

Abstract
Vascular endothelial growth factor, which is critical for blood vessel formation, is regulated by hypoxia inducible transcription factors (HIFs). A component of the E3 ubiquitin ligase complex, von Hippel-Lindau (VHL) facilitates oxygen-dependent polyubiquitination and proteasomal degradation of HIFα subunits. Hypothesizing that deletion of podocyte VHL would result in HIFα hyperstabilization, we crossed podocin promoter-Cre transgenic mice, which express Cre recombinase in podocytes beginning at the capillary loop stage of glomerular development, with floxed VHL mice. Vascular patterning and glomerular development appeared unaltered in progeny lacking podocyte VHL. However, urinalysis showed increased albumin excretion by 4 weeks when compared with wild-type littermates with several sever cases (>1000 μg/ml). Many glomerular ultrastructural changes were seen in mutants, including focal subendothelial delamination and widespread podocyte foot process broadening, and glomerular basement membranes (GBMs) were significantly thicker in 16-week-old mutants compared with controls. Moreover, immunoelectron microscopy showed ectopic deposition of collagen α1α2α1(IV) in GBM humps beneath podocytes. Significant increases in the number of Ki-67-positive mesangial cells were also found, but glomerular WT1 expression was significantly decreased, signifying podocyte death and/or de-differentiation. Indeed, expression profiling of mutant glomeruli suggested a negative regulatory feedback loop involving the HIFα prolyl hydroxylase, Egln3. In addition, the brain oxygen-binding protein, Neuroglobin, was induced in mutant podocytes. We conclude that podocyte VHL is required for normal maintenance of podocytes, GBM composition and ultrastructure, and glomerular barrier properties.
The basement membrane that lies between endothelial cells and podocytes of the vertebrate glomerular capillary is an indispensible component of the glomerular filtration barrier. This glomerular basement membrane (GBM), along with the endothelial glycocalyx, and the podocyte slit diaphragms, provides both a size and charge-selective barrier to plasma proteins.1 Filtered components pass into the tubule system, which selectively reabsorbs certain molecules and water, thereby condensing the urine for excretion.
The components of the GBM are similar with those of basement membranes elsewhere and include type IV collagen, laminins, entactin/nidogen, and proteoglycans.2 In contrast to most other basement membranes, however, GBM laminins and collagen IV undergo developmental substitutions where isoforms synthesized during initial glomerulogenesis are replaced with new isoforms during glomerular maturation. Immature podocytes and endothelial cells jointly synthesize laminin 111, which is replaced by laminin 521 beginning at the S-shaped stage of nephron development.3 Similarly, a network of collagen α1α2α1(IV) is synthesized by both podocytes and endothelial cells in early comma and S-shaped glomeruli. Beginning at the capillary loop stage, podocytes alone synthesize the mature collagen α3α4α5(IV), which replaces the α1α2α1(IV) network.4
Why the GBM undergoes laminin and collagen IV isoform transitioning during development is uncertain, but it may be necessary for endothelial cells and podocytes to achieve their highly differentiated states and for the full acquisition of glomerular barrier properties. Indeed, errors in glomerular morphology and function are invariably seen if adult isoforms of laminins and collagen IV fail to be expressed properly. For example, human LAMB2 mutations result in congenital nephrosis with mesangial sclerosis, ocular anomalies, and neuromuscular junction defects, a condition known as Pierson syndrome.5,6 Similarly, mice with deletions of the Lamb2 gene have neuromuscular junction deficits, diffuse podocyte foot process effacement, and die of renal failure by 6 weeks of age.7 In human Alport syndrome where the COL4A3, COL4A4, or COL4A5 genes are mutated, the GBM has a characteristic basket weave, or “moth-eaten” appearance, a stable network of collagen α3α4α5(IV) does not form, and most patients eventually progress to renal failure.8 In Alport mouse models, the deletion of collagen α3(IV)9,10,11 results not only in an absence of the mature collagen α3α4α5(IV) network, but the reappearance of immature laminin α1 and β1 chains, along with increased expression of laminin α5.12,13 Similar dysregulation of GBM protein expression has been observed in a canine model of Alport disease, as well as in Alport patients.14 In contrast, heterozygous Col4α3+/− mice have thinner GBMs and resemble human COL4A3 carriers with thin basement membrane nephropathy.15 Many other renal diseases also display GBM modifications during the course of progression to fibrosis, including the thickened GBM of mesangial sclerosis16 and diabetic nephropathy.17
The tight regulation of GBM components led us to question what transcription factor systems might play a role in either the normal developmental isoform substitutions or the re-expression of matrix components during disease. The hypoxia-inducible factors (HIFs) are expressed in glomeruli of developing kidney18 and also have been shown to be induced in some models of renal injury.19 The HIFα subunits, and their ability to heterodimerize with HIFβ, are regulated by oxygen availability.20 In normoxia, prolyl hydroxylases modify key residues of HIFα subunits, targeting them for polyubiquitination by the von Hippel-Lindau (VHL) protein, and subsequent degradation in the proteasome. In hypoxia, prolyl hydroxylation of HIFα is blocked, and stable HIFα subunits dimerize with HIFβ in the nucleus, resulting in transcription of HIF-responsive genes such as vascular endothelial growth factor (VEGF). Impaired VHL activity results in an accumulation of HIF subunits and increases in HIF-mediated gene expression. VHL disease is characterized by highly vascularized tumors such as hemangioblastomas, and increased incidence of renal clear cell (RCC) carcinoma.21 Studies in RCC cell lines have shown that VHL also plays an important role in the synthesis of extracellular matrix in addition to its ligase activity.22 We chose to selectively delete VHL in podocytes, the epithelial cell of the glomerulus, which synthesizes the mature GBM type IV collagen network.
Materials and Methods
Animals and Genotyping
Podocin-Cre mice23 were mated with a Cre reporter strain (B6.129S4-Gt(ROSA)26Sortm1Sor/J) purchased from Jackson Laboratories (Bar Harbor, ME). Dissected kidneys from newborn mice were fixed overnight in freshly prepared 0.2% paraformaldehyde in 0.1 M piperazine-N,N′-bis[2-ethanesulfonic acid], pH 6.9, at 4°C and were processed for β-galactosidase activity as previously described.24
Mice harboring the floxed VHL allele (fVHL)25 were purchased from Jackson Laboratories (Bar Harbor, ME) and crossed with the Pod-Cre strain. Progeny from matings were genotyped at 2 weeks of age by using the following primers: for detection of Cre recombinase: 5′-AATGCTTCTGTCCGTTTG-3′ and 5′-GGATTAACATTCTCCCACC-3′ and for detection of fVHL: 5′- CTCAGGTCATCTTCTGCAACC-3′ and 5′-TCTGTCTTGGCCTCCTGAGT-3′. Mice for analysis had at least one copy of Cre recombinase and were homozygous for fVHL (Pod-Cre fVHL). Littermates serving as controls were either homozygous wild-type for fVHL, or did not contain the Cre transgene (wt).
For detection of the recombined VHL allele, genomic DNA was isolated by using the Qiagen DNeasy Tissue kit (Valencia, CA) from tail, kidney, heart, and liver and was amplified with the following primers: 5′-CTGGTACCCACGAAACTGTC-3′, 5′-CTAGGCACCGAGCTTAGAGGTTTGCG-3′, and 5′-CTGACTTCCACTGATGCTTGTCACAG-3′.26 The wild-type VHL allele was detected as a ∼290-bp band, the fVHL allele (VHL flanked with two LoxP sites) appeared as a ∼460-bp band, and the recombined VHL allele migrated as a ∼260-bp band.
Urine and Blood Collection and Analysis
At monthly intervals beginning at 4 weeks of age, mice were placed in metabolic cages, and urine was collected for 6 hours. Six microliters of urine in loading buffer was run on precast polyacrylamide gels and stained with BioSafe coomassie (BioRad, Hercules, CA). Bovine serum albumin standards (5 μg and 0.5 μg) were run on each gel to provide a size marker for albumin. Based on the gel analysis, urine was diluted 1:10 to 1:1000 for total albumin determination by using an enzyme-linked immunosorbent assay Albuwell kit for mice (Exocell, Philadelphia, PA). Total protein and creatinine were quantified from the remaining urine samples by using an autoanalyzer (Physician’s Reference Laboratory, LLC, Overland Park, KS). Blood was collected by cardiac puncture, and blood urea nitrogen levels were quantified from serum (Physician’s Reference Laboratory).
Tissue Collection and Analysis
Mice were anesthetized with halothane and sacrificed by cervical dislocation. For routine histology, kidneys were fixed in 4% paraformaldehyde and dehydrated in graded ethanols before embedding in paraffin. Five micron-thick sections were stained with either H&E or the PAS reagent.
For immunofluorescence microscopy, deparaffinized sections of 4-week-old kidneys were boiled in antigen unmasking solution (Vector Laboratories, Burlingame, CA) and blocked with 1 M NH4Cl. Slides were double labeled with either a 1:10 dilution of rat anti-Ki-67 (Dako, Carpinteria, CA) and a 1:10 dilution of rabbit anti-WT1 (Santa Cruz Biotechnology, Santa Cruz, CA) or with a 1:100 dilution of mouse anti-synaptopodin (Biodesign, Carmel, NY) and a 10 μg/ml concentration of rabbit anti-Neuroglobin (Sigma, St. Louis, MO) or with a 1:100 dilution of rabbit anti-nephrin27 and a 1:100 dilution of mouse anti-synaptopodin for 1 hour. After three rinses in PBS, Alexa-conjugated secondary antibodies (Molecular Probes, Eugene, OR) were applied for 1 hour. Slides were rinsed in PBS and mounted in Prolong Gold plus 4′,6-diamidino-2-phenylindole (Molecular Probes).
