

Abstract
Glomerular endothelial cell injury is a key component of a variety of diseases. Factors involved in glomerular endothelial cell repair are promising therapeutic agents for such diseases. Platelet-derived growth factor (PDGF)–C has pro-angiogenic properties; however, nothing is known about such functions in the kidney. We therefore investigated the consequences of either PDGF-C infusion or inhibition in rats with mesangioproliferative glomerulonephritis, which is accompanied by widespread glomerular endothelial cell damage. We also assessed the role of PDGF-C in a mouse model of thrombotic microangiopathy as well as in cultured glomerular endothelial cells. PDGF-C infusion in nephritic rats significantly reduced mesangiolysis and microaneurysm formation, whereas glomerular endothelial cell area and proliferation increased. PDGF-C infusion specifically up-regulated glomerular fibroblast growth factor-2 expression. In contrast, antagonism of PDGF-C in glomerulonephritis specifically reduced glomerular endothelial cell area and proliferation and increased mesangiolysis. Similarly, PDGF-C antagonism in murine thrombotic microangiopathy aggravated the disease and reduced glomerular endothelial area. In conditionally immortalized glomerular endothelial cells, PDGF-C was mitogenic and induced a 27-fold up-regulation of fibroblast growth factor-2 mRNA. PDGF-C also exerted indirect pro-angiogenic effects, since it induced endothelial cell mitogens and pro-angiogenic factors in mesangial cells and macrophages. These results identify PDGF-C as a novel, potent pro-angiogenic factor in the kidney that can accelerate capillary healing in experimental glomerulonephritis and thrombotic microangiopathy.
Glomerular endothelial cell injury is a key feature of many human glomerular diseases, including pre-eclampsia, hemolytic uremic syndrome, lupus nephritis, most types of vasculitides, and many glomerulonephritides, as well as renal transplant rejection.1 Glomerular endothelial cell damage also characterizes conditions associated with glomerular hypertension and hyperperfusion.2 Maintaining an endothelial cell homeostasis in the glomerulus is essential for renal function.3,4 Consequently, knowledge about factors that maintain the integrity of the glomerular capillary wall may help to understand the pathophysiology of progressive renal disease.
Platelet-derived growth factor (PDGF-C) is a specific ligand of the PDGFR-α chain, which is released as an inactive, latent homodimer, and extracellular cleavage of the N-terminal “complement C1r/C1a, Uegf, BMP1” (CUB) domain is required for receptor binding and activation.5 PDGF-C has recently been identified as a potent pro-angiogenic factor.6,7,8 Nothing is known so far about the role of PDGF-C in renal endothelial homeostasis. The identification of a pro-angiogenic cytokine that lacks systemic side effects such as vascular endothelial growth factor (VEGF)-induced capillary leakage could have a significant clinical impact on the treatment of glomerular disease associated with endothelial cell injury.
In the present study, the role of PDGF-C in capillary repair processes was studied in vitro as well as in models of secondary and primary glomerular endothelial cell damage in vivo. In a rat anti-Thy1.1 model of mesangioproliferative glomerulonephritis (GN), severe glomerular endothelial cell damage with subsequent repair occurs secondary to mesangial cell damage.9,10,11,12,13 We also studied a mouse model of thrombotic microangiopathy (TMA) in which microvascular endothelial cells are selectively damaged via renal arterial perfusion of lectin concanavalin A (conA) followed by rabbit anti-conA antibody.14
Materials and Methods
In Vivo Experiments
All animal experiments were approved by the local review boards. Animals were held in rooms with constant temperature and humidity, 12 hours/12 hours light cycles, and had ad libitum access to drinking water and food.
Experimental anti-Thy1.1 glomerulonephritis was induced in 30 (PDGF-C infusion) and 20 (PDGF-C inhibition) male Wistar rats weighing 200 g (Charles River, Sulzfeld, Germany) by injection of 1 mg/kg monoclonal anti-Thy1.1 antibody (clone OX-7; European Collection of Animal Cell Cultures, Salisbury, England) as described previously.10,15 For the PDGF-C infusion experiments, ALZET micro-osmotic pumps (model 1003D; pump rate 1.00 μl/h for 3 days, DURECT Corpor., Cupertino, CA) were implanted i.p. on day 1 (24 hours after disease induction). The pumps were filled with either active recombinant human PDGF-C (lacking the “complement C1r/C1a, Uegf, BMP1” (CUB) domain, n = 15; 14,6 μg/day/rat) or vehicle (PBS, n = 15). For the PDGF-C inhibition experiments, rats were injected i.p. on day 1 (24 hours after disease induction) with either sheep polyclonal PDGF-C-neutralizing antibody (5 mg/kg body weight; n = 10; kindly provided by Dr. Arne Östman, Karolinska Institute, Stockholm, Sweden)16 or irrelevant sheep IgG (5 mg/kg body weight; n = 10).
In rats with anti-Thy1.1 GN, on day 4, blood pressure was measured by the tail-cuff method, using a programmed sphygmomanometer (Softron Co., Tokyo, Japan). Subsequently, rats were placed in metabolic cages for 24-hour urine collection. Following sacrifice of all rats on day 5, serum samples and renal tissues for histological evaluations were collected and glomeruli were isolated by using a standard sieving technique.17
TMA was induced in 12 male C57Bl/6 mice weighing 22 g (Charles River, Sulzfeld, Germany) as described previously.14 In short, via paravertebral flank incision under a Zeiss OPMI 1-FC operation microscope, the left renal artery was cannulated with a 31-G microcannula (FST, Heidelberg, Germany). The left kidney was then sequentially perfused with 150 μl of PBS to remove blood cells from the kidney, followed by 200 μg of conA, a specific endothelial cell glycoprotein-binding lectin (Sigma-Aldrich, Taufkirchen, Germany) and rabbit-anti-conA antibody, which results in immune complex formation and complement activation on the endothelial surface. After perfusion, the abdominal aorta was clamped for vessel closure for <3 minutes followed by unclamping and visual control of kidney perfusion and absence of bleeding. Next, the mice were randomized to receive either sheep polyclonal PDGF-C-neutralizing antibody (50 mg/kg body weight; n = 6) or irrelevant sheep IgG (50 mg/kg body weight; n = 6) via i.p. injection at 4, 24 and 48 hours after disease induction. A similar treatment (anti-PDGF-C antibody, n = 6; and control IgG, n = 6) was performed in sham animals in which renal perfusion with rabbit-anti-conA was substituted with PBS. The antibody concentration was chosen based on our previous results.16 All mice were placed in metabolic cages for 24-hour urine collection on day 3 after disease induction. Following sacrifice on day 4, serum samples and renal tissues for histological evaluations were collected.
Recombinant human PDGF-C was prepared as described previously.16,18,19 The anti-PDGF-C sheep serum was described recently.16 For the present study, we affinity-purified IgG fractions from the sera using an IgG purification kit (Affiland, Liege, Belgium).
Renal Morphology
Tissue for light microscopy and immunohistochemistry was fixed in methyl Carnoy’s solution or formalin and embedded in paraffin. Four μm sections were stained with periodic acid-Schiff’s reagent and counterstained with hematoxylin. All histomorphological analyses were done in a blinded manner. In rats approximately 100 (range, 80 to 120) and in mice 30 to 40 consecutive cortical glomeruli were counted.
In periodic acid-Schiff-stained sections, the total number of endothelial and nonendothelial mitotic figures was counted as described previously.10 Mesangiolysis was graded on a semiquantitative scale as follows: 0 = no mesangiolysis, 1 = segmental mesangiolysis, 2 = global mesangiolysis, 3 = microaneurysm. The number of glomeruli exhibiting microaneurysms was counted as described previously.10 In TMA mice, percentages of glomeruli exhibiting injury, ie, necrosis, thrombosis, mesangiolysis, or microaneurysms, were counted.
