evidence of AML. This suggests that this leukemia occurred de novo after transplantation. A reanalysis of a bone marrow engraftment assay performed on a peripheral blood specimen two months before frank recurrence was performed. No samples were submitted for histological examination at that time. As shown in Figure 4, a very subtle allelic imbalance can be seen for the D7S820 marker in the CD15+ fraction when compared with the CD3+ fraction or the unfractionated sample, and to the historical samples from this patient (Figure 1). Although the imbalance can be appreciated retrospectively, without knowledge of the ultimate chromosomal abnormalities present in this patient's donor cell leukemia, and without evidence of any histological changes, these findings could only be interpreted as complete engraftment by donor cells. Even with this knowledge, given the potential range of ‘normal’ allelic ratios for the D7S820 marker, it would be difficult to call this definitive evidence of a partial loss of chromosome 7.
Figure 4.
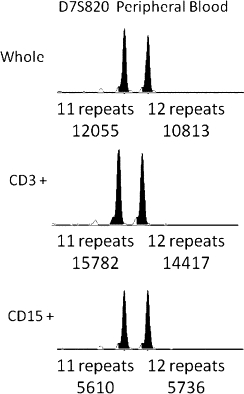
Bone marrow engraftment analysis 2 months before clinical recurrence. Shown are capillary electropherograms of PCR amplification products for a single chromosome 7 short tandem repeat marker (D7S820). A fractionated peripheral blood sample is shown. Whole, CD3+, and CD15+ denote an unfractionated sample, a CD3 selected whole blood fraction (T-cells), and a CD15 selected whole blood fraction (myeloid cells), respectively. Below each peak, the number of repeats and integrated fluorescence intensity are provided.
Discussion
Here we report a case of donor cell leukemia after UCBT that was detected by a laboratory-developed bone marrow engraftment analysis assay that uses PCR of STRs. This assay is widely used by many diagnostic laboratories and is comparable to identity testing as used in forensic or paternity testing. It is a highly sensitive technique, compared with FISH analysis for the X and Y chromosomes, with the added benefit of having utility in sex-matched transplants. In this assay, multiple STR alleles from different chromosomes are analyzed to identify informative alleles with unique repeat lengths in either the donor or recipient cells. The proportion of donor and recipient alleles is presumed to reflect the proportion of donor and recipient cells in the patient. The primary purpose of engraftment analysis after transplant is to monitor engraftment status, looking specifically for graft failure and/or disease relapse.25 However, much more information can be gained from a simple engraftment analysis. A new allelic imbalance or loss in the donor cell population may suggest a donor-derived process like DCL, as was seen in the case presented here.
This case also highlights some of the subtleties and confounding issues in bone marrow engraftment analysis in the context of hematological malignancies and dysplastic processes using PCR of STRs (particularly STRs on chromosome 5 such as D5S818 or chromosome 7 such as D7S820). A subset of AML and myelodysplastic syndrome has characteristic chromosomal abnormalities including recurrent loss of either all or just the long arms of chromosomes 5 and 7. Of particular relevance to this case is the loss of chromosome 7 that can be seen in acute myeloid leukemia and therapy-related acute myeloid leukemia.26 While in the context of this case a loss of heterozygosity aided in the correct diagnosis, LOH can confound engraftment calculations in other contexts. For example, in a patient receiving a transplant for a myelodysplastic syndrome with a loss of 7q, using markers on chromosome 7 for evaluating engraftment status could lead to an artificially high calculation for percent donor. In the worst case, if a marker on chromosome 7 was used as the sole informative allele in such a patient, this could lead to an interpretation of complete engraftment in a patient with no engraftment at all. This is further complicated by therapy-related MDS that can occur de novo in recipient cells after transplantation and has a high incidence of del(5) and del(7). This can change informative STR markers on chromosomes 5 and 7 to markers that should be excluded from further calculations. While less of an issue in the transplant setting, primer site polymorphisms can also lead to lack of amplification of an allele. Although this is an infrequent occurrence, it is of critical importance when using STR-based assays for forensic identity and paternity testing.27 While it is difficult to anticipate every potential confounding factor in an engraftment analysis, close correlation with concurrent or diagnostic histological, other laboratory, and clinical findings, and careful selection of markers used in the actual calculation of percent engraftment is an integral part of accurate interpretation.