To quantify Ki-67 and WT1 labeling, a total of 10 images containing unique glomeruli per animal were captured for each label. The number of podocytes (WT1-positive cells within glomeruli) and proliferating cells (Ki-67 positive cells within glomeruli) were averaged for five wild-type mice, five nonproteinuric Pod-Cre fVHL mice, and two proteinuric Pod-Cre fVHL mice. Statistical analysis (analysis of variance) was performed by using InStat (GraphPad Software, San Diego, CA).
Kidneys were also fixed and processed for electron microscopy as previously described.28 GBM thickness at 16 weeks of age in three Pod-Cre fVHL mice and two wild-type mice was determined by using the orthogonal intercept method.29 Nine digital images at 14,000 × magnification were captured per animal (three capillary loops per glomerulus, three glomeruli per animal) by using a JEOL 100CX electron microscope. Digital images were opened in Photoshop CS (Adobe Systems Inc, San Jose, CA), and a grid mask with 16 intercepts was applied to each image. GBMs were measured in pixels at each intercept from the basal endothelial to the basal podocyte plasma membranes. Pixels were converted to nanometers by use of a calibration grating replica (Electron Microscopy Sciences, Fort Washington, PA).
For postfixation immunoelectron microscopy, 2-mm wedges of kidney cortices were fixed with 1% paraformaldehyde and 0.05% glutaraldehyde in 0.1 M sodium phosphate buffer, pH 7.3, for 1.5 hours on ice. Tissues were washed, equilibrated with 30% sucrose in buffer, and snap frozen in tissue freezing medium (Triangle Biomedical Sciences, Durham, NC) by using isopentane chilled in a dry ice-acetone bath. Frozen sections (30-μm thick) were collected on Thermanox coverslips (Miles Laboratories, Inc., Naperville, IL), and then air dried at room temperature. Sections were blocked for 30 minutes each in 0.5 M ammonium chloride in PBS and then with 5% goat serum and 0.1% bovine serum albumin in PBS. Sections were then immunolabeled with goat anti-α1α2α1 collagen IV (Southern Biotech, Birmingham, AL; 20 μg/ml in PBS) for 1 hour, and washed with PBS. Sections were then treated with rabbit anti-goat IgG-horseradish peroxidase (HRP; MP Biomedicals, Irvine, CA; 50 μg/ml in PBS) for 1 hour, washed, refixed in Karnovsky’s fixative, developed for peroxidase histochemistry, and processed for electron microscopy as described previously.28
Glomerular Isolation and Real-Time RT-PCR
Glomeruli were isolated from Pod-Cre fVHL mice through the injection of magnetic beads, which become trapped within glomerular capillaries.30 Briefly, mice were anesthetized with 1 mg/10 grams body weight ketamine and 0.15 mg/10 grams body weight of xylazine. Blood was washed from the animals by perfusion of the heart with HBSS followed by intracardiac injection of 2 × 106 Dynabeads M-450/ml in HBSS (Invitrogen, Carlsbad, CA). Kidneys were removed and minced on ice, followed by digestion at 37°C with 1 mg/ml collagenase and 100 U/ml DNase I for 30 minutes. Digested kidneys were filtered twice with 100 micron Falcon cell strainers, and tissue was pelleted by gentle centrifugation (200 g, 5 minutes). Glomeruli were isolated for three rounds by using a DynaMag-2 magnetic particle concentrator (Invitrogen), resuspending in HBSS after each collection. The glomerular preparations were briefly spun, frozen immediately on dry ice, and stored at −80°C.
Total RNA was isolated from glomeruli by using the RNeasy Micro kit (Qiagen). Samples (10 ng/μl) were amplified by using the QuantiTect SYBR Green RT-PCR kit (Qiagen) and appropriate, gene-specific primers (Table 1). Real-time RT-PCR was performed by using an iCycler (BioRad). The primer sets were validated for efficiency by the comparative cycle threshold method31 by using standard curve analysis. RT-PCR products were sequenced and verified with the Basic Local Alignment Search Tool (http://www.ncbi.nlm.nih.gov/BLAST; last accessed May 3, 2010) and/or analyzed on agarose gels to confirm size. After amplification, all products appeared as single peaks on melt curve analysis.
Table 1.
List of Primers for Quantitative Real-Time RT-PCR Experiments
| Gene symbol accession | Primer designation | Primer sequence | Product length, bp |
|---|---|---|---|
| Col4a1 NM_009931.1 | Col4a1S1 forward | 5′-CTGGCACAAAAGGGACGAG-3′ | 238 |
| Col4a1S1 reverse | 5′-ACGTGGCCGAGAATTTCACC-3′ | ||
| Col4a2 NM_009932.2 | Col4a2S2 forward | 5′-TGCTACCCGGAGAAAGGAG-3′ | 106 |
| Col4a2S2 reverse | 5′-CTTTGCGGCCCTGTAGTCC-3′ | ||
| Col4a3 NM_007734.1 | Col4a3S7 forward | 5′-GGGACATGTAACTACTACTCAAACTCC-3′ | 88 |
| Col4a3S7 reverse | 5′-TCACAGTTGATGGAATAGGTTTTCT-3′ | ||
| Col4a4 NM_007735.1 | Col4a4S4 forward | 5′-CTGGCTTGAAGGGAGACCT-3′ | 69 |
| Col4a4S4 reverse | 5′-CTCCTGCATCACCAGGAAGT-3′ | ||
| Col4a5 NM_007736.2 | Col4a5 forward | 5′-GGAGAACGGGGGTTTCCAG-3′ | 247 |
| Col4a5 reverse | 5′-CTCCCTTGGTTCCATTGCATC-3′ | ||
| Cxcl12 NM_013655.3 | Cxcl12 forward | 5′-CAAGGTCGTCGCCGTGCTG-3′ | 122 |
| Cxcl12 reverse | 5′-CGTTGGCTCTGGCGATGTGG-3′ | ||
| Cxcr4 NM_009911 | Cxcr4 forward | 5′-CAGAGGCCAAGGAAACTGCT-3′ | 101 |
| Cxcr4 reverse | 5′-CTGACGTCGGCAAAGATGAA-3′ | ||
| Cyclophilin NM_008907 | cyclophilin forward | 5′-CAGACGCCACTGTCGCTTT-3′ | 132 |
| cyclophilin reverse | 5′-TGTCTTTGGAACTTTGTCTGCAA-3′ | ||
| Egln3 NM_NM_028133.1 | Egln3 forward | 5′-TGTCTGGTACTTCGATGCTGA-3′ | 85 |
| Egln3 reverse | 5′-AGCAAGAGCAGATTCAGTTTTTC-3′ | ||
| Epas1 NM_001430 | PPM03309A-200 | Referenceposition457–477 | 164 |
| Hif1a NM_001530 | PPM03799B-200 | Referenceposition700–720 | 160 |
| Hif3a NM_152794 | PPM05268B-200 | Referenceposition1998–2018 | 180 |
| Lama1 NM_008480.2 | Lama1P1 forward | 5′-TGTAGATGGCAAGGTCTTATTTCA-3′ | 261 |
| Lama1P1 reverse | 5′-CTCAGGCAGTTCTGTTTGATGT-3′ | ||
| Lama5 NM_001081171.1 | Lama5P2 forward | 5′-ACCCAAGGACCCACCTGTAG-3′ | 169 |
| Lama5P2 reverse | 5′-TCATGTGTGCGTAGCCTCTC-3′ | ||
| Lamb1-1 NM_008482.2 | Lamb1-1P66 forward | 5′-GGCAAACTGCAAAGTCTCG-3′ | 61 |
| Lamb1-1P66 reverse | 5′-CTGGAGGTGTTCCACAGGTC-3′ | ||
| Lamb2 NM_008483.2 | Lamb2S2 forward | 5′-GTGTGGCTTGCATAGCCCT-3′ | 122 |
| Lamb2S2 reverse | 5′-TCCGATGACTATTTGGGTTGTCT-3′ | ||
| Ngb NM_022414 | PPM28801A-200 | Referenceposition759–781 | 163 |
| Vegf NM_03376 | PPM03041E-200 | Referenceposition2467–2487 | 191 |
| Vhlh NM_000551 | PPM27529A-200 | Referenceposition2474–2493 | 190 |
Cxcr4 and Cxcl1233 and cyclophilin56 primers were designed based on prior published work. All other primers were designed by using Primer Bank (http://pga.mgh.harvard.edu/primerbank/; last accessed May 3, 2010) or the Roche Universal Probe Library (Roche Applied Science, Indianapolis, IN). Epas1, Hif1α, Hif3α, Ngb, Vegf, and Vhlh primers were purchased from SABiosciences (Frederick, MD) and sequences are unavailable; shown are the reference position obtained from SABiosciences for the amplicon. All amplified products resulted in the appropriate size band on agarose gels.
Microarray Analysis
Three 6-week-old Pod-Cre fVHL female mice and three wild-type control mice, all with normal levels of urinary albumin (1 to 17 μg/ml) were selected for glomerular isolation. Glomeruli were harvested, and RNA was purified by using Qiagen RNA spin columns. RNA integrity was assessed by using an Agilent Bioanalyzer before hybridization on separate Affymetrix GeneChips (Mouse Genome 430 2.0 Array, Santa Clara, CA). Signal values were analyzed by the Affymetrix GeneChip Operating Software, and normalized by using robust multi-array analysis (Partek, St. Louis, MO). The most likely affected intracellular networks were generated through the use of Ingenuity Pathways Analysis (Ingenuity Systems, Redwood City, CA).
