

Abstract
Harlequin ichthyosis (HI) is caused by loss-of-function mutations in the keratinocyte lipid transporter ABCA12. The patients often die in the first 1 or 2 weeks of life, although HI survivors’ phenotypes improve within several weeks after birth. In order to clarify the mechanisms of phenotypic recovery, we studied grafted skin and keratinocytes from Abca12-disrupted (Abca12−/−) mice showing abnormal lipid transport. Abca12−/− neonatal epidermis showed significantly reduced total ceramide amounts and aberrant ceramide composition. Immunofluorescence and immunoblotting of Abca12−/− neonatal epidermis revealed defective profilaggrin/filaggrin conversion and reduced protein expression of the differentiation-specific molecules, loricrin, kallikrein 5, and transglutaminase 1, although their mRNA expression was up-regulated. In contrast, Abca12−/− skin grafts kept in a dry environment exhibited dramatic improvements in all these abnormalities. Increased transepidermal water loss, a parameter representing barrier defect, was remarkably decreased in grafted Abca12−/− skin. Ten-passage sub-cultured Abca12−/− keratinocytes showed restoration of intact ceramide distribution, differentiation-specific protein expression and profilaggrin/filaggrin conversion, which were defective in primary-cultures. Using cDNA microarray analysis, lipid transporters including four ATP-binding cassette transporters were up-regulated after sub-culture of Abca12−/− keratinocytes compared with primary-culture. These results indicate that disrupted keratinocyte differentiation during the fetal development is involved in the pathomechanism of HI and, during maturation, Abca12−/− epidermal keratinocytes regain normal differentiation processes. This restoration may account for the skin phenotype improvement observed in HI survivors.
Harlequin ichthyosis (HI) (OMIM 242500) is one of the most severe genetic skin disorders, and its clinical features at birth include severe ectropion, eclabium, flattening of the ears, and large thick plate-like scales over the entire body.1 Infants affected with HI frequently die within the early neonatal period, although an increasing survival rate for HI newborns has recently been highlighted.2 In 2005, we and another independent research group identified mutations in the ATP-binding cassette transporter A12 (ABCA12) gene as the cause of HI.3,4 We previously demonstrated that a severe ABCA12 deficiency causes defective lipid transport in lamellar granules in the upper spinous and granular layer keratinocytes, resulting in malformation of intercellular lipid layers at the granular/cornified layer interface and epidermal lipid barrier disruption resulting in HI phenotype.3 We recently generated Abca12-disrupted (Abca12−/−) mice by targeting Abca12, which closely reproduced the human HI phenotype and died soon after birth.5 We tried systemic retinoid administration to the pregnant female mice as a form of fetal therapy, although no therapeutic effect was obtained in the Abca12−/− newborns after treatment.5 After our publication, Zuo et al6 also reported another Abca12 knockout mouse, whose skin showed similar features to our model mice.
Previously, we demonstrated severe skin barrier defects in Abca12−/− mice and suggested that “barrier insufficiency” plays an important role in HI phenotype expression.5 However, “the barrier insufficiency” theory fails to completely explain the pathomechanism of the HI phenotype. HI fetuses show a HI phenotype even in utero, where skin barrier protection against a dry environment is not required. In addition, the skin phenotype of HI long-term survivors maintained in a dry environment shows a dramatic improvement within several weeks after birth where they require a normal skin barrier function. Thus, we suspected that other unknown mechanisms are involved in HI and the formation HI survivors’ skin phenotypes. To date there have been no reports which have compared the skin phenotypes in fatally affected HI neonates and survivors, and the exact mechanism of HI survivors’ skin phenotype improvement has yet to be clarified. Thus, we have carefully analyzed the keratinization process of neonatal versus grafted HI model mice skin and primary versus subcultured Abca12−/− keratinocytes instead of human HI neonatal and survivors’ skin. Initially, we investigated the distribution and amounts/composition of lipids, and expression of differentiation-specific molecules in neonatal HI model mice skin. Then, we studied the alteration of them in grafted HI model mouse skin transplanted onto severe combined immunodeficient (SCID) mice. In addition, we performed keratinocyte culture experiments including immunostaining and Western blotting using primary/subcultured Abca12−/− keratinocytes to confirm the results of the neonatal and grafted skin experiments. Further, we analyzed the whole gene expression profile of primary versus subcultured Abca12−/− keratinocytes using cDNA microarray methods. Finally, we conducted therapeutic trials on primary-cultured Abca12−/− keratinocytes and grafted HI model mice skin with retinoids.
Materials and Methods
Animals
All animal studies were reviewed and approved by the Animal Use and Care Committee of the Hokkaido University Graduate School of Medicine. C57BL/6 strain mice and SCID mice were purchased from Clea (Tokyo, Japan). All animals used for this study were maintained under pathogen-free conditions.