Donor cell leukemia after any cellular transplant is rare. This rarity makes DCL an infrequently considered diagnosis in patients with recurrent disease after transplant for leukemia. The estimated incidence of DCL is approximately 0.1% (14/10489 from the EBMT data).11 Ruiz-Arguelles et al report a 5% incidence in their experience, but this number is based on fewer cases (2/40).12 The first case of DCL was reported in 1971, and the first cases of DCL after UCBT were reported in 2005.16,20 Donor cell leukemia may be more frequent after cord blood transplantation than after transplantation with peripheral blood stem cells or bone marrow. Based on the experience of the Tokyo Cord Blood Bank, the incidence of DCL after UCBT may be closer to 1% (4/478).14 This is higher than the de novo incidence of AML in the age matched general population and may have implications for disease etiology as discussed below.
This case is one of 11 cases of donor cell leukemia after UCBT reported in the literature (Table 2).13,14,15,16,17,19,20,21 Eight of these cases occurred in adults, and three in children. Six cases occurred in males and five in females, with ages ranging from 1 to 58 years. One of the pediatric patients developed a transient myelodysplastic syndrome with monosomy 7, and the other pediatric case was transplanted for acute lymphoblastic leukemia, not AML. Thus, the case presented here is unique. Seven cases of DCL manifested as AML: three as FAB M5, two as M2, and two not further specified. Of all 11 cases, three were AML before and after transplant.
Table 2.
UCBT Donor-Derived Leukemia
| Case | Age/sex | Year | Pre-TX | DCL | Time (months) | HLA mismatches | Reference |
|---|---|---|---|---|---|---|---|
| 1 | 1/M | 2005 | LCH | AML (NOS) | 40 | 2 | 20 |
| 2 | 58/M | 2007 | AML (NOS) | T-LGL | u | 2 | 17 |
| 3 | 32/F | 2005 | AML (M0) | AML (M2) | 11 | 2 | 19 |
| 4 | 31/M | 2007 | CHL | AML (M5a) | 16 | 2 | 21 |
| 5 | 32/F | 2008 | AML (M2) | AML (NOS) | 15 | u | 14 |
| 6 | 30/M | 2008 | CHL | AML (M5) | 16 | u | 14 |
| 7 | 57/F | 2003 | ATCL | AML (M2) | 8 | 1 | 16 |
| 8 | 41/M | 2005 | ALL + NK/T | CMPD | 9 | u | 15 |
| 9 | 5/F | 2002 | t-AML | MDS−7 | 3 | 1 | 13 |
| 10 | 34/M | 2008 | ALL | MDS | 10 | 1 | 18 |
| 11 | 3/F | 2007 | AML (M5a) | AML (M5a) | 14 | u | Current case |
ALL indicates acute lymphoblastic leukemia; AML, acute myeloid leukemia; ATCL, adult T-cell lymphoma; CHL, classical Hodgkin lymphoma; CMPD, chronic myeloproliferative disease; LCH, Langerhans cell histiocytosis; MDS, myelodysplastic syndrome; NK/T, NK/T-cell lymphoma; NOS, not otherwise specified; M0, M2, M5, M5a, AML FAB subtypes; t-AML, therapy-related AML; T-LGL, T-cell large granular lymphocytic leukemia; u, unknown.
The DCL in this patient had monosomy 7. Of the eight cases with reported karyotypes, six had abnormalities involving chromosome 7, most commonly monosomy 7 (4/6). Monosomy 7 has been identified sporadically after bone marrow transplant, in association with G-CSF administration, is one of the therapy-related chromosomal abnormalities and is associated with de novo MDS.28,29 In therapy-related AML or MDS, cases with chromosome 7 abnormalities are associated with exposure to alkylating agents or radiation therapy and a latency of 5–10 years.26 In this case, the time from initial chemotherapy to diagnosis of DCL was 2.5 years. However, chemotherapeutic treatment of this patient had been completed before transplantation.