Western Blotting
Glomerular isolates from 6-week-old Pod-Cre fVHL and wild-type littermates were homogenized on ice in H buffer (40 mmol/L Tris pH 7.5, 15 mmol/L NaCl, 2 mmol/L CaCl2, 1× protease inhibitors [Sigma]) and were extracted for 10 minutes on a rocker at 4°C, followed by two freeze/thaw cycles. For Hif1α detection, kidney cortex was extracted in radioimmunoprecipitation assay buffer (50 mmol/L Tris, pH 8.0, 150 mmol/L NaCl, 1.0% NP-40, 0.4% sodium deoxycholate, 0.1% SDS, 1× protease inhibitors). Insoluble material was pelleted by centrifugation for 20 minutes at 12,000 × g. Supernatants were boiled for 5 minutes in SDS sample buffer with dithiothreitol, and proteins were separated by electrophoresis in a 12% Tris-HCl precast BioRad gel. Proteins were then electrotransferred to polyvinylidene difluoride. Antigens were detected by using a SNAP ID protein detection system (Millipore, Billerica, MA) according to the manufacturer’s instructions by using rabbit anti-Neuroglobin (Sigma) and rabbit anti-Egln3 (Novus Biologicals, Littleton, CO) or by incubation overnight at 4°C with mouse anti-HIF1α (Millipore). Secondary anti-rabbit or anti-mouse HRP antibodies (GE Health care, Piscataway, NJ) were detected by using chemiluminescence. Blots were stripped and reprobed with anti-mouse β actin (Sigma).
Results
To investigate possible roles of VHL in glomerular development and function, VHL was selectively deleted from podocytes by using Cre-LoxP technology. The podocin promoter, which has been previously characterized and shown to express exclusively in podocytes,23 was used to excise fVHL by driving the expression of Cre recombinase (Pod-Cre). To verify podocyte-specific recombination, Pod-Cre mice were bred with a floxed Rosa26 reporter mouse. As shown in Figure 1A, β-galactosidase activity was observed only in podocytes beginning at the early capillary loop stage of glomerular development, and podocyte expression persisted in maturing glomeruli. Podocytes of all Pod-Cre+ animals (n = 5) were positive, and all control littermates lacking Cre recombinase were negative for β-galactosidase, as expected (n = 4).
Figure 1.

The Podocin-Cre strain recombines floxed alleles in podocytes of the kidney. A: Pod-Cre mice were mated to the floxed LacZ (β-galactosidase) reporter mouse R26R, and progeny were genotyped for Cre. Both Cre+ and Cre− kidneys were developed for β-galactosidase activity. Blue, β-galactosidase positive cells are confined to podocytes of developing glomeruli, beginning at the early capillary loop stage (inset, arrowheads), exclusively in Cre+ kidneys (n = 5). No β-galactosidase positive cells were seen in Cre− kidneys (n = 4), or in late S-shaped developing glomeruli (inset, arrow). B: Pod-Cre mice were crossed to fVHL mice generating a podocyte-specific deletion of VHL. Genomic DNA was isolated from Pod-Cre fVHL tissues (K = kidney, H = heart, L = liver, and T = tail). Primers flanking the floxed VHL locus amplified the nonrecombined VHL locus (460 bp) in all tissues. The smaller, recombined product (260 bp) is only seen in kidney tissue. C: Glomerular development is normal in Pod-Cre fVHL mice. Pod-Cre × fVHL litters were screened for amplification of Cre and presence of the floxed VHL locus. Animals that were either homozygous wild-type at the VHL locus, or were Cre-negative, served as wild-type littermate controls (wild-type, left). Pod-Cre fVHL mice were both Cre-positive and homozygous for the fVHL locus (Pod-Cre fVHL). At postnatal day five, kidney morphology of H&E paraffin sections shows normal capillary loop stage glomeruli in both wild-type and Pod-Cre fVHL (top). Mature glomeruli also appear normal in both wild-type and Pod-Cre fVHL animals (bottom) with red blood cells present in capillary loops (arrows).
Pod-Cre mice were crossed with floxed VHL mice,25 and genomic DNA was isolated from kidney, heart, liver, and tail tissues. To confirm that the VHL locus could be efficiently recombined by Pod-Cre, PCR was performed on genomic DNA by using VHL-specific primers that flanked the floxed insertion site. A predicted 260 bp recombined allele was seen only in genomic samples from kidney (Figure 1B). Because recombination was confined to podocytes, representing only a small proportion of total kidney cells, kidney samples also contained the 460 bp nonrecombined product, which was the only product seen in samples from the heart, liver, and tail (Figure 1B). Based on the Cre reporter expression pattern and PCR amplification findings, we therefore concluded that recombination of the VHL locus was occurring solely in renal podocytes.
Given that VHL excision was occurring in early capillary loop stage glomeruli, we examined kidneys from 5-day-old Pod-Cre fVHL mice for possible developmental abnormalities in vascular patterning. However, routine histological analysis of Pod-Cre fVHL mice at this age did not show any morphological differences from wild-type littermate controls (Figure 1C). All mice showed normal comma and S-shaped nephric figures, as well as capillary loop stage and maturing stage glomeruli containing erythrocytes (Figure 1C). There also were no abnormalities evident in peritubular microvessels or larger arterioles and veins.
Though no striking developmental phenotype was uncovered in infant Pod-Cre fVHL mice, polyacrylamide gel electrophoresis showed that urinary albumin excretion was elevated in many animals at 4 weeks of age, and markedly so in some (Figure 2A). Albuminuria, quantified by enzyme-linked immunosorbent assay in 43 Pod-Cre fVHL mice, was statistically significantly greater than 21 wild-type controls (P < 0.03), and there was no gender bias (Figure 2B). Pod-Cre fVHL mice with highest urinary albumin levels displayed classic signs of end stage renal failure, with transient edema, followed by wasting and death beginning at ∼6 months of age. Blood urea nitrogen levels measured from these severely nephrotic mice were sixfold greater than blood urea nitrogen levels from Pod-Cre fVHL littermates with lower levels of albumin (Figure 2C). Analysis of end-stage renal morphology in heavily proteinuric Pod-Cre fVHL mice showed interstitial fibrosis, dilated tubules containing proteinaceous casts and cellular debris (Figure 3A), and severely fibrotic glomeruli (Figure 3D). To determine the earliest glomerular phenotype, Pod-Cre fVHL mice with severe albuminuria were sacrificed at 4 weeks of age. These mice had glomeruli with larger capillary lumens (Figure 3B), increased mesangial matrix (Figure 3, B and C), and cellular crescents (Figure 3C).
Figure 2.

Podocyte-specific deletion of VHL results in a range of proteinuria. A: Urine was collected from 4-week-old litters resulting from Pod-Cre × fVHL crosses, separated by polyacrylamide gel electrophoresis and stained with Coomassie blue alongside bovine serum albumin standards (lane 1 = 5 μg; lane 2 = 0.5 μg). Urine from wild-type mice (lane 4) does not contain albumin, whereas urine from Pod-Cre fVHL animals show varying levels of albumin (lanes 3, 5, and 6–8). Some Pod-Cre fVHL urine (lane 9) looks similar to wild-type animals and contains no detectable albumin. The arrow marks the ∼68 kDa albumin band. B: Urinary albumin was quantified by using Albuwell enzyme-linked immunosorbent assays in 64 Pod-Cre × fVHL progeny (n = 21 wild-type; n = 43 Pod-Cre fVHL). Nonparametric t-tests were significant for both female and male data (*P = 0.03). C: Blood was isolated from 33- to 41-week-old Pod-Cre × fVHL mice, and blood urea nitrogen levels were quantified and plotted against urinary albumin. Elevated blood urea nitrogen was associated with only the highest levels of albuminuria (>1000 μg/ml).
Figure 3.

Phenotypic analysis of Pod-Cre fVHL mice. A–D: PAS staining of paraffin sections of massively proteinuric Pod-Cre fVHL mice at four or 25 weeks. A: Entire kidney is fibrotic, and tubules and ducts are packed with proteinaceous material in 25-week-old Pod-Cre fVHL mice. B: Proteinuric Pod-Cre fVHL kidney at four weeks of age with dilated glomerular capillary lumen (arrow) and mesangial matrix expansion and hypercellularity (arrowhead). C: Proteinuric Pod-Cre fVHL at 4 weeks showing mesangial hypercellularity and glomerular crescent. D: Proteinuric Pod-Cre fVHL at 25 weeks showing a completely fibrotic glomerulus. E: PAS staining of paraffin sections of a four-week-old nonproteinuric Pod-Cre fVHL kidney shows occasional dilated tubules (arrows). Inset shows hypercellular mesangium with increased PAS+ matrix accumulation and prominent GBM. F: Kidney sections show labeling pattern of WT1 in podocytes (pink, arrows) and Ki-67 in mesangium (green, arrowheads) in nonproteinuric Pod-Cre fVHL mice. Slides were counterstained with 4′,6-diamidino-2-phenylindole. Numbers of Ki-67-positive (G) and WT1-positive cells (H) were counted in at least 10 glomeruli per animal (W = wild-type [n = 5], N = nonproteinuric [n = 5], and P = proteinuric [n = 2]). G: Significantly more Ki-67-positive cells are observed in 4-week-old proteinuric Pod-Cre fVHL glomeruli compared with wild-type (*P < 0.05). H: Conversely, fewer WT1-positive cells per glomerulus are observed in four-week-old Pod-Cre fVHL compared with four-week-old wild-type (**P < 0.01 proteinuric versus wild-type; *P < 0.05 nonproteinuric versus wild-type). I–N: Kidneys at eight weeks of age (I–K) or 41 weeks (L–N) were labeled with anti-synaptopodin antibody and counterstained with 4′,6-diamidino-2-phenylindole. In contrast to wild-type synaptopodin labeling (I and L), Pod-Cre fVHL glomeruli have decreased synaptopodin labeling in wide, central regions (J and M, arrow). Some Pod-Cre fVHL glomeruli completely lack synaptopodin labeling on one half of the glomerulus (K and N, dashed circles). O and P: Kidneys at eight weeks of age were labeled with anti-nephrin antibody. Pod-Cre fVHL glomeruli (P) have decreased nephrin labeling compared with wild-type (O). Original magnification: ×100 (A and E); ×200 (inset of E); ×400 (B, C, and D).