Immunohistochemistry
Four-μm sections of methyl Carnoy’s- or formalin-fixed and paraffin-embedded renal tissues were processed as previously described.10,15 All stainings, except for VEGF and CD31, were done in methyl Carnoy’s-fixed tissues. VEGF staining was done on formalin-fixed tissues with heat antigen retrieval in citrate buffer (pH = 6.0). CD31 staining was performed on frozen sections. For both PDGFR stainings, antigen retrieval with the Trilogy system (Cell-Marque Corp., Rocklin, CA) was performed. Primary antibodies included a murine monoclonal antibody (mAb) (clone 1A4) to α-smooth muscle actin (SMA; DAKO A/S, Glostrup, Denmark) or the same antibody directly conjugated with alkaline phosphatase (Sigma, St. Louis, MO), a murine mAb (clone JG12) to rat endothelial cells (Bender MedSystems GmbH, Vienna, Austria), a directly biotin-conjugated rat anti-mouse CD31 mAb (BD Biosciences, Heidelberg, Germany), rabbit anti-human fibrinogen polyclonal Ab (DAKO A/S, Glostrup, Denmark), a murine mAb (clone ED1) to CD68 present on monocytes, macrophages, and dendritic cells (Serotec Ltd., Oxford, UK), a goat polyclonal Ab raised against human PDGFR-α, a murine mAb raised against human PDGFR-β and goat polyclonal Ab raised against rat VEGF (last three Abs from R&D, MN), plus appropriate negative controls as described previously.15 To assess the numbers of infiltrating monocytes/macrophages in glomeruli, ED1-positive cells in 100 consecutive glomeruli were counted. The point-counting method was used to determine the area of glomerular endothelial cells (JG12 stain) as follows: a grid composed of 100 dots was superimposed on 25 consecutive glomeruli (magnification 1000-fold), and the number of dots overlying the stained areas and the number of dots overlying the glomerular tuft were counted. From these data, glomerular tuft area and % of JG12 positively stained area were calculated. Glomerular or interstitial area positively stained for α–SMA and VEGF were evaluated by computer-based morphometry using the analySIS v3.1 software in 50 glomeruli (Soft Imaging System GmbH, Münster, Germany). In the mice, glomerular area positive for fibrinogen (at ×400 magnification) or CD31 (at ×600 magnification) was counted semiquantitatively using a score (0 to 3) expressing positively stained glomerular area: 0 = <5%; 1 = 5 to 25%; 2 = 25 to 50%; 3 = >50%.
Immunohistochemical Double Stainings
Identification of Proliferating Cells
To identify proliferating cell nuclear antigen (PCNA)-positive mesangial cells, sections were stained with mAb against PCNA (Oncogene, MA) and visualized as brown diaminobenzidine product followed by α-SMA staining visualized by Vector Blue substrate kit resulting in blue color. For PCNA-positive endothelial cells, sections were stained first with JG12 antibody (brown diaminobenzidine staining) followed by PCNA staining (blue Vector Blue substrate kit staining). For identification of PCNA-positive monocytes/macrophages, sections were first stained for PCNA (brown diaminobenzidine staining) followed by ED-1 staining (blue Vector Blue substrate kit staining). Double-positive cells were identified by positive nuclear staining for PCNA completely surrounded by cytoplasm positive for α–SMA, JG12, or ED-1. For each double staining, approx. 100 consecutive glomeruli were counted; results are given as double-positive cells per glomerulus.
Glomerular RNA Extraction and Analyses
Total RNA was extracted from isolated rat glomeruli or mice renal cortex using the RNeasy Mini Kit (Qiagen, Hilden, Germany). Complementary DNA (cDNA) syntheses and real-time quantitative PCRs were performed as previously described.15 Sequences of primers and probes used are shown in Table 1. Each sample was normalized to glyceraldehyde-3-phosphate dehydrogenase. Results are given as fold change in mRNA expression in PDGF-C-infused or PDGF-C -antagonized animals or PDGF-C-stimulated cells relative to PBS-infused or irrelevant sheep IgG-treated animals or unstimulated cells (set as 1). Due to technical reasons, in the in vivo infusion experiment, the analyses were performed with n = 12 per group.
Table 1.
Primers and Probes Used for Real-Time Reverse Transcriptase PCR to Detect Human (A), rat (B), or Mouse (C) mRNA Expression
| Forward primer | Reverse primer | Taqman probe | |
|---|---|---|---|
| Human gene | |||
| GAPDH | 5′-AGCCACATCGCTCAGACACC-3′ | 5′-GCGCCCAATACGACCAAA-3′ | 5′-CCGTTGACTCCGACCTTCACCTTCC-3′ |
| VEGF | 5′-TTGCTGCTCTACCTCCACCAT-3′ | 5′-TCTGCCCTCCTCCTTCTGC-3′ | SYBR Green Kit |
| VEGFR-1 | 5′-CCACTTGACACTTTGATCCCTG-3′ | 5′-TTGTACGTTGCATTTGATATGATGA-3′ | SYBR Green Kit |
| VEGFR-2 | 5′-ATCCACTGGTATTGGCAGTTGG-3′ | 5′-GGGTATGGGTTTGTCACTGAGAC-3′ | SYBR Green Kit |
| Ang-1 | 5′-TGCAAATGTGCCCTCATGTTA-3′ | 5′-TCCCGCAGTATAGAACATTCCA-3′ | SYBR Green Kit |
| Ang-2 | 5′-TCCTCCTGCCAGAGATGGAC-3′ | 5′-CTGCACAGCATTGGACACGTA-3′ | SYBR Green Kit |
| FGF-2 | 5′-CATCAAGCTACAACTTCAAGCAGAA-3′ | 5′-GCCAGTAATCTTCCATCTTCCTTC-3′ | SYBR Green Kit |
| TSP-1 | 5′-AATGAGCTGAGGCGGCC-3′ | 5′-AGCTATCAACAGTCCATTCCTCG-3′ | SYBR Green Kit |
| PDGF-A | 5′-GGAGGAAGAGAAGCATCGAGG-3′ | 5′-CGACCTGACTCCGAGGAATCT-3′ | 5′-CCGCTGTCTGCAAGACCAGGACG-3′ |
| PDGF-B | 5′-AGGAGGGAGACTGTGGTAGGG-3′ | 5′-GTGGACTTTGGGAAATGGAG-3′ | 5′-CATGTGGACAGCAGTGTTGCCTCCCT-3′ |
| PDGF-C | 5′-GCCTCTTCGGGCTTCTCC-3′ | 5′-TGAGGATCTTGTACTCCGTTCTGTT-3′ | 5′-CCGGCCAGAGACGAGGGACTCA-3′ |
| PDGF-D | 5′-TCATACCATGACCGGAAGTCAA-3′ | 5′-TGGGAGTGCAACTGTAACGCT-3′ | 5′-TTGACCTGGATAGGCTCAATGATGATGCC-3′ |
| PDGFR-α | 5′-CCCTTGGTGGCACCCC-3′ | 5′-CGGTACCCACTCTTGATCTTATTGT-3′ | SYBR Green Kit |
| PDGFR-β | 5′-ATGCCTTACCACATCCGCTC-3′ | 5′-ACATTGCAGGTGTAGGTCCCC-3′ | SYBR Green Kit |
| Rat gene | |||
| GAPDH | 5′-ACAAGATGGTGAAGGTCGGTG-3′ | 5′-AGAAGGCAGCCCTGGTAACC-3′ | 5′-CGGATTTGGCCGTATCGGACGC-3′ |
| MCP-1 | 5′-CCAGATGCAGTTAATGCCCC-3′ | 5′-TCTCCAGCCGACTCATTGG-3′ | SYBR Green Kit |
| RANTES | 5′-ACTCCCTGCTGCTTTGCCT-3′ | 5′-GTGTAAAAATACTCCTTCACGTGGG-3′ | SYBR Green Kit |
| VEGF | 5′-GTACCTCCACCATGCCAAGTG-3′ | 5′-ATGGGCTTTCTGCTCCCCT-3′ | 5′-CCCAGGCTGCACCCACGACAG-3′ |
| VEGFR-1 | 5′-CCCGAATCCATCTTTGACAAG-3′ | 5′-TGGAGAACCCCCTAAGGAAAA-3′ | 5′-ACACTCCGTAGGACCACACGTCACTCTTG-3′ |
| VEGFR-2 | 5′-GCAGACGCTGACATGCACA-3′ | 5′-GGGCCTGTAGGAGCATGCT-3′ | 5′-CCAACCCTCCCCTGCACCACATC-3′ |
| Ang-1 | 5′-GCAAATGCGCCCTTATGC-3′ | 5′-CCGTTTAGATTGGAAGGGCC-3′ | SYBR Green Kit |
| Ang-2 | 5′-GACTTCCAGAGAACGTGGAAAGA-3′ | 5′-TTGCCCAGCCAATACTCTCC-3′ | SYBR Green Kit |
| FGF-2 | 5′-GTGGACGGCGTCCGG-3′ | 5′-CAACTCCTCTCTCTTCTGCTTGG-3′ | SYBR Green Kit |
| TSP-1 | 5′-GCAGAGCTGGATGTTCCCAT-3′ | 5′-TTGCGACTCGGAGCCTG-3′ | SYBR Green Kit |
| PDGF-A | 5′-TTCTTGATCTGGCCCCCAT-3′ | 5′-TTGACGCTGCTGGTGTTACAG-3′ | 5′-CAGTGCAGCGCTTCACCTCCACA-3′ |
| PDGF-B | 5′-GCAAGACGCGTACAGAGGTG-3′ | 5′-GAAGTTGGCATTGGTGCGA-3′ | 5′-TCCAGATCTCGCGGAACCTCATCG-3′ |
| PDGF-C | 5′-CAGCAAGTTGCAGCTCTCCA-3′ | 5′-GACAACTCTCTCATGCCGGG-3′ | 5′-CGACAAGGAGCAGAACGGAGTGCAA-3′ |
| PDGF-D | 5′-ATCGGGACACTTTTGCGACT-3′ | 5′-GTGCCTGTCACCCGAATGTT-3′ | 5′-TTGCGCAATGCCAACCTCAGGAG-3′ |
| PDGFR-α | 5′-GCCACGAAAGAGGTCAAGGA-3′ | 5′-GCCTGATCTGGACGAAGCC-3′ | 5′-TGAAGACAGTCACCATTTCTGTTCACGAGAA-3′ |
| PDGFR-β | 5′-AATGACCACGGCGATGAGA-3′ | 5′-TCTTCCAGTGTTTCCAGCAGC-3′ | 5′-CATCAACGTTACTGTGATCGAAAATGGCTATG-3′ |
| Mouse gene | |||
| GAPDH | 5′-GGCAAATTCAACGGCACAGT-3′ | 5′-AGATGGTGATGGGCTTCCC-3′ | SYBR Green Kit |
| VEGF | 5′-TCTTCAAGCCGTCCTGTGTG-3′ | 5′-CTCCAGGGCTTCATCGTTACA-3′ | SYBR Green Kit |
| FGF-2 | 5′-CCGCGTGGATGGCGT-3′ | 5′-CCTCTCTCTTCTGCTTGGAGTTG-3′ | SYBR Green Kit |
| PDGF-C | 5′-CTGGTGTGGAGATTAGTTGCAGTAG-3′ | 5′-CCAGCCCAAATCTCTCATCAA-3′ | 5′-TGAAAATGTGCGGATCCAGCTGACA-3′ |
Ang, angiopoietin; FGF-2, fibroblast growth factor-2; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; MCP-1, monocyte chemotactic protein 1 (chemokine CCL2); PDGF, platelet-derived growth factor; PDGFR, receptor for PDGF; RANTES, regulated upon activation, normally T-expressed, and presumably secreted (chemokine CCL5); TSP-1, thrombospondin-1; VEGF, vascular endothelial growth factor; VEGFR, receptor for VEGF.