There does not appear to be a relationship between the lineage of the original disease and the lineage of the DCL in the cohort that includes PBPC, BMT, and UCBT patients. Of the 41 cases enumerated in the review by Ruiz-Argüelles et al, only four were AML to AML, and one was AML to MDS/AML.10 The exact phenotypes of the previously reported cases are not specified, and it is not known how many may have been thought of as recurrent. However, immunophenotypic or cytogenetic evolution is not a reliable indication of a de novo process such as DCL. In fact, approximately 50% of recurrent AML will have immunophenotypic differences from presentation, and two-thirds of cases that present with an abnormal karyotype will have a change or evolution of their cytogenetic abnormality at the time of relapse.23,24 Another confounding factor in the diagnosis of DCL is therapy-related leukemia (t-AML). In the context of immunophenotypic drift or novel cytogenetic abnormalities t-AML is much higher on the differential diagnosis than DCL. In fact, in the case presented here, in the absence of the karyotype (presence of the Y chromosome) and engraftment studies documenting a donor-derived process, the immunophenotypic changes, loss of the t(11;19)(q23;p13) translocation and the new occurrence of monosomy 7 may have suggested a therapy-related de novo process.
Because of its rarity, the prognosis of DCL has not yet been established. The heterogeneity of the cases is not limited to the lineage of the original disease or DCL, but also includes the nature of the cellular transplant, and time between transplant and development of DCL. The longest reported interval is 13.7 years, while other cases occurred within 2 months of transplant.10 For prognostic and treatment purposes, it is not clear whether DCL should be considered as new leukemia, therapy-related leukemia, or recurrent leukemia. The prognosis of therapy related (t-AML) is poor (5-year survival <10%) and is even worse for those with chromosome 7 abnormalities (median survival <1 year).26 The prognosis of de novo AML depends on patient age and karyotype; the 5-year overall survival ranges from 11% for cases with unfavorable cytogenetics to 55% for cases with favorable cytogenetics.30 The overall 5-year survival for pediatric AML is 54.1%.31 Of the 11 cases of DCL following UCBT, outcomes were reported for eight. Two of the patients were alive at the time of reporting (one was the case of transient MDS that resolved, and one was disease-free after a subsequent transplant).13,15 The other six patients died; the longest reported survival is 13 months. Based on these few cases, the prognosis appears more similar to that of recurrent or t-AML than de novo AML.
Numerous mechanisms for the genesis of DCL have been postulated, implicating both the donor and the recipient, including occult leukemia in the donor, malignant predisposition of the donor cells, transfection and transformation of donor cells by host DNA either genomic or viral, impaired host immune surveillance, drug toxicity, and a leukemogenic milieu in the host.10,32 Although clones with the TEL/AML1 and AML/ETO fusion have been detected in presumably normal cord blood samples, providing potential weight for a donor cause, no cases of leukemia have been reported in any of the donors that have had clinical follow-up.33 Using the seeds and soil analogy, the cord blood cells are relatively young and pristine, while the soil has definitely been poisoned. Intuitively, recipient factors, like those elaborated above, seem the more likely culprit. However, because of the rarity and heterogeneity of cases of DCL, the causal mechanism(s) may prove elusive.