Although several Pod-Cre fVHL mice progressed to end-stage renal disease, the majority (54%, males and females) had albumin levels indistinguishable from wild-type mice (wild-type albumin range = 2.9 to 29.7 μg/ml). These nonproteinuric Pod-Cre fVHL mice survived to more than 1 year of age without overt health problems. Nevertheless, light microscopy of 4-week-old nonproteinuric Pod-Cre fVHL kidneys revealed proteinaceous casts in dilated medullary collecting ducts (Figure 3E, arrows) and slightly increased PAS-positive mesangial matrix (Figure 3E, inset). Sections from Pod-Cre fVHL mice were double labeled with Ki-67 to identify proliferating cells (Figure 3F, arrowheads) and WT1 to mark podocytes (Figure 3F, arrows). Ten individual glomeruli were imaged from each animal, and stained cells were blindly counted and averaged (n = 5 for wild-type and nonproteinuric Pod-Cre fVHL; n = 2 for proteinuric Pod-Cre fVHL). There was a significant increase in the number of Ki-67-positive cells within glomeruli of proteinuric Pod-Cre fVHL mice (Figure 3G; 2.2 cells/glomeruli vs 0.9 cells/glomeruli wild-type, P < 0.05). The proliferating cells appeared to be mesangial cells, as only a single Ki-67-positive cell was also shown to be WT1-positive. Proteinuric Pod-Cre fVHL mice also had a significant decrease in the number of podocytes, containing an average of 6.5 WT1-positive cells within glomeruli compared with 9.4 WT1-positive cells within glomeruli of wild-type littermates (Figure 3H; P < 0.01). Nonproteinuric mice also showed a significant decrease in the number of WT1-positive cells within glomeruli compared with wild-type (Figure 3H; 8.4 cells/glomeruli vs 9.4 cells/glomeruli wild-type, P < 0.05). The increase in proliferating cells seen in proteinuric glomeruli therefore correlated with a decrease in WT1 podocyte marker expression.
We also labeled glomeruli with anti-synaptopodin antibodies and found that proteinuric Pod-Cre fVHL glomeruli displayed a simplified synaptopodin pattern with linear deposition located in the outermost podocyte layer of the capillary tuft (Figure 3, compare I and L to J and M). Wide expanses of synaptopodin negative regions were seen at both 8 and 33 weeks (Figure 3, J and M, arrows) in more central regions of the capillary tuft. Nonproteinuric Pod-Cre fVHL glomeruli had a similar pattern of synaptopodin labeling. Also, we found proteinuric Pod-Cre fVHL glomeruli that either had no synaptopodin labeling or expressed a simplified pattern in only a portion of the tuft (Figure 3, K and N). Focal regions devoid of synaptopodin were packed with nuclei and probably represented severely fibrotic areas. As compared with wild-type controls, Pod-Cre fVHL glomeruli also had reduced labeling with anti-nephrin antibodies (Figure 3, O and P).
Given that Pod-Cre fVHL mice had altered podocyte numbers and evidence of mesangial proliferation, we undertook an ultrastructural evaluation to determine what glomerular lesions preceded the increased capillary lumen evident in proteinuric mice. Ultrastructural abnormalities detected in glomerular capillary loops at 4 weeks of age of all nonproteinuric Pod-Cre fVHL mice examined included podocyte foot process broadening (Figure 4A, arrowheads), subendothelial delamination (Figure 4, A and B, arrows), and irregular GBM thickenings with numerous subepithelial hump formations (Figure 4, B and C, double arrows).
Figure 4.

Four-week-old nonproteinuric Pod-Cre fVHL mice display podocyte foot process effacement and GBM irregularities. A: Widespread foot process effacement is indicated by the arrowheads. Incompletely fused or fragmented GBM appears on the subendothelial side of the capillary loop (arrow). B: Abnormal subepithelial thickenings are shown by the double arrows, and a large area of lacey subendothelial matrix projections is also shown (arrow). C: Another example of a typical subepithelial “hump” of GBM (double arrows). En = endothelium; Po = podocyte.
In 16-week-old nonproteinuric Pod-Cre fVHL mice, GBM alterations persisted (Figure 5, A and B), and the overall thickness of GBM increased to almost 100 nanometers more than control littermates (Figure 5C). We surveyed glomerular matrix proteins by quantitative real- time RT-PCR of isolated glomerular RNA from nonproteinuric Pod-Cre fVHL and littermate controls. There were significant increases in the mRNAs encoding Col4a1, Col4a2, and Col4a5 (Figure 5D), as well as Lamb1 and Lama5 (Figure 5E) in samples from Pod-Cre fVHL mice. Because glomerular matrix proteins were apparently being synthesized at a higher rate in nonproteinuric Pod-Cre fVHL mice, we performed postfixation immunoelectron microscopy to define the cellular localization for collagen α1α2α1(IV), which is normally confined mainly to the mesangium in adult glomeruli. In addition to the mesangial expression seen in wild-type littermate controls (Figure 6D), there was excessive subendothelial GBM expression of α1α2α1(IV) in Pod-Cre fVHL glomeruli (Figure 6, A–C), which has been reported previously in human membranous nephropathy.32 However, anti-collagen α1α2α1(IV) immunolabeling was also observed immediately beneath podocyte foot processes overlying many of the subepithelial humps typical of Pod-Cre fVHL GBM (Figure 6, B and C, double arrows). Because the samples were processed with light fixation, which preserves intracellular antigens as well, we also noted HRP reaction product within intracellular compartments in both endothelial cells (Figure 6C, arrowhead) and podocytes (Figure 6A, arrow), indicating that both cell types were synthesizing collagen α1(IV) and α2(IV) chains.
Figure 5.
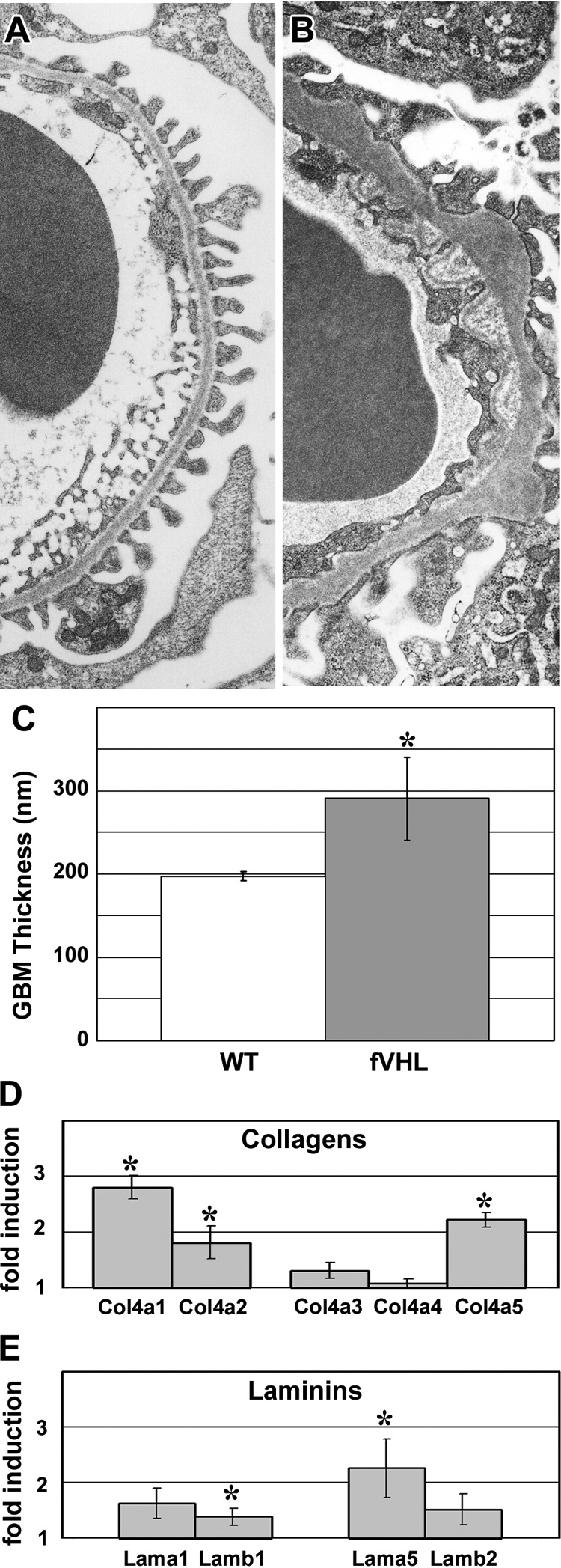
Sixteen-week-old Pod-Cre fVHL mice have thickened GBM. A: GBM of a 16-week-old wild-type littermate control shows regular podocyte foot process registration adjacent to the entire length of capillary loop GBM. B: 16-week-old mildly proteinuric Pod-Cre fVHL mouse GBM showing overall thickening of the GBM, along with subepithelial “humps” and subendothelial projections. C: Using the orthoganol intercept method, GBM width averaged 291 nm in 16-week-old nonproteinuric Pod-Cre fVHL kidneys, which was almost 100 nm wider than wild-type littermate controls (*P < 0.05). D and E: Glomeruli were isolated from six-week-old nonproteinuric Pod-Cre fVHL mice and littermate controls. Levels of collagen IV (D) and laminin (E) mRNAs were determined by real-time quantitative RT-PCR, and were expressed as ratios (Pod-Cre fVHL/wild-type). Significant increases were seen in Col4a1, Col4a2, and Col4a5, as well as Lamb1 and Lama5 (*P < 0.05).