Miscellaneous Measurements
Serum and urine biochemistry were performed by an autoanalyzer. Quantikine rat VEGF and DuoSet human fibroblast growth factor (FGF)-2 ELISA kits (R&D) were used according to the manufacturer’s instructions to determine the VEGF concentrations in serum and FGF-2 concentrations in glomerular lysates or serum. Glomerular FGF-2 concentration was normalized to protein content measured by a bicinchoninic acid protein assay kit (Pierce, Rockford, IL, USA), according to manufacturer’s instructions.
In Vitro Experiments
Conditionally immortalized human glomerular endothelial cells (CiGEnC) were obtained, characterized, described, and cultured with the EGM-2MV medium (“hunger medium”) containing fetal bovine serum, epidermal growth factor, VEGF, FGF-2, R3-IGF-1, hydrocortisone, ascorbic acid, and gentamicin/amphotericin-B (“full medium”; Lonza, Walkersville, MD) as described in detail previously.20 Primary human mesangial cells were obtained from Biowithaker (Verviers, Belgium) and were maintained as described.21 The THP-1 monocytic cell line derived from a patient with acute myelomonocytic leukemia was obtained from ATCC (Manassas, VA) and cultured according to ATCC instructions. THP-1 cells were differentiated to macrophage-like cells according to a standard protocol with 100 nmol/L phorbol-12-myristat-13-acetate (Sigma) for 48 hours. These cells are widely used as a model of human monocytic cells.22
Detection of PDGFR-α
For detection of PDGFR-α mRNA expression, CiGEnC were seeded into 75 mm2 flasks, grown to subconfluency at 33°C and then either harvested directly for the isolation of RNA with consecutive reverse transcription PCR, or after being transferred to 37°C for 3, 5, and 7 days, as described previously.20 For immunocytochemistry, CiGEnC were seeded on glass cover slides in 6-well plates, grown to subconfluency at 33°C and then transferred to 37°C for 5 days to fully differentiate. The cover slides were then removed, dried, and fixed in 50% methanol/50% ethanol for 10 minutes, dried again, and stained with goat anti-human PDGFR-α Ab and Cy2-coupled rabbit anti-goat secondary Ab. The stained cover slides were examined using a laser scanning microscope.
Detection of Angiogenic Molecules
CiGEnC were seeded into 75 mm2 flasks, grown to subconfluency at 33°C, transferred to 37°C for 3 days, deprived of growth factors and serum for 24 hours and then stimulated with PDGF-C or left unstimulated for 24 hours. For stimulation experiments in full medium, CiGEnC were left for 4 days at 37°C and then stimulated for 24 hours with PDGF-C in full medium. At day 5 after transfer to 37°C all cells were harvested, RNA isolated and real-time reverse transcription PCR performed as described.21 Human mesangial cells and THP-1 monocytic cells were seeded into 75 mm2 flasks, grown to subconfluency and serum deprived for 24 hours with subsequent stimulation. Macrophages were not serum deprived but directly stimulated after 48 hours of differentiation with phorbol-12-myristat-13-acetate. All cells were either unstimulated (left on hunger medium) or stimulated with 5 ng/ml PDGF-C for 24 hours.
Proliferation Assays
CiGEnC were seeded in 96-well plates (Nalge Nunc, Naperville, IL) and grown to subconfluency at 33°C. Cells were then either directly growth arrested for 24 hours and stimulated for 24 hours in 33°C, or transferred to 37°C for 3 days, deprived of serum and growth factors (hunger medium) for 24 hours and then stimulated for 24 hours. Stimulations were identical for cells in 33°C and 37°C and consisted of the following: hunger medium, 5 ng/ml PDGF-C in hunger medium, full medium, 5 ng/ml PDGF-C in full medium, and conditioned media from unstimulated or PDGF-C-stimulated (5 ng/ml) cells (CiGEnC, mesangial cells, monocytes and macrophages). DNA synthesis, a widely used surrogate of cell proliferation in vitro, was determined by a 5-bromo-2′-deoxyuridine incorporation assay according to the manufacturer’s instructions (Roche, Mannheim, Germany). 5-Bromo-2′-deoxyuridine was added for 16 hours to CiGEnC at 37°C and for 4 hours to CiGEnC at 33°C.
All in vitro experiments were performed in triplicate, apart from proliferation assays which were performed in quadruplicate.
Statistical Analyses
All values are expressed as means ± SD. In scatter plots each point represents data of one animal. Data analyses were performed using Mann-Whitney U-test or Student’s t-test where appropriate. For multiple group comparison in vitro, analysis of variance with posthoc Scheffé test was applied. To confirm the difference in the number of animals that doubled blood urea nitrogen, one-tailed χ2 test was applied. Statistical significance was defined as P < 0.05.
Results
In Vivo Results
PDGF-C Infusion Accelerates Glomerular Endothelial Repair
In rats with anti-Thy1.1 GN, we infused PDGF-C via micro-osmotic pumps from day 1 to 3 after disease induction. At sacrifice on day 5, the mesangiolysis score, an indicator of damage to the glomerular endothelium and mesangium, was reduced by 30% in PDGF-C-treated rats (Figure 1A). PDGF-C-infused rats exhibited 61% fewer microaneurysms (Figures 1, B, F, and G) and a 32% increase in glomerular endothelial area (Figures 1, C, H, and I; 42 ± 9 vs. 32 ± 5% JG12 positive glomerular area in PDGF-C infused versus PBS rats, P < 0.01). Enhanced glomerular repair was also evidenced by lesser expansion of the glomerular tuft area in PDGF-C-infused rats (88 ± 10 vs. 80 ± 11 μm2, PBS versus PDGF-C-infused rats; P < 0.05).
Figure 1.

PDGF-C accelerates glomerular endothelial repair in anti-Thy1.1 GN. PDGF-C infusion reduced the mesangiolysis score (A), the number of glomeruli with microaneurysms (B, F, G; arrows (in F and G) point to glomeruli with microaneurysms) and increased the glomerular endothelial area (C, H, I). These effects were not mediated via VEGF, since we observed no difference between the groups in glomerular VEGF protein expression (D). The pro-angiogenic FGF-2 transcript was markedly up-regulated in rats treated with PDGF-C (E). All graphs in A–D show individual values of each animal (PBS group as triangles; PDGF-C group as rectangles); the line depicts the mean value for each group. Graph E shows fold increase in individual mRNA transcripts in PDGF-C-treated animals, as compared with PBS-treated animals. Original magnifications ×100 (F, G) or ×400 (H, I). *P < 0.05; **P < 0.01.
PDGF-C infusion resulted in an 82% rise in endothelial mitoses, whereas non-endothelial mitoses were not affected (Figure 2A). To independently verify these findings, we counted glomerular cells with double positivity for the proliferation marker PCNA and either the endothelial marker JG12 or the mesangial cell marker α–SMA. PCNA/JG12 double-stained cells increased in glomeruli of PDGF-C-infused rats (Figure 2, B and F; P = 0.055), whereas PCNA/α-SMA double staining showed no difference between both treatment groups (Figure 2, C and E). Finally, the glomerular α-SMA-positive area, a marker of activated mesangial cells (Figure 2, D and G) as well as renal tubulointerstitial α-SMA expression, which labels myofibroblasts, was not different between the groups (data not shown). Mitoses in peritubular capillaries or larger vessels were rare and similar in both groups (data not shown).