In our case it appears that the leukemia arose after transplantation because the new genetic abnormality, monosomy 7, was not present in the umbilical cord blood sample. The evaluation of monosomy 7 was done using FISH. Two hundred nuclei were scored, and 97.5% of cells had a normal signal pattern. This is within the range of normal for this probe set, and we interpret this finding as ‘normal.’ However, FISH is not ideal for this analysis because of the potential for probe overlap in a single cell appearing as a deletion. For deletion probe sets, the deletion would have to be present in at least 3% of cells to be considered abnormal. For clinical testing, we are even hesitant to call these borderline cases abnormal because they are so close to the upper limit of what we have seen in our validation study database. However, FISH is the most sensitive assay we have at our disposal for this evaluation. This approach is more sensitive than our STR-based engraftment assay. Typically for STR-based assays for loss of heterozygosity, LOH must be present in >5 to 10% of cells (at least). The engraftment assay for this donor sample is presented in Figure 1. No allelic imbalance is seen in this sample using this method. We currently do not have a method for detecting loss of chromosome 7 at the very low levels of 1 in 1000 to 10,000. Detecting a loss at this level would be very difficult (essentially detecting the difference between on average 2 copies of chromosome 7 and on average 1.999–1.9999 copies of chromosome 7). With these limitations, it remains a possibility that the abnormal clone was present in the umbilical cord blood sample at a level below our detection limit, and that this clone flourished in the transplant recipient but not in the donor.
A protocol for donor follow-up has not been established and will likely depend on the still unknown pathogenic mechanisms. If it were shown that donor cell leukemia is attributable only to host factors and not to occult leukemia or other donor pathologies, then there would not seem to be a rational basis for following the donors. If there were any implication of the donor cells, then following the donor would seem necessary. However, until the pathogenesis is known, the issue of donor follow-up is left to the individual clinicians. In this case, there has been clinical follow-up of the donor, who is healthy.
Recurrence of AML is far more common than DCL, but DCL may not be quite so rare as it now seems. Depending on one's perspective (molecular or hematopathologist), DCL can appear as complete engraftment or as recurrent or t-AML. The allelic changes on engraftment studies can be subtle, as in this case, which emphasizes the importance of well-performed and carefully interpreted studies correlated with other laboratory and clinical findings.34,35 Unless DCL is thought of as a possibility and specifically looked for, it will likely remain under-diagnosed.
At this time, the prognosis appears poor, and the distinction between DCL and recurrent or t-AML may not seem relevant clinically. The detection and reporting of additional cases will provide more information on prognosis and possible treatment outcomes and may provide further clues regarding leukemogenesis. For optimal detection, the use of sensitive molecular techniques in combination with histology is recommended. The most compelling feature of this case is the combination of the histological “recurrence” with the abnormal engraftment study, which was subsequently confirmed by karyotype and FISH. Together, the histology and the molecular studies paint a coherent picture of donor cell leukemia and emphasize the need for correlation between these two arenas.