Figure 6.

Podocytes and endothelial cells synthesize collagen α1α2α1(IV) in adult Pod-Cre fVHL mice. A–C: 37-week-old Pod-Cre fVHL proteinuric kidney tissue was lightly fixed, and frozen sections were labeled with anti-collagen α1α2α1(IV) antibodies, followed by indirect immunoperoxidase as described in Materials and Methods. Intense HRP reaction product is seen along the subendothelial portion of the GBM (A–C). In addition, Pod-Cre fVHL animals displayed HRP reaction product in subepithelial humps immediately beneath effaced podocyte foot processes (double arrows, B and C). HRP reaction product was also seen intracellularly within endothelial cells (arrowhead, A and C) and podocytes (arrow, A). In control tissue (D), HRP reaction product is mainly confined to the mesangial matrix; in areas, subendothelial segments of GBM are also weakly labeled. En = endothelium; Po = podocyte; M = mesangium.
Quantitative real-time RT-PCR of isolated glomerular RNA from nonproteinuric Pod-Cre fVHL and littermate controls showed that VEGF mRNA, a known HIF target, showed a small increase at 6 weeks of age (Figure 7A). HIF1α mRNA was also slightly increased, as was the chemokine Cxcr4, which has been previously implicated in a similar model of podocyte-specific VHL deletion.33 No changes were detected in the level of Hif2α, Hif3α, or Cxcl12 (a Cxcr4 ligand). Western blotting confirmed the presence of more HIF1α protein in Pod-Cre fVHL kidney cortex (Figure 7B).
Figure 7.

A: Glomerular mRNAs were isolated from six-week-old Pod-Cre fVHL (n = 4) and wild-type (n = 3) littermate controls. VEGF, Hif1α, and Cxcr4 primers amplified significantly more product by quantitative real-time RT-PCR in Pod-Cre fVHL glomeruli (gray bars) compared with wild-type littermates (white bars), whereas no change was observed with Hif2α, Hif3α, or Cxcl12 primers. *P <0.02. B: Western blotting with equal amounts of extracts of kidney cortex show increased Hif1α labeling in two Pod-Cre fVHL mice compared with wild-type (wt). Actin immunoblotting (lower blot) was used as a loading control. C: Quantitative real-time RT-PCR shows a 100-fold increase of Egln3 in Pod-Cre fVHL glomerular RNAs. *P <0.00006. D: Western blotting with equal amounts of glomerular extracts from a wt or two Pod-Cre fVHL mice by using Egln3 antibody. Actin immunoreactivity was used as a loading control. E: Quantitative real-time RT-PCR of glomerular RNAs shows ∼18-fold increase of Ngb. *P <0.0001. F: Western blotting with equal amounts of glomerular extracts from a wt or two Pod-Cre fVHL mice by using an Ngb antibody. Actin immunoreactivity was used as a loading control. G and H: Paraffin sections were double labeled with anti-Ngb and anti-synaptopodin antibodies. Synaptopodin labeling (red) is seen in both wild-type (G) and Pod-Cre fVHL glomeruli (H), but Ngb labeling (green, arrows) is only present in presumptive podocytes of Pod-Cre fVHL glomeruli (H). I–N: Serial paraffin sections of Pod-Cre fVHL kidneys were double labeled with anti-nephrin and anti-synaptopodin (I–K), or with anti-Ngb and anti-synaptopodin (L–N). Simplified nephrin (green) and synaptopodin (red) expression (I, J, M) with podocyte-specific Ngb labeling (green, L) is seen in some glomeruli. Other, presumably fibrotic glomeruli (I–N, dashed circles), completely lack all three markers.
In view of VHL’s role in degradation of the HIFα transcription factor subunit, experimental deletion of VHL could affect transcription of many genes. Additionally, we sought to identify gene products that rendered many Pod-Cre fVHL mutant mice insusceptible to proteinuria. Hence, global changes in transcription between nonproteinuric Pod-Cre fVHL mice and wild-type littermates were assessed by profiling glomerular RNA by using Affymetrix 3′ expression arrays (n = 3 for each genotype, 6 weeks of age). (The complete data set discussed here has been deposited in the National Center for Biotechnology Information Gene Expression Omnibus34 and is accessible through Series accession number GSE20235 [http://www.ncbi.nlm.nih.gov/geo/]; last accessed on May 3, 2010.) Ingenuity Pathways Analysis software was used to scrutinize the microarray data because ∼2099 probe sets displayed fold changes with statistically relevant P values (P < 0.05). The threshold for core Ingenuity Pathway Analysis was set at a fold change greater than ± 1.5, with a P value of <0.05. The top two canonical networks identified in this dataset were glycolysis and gluconeogenesis, and HIF1α signaling. The associated genes, fold changes, and P values are shown in Table 2. These data suggest that deletion of VHL in the podocytes activated at least portions of the hypoxia response cascade.
Table 2.
Microarray Analysis of Isolated Glomeruli Taken at 6 Weeks of Age from Nonproteinuric Pod-Cre fVHL and Wild-Type Littermates
| Gene symbol | Gene name | Fold change | P |
|---|---|---|---|
| Glycolysis and gluconeogenesis | |||
| Ldha | Lactate dehydrogenase A | 1.56 | 0.003 |
| Pfkl | Phosphofructokinase, liver, B-type | 1.77 | 0.003 |
| Pkm2 | Pyruvate kinase, muscle | 2.14 | 0.00004 |
| Tpi1 | Triosephosphate isomerase 1 | 2.42 | 0.00002 |
| HIF1α signaling | |||
| Egln3 | EGL nine homolog 3 | 45.4 | 0.0002 |
| Ldha | Lactate dehydrogenase A | 1.56 | 0.003 |
| Mmp2 | Matrix metallopeptidase 2 | 1.54 | 0.05 |
| Slc2a1 | Solute carrier family 2 (facilitated glucose transporter), member 1 | 1.72 | 0.01 |
The genes listed are all increased in Pod-Cre fVHL mutant glomeruli (fold change) and are members of the top two networks identified in this dataset by Ingenuity Pathways Analysis.
One of the most abundantly up-regulated genes from the microarray was Egln3, a prolyl hydroxylase that normally serves to modify two available proline residues in the N-terminal oxygen-dependent degradation domain of HIF-1α. Quantitative real-time RT-PCR showed that Egln3 mRNA was increased ∼100-fold in Pod-Cre fVHL glomeruli compared with those from wild-type littermates (Figure 7C). Western blotting of glomerular extracts also showed a strong band at the predicted 28 kDa size from two individual Pod-Cre fVHL mice, compared with a much weaker band seen in a wild-type littermate (Figure 7D).
Another particularly interesting finding in the microarray data was a 15-fold increase of neuroglobin (Ngb), an ancient, highly conserved oxygen-binding heme protein originally discovered in brain35 and not previously associated with kidney. Quantitative real-time RT-PCR verified a ∼17-fold increase in Ngb mRNA in isolated Pod-Cre fVHL glomeruli (Figure 7E). Ngb protein was also increased in Pod-Cre fVHL glomeruli, as analyzed by Western blotting (Figure 7F). Immunofluorescence microscopy showed that Ngb immunolabeling was not seen in wild-type glomeruli, but strong Ngb immunolabeling was seen in Pod-Cre fVHL tissue (Figure 7, G and H). Specifically, the Ngb signal co-localized with the podocyte-specific markers, anti-nephrin and anti-synaptopodin (Figure 7, I–N), signifying that Ngb expression was up-regulated specifically in podocytes. Interestingly, in what appeared to be severely fibrotic glomeruli, there was a complete absence of immunolabeling for Ngb, and loss of the podocyte differentiation markers, synaptopodin, and nephrin as well (Figure 7, I–N, dashed circles).
We also attempted to isolate glomeruli from heavily proteinuric Pod-Cre fVHL mice. Our preparations were insufficiently enriched for glomeruli, however, probably because of inadequate perfusion of Dynabeads into the fibrotic and occluded capillary tufts.
Discussion
This model of podocyte-specific deletion of VHL has maximal penetrance (all progeny have a phenotype), but with varying expressivity ranging from mild (nonproteinuric but with GBM ultrastructural changes) to severe (heavily proteinuric with fibrotic glomeruli). The most severe phenotype resulted in end stage renal failure beginning ∼6 months of age. However, all Pod-Cre fVHL animals displayed mesangial hypercellularity, changes in basement membrane gene expression and matrix deposition, marked increases in GBM thickness, podocyte foot process effacement, and cell loss. We conclude that podocyte VHL is necessary for mesangial homeostasis, maintenance of the podocyte, normal structure and composition of the GBM, and maintenance of glomerular barrier properties.