Figure 2.

PDGF-C induces glomerular endothelial cell proliferation but does not affect mesangial cell proliferation or activation. PDGF-C infusion augmented proliferation of glomerular endothelial cells but not of non-endothelial cells as assessed by counting mitotic figures (A). Numbers of JG12 (brown)/PCNA (blue) double-positive-stained glomerular cells in the PDGF-C-treated group increased in comparison with rats receiving PBS only (B and F, P = 0.055). Proliferating mesangial cells were identified by PCNA (brown)/α-SMA (blue) double staining (C and E) and showed no differences between the groups. Glomerular α-SMA expression, a marker of activated mesangial cells, was not affected by PDGF-C treatment (D and G). Arrows point to double-positive cells. All graphs in A–D show individual values per animal (PBS group as triangles; PDGF-C group as rectangles); the line depicts the mean value for each group. Original magnifications ×400. *P < 0.05; **P < 0.01.
PDGF-C Infusion in Early anti-Thy1.1 GN Specifically Up-Regulates FGF-2
To assess potential mechanisms underlying the PDGF-C effects, we investigated the expression of angiogenic systems. In both groups, which were treated either by PDGF-C or by PBS infusion, systemic levels of VEGF protein were below the detection level, ie, below 8.4 pg/ml. By immunohistochemistry, glomerular expression of VEGF protein was similar in both groups (see Supplemental Figure 1A at http://ajp.amjpathol.org) as was the glomerular transcript expression of VEGF, its receptors VEGFR-1 and −2, angiopoietin-1 and −2 (Ang-1, Ang-2), and thrombospondin-1 (see Supplemental Figure 1B at http://ajp.amjpathol.org and data not shown). Tubulointerstitial VEGF protein expression was also identical between the groups, as evaluated by immunohistochemistry (data not shown). The glomerular transcript expression of either PDGFR chain or PDGF isoform was not affected (data not shown). In contrast, PDGF-C infusion markedly up-regulated glomerular FGF-2 transcripts (Figure 1E) and glomerular FGF-2 protein increased significantly (158 ± 39 vs. 130 ± 27 pg FGF-2/mg protein in PDGF-C infused versus PBS group, P < 0.05). Systemic FGF-2 levels were below the detection limit in all animals (data not shown).
PDGF-C Reduces Glomerular Infiltration of Monocytes/Macrophages but Augments their Proliferation
Compared with PBS treatment, the number of glomerular ED-1-positive monocytes/macrophages decreased significantly on PDGF-C infusion (Figure 3A), possibly as a result of reduced mesangiolytic damage. Glomerular mRNA expression of the chemokines monocyte chemotactic protein-1 and regulated on activation normal T cell expressed and secreted (RANTES), however, remained unchanged (data not shown). Quantification of PCNA/ED-1 double staining revealed no difference between either treatment (Figure 3B), implying that the proportion of proliferating ED-1-positive cells in PDGF-C-infused rats was about 44% higher than in the controls (Figure 3C). This is in line with data suggesting that PDGF-C acts as a potent mitogen for monocytes.22 The number of infiltrating monocytes/macrophages was not significantly lower in the tubulointerstitium of PDGF-C infused rats (5.6 ± 2.3 vs. 7.4 ± 2.9 ED1+ cells per view field in PDGF-C infused versus PBS rats, P = 0.09).
Figure 3.

Effect of PDGF-C infusion on glomerular monocytes/macrophages (Mo/Ma). PDGF-C infusion significantly decreased glomerular Mo/Ma infiltration (A; ED-1 immunohistochemistry). We found no difference in the number of proliferating glomerular Mo/Ma, as quantified by PCNA/ED-1 double staining (B). However, the relative proportion of proliferating Mo/Ma among glomerular Mo/Ma (ratio calculated from A and B) was significantly higher in PDGF-C-infused rats (C). Graphs A–C show individual values of each animal (PBS group as triangles; PDGF-C group as rectangles), the line depicts the mean value for each group. *P < 0.05; **P < 0.01.
Effects of PDGF-C Infusion on Blood Pressure and Renal Function
PDGF-C infusion was well tolerated and no rat exhibited signs of toxicity or died during the experiment. As expected, in this model body weight, blood pressure, proteinuria and serum creatinine levels as well as creatinine clearance remained normal at day 5, and no differences between PDGF-C- and PBS-infused rats were observed (Table 2). In the PDGF-C group, one-third (5 of 15) of the rats doubled their blood urea nitrogen compared with two-thirds (10 of 15) in the PBS group (P < 0.05).
Table 2.
Renal Functional Parameters, Blood Pressure, and Body Weight Were Not Affected by PDGF-C Infusion in Anti-Thy1.1 GN
| PBS (n = 15) | PDGF-C (n = 15) | |
|---|---|---|
| Serum creatinine [μmol/L] | 45 ± 8 | 47 ± 11 |
| BUN [mmol/L] | 16 ± 6 | 13 ± 3 |
| Cl.crea/100g BW [ml/min/100g] | 0.35 ± 0.11 | 0.34 ± 0.10 |
| Diuresis [ml/d] | 14 ± 12 | 14 ± 8 |
| Proteinuria [mg/d] | 126 ± 40 | 122 ± 45 |
| u-prot to u-crea ratio [AU] | 0.33 ± 0.11 | 0.30 ± 0.10 |
| Systolic blood pressure [mmHg] | 119 ± 10 | 117 ± 9 |
| Body weight day 0 [g] | 179 ± 19 | 183 ± 19 |
| Body weight day 5 [g] | 175 ± 17 | 182 ± 18 |
Data are means ± SD, all differences between the nephritic groups (PBS and PDGF-C) are not statistically significant (P > 0.05).
BUN, blood urea nitrogen; Cl.crea/100g BW, creatinine clearance per 100g body weight; u-prot to u-crea ratio, urinary protein to urinary creatinine ratio.
Inhibition of PDGF-C in Anti-Thy1.1 GN Delays Glomerular Endothelial Recovery
We next injected rats with anti-Thy1.1 nephritis either with PDGF-C-neutralizing antibodies or irrelevant IgG on day 1 following disease induction and once again sacrificed all rats on day 5.
Inhibition of PDGF-C increased mesangiolysis, but the number of microaneurysms was not different (Figure 4, A and B). PDGF-C inhibition also resulted in a significant reduction of the glomerular endothelial area (−40%; Figure 4C; 15 ± 5 vs. 26 ± 7% JG12 positive glomerular area in PDGF-C antibody versus IgG, P < 0.01), glomerular endothelial mitotic figures (−60%; Figure 4D) and numbers of PCNA/JG12 double-positive cells (Figure 4E). Blocking PDGF-C had no effect on glomerular non-endothelial cell proliferation (Figure 4D), the number of PCNA/α–SMA double-positive cells or the glomerular area stained positively for α–SMA (data not shown). Glomerular infiltration and proliferation of monocytes/macrophages was not different between the groups (data not shown).
Figure 4.

Inhibition of PDGF-C confirms the pro-angiogenic role of PDGF-C in glomerular capillary repair. Rats treated with the PDGF antibody exhibited a significantly reduced mesangiolysis score (A), whereas the number of glomeruli exhibiting microaneurysms was not significantly, different (B). The group treated with the PDGF-C antibody exhibited a significantly reduced endothelial area positive for JG12 (C), as well as endothelial mitotic figures (D) and cells double positive for the endothelial cell marker JG12 and the proliferation marker PCNA (E). Graphs A–E show individual values of each animal (IgG group as triangles; PDGF-C antibody group as rectangles); the line depicts the mean value for each group. *P < 0.05; **P < 0.01.
PDGF-C inhibition led to a 2.2- and 1.6-fold increase in glomerular mRNA expression of the anti-angiogenic molecules thrombospondin-1 and Ang-2, respectively (both P < 0.05, see Supplemental Figure 1C at http://ajp.amjpathol.org), but had no effect on VEGF, VEGFR or FGF-2 (see Supplemental Figure 1C at http://ajp.amjpathol.org). Inhibition of PDGF-C had no effect on renal function, proteinuria, body weight, or blood pressure (data not shown).
PDGF-C Inhibition in Mice Aggravates Thrombotic Microangiopathy
To further assess the role of PDGF-C in capillary healing, mice with TMA were injected with PDGF-C-neutralizing antibodies or irrelevant IgG at 4 hours, 24 hours and 48 hours after disease induction. Inhibition of PDGF-C resulted higher weight loss on day 4 (Figure 5A). PDGF-C antibody treatment significantly increased serum creatinine (Figure 5B). Blood urea nitrogen also increased in this group but without statistical significance (10.1 ± 1.9 vs. 8.8 ± 2.6 g PDGF-C antibody versus IgG, n.s.). Proteinuria and albuminuria were not different (data not shown).