References
- 1.Rocha V, Labopin M, Sanz G, Arcese W, Schwerdtfeger R, Bosi A, Jacobsen N, Ruutu T, de Lima M, Finke J, Frassoni F, Gluckman E. Transplants of umbilical-cord blood or bone marrow from unrelated donors in adults with acute leukemia. N Engl J Med. 2004;351:2276–2285. doi: 10.1056/NEJMoa041469. [DOI] [PubMed] [Google Scholar]
- 2.Kurtzberg J, Prasad VK, Carter SL, Wagner JE, Baxter-Lowe LA, Wall D, Kapoor N, Guinan EC, Feig SA, Wagner EL, Kernan NA. Results of the Cord Blood Transplantation Study (COBLT): clinical outcomes of unrelated donor umbilical cord blood transplantation in pediatric patients with hematologic malignancies. Blood. 2008;112:4318–4327. doi: 10.1182/blood-2007-06-098020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martin PL, Carter SL, Kernan NA, Sahdev I, Wall D, Pietryga D, Wagner JE, Kurtzberg J. Results of the cord blood transplantation study (COBLT): outcomes of unrelated donor umbilical cord blood transplantation in pediatric patients with lysosomal and peroxisomal storage diseases. Biol Blood Marrow Transplant. 2006;12:184–194. doi: 10.1016/j.bbmt.2005.09.016. [DOI] [PubMed] [Google Scholar]
- 4.MacMillan ML, Weisdorf DJ, Brunstein CG, Cao Q, DeFor TE, Verneris MR, Blazar BR, Wagner JE. Acute graft-versus-host disease after unrelated donor umbilical cord blood transplantation: analysis of risk factors. Blood. 2009;113:2410–2415. doi: 10.1182/blood-2008-07-163238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rocha V, Kabbara N, Ionescu I, Ruggeri A, Purtill D, Gluckman E. Pediatric related and unrelated cord blood transplantation for malignant diseases. Bone Marrow Transplant. 2009;44:653–659. doi: 10.1038/bmt.2009.291. [DOI] [PubMed] [Google Scholar]
- 6.Barker JN, Martin PL, Coad JE, DeFor T, Trigg ME, Kurtzberg J, Weisdorf DJ, Wagner J. Low incidence of Epstein-Barr virus-associated posttransplantation lymphoproliferative disorders in 272 unrelated-donor umbilical cord blood transplant recipients. Biol Blood Marrow Transplant. 2001;7:395–399. doi: 10.1053/bbmt.2001.v7.pm11529490. [DOI] [PubMed] [Google Scholar]
- 7.Gong JZ, Bayerl MG, Sandhaus LM, Sebastian S, Rehder CW, Routbort M, Lagoo AS, Szabolcs P, Chiu J, Comito M, Buckley PJ. Posttransplant lymphoproliferative disorder after umbilical cord blood transplantation in children. Am J Surg Pathol. 2006;30:328–336. doi: 10.1097/01.pas.0000188030.63706.e7. [DOI] [PubMed] [Google Scholar]
- 8.Chadburn A, Suciu-Foca N, Cesarman E, Reed E, Michler RE, Knowles DM. Post-transplantation lymphoproliferative disorders arising in solid organ transplant recipients are usually of recipient origin. Am J Pathol. 1995;147:1862–1870. [PMC free article] [PubMed] [Google Scholar]
- 9.Capello D, Rasi S, Oreste P, Veronese S, Cerri M, Ravelli E, Rossi D, Minola E, Colosimo A, Gambacorta M, Muti G, Morra E, Gaidano G. Molecular characterization of post-transplant lymphoproliferative disorders of donor origin occurring in liver transplant recipients. J Pathol. 2009;218:478–486. doi: 10.1002/path.2555. [DOI] [PubMed] [Google Scholar]
- 10.Ruiz-Arguelles GJ, Ruiz-Arguelles A, Garces-Eisele J. Donor cell leukemia: a critical review. Leuk Lymphoma. 2007;48:25–38. doi: 10.1080/10428190601003462. [DOI] [PubMed] [Google Scholar]
- 11.Hertenstein B, Hambach L, Bacigalupo A, Schmitz N, McCann S, Slavin S, Gratwohl A, Ferrant A, Elmaagacli A, Schwertfeger R, Locasciulli A, Zander A, Bornhauser M, Niederwieser D, Ruutu T. Development of leukemia in donor cells after allogeneic stem cell transplantation–a survey of the European Group for Blood and Marrow Transplantation (EBMT) Haematologica. 2005;90:969–975. [PubMed] [Google Scholar]