In addition to the work described here, two earlier studies have selectively inactivated fVHL in podocytes, using the mouse and human podocin promoter, respectively, to drive expression of Cre recombinase.33,36 In all three studies, however, glomerular development and kidney microvascular formation proceeded normally.33,36 This finding is somewhat surprising because an absence of VHL in podocytes would be expected to result in an overabundance of active HIFα subunits, which in turn should lead to overexpression of HIF target genes, possibly dysregulating glomerulogenesis. In particular, precise levels of the HIF target, VEGF, have been shown to be critical for normal glomerular development.37,38 For example, mice that are podocyte-specific heterozygous for VEGF-A suffer proteinuria and endotheliosis at 2.5 weeks of age and podocyte-specific homozygous nulls die at birth.37 In another case, reduced levels of VEGF leads to death at 3 weeks and loss of mesangial cells.38 On the other hand, podocyte-specific overexpression of VEGF-164 results in a phenotype resembling collapsing glomerulopathy.37 Given that apparently stable HIFα subunits are found in the nucleus of immature podocytes,18 perhaps endogenous VHL is functionally inactive during glomerulogenesis. Alternatively, because Cre-mediated excision driven by the podocin promoter first occurs at the capillary loop stage and thereafter, this may be too late in nephron development to affect developmental HIF- and/or VHL-dependent activities.
A problem we encountered during the initial analysis of our Pod-Cre fVHL crosses was the range of phenotypes. Although Brukamp et al36 analyzed relatively small numbers of Pod-Cre fVHL mice, they also observed a diverse range and frequency of phenotypes. In their study, 1 of 11 (9%) animals died of renal failure, and two others displayed slowly progressive renal disease. They also reported subtle glomerular changes in several mice with normal urine protein levels. Using a transgenic mouse expressing Cre under control of the mouse podocin promoter, Ding et al33 described the onset of rapidly progressive glomerulonephritis (RPGN) beginning at 4 weeks of age resulting in renal failure 2 to 3 weeks later in all animals. Up-regulation of both the chemokine receptor Cxcr4 and its ligand Cxcl12 (SDF-1) were implicated as a likely pathogenic mechanism of RPGN. Using quantitative real-time RT-PCR of isolated glomerular RNA, we evaluated Cxcr4 and Cxcl12 expression in our Pod-Cre fVHL mice and found an increase of Cxcr4 mRNA, but no change in message for the Cxcl12 ligand. Perhaps this chemokine system plays a heightened role in the RPGN model due to differences in environmental factors or in genetic backgrounds of the mice. Additionally, the murine podocin promoter used in the RPGN model may have resulted in more robust Cre expression than the human podocin promoter used in our study and that by Brukamp et al,36 which could also account for the differences in the incidence and severity of phenotype. Regardless, we did not observe any Pod-Cre fVHL mouse in our colony that progressed to renal failure in the relatively brief 6- to 7-week timeframe. We did note dilated capillary lumens in severely affected mice at 4 weeks of age, however, which is similar to the histology described by Ding et al,33 before the onset of RPGN.
In normal developing glomeruli, a network of collagen α1α2α1(IV) is found in GBMs of early nephrons, but as glomeruli mature, this network is replaced by collagen α3α4α5(IV) by unknown mechanisms.2 Our study presented here contains several pieces of evidence showing that GBM assembly was abnormal in Pod-Cre fVHL mice. First, our quantitative real time RT-PCR results with isolated glomeruli from 6-week-old mutants showed transcriptional up-regulation of many basement membrane genes, including both Col4a1 and Col4a2. Second, ultrastructural morphometry showed that GBMs in Pod-Cre fVHL mice were abnormally thickened. Third, immunoelectron microscopy clearly demonstrated that the abnormal subepithelial GBM protrusions of Pod-Cre fVHL glomeruli contained considerable collagen α1α2α1(IV) immediately beneath podocytes, and distinct from the minimal subendothelial labeling seen in wild-type littermate controls. Fourth, we also observed that both endothelial cells and podocytes of mutant glomeruli contained intracellular labeling for collagen α1α2α1(IV), indicating that the immature GBM collagen IV network was being ectopically re-expressed.
How might the deletion of podocyte VHL result in the ultrastructural and compositional changes we observed in the GBM? VHL has multiple functions in addition to the classic ligase function leading to HIF degradation in normal oxygen conditions. One of these HIF-independent functions is regulation of extracellular matrix synthesis, although details of how this occurs are only beginning to emerge. VHL-negative cell lines fail to form a fibronectin rich extracellular matrix in vitro.39,40 If VHL is restored in RCC lines by using variants that have been shown to retain HIF binding and degradation functions, matrix is still not assembled normally.41 Based on these studies, we would have predicted that lack of VHL in podocytes should result in a thinner or perhaps “lacy” GBM such as that seen in Alport mice,9,15 especially in view of the fact that podocytes are entirely responsible for synthesis of collagen α3α4α5(IV) of mature GBM.4 Why the lack of podocyte VHL leads to apparently increased matrix is therefore unclear. Because we observed transcriptional up-regulation of HIF-1α and VEGF, perhaps overexpression of VEGF is linked to the observed dysregulation of matrix synthesis. Thickened GBM is a common feature of diabetic nephropathy where high glucose is responsible for triggering increased VEGF production.17,42 The effects of high glucose in boosting VEGF production43 and subsequent collagen synthesis (Col4α3) has also been shown previously in cultured podocytes.44
Recently, two groups have revealed independently that VHL, a cytoplasmic protein, interacts with collagen α1(IV)45 and α2(IV)45,46 polypeptide chains, which are in the secretory pathway. This interaction is reported to be HIF-independent and not reliant on the ligase function of VHL, but does require hydroxylation of collagen. Moreover, immunoprecipitation and enzyme digestion studies have revealed that VHL appears to bind to the N-terminus of collagen α2(IV) on the cytoplasmic surface of the endoplasmic reticulum, with the C-terminus of the collagen polypeptide projecting into the endoplasmic reticulum lumen.46 Additionally, RCC cells containing mutated VHL assemble abnormal collagen IV networks in vitro or when injected into mice in vivo, suggesting that VHL participates in the synthesis, triple helical assembly, and/or secretion of collagen IV.46
Taking all of the evidence together, we speculate that in the glomerulus, VHL participates directly or indirectly in remodeling of the GBM collagen IV network. Further, we hypothesize that lack of VHL results in re-expression and ectopic assembly of α1α2α1(IV) collagen by mature endothelial cells and podocytes. This abnormal GBM may then have signaled podocytes to undergo foot process effacement, and detach or dedifferentiate, explaining the decreased WT1 labeling seen in Pod-Cre fVHL glomeruli. These pathogenic events may have also secondarily induced the observed mesangial expansion. In addition, given that our microarray indicated an increase in the mRNA for the matrix metalloprotease MMP-2, altered extracellular matrix degradation may have played a significant role in the Pod-Cre fVHL GBM phenotype. Without question, further experimental work is necessary to verify these possibilities. Nevertheless, because in our experiments fVHL was deleted only in podocytes, these cells must somehow have communicated across the GBM to incite collagen α1α2α1(IV) synthesis by the endothelium as well. Additional experiments with these Pod-Cre fVHL mutants may shed more light on basic mechanisms regulating GBM type IV collagen network assembly and podocyte-endothelium cross talk.
By microarray profiling, we found two unique genes that were massively up-regulated in nonproteinuric Pod-Cre fVHL glomeruli: Egln3 and Ngb. The Egln3 gene contains a functional hypoxia response element that is induced by HIF1, and probably comprises an autoregulatory feedback loop for the hypoxia response cascade.47 In addition to initiating HIFα proteasomal degradation, Egln3 has also been implicated as a pro-apoptotic agent in some cells,48 and its overexpression in Pod-Cre fVHL podocytes may have contributed to the loss of WT1-positive cells that we observed. Additional work will be necessary to confirm this possibility, however.
Neuroglobin, a novel tissue globin first discovered in brain,35 was also highly up-regulated in Pod-Cre fVHL glomeruli. Neuroglobin has been shown to be induced by hypoxia,49,50,51 and perhaps the increase in glomerular HIF1α was responsible for the increased Ngb expression. Our immunolocalization studies revealed that Ngb was expressed specifically in podocytes of Pod-Cre fVHL mutants and completely absent in wild-type mice. This marks the first description of endogenous Ngb expression outside of the brain and provides yet another similarity between the kidney podocyte and neurons.52,53 The physiological function(s) of Ngb is uncertain, but evidence exists indicating a role for this highly conserved globin protein in oxygen storage and/or delivery. In particular, Ngb has been implicated in supplying oxygen to the electron transport chain in mitochondria, detoxifying reactive oxygen or nitrogen species, removing excess nitric oxide at normoxia or producing sufficient nitric oxide at hypoxia, acting as a signal transducer through interaction with heterotrimeric G proteins, and reducing cytochrome c to prevent hypoxia-induced apoptosis.54 Additionally, neuronal Ngb expression is induced by experimental cerebral ischemia and protects cultured neurons from hypoxia in vitro.49 Further, transgenic mice that widely overexpress Ngb through the β-actin promoter are resistant to experimental ischemia induced in the brain and heart.55 Exactly why Ngb was up-regulated in nonproteinuric Pod-Cre fVHL podocytes is unclear, but we are intrigued by the finding that the pattern of Ngb expression correlated precisely with that for the podocyte differentiation markers, synaptopodin and nephrin. Additionally, an absence of Ngb in glomeruli of Pod-Cre fVHL mutants paralleled the loss of these proteins. We therefore wonder whether Ngb can play a protective role in the glomerulus, and we are currently pursuing this hypothesis.
Acknowledgments
We thank Eileen Roach (deceased) for help with morphometry and the Kansas University Medical Center-Microarray Facility for generating array data sets.