Figure 5.

Inhibition of PDGF-C aggravates thrombotic microangiopathy (TMA) in mice. Mice treated with PDGF-C antibody had significantly higher body weight loss (A), serum creatinine (B) and glomeruli exhibiting injury, ie, necrosis, thrombosis, mesangiolysis, or microaneurysms (C), as well as fibrinogen deposition (D). PDGF-C inhibition significantly reduced endothelial area positive for CD31 (E). Corresponding pictures of periodic acid-Schiffs, fibrinogen, and CD31 stainings of glomeruli from mice with TMA treated with PDGF-C-neutralizing antibody (G, I, K) or control IgG (F, H, J) are shown. In sham-treated animals, all assessed parameters were within the normal range and were not affected by PDGF-C inhibition (L–Q). Graphs A–E show individual values of each animal (IgG group as triangles; PDGF-C antibody group as rectangles); the line depicts the mean value for each group. Original magnifications ×400. Glom. – glomerular, s-crea. – serum creatinine. *P < 0.05; **P < 0.01.
In PDGF-C antibody-treated TMA mice, significantly more glomeruli (+170%) exhibited segmental or global necrosis, thrombosis, mesangiolysis, and microaneurysms (Figure 5, C, F, and G). Glomerular fibrinogen also increased by 77% in the PDGF-C antibody group (Figure 5, D, H, and I). No significant differences were found in mesangiolysis score (1.31 ± 0.42 vs. 1.05 ± 0.44 PDGF-C antibody versus IgG) and microaneurysms (11.5 ± 12.7 vs. 1.2 ± 0.44% PDGF-C antibody versus IgG). Importantly, glomerular endothelial area was significantly reduced by PDGF-C inhibition (Figure 5, E, J, and K; 1.83 ± 0.53 vs. 2.48 ± 0.25% CD31 positive glomerular area in PDGF-C antibody versus IgG, P < 0.01). No glomerular α–SMA expression was detected in TMA mice (data not shown). The renal cortical mRNA expression of PDGF-C was significantly reduced by 39% in the antibody treated TMA mice (0.62 ± 0.35 vs. 1.00 ± 0.23 relative mRNA expression in PDGF-C antibody versus IgG, P < 0.05). No significant differences were found in the FGF-2 gene expression (0.29 ± 0.27 vs. 1.00 ± 0.96 relative mRNA expression in PDGF-C antibody versus IgG, P = 0.09) or the VEGF mRNA levels (0.75 ± 0.26 vs. 1.00 ± 0.21 relative mRNA expression in PDGF-C antibody versus IgG, P = 0.08).
In healthy sham animals, body weight, renal function, histology, and mRNA expression of VEGF, FGF-2, and PDGF-C in renal cortex were normal and did not differ between mice receiving PDGF-C antibody or irrelevant IgG (Figure 5, L–Q).
In Vitro Results
Potential mechanisms of PDGF-C-induced glomerular endothelial alterations were analyzed in conditionally immortalized human glomerular endothelial cells (CiGEnC) with a temperature-sensitive SV40LT construct.20 This construct allows proliferation at 33°C, whereas at 37°C the construct is inactive and CiGEnC differentiate and exhibit typical glomerular endothelial features (eg, fenestrae, albeit at much lower density as in vivo) and a low proliferation rate.20
CiGEnC Express the PDGF-C Receptor PDGFR-α
We first verified PDGFR-α mRNA and protein expression in CiGEnC (Figure 6, A and B), which is consistent with previous reports describing PDGFR-α on microvascular endothelium.8,23,24 As compared with CiGEnC proliferating at 33°C, PDGFR-α transcript expression increased about threefold during endothelial differentiation at 37°C (Figure 6A). CiGEnC also expressed PDGFR-β transcripts, which, however, did not change during differentiation (data not shown).
Figure 6.

Expression of PDGFR-α on human glomerular endothelial cells in vitro and their proliferative response to PDGF-C and PDGF-C-conditioned media. PDGFR-α mRNA and protein expression were detected in CiGEnC by reverse transcription PCR (A) and immunofluorescence (B). PDGFR-α transcript expression increased with time of CiGEnC differentiation and reached its peak on day 5. PDGF-C alone did not induce CiGEnC proliferation under permissive (D) or nonpermissive temperatures (C). When added to full medium, PDGF-C induced marked proliferation of differentiated CiGEnC (C). This effect was not observed in undifferentiated cells (D). Conditioned medium from mesangial cells or macrophages stimulated with 5 ng/ml PDGF-C induced proliferation of CiGEnC at the permissive and the nonpermissive temperature (E–H). *P < 0.05 vs. unstimulated cells, **P < 0.05 vs. full medium.
PDGF-C-Induced Proliferation of CiGEnC Depends on the Presence of Additional Factors
When added alone, PDGF-C did not affect CiGEnC DNA synthesis at the permissive or nonpermissive temperature (Figure 6, C and D). In differentiated CiGEnC, addition of full medium (containing eg, VEGF and FGF-2) induced a 51% increase in DNA synthesis (P = 0.07), whereas full medium plus PDGF-C induced a 303% increase (Figure 6C). This synergistic effect was lost in proliferating CiGEnC at the permissive temperature, where full medium alone induced maximum CiGEnC proliferation (Figure 6D).
Conditioned Media from PDGF-C-Stimulated Mesangial Cells and Macrophage-Like Cells Induce CiGEnC Proliferation
We next analyzed the proliferation of CiGEnC stimulated with conditioned media from CiGEnC, human mesangial cells, human monocyte-like (THP-1) or human adherent macrophage-like (differentiated THP-1) cells that had been stimulated with PDGF-C. Conditioned medium from both PDGF-C-stimulated mesangial cells and differentiated THP-1 cells induced a significant increase in CiGEnC DNA synthesis in both differentiated and proliferating CiGEnC (Figure 6, E–H). Medium from PDGF-C- stimulated CiGEnC or from unstimulated THP-1 cells failed to affect CiGEnC DNA synthesis (data not shown).
PDGF-C Regulates mRNA Expression of Angiogenic Factors in Mesangial Cells and Macrophage-Like Cells
We next stimulated CiGEnC, human mesangial cells, THP-1 and differentiated THP-1 cells with PDGF-C and analyzed the transcripts of various pro- and anti-angiogenic molecules. In mesangial cells, PDGF-C significantly induced the expression of FGF-2 (1.4-fold, P < 0.05) and Ang-2 (1.4-fold, P < 0.05). PDGF-C-stimulated differentiated THP-1 cells down-regulated Ang-2 expression by 70% (P < 0.05). FGF-2- and Ang-2-transcripts were increased in THP-1 cells on PDGF-C stimulation by 2.7- and 1.6-fold, respectively (both P < 0.05). In comparison with nonstimulated cells, PDGF-C up-regulated the mRNA of PDGFR-α and -β chains in CiGEnC 1.3- and 1.7-fold (both P < 0.05). We found no regulation of VEGF, its receptors, Ang-1, thrombospondin-1, or the PDGF ligands by PDGF-C in any of the cells tested.
PDGF-C Induces a Pro-Angiogenic Phenotype in CiGEnC in Full Medium
PDGF-C induced prominent CiGEnC proliferation when added to full medium. Therefore, we analyzed the message of angiogenic molecules in these cells. In concert with other factors from the full medium, PDGF-C potently induced FGF-2 (27-fold), and to lesser degrees, VEGF (3.5-fold), Ang-1 (1.1-fold), and PDGF-C itself (1.2-fold, P < 0.05) (Figure 7). Both PDGFR-α and -β were also significantly up-regulated by 3.9-fold and 5.7-fold, respectively (Figure 7).
Figure 7.