- 12.Ruiz-Arguelles GJ, Ruiz-Delgado GJ, Garces-Eisele J, Ruiz-Arguelles A, Perez-Romano B, Reyes-Nunez V. Donor cell leukemia after non-myeloablative allogeneic stem cell transplantation: a single institution experience. Leuk Lymphoma. 2006;47:1952–1955. doi: 10.1080/10428190600693099. [DOI] [PubMed] [Google Scholar]
- 13.Sevilla J, Querol S, Molines A, Gonzalez-Vicent M, Balas A, Carrio A, Estella J, Angel Diaz M, Madero L. Transient donor cell-derived myelodysplastic syndrome with monosomy 7 after unrelated cord blood transplantation. Eur J Haematol. 2006;77:259–263. doi: 10.1111/j.1600-0609.2006.00716.x. [DOI] [PubMed] [Google Scholar]
- 14.Nagamura-Inoue T, Kodo H, Takahashi TA, Mugishima H, Tojo A, Asano S. Four cases of donor cell-derived AML following unrelated cord blood transplantation for adult patients: experiences of the Tokyo Cord Blood Bank. Cytotherapy. 2007;9:727–728. doi: 10.1080/14653240701466339. [DOI] [PubMed] [Google Scholar]
- 15.Mitsui H, Nakazawa T, Tanimura A, Karasuno T, Hiraoka A. Donor cell-derived chronic myeloproliferative disease with t(7;11)(p15;p15) after cord blood transplantation in a patient with Philadelphia chromosome-positive acute lymphoblastic leukemia. Int J Hematol. 2007;86:192–195. doi: 10.1532/IJH97.06162. [DOI] [PubMed] [Google Scholar]
- 16.Matsunaga T, Murase K, Yoshida M, Fujimi A, Iyama S, Kuribayashi K, Sato T, Kogawa K, Hirayama Y, Sakamaki S, Kohda K, Niitsu Y. Donor cell derived acute myeloid leukemia after allogeneic cord blood transplantation in a patient with adult T-cell lymphoma. Am J Hematol. 2005;79:294–298. doi: 10.1002/ajh.20349. [DOI] [PubMed] [Google Scholar]
- 17.Kusumoto S, Mori S, Nosaka K, Morita-Hoshi Y, Onishi Y, Kim SW, Watanabe T, Heike Y, Tanosaki R, Takaue Y, Tobinai K. T-cell large granular lymphocyte leukemia of donor origin after cord blood transplantation. Clin Lymphoma Myeloma. 2007;7:475–479. doi: 10.3816/clm.2007.n.031. [DOI] [PubMed] [Google Scholar]
- 18.Konuma T, Ooi J, Takahashi S, Tomonari A, Tsukada N, Kato S, Sato A, Monma F, Hongo E, Uchimaru K, Tojo A, Asano S. Donor cell-derived myelodysplastic syndrome after cord blood transplantation. Bone Marrow Transplant. 2009;43:429–431. doi: 10.1038/bmt.2008.344. [DOI] [PubMed] [Google Scholar]
- 19.Ando T, Yujiri T, Mitani N, Takeuchi H, Nomiyama J, Suguchi M, Matsubara A, Tanizawa Y. Donor cell-derived acute myeloid leukemia after unrelated umbilical cord blood transplantation. Leukemia. 2006;20:744–745. doi: 10.1038/sj.leu.2404121. [DOI] [PubMed] [Google Scholar]
- 20.Fraser CJ, Hirsch BA, Dayton V, Creer MH, Neglia JP, Wagner JE, Baker KS. First report of donor cell-derived acute leukemia as a complication of umbilical cord blood transplantation. Blood. 2005;106:4377–4380. doi: 10.1182/blood-2005-06-2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hamaki T, Kajiwara K, Kami M, Murashige N, Funaki M, Harima A, Kogure K, Yamada K, Kodo H, Kouzai Y. Donor cell-derived acute monoblastic leukemia involving MLL gene translocation in an adult patient who received umbilical cord blood transplantation. Bone Marrow Transplant. 2008;41:91–92. doi: 10.1038/sj.bmt.1705836. [DOI] [PubMed] [Google Scholar]
- 22.Ravindranath Y, Chang M, Steuber CP, Becton D, Dahl G, Civin C, Camitta B, Carroll A, Raimondi SC, Weinstein HJ. Pediatric Oncology Group (POG) studies of acute myeloid leukemia (AML): a review of four consecutive childhood AML trials conducted between 1981 and 2000. Leukemia. 2005;19:2101–2116. doi: 10.1038/sj.leu.2403927. [DOI] [PubMed] [Google Scholar]