Footnotes
Address reprint requests to Dale R. Abrahamson, Ph.D., Department of Anatomy and Cell Biology, University of Kansas Medical Center, Mail Stop 3038, 3901 Rainbow Blvd, Kansas City, KS 66160. E-mail: dabrahamson@kumc.edu.
Supported by National Institutes of Health grants DK052483, DK065123, and RR024214. The KUMC Microarray Facility is supported by the University of Kansas School of Medicine, Kansas University Medical Center Biotechnology Support Facility, the Smith Intellectual and Developmental Disabilities Research Center (NIH grant HD02528), and the Kansas IDeA Network of Biomedical Research Excellence (NIH grant RR016475).
The author P.B. Freeburg is deceased.
We dedicate this work to the memory of our friends and colleagues, Dr. Paul B. Freeburg, who passed away September 2005, and Eileen Roach, who passed away December 2009.
References
- Haraldsson B, Nyström J, Deen WM. Properties of the glomerular barrier and mechanisms of proteinuria. Physiol Rev. 2008;88(2):451–487. doi: 10.1152/physrev.00055.2006. [DOI] [PubMed] [Google Scholar]
- Miner JH, Abrahamson DR. Alpern RJ, Hebert SC, editors. Burlington: Academic Press,; Molecular and cellular mechanisms of glomerular capillary development. Seldin and Giebisch’s the kidney, vol 1–2, ed 4physiology and pathophysiology. 2007:pp 691–706. [Google Scholar]
- St. John PL, Abrahamson DR. Glomerular endothelial cells and podocytes jointly synthesize laminin-1 and -11 chains. Kidney Int. 2001;60(3):1037–1046. doi: 10.1046/j.1523-1755.2001.0600031037.x. [DOI] [PubMed] [Google Scholar]
- Abrahamson DR, Hudson BG, Stroganova L, Borza DB, St John PL. Cellular origins of type IV collagen networks in developing glomeruli. J Am Soc Nephrol. 2009;20(7):1471–1479. doi: 10.1681/ASN.2008101086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenker M, Aigner T, Wendler O, Tralau T, Muntefering H, Fenski R, Pitz S, Schumacher V, Royer-Pokora B, Wuhl E, Cochat P, Bouvier R, Kraus C. Human laminin beta2 deficiency causes congenital nephrosis with mesangial sclerosis and distinct eye abnormalities. Hum Mol Genet. 2004;13:2625–2632. doi: 10.1093/hmg/ddh284. [DOI] [PubMed] [Google Scholar]
- Wuhl E, Kogan J, Zurowska A, Matejas V, Vandervoorde RG, Aigner T, Wendler O, Lesniewska I, Bouvier R, Reis A, Cochat P, Zenker M. Neurodevelopmental deficits in Pierson (microcoria-congenital nephrosis) syndrome. Am J Med Genet A. 2007;143:311–319. doi: 10.1002/ajmg.a.31564. [DOI] [PubMed] [Google Scholar]
- Noakes PG, Miner JH, Gautam M, Cunningham JM, Sanes JR, Merlie JP. The renal glomerulus of mice lacking s-laminin/laminin b2: nephrosis despite molecular compensation by laminin b1. Nat Genet. 1995;10:400–406. doi: 10.1038/ng0895-400. [DOI] [PubMed] [Google Scholar]
- Kashtan CE. Familial hematurias: what we know and what we don’t. Pediatr Nephrol. 2005;20:1027–1035. doi: 10.1007/s00467-005-1859-z. [DOI] [PubMed] [Google Scholar]
- Cosgrove D, Meehan DT, Grunkemeyer JA, Kornak JM, Sayers R, Hunter WJ, Samuelson GC. Collagen COL4A3 knockout: a mouse model for autosomal Alport syndrome. Genes Dev. 1996;10(23):2981–2992. doi: 10.1101/gad.10.23.2981. [DOI] [PubMed] [Google Scholar]
- Miner JH, Sanes JR. Molecular and functional defects in kidneys of mice lacking alpha collagen 3(IV): implications for Alport syndrome. J Cell Biol. 1996;135:1403–1413. doi: 10.1083/jcb.135.5.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Phillips CL, Killen PD, Hlaing T, Harrison WR, Elder FF, Miner JH, Overbeek PA, Meisler MH. Insertional mutation of the collagen genes Col4a3 and Col4a4 in a mouse model of Alport syndrome. Genomics. 1999;61:113–124. doi: 10.1006/geno.1999.5943. [DOI] [PubMed] [Google Scholar]
- Abrahamson DR, Prettyman AC, Robert B, St John PL. Laminin-1 re-expression in Alport mouse glomerular basement membranes. Kidney Int. 2003;63:826–834. doi: 10.1046/j.1523-1755.2003.00800.x. [DOI] [PubMed] [Google Scholar]
- Abrahamson DR, Isom K, Roach E, Stroganova L, Zelenchuk A, Miner JH, St John PL. Laminin compensation in collagen α3(IV) knockout (Alport) glomeruli contributes to permeability defects. J Am Soc Nephrol. 2007;18(9):2465–2472. doi: 10.1681/ASN.2007030328. [DOI] [PubMed] [Google Scholar]
- Kashtan CE, Kim Y, Lees GE, Thorner PS, Virtanes I, Miner JH. Abnormal glomerular basement membrane laminins in murine, canine, and human Alport syndrome: aberrant laminin alpha2 deposition is species independent. J Am Soc Nephrol. 2001;12(2):252–260. doi: 10.1681/ASN.V122252. [DOI] [PubMed] [Google Scholar]
- Beirowski B, Weber M, Gross O. Chronic renal failure and shortened lifespan in COL4A3+/− mice: an animal model for thin basement membrane nephropathy. J Am Soc Nephrol. 2006;17(7):1986–1994. doi: 10.1681/ASN.2005101044. [DOI] [PubMed] [Google Scholar]
- Yang Y, Zhang SY, Sich M, Béziau A, van den Heuvel LP, Gubler MC. Glomerular extracellular matrix and growth factors in diffuse mesangial sclerosis. Pediatr Nephrol. 2001;16(5):429–438. doi: 10.1007/s004670100563. [DOI] [PubMed] [Google Scholar]
- Wang Z, Jiang T, Li J, Proctor G, McManaman JL, Lucia S, Chua S, Levi M. Regulation of renal lipid metabolism, lipid accumulation, and glomerulosclerosis in FVBdb/db mice with type 2 diabetes. Diabetes. 2005;54(8):2328–2335. doi: 10.2337/diabetes.54.8.2328. [DOI] [PubMed] [Google Scholar]
- Freeburg PB, Robert B, St John PL, Abrahamson DR. Podocyte expression of hypoxia-inducible factor (HIF)-1 and HIF-2 during glomerular development. J Am Soc Nephrol. 2003;14(4):927–938. doi: 10.1097/01.asn.0000059308.82322.4f. [DOI] [PubMed] [Google Scholar]
- Heyman SN, Khamaisi M, Rosen S, Rosenberger C. Renal parenchymal hypoxia, hypoxia response and the progression of chronic kidney disease. Am J Nephrol. 2008;28(6):998–1006. doi: 10.1159/000146075. [DOI] [PubMed] [Google Scholar]
- Mole DR, Ratcliffe PJ. Cellular oxygen sensing in health and disease. Pediatr Nephrol. 2008;23(5):681–694. doi: 10.1007/s00467-007-0632-x. [DOI] [PubMed] [Google Scholar]
- Kaelin WG., Jr The von Hippel-Lindau tumour suppressor protein: O2 sensing and cancer. Nat Rev Cancer. 2008;8(11):865–873. doi: 10.1038/nrc2502. [DOI] [PubMed] [Google Scholar]
- Nyhan MJ, O'Sullivan GC, McKenna SL. Role of the VHL (von Hippel-Lindau) gene in renal cancer: a multifunctional tumour suppressor. Biochem Soc Trans. 2008;36(pt 3):472–478. doi: 10.1042/BST0360472. [DOI] [PubMed] [Google Scholar]
- Moeller MJ, Sanden SK, Soofi A, Wiggins RC, Holzman LB. Podocyte-specific expression of cre recombinase in transgenic mice. Genesis. 2003;35(1):39–42. doi: 10.1002/gene.10164. [DOI] [PubMed] [Google Scholar]
- Steenhard BM, Isom KS, Cazcarro P, Dunmore JH, Godwin AR, St John PL, Abrahamson DR. Integration of embryonic stem cells in metanephric kidney organ culture. J Am Soc Nephrol. 2005;16(6):1623–1631. doi: 10.1681/ASN.2004070584. [DOI] [PubMed] [Google Scholar]
- Haase VH, Glickman JN, Socolovsky M, Jaenisch R. Vascular tumors in livers with targeted inactivation of the von Hippel-Lindau tumor suppressor. Proc Natl Acad Sci USA. 2001;98(4):1583–1588. doi: 10.1073/pnas.98.4.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biju MP, Neumann AK, Bensinger SJ, Johnson RS, Turka LA, Haase VH. Vhlh gene deletion induces Hif-1-mediated cell death in thymocytes. Mol Cell Biol. 2004;24(20):9038–9047. doi: 10.1128/MCB.24.20.9038-9047.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzman LB, St John PL, Kovari IA, Verma R, Holthofer H, Abrahamson DR. Nephrin localizes to the slit pore of the glomerular epithelial cell. Kidney Int. 1999;56(4):1481–1491. doi: 10.1046/j.1523-1755.1999.00719.x. [DOI] [PubMed] [Google Scholar]
- Abrahamson DR, St John PL. Loss of laminin epitopes during glomerular basement membrane assembly in developing mouse kidneys. J Histochem Cytochem. 1992;40(12):1943–1953. doi: 10.1177/40.12.1280666. [DOI] [PubMed] [Google Scholar]