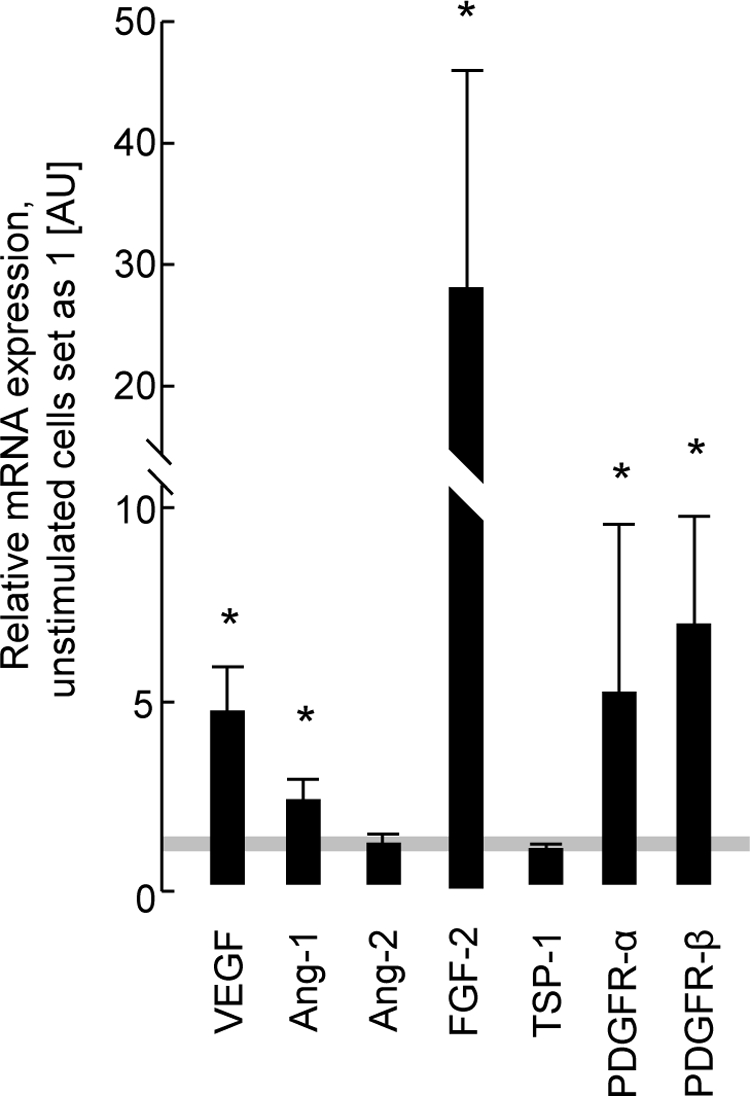
Transcript expression of angiogenic molecules in PDGF-C-stimulated glomerular endothelial cells (CiGEnC) cultured with full medium. In cooperation with other molecules contained in the full medium (EGM-2MV), PDGF-C induced a strong expression of pro-angiogenic molecules, mainly of FGF-2. *P < 0.05 vs. cells with full growth EGM-2MV medium only.
Discussion
In the present study, we investigated the role of PDGF-C in primary and secondary glomerular endothelial cell damage. Following anti-Thy1.1 antibody-mediated lysis of the mesangium in rats, glomerular endothelial cell damage occurs as a secondary event. Glomerular repair involves endothelial cell recovery and proliferation as well as mesangial cell proliferation.9,10 Endothelial recovery is a critical step in the resolution of the disease in this model, and molecules like VEGF, FGF-2, inducible nitric oxide synthase, or cyclooxygenase-2 are crucially involved.9,10,11,12,13 Initiation and duration of treatments affecting PDGF-C levels were chosen to cover the “therapeutic” phase of endothelial recovery between days 1 and 4 after disease induction. In the second model, conA/anti-conA induced experimental TMA in the mouse within 4 hours after disease induction, characterized by an acute decrease in glomerular cell counts, glomerular capillary rarefaction, direct endothelial cell damage and infiltration of inflammatory cells.14 The period of PDGF-C inhibition in our study was again chosen to cover subsequent glomerular regenerative responses between 4 hours and day 3. Since our human recombinant PDGF-C did not stimulate mouse cells (unpublished data), we abstained from PDGF-C infusion in mice with TMA.
A major novel finding of the present study is the identification of PDGF-C as a key pro-angiogenic factor in the regenerative processes that follows both primary or secondary glomerular endothelial cell damage. Thus, in vivo PDGF-C infusion in rats with anti-Thy1.1 nephritis markedly accelerated glomerular capillary healing, whereas PDGF-C inhibition disrupted it. Similarly, in experimental TMA, PDGF-C inhibition aggravated the disease and hindered endothelial recovery.
It was previously reported that cultured microvascular endothelium expresses the PDGF-C receptor PDGFR-α at low levels and that PDGF-C induces migration, but not proliferation, in these cells.8,23,24 In agreement with this, we detected expression of PDGFR-α on human glomerular endothelial cells (CiGEnC) and an absent proliferative response of these cells on PDGF-C stimulation. However, in full medium, PDGF-C induced a more than threefold proliferation of differentiated CiGEnC, suggesting that PDGF-C act in concert with or via the induction of other pro-angiogenic factors. Expression profiles of angiogenic molecules corroborated this assumption: in full medium PDGF-C induced a prominent pro-angiogenic switch with pronounced up-regulation of FGF-2, and to a lesser degree, VEGF transcripts. These observations were confirmed in our nephritic rats, where PDGF-C infusion led to a selective increase of glomerular FGF-2 transcript and protein expression whereas other angiogenic factors were not affected. Taking the in vivo and in vitro data together, PDGF-C appears to induce proliferation of glomerular endothelial cells in cooperation with or via the induction of FGF-2.
FGF-2 has previously been demonstrated to be mitogenic for glomerular endothelial cells25,26 and to play a role in glomerular capillary repair in anti-Thy1.1 GN.9 In these studies, administration of FGF-2-neutralizing antibodies in anti-Thy1.1 GN significantly reduced endothelial cell proliferation.9 Our data are similar to observations obtained with PDGF-B, where only in synergism with FGF-2 did PDGF-B induce neovascularization in corneal pocket assays, after myocardial infarction or hind limb ischemia.26,27,28 Again, similar to our data, such a synergistic effect appeared to be independent of VEGF.26 Furthermore, in vitro, after pretreatment of bovine capillary endothelial cells with PDGF-B, subsequent stimulation with FGF-2 induced proliferation, whereas pretreatment with FGF-2 and subsequent PDGF-BB stimulation induced migration.28 On the other hand, it is unlikely that all of the observed effects were mediated by FGF-2. We have previously shown that FGF-2 release mediates cytotoxicity and early glomerular damage in the anti-Thy 1.1 nephritis, ie, the opposite of what we would assume based on the present results.29 However, fully clarifying this interaction will be the topic of future studies.
Cross talk between various glomerular cell types exerts important roles in both the healthy condition and disease.3,4,30,31,32 We previously showed that PDGF-C protein is produced by mesangial cells, and is up-regulated during the course of anti-Thy1.1 nephritis, with this up-regulation localizing to mesangial regions.33 In the context of the present study, these data point to the importance of a mesangial-endothelial cross talk in driving capillary repair. In addition to mesangial cells, macrophages also infiltrate the mesangium in anti-Thy1.1 GN. Macrophages are important regulators of angiogenesis, and can acquire both pro- and anti-angiogenic phenotypes depending on the microenvironment and the clues therein.34,35 Pro-angiogenically polarized macrophages produce a multitude of angiogenic molecules, including FGF-2 and VEGF.34,35 The anti-angiogenic protein Ang-2 was detected in macrophages in plaques from patients with atherosclerosis, and stimulation of macrophages with VEGF induced Ang-2 production.36,37 In our in vitro experiments, PDGF-C-stimulated macrophage-like THP-1 cells exhibited a pro-angiogenic phenotype associated with a significant down-regulation of Ang-2 in them. In vivo, inhibition of PDGF-C in anti-Thy1.1 GN resulted in increased glomerular Ang-2 mRNA expression. Thus, in addition to the cooperation with FGF-2 (see above), another pro-angiogenic action of PDGF-C may involve down-regulation of anti-angiogenic molecules such as Ang-2 in macrophages. Collectively, our in vitro data suggest that the pro-angiogenic actions of PDGF-C on glomerular endothelial cells require one or several co-factors and that an autocrine production of angiogenic factors (eg, VEGF or FGF-2) augments this effect. The participating co-factor(s) as well as the paracrine factor(s) produced by mesangial cells and differentiated THP-1 cells remain to be identified. Although our data point to a possible role of FGF-2, further studies are required to validate this finding.
In addition, we showed that glomerular infiltration of monocytes/macrophages was decreased in nephritic rats infused with PDGF-C. PDGF-C acts as a chemotactic factor.16 Since we implanted PDGF-C pumps i.p., the highest PDGF-C concentration was in the peritoneal cavity (forming a chemotactic gradient). This gradient may have competed with the chemokines which normally drive monocytes/macrophages influx into glomeruli of nephritic rats.
Apart from its actions on endothelial cells, PDGF-C also induces proliferation and migration of vascular smooth muscle cells and pericytes, leading to new vessel sprouts.6,7,8 In vitro, PDGF-C stimulated the proliferation of rat33 and human (unpublished data) glomerular “pericytes”—the mesangial cells. These cells constitute the most prominent glomerular cell type expressing PDGFR-α.38,39,40 However, in the present study, we could not detect any influence of either PDGF-C infusion or inhibition on mesangial expansion, activation, or proliferation. Our data are in line with the in vivo transfection of the liver with a PDGF-C expression vector, which induced no renal pathology, whereas PDGF-D induced massive mesangioproliferative GN.41 Several reasons may account for this finding. First, mesangial cell proliferation and activation peak in later phases of anti-Thy1.1 GN, thus we may have missed such effects. However, in unpublished studies from our group, inhibition of PDGF-C also had no effect on mesangial cell proliferation on day 7 of anti-Thy1.1 GN. Second, even though the above data33 were generated with primary mesangial cells, the two-dimensional cell culture conditions may not adequately reflect the complex and dynamic three-dimensional situation in the glomerulus. Third, the PDGFR-α signaling plays a minor role compared with the prominent PDGFR-β pathway in mesangial cell proliferation and activation toward a “myofibroblast-like” phenotype.38 For example, while PDGF-B deficient mice completely lack a mesangium,42 the glomerular morphology is normal in PDGF-A-, -C-, or PDGFR-α-deficient mice.19,43 Our present data thus further support the notion that signaling through PDGFR-α is not an important event in driving proliferation of mesangial cells or their activation toward a “myofibroblast”-like phenotype in vivo.