- 23.Estey EKM, Pierce S, Stass S. Change in karyotype between diagnosis and first relapse in acute myelogenous leukemia. Leukemia. 1995;9:972–976. [PubMed] [Google Scholar]
- 24.Hur M, Chang YH, Lee DS, Park MH, Cho HI. Immunophenotypic and cytogenetic changes in acute leukaemia at relapse. Clin Lab Haematol. 2001;23:173–179. doi: 10.1046/j.1365-2257.2001.00389.x. [DOI] [PubMed] [Google Scholar]
- 25.Vnencak-Jones CL. Bone marrow engraftment studies. Curr Protoc Hum Genet. 2009;Chapter 9:Unit 9 17. doi: 10.1002/0471142905.hg0917s62. [DOI] [PubMed] [Google Scholar]
- 26.Vardiman JW AD, Brunning RD, Larson RA, Matutes E, Baumann I, Thiele J. Therapy-related myeloid neoplasms. In: Campo E, Swerdlow SH, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW, editors. WHO classification of tumours of haematopoietic and lymphoid tissues. International Agency for Research on Cancer; Lyon: 2008. pp. 127–129. [Google Scholar]
- 27.Budowle B, Masibay A, Anderson SJ, Barna C, Biega L, Brenneke S, Brown BL, Cramer J, DeGroot GA, Douglas D, Duceman B, Eastman A, Giles R, Hamill J, Haase DJ, Janssen DW, Kupferschmid TD, Lawton T, Lemire C, Llewellyn B, Moretti T, Neves J, Palaski C, Schueler S, Sgueglia J, Sprecher C, Tomsey C, Yet D. STR primer concordance study. Forensic Sci Int. 2001;124:47–54. doi: 10.1016/s0379-0738(01)00563-1. [DOI] [PubMed] [Google Scholar]
- 28.Lang Z, Dinndorf P, Ladisch S, Bayever E, Reaman G. Chromosomal transformation in donor cells following allogeneic bone marrow transplantation. Bone Marrow Transplant. 2004;33:1253–1256. doi: 10.1038/sj.bmt.1704450. [DOI] [PubMed] [Google Scholar]
- 29.Sloand EM, Yong AS, Ramkissoon S, Solomou E, Bruno TC, Kim S, Fuhrer M, Kajigaya S, Barrett AJ, Young NS. Granulocyte colony-stimulating factor preferentially stimulates proliferation of monosomy 7 cells bearing the isoform IV receptor. Proc Natl Acad Sci U S A. 2006;103:14483–14488. doi: 10.1073/pnas.0605245103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Slovak ML, Kopecky KJ, Cassileth PA, Harrington DH, Theil KS, Mohamed A, Paietta E, Willman CL, Head DR, Rowe JM, Forman SJ, Appelbaum FR. Karyotypic analysis predicts outcome of preremission and postremission therapy in adult acute myeloid leukemia: a Southwest Oncology Group/Eastern Cooperative Oncology Group Study. Blood. 2000;96:4075–4083. [PubMed] [Google Scholar]
- 31.Horner MJ RL, Krapcho M, Neyman N, Aminou R, Howlader N, Altekruse SF, Feuer EJ, Huang L, Mariotto A, Miller BA, Lewis DR, Eisner MP, Stinchcomb DG, Edwards BK, editors. SEER Cancer Statistics Review, 1975–2006. National Cancer Institute; Bethesda MD: 2007. [Google Scholar]
- 32.Cooley LD, Sears DA, Udden MM, Harrison WR, Baker KR. Donor cell leukemia: report of a case occurring 11 years after allogeneic bone marrow transplantation and review of the literature. Am J Hematol. 2000;63:46–53. doi: 10.1002/(sici)1096-8652(200001)63:1<46::aid-ajh11>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 33.Greaves MF. Cord blood donor cell leukemia in recipients. Leukemia. 2006;20:1633–1634. doi: 10.1038/sj.leu.2404293. [DOI] [PubMed] [Google Scholar]
- 34.Schichman SA, Lin P, Gilbrech LJ, Gray PS, Wilson CS, Sawyer JR. Bone marrow transplant engraftment analysis with loss of an informative allele. J Mol Diagn. 2002;4:230–232. doi: 10.1016/S1525-1578(10)60708-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dunn T, Allen R, Bates F, Kurkjian C, Kamble R, Kharfan-Dabaja M. Cytogenetic changes associated with myelodysplastic syndrome affecting bone marrow engraftment analysis. J Mol Diagn. 2006;8:288–294. doi: 10.2353/jmoldx.2006.050097. [DOI] [PMC free article] [PubMed] [Google Scholar]