- Jensen EB, Gundersen HJ, Osterby R. Determination of membrane thickness distribution from orthogonal intercepts. J Microsc. 1979;115(1):19–33. doi: 10.1111/j.1365-2818.1979.tb00149.x. [DOI] [PubMed] [Google Scholar]
- Takemoto M, Asker N, Gerhardt H, Lundkvist A, Johansson BR, Saito Y, Betsholtz C. A new method for large scale isolation of kidney glomeruli from mice. Am J Pathol. 2002;161(3):799–805. doi: 10.1016/S0002-9440(10)64239-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Zhang YZ, Lee HS. Quantitative changes in the glomerular basement membrane components in human membranous nephropathy. J Pathol. 1997;183(1):8–15. doi: 10.1002/(SICI)1096-9896(199709)183:1<8::AID-PATH1079>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Ding M, Cui S, Li C, Jothy S, Haase V, Steer BM, Marsden PA, Pippin J, Shankland S, Rastaldi MP, Cohen CD, Kretzler M, Quaggin SE. Loss of the tumor suppressor Vhlh leads to upregulation of Cxcr4 and rapidly progressive glomerulonephritis in mice. Nat Med. 2006;12(9):1081–1087. doi: 10.1038/nm1460. [DOI] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, Lash AE. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30(1):207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burmester T, Weich B, Reinhardt S, Hankeln T. A vertebrate globin expressed in the brain. Nature. 2000;407(6803):520–523. doi: 10.1038/35035093. [DOI] [PubMed] [Google Scholar]
- Brukamp K, Jim B, Moeller MJ, Haase VH. Hypoxia and podocyte-specific Vhlh deletion confer risk of glomerular disease. Am J Physiol Renal Physiol. 2007;293(4):F1397–F1407. doi: 10.1152/ajprenal.00133.2007. [DOI] [PubMed] [Google Scholar]
- Eremina V, Sood M, Haigh J, Nagy A, Lajoie G, Ferrara N, Gerber HP, Kikkawa Y, Miner JH, Quaggin SE. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J Clin Invest. 2003;111(5):707–716. doi: 10.1172/JCI17423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eremina V, Cui S, Gerber H, Ferrara N, Haigh J, Nagy A, Ema M, Rossant J, Jothy S, Miner JH, Quaggin SE. Vascular endothelial growth factor a signaling in the podocyte-endothelial compartment is required for mesangial cell migration and survival. J Am Soc Nephrol. 2006;17(3):724–735. doi: 10.1681/ASN.2005080810. [DOI] [PubMed] [Google Scholar]
- Ohh M, Yauch RL, Lonergan KM, Whaley JM, Stemmer-Rachamimov AO, Louis DN, Gavin BJ, Kley N, Kaelin WG, Jr, Iliopoulos O. The von Hippel-Lindau tumor suppressor protein is required for proper assembly of an extracellular fibronectin matrix. Mol Cell. 1998;1(7):959–968. doi: 10.1016/s1097-2765(00)80096-9. [DOI] [PubMed] [Google Scholar]
- Bluyssen HA, Lolkema MP, van Beest M, Boone M, Snijckers CM, Los M, Gebbink MF, Braam B, Holstege FC, Giles RH, Voest EE. Fibronectin is a hypoxia-independent target of the tumor suppressor VHL. FEBS Lett. 2004;556(1–3):137–142. doi: 10.1016/s0014-5793(03)01392-9. [DOI] [PubMed] [Google Scholar]
- Kurban G, Hudon V, Duplan E, Ohh M, Pause A. Characterization of a von Hippel Lindau pathway involved in extracellular matrix remodeling, cell invasion, and angiogenesis. Cancer Res. 2006;66(3):1313–1319. doi: 10.1158/0008-5472.CAN-05-2560. [DOI] [PubMed] [Google Scholar]
- Ku CH, White KE, Dei Cas A, Hayward A, Webster Z, Bilous R, Marshall S, Viberti G, Gnudi L. Inducible overexpression of sFlt-1 in podocytes ameliorates glomerulopathy in diabetic mice. Diabetes. 2008;57(10):2824–2833. doi: 10.2337/db08-0647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iglesias-de la Cruz MC, Ziyadeh FN, Isono M, Kouahou M, Han DC, Kalluri R, Mundel P, Chen S. Effects of high glucose and TGF-beta1 on the expression of collagen IV and vascular endothelial growth factor in mouse podocytes. Kidney Int. 2002;62(3):901–913. doi: 10.1046/j.1523-1755.2002.00528.x. [DOI] [PubMed] [Google Scholar]
- Chen S, Kasama Y, Lee JS, Jim B, Marin M, Ziyadeh FN. Podocyte-derived vascular endothelial growth factor mediates the stimulation of alpha3(IV) collagen production by transforming growth factor-beta1 in mouse podocytes. Diabetes. 2004;53(11):2939–2949. doi: 10.2337/diabetes.53.11.2939. [DOI] [PubMed] [Google Scholar]
- Grosfeld A, Stolze IP, Cockman ME, Pugh CW, Edelmann M, Kessler B, Bullock AN, Ratcliffe PJ, Masson N. Interaction of hydroxylated collagen IV with the von hippel-lindau tumor suppressor. J Biol Chem. 2007;282(18):13264–13269. doi: 10.1074/jbc.M611648200. [DOI] [PubMed] [Google Scholar]
- Kurban G, Duplan E, Ramlal N, Hudon V, Sado Y, Ninomiya Y, Pause A. Collagen matrix assembly is driven by the interaction of von Hippel-Lindau tumor suppressor protein with hydroxylated collagen IV alpha 2. Oncogene. 2008;27(7):1004–1012. doi: 10.1038/sj.onc.1210709. [DOI] [PubMed] [Google Scholar]
- Minamishima YA, Moslehi J, Padera RF, Bronson RT, Liao R, Kaelin WG., Jr A feedback loop involving the Phd3 prolyl hydroxylase tunes the mammalian hypoxic response in vivo. Mol Cell Biol. 2009;29(21):5729–5741. doi: 10.1128/MCB.00331-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Nakamura E, Yang H, Wei W, Linggi MS, Sajan MP, Farese RV, Freeman RS, Carter BD, Kaelin WG, Jr, Schlisio S. Neuronal apoptosis linked to EglN3 prolyl hydroxylase and familial pheochromocytoma genes: developmental culling and cancer. Cancer Cell. 2005;8(2):155–167. doi: 10.1016/j.ccr.2005.06.015. [DOI] [PubMed] [Google Scholar]
- Sun Y, Jin K, Mao XO, Zhu Y, Greenberg DA. Neuroglobin is up-regulated by and protects neurons from hypoxic-ischemic injury. Proc Natl Acad Sci USA. 2001;98(26):15306–15311. doi: 10.1073/pnas.251466698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li RC, Lee SK, Pouranfar F, Brittian KR, Clair HB, Row BW, Wang Y, Gozal D. Hypoxia differentially regulates the expression of neuroglobin and cytoglobin in rat brain. Brain Res. 2006;1096(1):173–179. doi: 10.1016/j.brainres.2006.04.063. [DOI] [PubMed] [Google Scholar]
- Schmidt-Kastner R, Haberkamp M, Schmitz C, Hankeln T, Burmester T. Neuroglobin mRNA expression after transient global brain ischemia and prolonged hypoxia in cell culture. Brain Res. 2006;1103(1):173–180. doi: 10.1016/j.brainres.2006.05.047. [DOI] [PubMed] [Google Scholar]
- Cohen CD, Doran PP, Blattner SM, Merkle M, Wang GQ, Schmid H, Mathieson PW, Saleem MA, Henger A, Rastaldi MP, Kretzler M. Sam68-like mammalian protein 2, identified by digital differential display as expressed by podocytes, is induced in proteinuria and involved in splice site selection of vascular endothelial growth factor. J Am Soc Nephrol. 2005;16(7):1958–1965. doi: 10.1681/ASN.2005020204. [DOI] [PubMed] [Google Scholar]
- Rastaldi MP, Armelloni S, Berra S, Calvaresi N, Corbelli A, Giardino LA, Li M, Wang GQ, Fornasieri A, Villa A, Heikkila E, Soliymani R, Boucherot A, Cohen CD, Kretzler M, Nitsche A, Ripamonti M, Malgaroli A, Pesaresi M, Forloni GL, Schlöndorff D, Holthofer H, D'Amico G. Glomerular podocytes contain neuron-like functional synaptic vesicles. FASEB J. 2006;20(7):976–978. doi: 10.1096/fj.05-4962fje. [DOI] [PubMed] [Google Scholar]
- Burmester T, Hankeln T. What is the function of neuroglobin? J Exp Biol. 2009;212:1423–1428. doi: 10.1242/jeb.000729. [DOI] [PubMed] [Google Scholar]
- Khan AA, Wang Y, Sun Y, Mao XO, Xie L, Miles E, Graboski J, Chen S, Ellerby LM, Jin K, Greenberg DA. Neuroglobin-overexpressing transgenic mice are resistant to cerebral and myocardial ischemia. Proc Natl Acad Sci USA. 2006;103(47):17944–17948. doi: 10.1073/pnas.0607497103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih SC, Robinson GS, Perruzzi CA, Calvo A, Desai K, Green JE, Ali IU, Smith LE, Senger DR. Molecular profiling of angiogenesis markers. Am J Pathol. 2002;161(1):35–41. doi: 10.1016/S0002-9440(10)64154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]