In conclusion, using models of primary and secondary glomerular endothelial cell damage, we demonstrate that PDGF-C is important for glomerular capillary repair. In vitro studies point toward complex PDGF-C effects on the glomerular endothelium including the cooperation of PDGF-C with other pro-angiogenic factors, eg, FGF-2, and down-regulation of anti-angiogenic proteins such as Ang-2. Finally, short-term PDGF-C infusion was well tolerated and did not induce other intrarenal effects, in particular mesangial cell proliferation or activation. Our study thus identifies PDGF-C as a potentially novel therapeutic approach to glomerular diseases with endothelial injury.
Acknowledgments
We thank Dr. Arne Östman (Karolinska Institute, Stockholm, Sweden) for kindly providing PDGF-C-neutralizing antibody. We also thank Gabriele Dietzel, Andrea Cosler, Lydia Zimmermanns, Claudia Schwarzenberger, and Sabrina Cabric for excellent technical assistance and Thomas Clahsen for his assistance at the laser scanning microscope.
Footnotes
Address reprint requests to Peter Boor, M.D., Ph.D., Division of Nephrology and Immunology, University Hospital Aachen, Pauwelsstr. 30, D-52074 Aachen, Germany. E-mail: boor@email.cz.
Supported by a stipend from the German Academic Exchange Service to P.B., a stipend from the Italian Society of Nephrology to L.V., a grant from the ICCR Erlangen to B.H. and C.H. (IZKF TP B14), grants from the German Research Foundation to T.O. and F.E. (SFB/TRR57 P14) and to J.F. and T.O. (SFB542, TP C7).
Supplemental material for this article can be found on http://ajp.amjpathol.org.
References
- Ballermann BJ. Neilson EG, Couser WG, editors. Philadelphia: Lippincott-Raven Publishers,; Endothelial responses to immune injury. 1997:pp 627–654. [Google Scholar]
- Lee LK, Meyer TW, Pollock AS, Lovett DH. Endothelial cell injury initiates glomerular sclerosis in the rat remnant kidney. J Clin Invest. 1995;96:953–964. doi: 10.1172/JCI118143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eremina V, Cui S, Gerber H, Ferrara N, Haigh J, Nagy A, Ema M, Rossant J, Jothy S, Miner JH, Quaggin SE. Vascular endothelial growth factor a signaling in the podocyte-endothelial compartment is required for mesangial cell migration and survival. J Am Soc Nephrol. 2006;17:724–735. doi: 10.1681/ASN.2005080810. [DOI] [PubMed] [Google Scholar]
- Eremina V, Sood M, Haigh J, Nagy A, Lajoie G, Ferrara N, Gerber HP, Kikkawa Y, Miner JH, Quaggin SE. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J Clin Invest. 2003;111:707–716. doi: 10.1172/JCI17423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reigstad LJ, Varhaug JE, Lillehaug JR. Structural and functional specificities of PDGF-C and PDGF-D, the novel members of the platelet-derived growth factors family. Febs J. 2005;272:5723–5741. doi: 10.1111/j.1742-4658.2005.04989.x. [DOI] [PubMed] [Google Scholar]
- Cao R, Brakenhielm E, Li X, Pietras K, Widenfalk J, Ostman A, Eriksson U, Cao Y. Angiogenesis stimulated by PDGF-CC, a novel member in the PDGF family, involves activation of PDGFR-alphaalpha and -alphabeta receptors. FASEB J. 2002;16:1575–1583. doi: 10.1096/fj.02-0319com. [DOI] [PubMed] [Google Scholar]
- Gilbertson DG, Duff ME, West JW, Kelly JD, Sheppard PO, Hofstrand PD, Gao Z, Shoemaker K, Bukowski TR, Moore M, Feldhaus AL, Humes JM, Palmer TE, Hart CE. Platelet-derived growth factor C (PDGF-C), a novel growth factor that binds to PDGF alpha and beta receptor. J Biol Chem. 2001;276:27406–27414. doi: 10.1074/jbc.M101056200. [DOI] [PubMed] [Google Scholar]
- Li X, Tjwa M, Moons L, Fons P, Noel A, Ny A, Zhou JM, Lennartsson J, Li H, Luttun A, Ponten A, Devy L, Bouche A, Oh H, Manderveld A, Blacher S, Communi D, Savi P, Bono F, Dewerchin M, Foidart JM, Autiero M, Herbert JM, Collen D, Heldin CH, Eriksson U, Carmeliet P. Revascularization of ischemic tissues by PDGF-CC via effects on endothelial cells and their progenitors. J Clin Invest. 2005;115:118–127. doi: 10.1172/JCI19189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iruela-Arispe L, Gordon K, Hugo C, Duijvestijn AM, Claffey KP, Reilly M, Couser WG, Alpers CE, Johnson RJ. Participation of glomerular endothelial cells in the capillary repair of glomerulonephritis. Am J Pathol. 1995;147:1715–1727. [PMC free article] [PubMed] [Google Scholar]
- Ostendorf T, Kunter U, Eitner F, Loos A, Regele H, Kerjaschki D, Henninger DD, Janjic N, Floege J. VEGF(165) mediates glomerular endothelial repair. J Clin Invest. 1999;104:913–923. doi: 10.1172/JCI6740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitahara M, Eitner F, Ostendorf T, Kunter U, Janssen U, Westenfeld R, Matsui K, Kerjaschki D, Floege J. Selective cyclooxygenase-2 inhibition impairs glomerular capillary healing in experimental glomerulonephritis. J Am Soc Nephrol. 2002;13:1261–1270. doi: 10.1681/ASN.V1351261. [DOI] [PubMed] [Google Scholar]
- Masuda Y, Shimizu A, Mori T, Ishiwata T, Kitamura H, Ohashi R, Ishizaki M, Asano G, Sugisaki Y, Yamanaka N. Vascular endothelial growth factor enhances glomerular capillary repair and accelerates resolution of experimentally induced glomerulonephritis. Am J Pathol. 2001;159:599–608. doi: 10.1016/S0002-9440(10)61731-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostendorf T, Van Roeyen C, Westenfeld R, Gawlik A, Kitahara M, De Heer E, Kerjaschki D, Floege J, Ketteler M. Inducible nitric oxide synthase-derived nitric oxide promotes glomerular angiogenesis via upregulation of vascular endothelial growth factor receptors. J Am Soc Nephrol. 2004;15:2307–2319. doi: 10.1097/01.ASN.0000136425.75041.9C. [DOI] [PubMed] [Google Scholar]
- Hohenstein B, Braun A, Amann KU, Johnson RJ, Hugo CP. A murine model of site-specific renal microvascular endothelial injury and thrombotic microangiopathy. Nephrol Dial Transplant. 2008;23:1144–1156. [Google Scholar]
- Ostendorf T, van Roeyen CR, Peterson JD, Kunter U, Eitner F, Hamad AJ, Chan G, Jia XC, Macaluso J, Gazit-Bornstein G, Keyt BA, Lichenstein HS, LaRochelle WJ, Floege J. A fully human monoclonal antibody (CR002) identifies PDGF-D as a novel mediator of mesangioproliferative glomerulonephritis. J Am Soc Nephrol. 2003;14:2237–2247. doi: 10.1097/01.asn.0000083393.00959.02. [DOI] [PubMed] [Google Scholar]
- Eitner F, Bucher E, van Roeyen C, Kunter U, Rong S, Seikrit C, Villa L, Boor P, Fredriksson L, Backstrom G, Eriksson U, Ostman A, Floege J, Ostendorf T. PDGF-C is a proinflammatory cytokine that mediates renal interstitial fibrosis. J Am Soc Nephrol. 2008;19:281–289. doi: 10.1681/ASN.2007030290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misra RP. Isolation of glomeruli from mammalian kidneys by graded sieving. Am J Clin Pathol. 1972;58:135–139. doi: 10.1093/ajcp/58.2.135. [DOI] [PubMed] [Google Scholar]
- Eitner F, Ostendorf T, Kretzler M, Cohen CD, Eriksson U, Grone HJ, Floege J. PDGF-C expression in the developing and normal adult human kidney and in glomerular diseases. J Am Soc Nephrol. 2003;14:1145–1153. doi: 10.1097/01.asn.0000062964.75006.a8. [DOI] [PubMed] [Google Scholar]
- Li X, Ponten A, Aase K, Karlsson L, Abramsson A, Uutela M, Backstrom G, Hellstrom M, Bostrom H, Li H, Soriano P, Betsholtz C, Heldin CH, Alitalo K, Ostman A, Eriksson U. PDGF-C is a new protease-activated ligand for the PDGF alpha-receptor. Nat Cell Biol. 2000;2:302–309. doi: 10.1038/35010579. [DOI] [PubMed] [Google Scholar]
- Satchell SC, Tasman CH, Singh A, Ni L, Geelen J, von Ruhland CJ, O'Hare MJ, Saleem MA, van den Heuvel LP, Mathieson PW. Conditionally immortalized human glomerular endothelial cells expressing fenestrations in response to VEGF. Kidney Int. 2006;69:1633–1640. doi: 10.1038/sj.ki.5000277. [DOI] [PubMed] [Google Scholar]
- van Roeyen CR, Ostendorf T, Denecke B, Bokemeyer D, Behrmann I, Strutz F, Lichenstein HS, Larochelle WJ, Pena CE, Chaudhuri A, Floege J. Biological responses to PDGF-BB versus PDGF-DD in human mesangial cells. Kidney Int. 2006;69:1393–1402. doi: 10.1038/sj.ki.5000332. [DOI] [PubMed] [Google Scholar]
- Wagsater D, Zhu C, Bjorck HM, Eriksson P. Effects of PDGF-C and PDGF-D on monocyte migration and MMP-2 and MMP-9 expression. Atherosclerosis. 2008;202:415–423. doi: 10.1016/j.atherosclerosis.2008.04.050. [DOI] [PubMed] [Google Scholar]
- Edelberg JM, Aird WC, Wu W, Rayburn H, Mamuya WS, Mercola M, Rosenberg RD. PDGF mediates cardiac microvascular communication. J Clin Invest. 1998;102:837–843. doi: 10.1172/JCI3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx M, Perlmutter RA, Madri JA. Modulation of platelet-derived growth factor receptor expression in microvascular endothelial cells during in vitro angiogenesis. J Clin Invest. 1994;93:131–139. doi: 10.1172/JCI116936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballermann BJ. Regulation of bovine glomerular endothelial cell growth in vitro. Am J Physiol. 1989;256:C182–C189. doi: 10.1152/ajpcell.1989.256.1.C182. [DOI] [PubMed] [Google Scholar]
- Cao R, Brakenhielm E, Pawliuk R, Wariaro D, Post MJ, Wahlberg E, Leboulch P, Cao Y. Angiogenic synergism, vascular stability and improvement of hind-limb ischemia by a combination of PDGF-BB and FGF-2. Nat Med. 2003;9:604–613. doi: 10.1038/nm848. [DOI] [PubMed] [Google Scholar]
- Hao X, Mansson-Broberg A, Gustafsson T, Grinnemo KH, Blomberg P, Siddiqui AJ, Wardell E, Sylven C. Angiogenic effects of dual gene transfer of bFGF and PDGF-BB after myocardial infarction. Biochem Biophys Res Commun. 2004;315:1058–1063. doi: 10.1016/j.bbrc.2004.01.165. [DOI] [PubMed] [Google Scholar]
- Nissen LJ, Cao R, Hedlund EM, Wang Z, Zhao X, Wetterskog D, Funa K, Brakenhielm E, Cao Y. Angiogenic factors FGF2 and PDGF-BB synergistically promote murine tumor neovascularization and metastasis. J Clin Invest. 2007;117:2766–2777. doi: 10.1172/JCI32479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floege J, Burg M, Hugo C, Gordon KL, Van Goor H, Reidy M, Couser WG, Koch KM, Johnson RJ. Endogenous fibroblast growth factor-2 mediates cytotoxicity in experimental mesangioproliferative glomerulonephritis. J Am Soc Nephrol. 1998;9:792–801. doi: 10.1681/ASN.V95792. [DOI] [PubMed] [Google Scholar]
- Eng E, Ballermann BJ. Diminished NF-kappaB activation and PDGF-B expression in glomerular endothelial cells subjected to chronic shear stress. Microvasc Res. 2003;65:137–144. doi: 10.1016/s0026-2862(03)00004-9. [DOI] [PubMed] [Google Scholar]
- Eng E, Holgren C, Hubchak S, Naaz P, Schnaper HW. Hypoxia regulates PDGF-B interactions between glomerular capillary endothelial and mesangial cells. Kidney Int. 2005;68:695–703. doi: 10.1111/j.1523-1755.2005.00448.x. [DOI] [PubMed] [Google Scholar]
- Sano H, Ueda Y, Takakura N, Takemura G, Doi T, Kataoka H, Murayama T, Xu Y, Sudo T, Nishikawa S, Nishikawa S, Fujiwara H, Kita T, Yokode M. Blockade of platelet-derived growth factor receptor-beta pathway induces apoptosis of vascular endothelial cells and disrupts glomerular capillary formation in neonatal mice. Am J Pathol. 2002;161:135–143. doi: 10.1016/s0002-9440(10)64165-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eitner F, Ostendorf T, Van Roeyen C, Kitahara M, Li X, Aase K, Grone HJ, Eriksson U, Floege J. Expression of a novel PDGF isoform, PDGF-C, in normal and diseased rat kidney. J Am Soc Nephrol. 2002;13:910–917. doi: 10.1681/ASN.V134910. [DOI] [PubMed] [Google Scholar]
- Dirkx AE, Oude Egbrink MG, Wagstaff J, Griffioen AW. Monocyte/macrophage infiltration in tumors: modulators of angiogenesis. J Leukoc Biol. 2006;80:1183–1196. doi: 10.1189/jlb.0905495. [DOI] [PubMed] [Google Scholar]
- Lamagna C, Aurrand-Lions M, Imhof BA. Dual role of macrophages in tumor growth and angiogenesis. J Leukoc Biol. 2006;80:705–713. doi: 10.1189/jlb.1105656. [DOI] [PubMed] [Google Scholar]
- Hubbard NE, Lim D, Mukutmoni M, Cai A, Erickson KL. Expression and regulation of murine macrophage angiopoietin-2. Cell Immunol. 2005;234:102–109. doi: 10.1016/j.cellimm.2005.06.001. [DOI] [PubMed] [Google Scholar]
- Post S, Peeters W, Busser E, Lamers D, Sluijter JP, Goumans MJ, de Weger RA, Moll FL, Doevendans PA, Pasterkamp G, Vink A. Balance between angiopoietin-1 and angiopoietin-2 is in favor of angiopoietin-2 in atherosclerotic plaques with high microvessel density. J Vasc Res. 2008;45:244–250. doi: 10.1159/000112939. [DOI] [PubMed] [Google Scholar]
- Floege J, Eitner F, Alpers CE. A new look at platelet-derived growth factor in renal disease. J Am Soc Nephrol. 2008;19:12–23. doi: 10.1681/ASN.2007050532. [DOI] [PubMed] [Google Scholar]
- Floege J, Hudkins KL, Davis CL, Schwartz SM, Alpers CE. Expression of PDGF alpha-receptor in renal arteriosclerosis and rejecting renal transplants. J Am Soc Nephrol. 1998;9:211–223. doi: 10.1681/ASN.V92211. [DOI] [PubMed] [Google Scholar]
- Floege J, Hudkins KL, Seifert RA, Francki A, Bowen-Pope DF, Alpers CE. Localization of PDGF alpha-receptor in the developing and mature human kidney. Kidney Int. 1997;51:1140–1150. doi: 10.1038/ki.1997.157. [DOI] [PubMed] [Google Scholar]
- Hudkins KL, Gilbertson DG, Carling M, Taneda S, Hughes SD, Holdren MS, Palmer TE, Topouzis S, Haran AC, Feldhaus AL, Alpers CE. Exogenous PDGF-D is a potent mesangial cell mitogen and causes a severe mesangial proliferative glomerulopathy. J Am Soc Nephrol. 2004;15:286–298. doi: 10.1097/01.asn.0000108522.79652.63. [DOI] [PubMed] [Google Scholar]
- Leveen P, Pekny M, Gebre-Medhin S, Swolin B, Larsson E, Betsholtz C. Mice deficient for PDGF B show renal, cardiovascular, and hematological abnormalities. Genes Dev. 1994;8:1875–1887. doi: 10.1101/gad.8.16.1875. [DOI] [PubMed] [Google Scholar]
- Ding H, Wu X, Bostrom H, Kim I, Wong N, Tsoi B, O'Rourke M, Koh GY, Soriano P, Betsholtz C, Hart TC, Marazita ML, Field LL, Tam PP, Nagy A. A specific requirement for PDGF-C in palate formation and PDGFR-alpha signaling. Nat Genet. 2004;36:1111–1116. doi: 10.1038/ng1415. [DOI] [PubMed] [Google Scholar]