Antibodies
Rabbit polyclonal affinity purified anti-mouse Abca12 antibody was raised in rabbits using a 14-amino acid sequence synthetic peptide (residues 2581 to 2594) derived from the mouse Abca12 sequence (XM001002308) as the immunogen.5 The other primary antibodies were rabbit anti-profilaggrin/filaggrin antibody (COVANCE, Princeton, NJ), rabbit anti-involucrin antibody (M-116; Santa Cruz Biotechnology, Santa Cruz, CA), rabbit anti-desmoglein 1 antibody (H-290; Santa Cruz Biotechnology), rabbit anti-mouse loricrin antibody (AF62; COVANCE), rabbit anti-kallikrein 5 antibody (ab7283; Abcam, Cambridge, UK), rabbit anti-glucosylceramide/ceramide antibody (Glycobiotech, Kukels, Germany), and mouse monoclonal anti-β actin antibody (Sigma Chemical Co., St. Louis, MO). Secondary antibodies used in the present study were as follows; Alexa Fluor 488-conjugated donkey anti-rabbit IgG (Invitrogen Corp., Carlsbad, CA), fluorescein isothiocyanate-conjugated goat anti-rabbit IgG (Jackson Immuno Research, West Grove, PA), horseradish peroxidase-conjugated goat anti-rabbit IgG or horseradish peroxidase-conjugated goat anti-mouse IgG (Invitrogen Corp.).
Generation of Abca12−/− Mouse
The procedure for generating Abca12−/− mice has been previously described.5 Briefly, we cloned mouse genomic DNA Abca12 fragments from the mouse 129Sv/Ev genomic library (Bacpac Resources Center, Children’s Hospital Oakland Research Institute, Oakland, CA). We subcloned a 10.6-kb fragment to make the targeting vector. We inserted the PGK/Neo cassette between 47 bp upstream of the exon 30 and 203 bp downstream of exon 30. We transfected the targeting vector by electroporation into 129Sv/Ev embryonic stem cells, then microinjected the correctly targeted embryonic stem cell line into blastocysts obtained from C57BL/6 mice to generate chimeric mice, which we then mated with C57BL/6 females. We crossed the heterozygotes with C57BL/6 over at least five generations, and then intercrossed them to generate the Abca12−/− mice. Genotyping was performed by PCR as described previously.5
Establishment of Abca12−/− Mice Keratinocyte Culture
Skin samples from Abca12−/− and wild-type mice were processed for primary keratinocyte culture, and cells were grown according to standard procedures in CnT-57 medium (Cellntec Advanced Cell Systems, Bern, Switzerland). For differentiation induction, culture medium was switched from CnT-57 medium to CnT-02 medium (Cellntec Advanced Cell Systems) and, 24 hours later, the calcium concentration was changed to 1.2 mmol/L. 48 hours later, we performed extractions of total RNA and protein from cultured cells. We established primary-cultured keratinocytes from two Abca12−/− mice and two wild-type mice.
Skin Grafting
In total, ten Abca12−/− and three wild-type neonates were sacrificed by anesthesia with ether inhalation, and their dorsal skin excised and transplanted onto SCID mice (Clea). Those skin grafts were fully adapted within 2 weeks after grafting. At 3 weeks after transplantation, the skin grafts were harvested for further analysis.
Extraction of Total RNA and Real-Time Reverse Transcriptase PCR Analysis
We separated the epidermis from whole skin samples of wild and Abca12−/− mice by 1 mol/L NaCl in sterile water at 4°C for 2 hours. We isolated total RNA from the epidermis using the Quick Gene RNA Tissue Kit SII (Fujifilm Corp, Tokyo, Japan). We also isolated total RNA from keratinocytes cultured from wild-type and Abca12−/− skin using the RNeasy mini kit (Qiagen Corp, Tokyo, Japan). RNA concentration was measured spectrophotometrically and samples were stored at −80°C until use for reverse transcriptase PCR. We reverse-transcribed RNA using Superscript II (Invitrogen Corp.) following the manufacturer’s instructions. Complementary DNA samples were analyzed by ABI prism 7000 sequence detection system (Applied Biosystems, Foster City, CA). Primers and probes specific for differentiation-specific protein genes including loricrin, kallikrein 5, transglutaminase 1, involucrin, filaggrin, and control house keeping genes, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and β-actin, were obtained from Taqman Gene Expression Assay (Applied Biosystems) (Probe ID; Mm01962650_s1, Mm01203811_ a1, Mm00498375_a1, Mm00515219_s1, Mm01716522_m1, Mm99999915_g1, and Mm00607939_s1). Differences between the mean CT values of loricrin, kallikrein 5, transglutaminase 1, involucrin, filaggrin and those of GAPDH or β-actin were calculated as: ΔCTAbca12−/− mice = CTloricrin (or other keratinization markers) − CTGAPDH (or other house keeping genes) and those of ΔCT for the Abca12+/+ asΔCT calibrator = CTloricrin (or other keratinization markers) − CTGAPDH (or other house keeping genes).
We could obtain the similar results from GAPDH and β-actin standard, thus we described the results of GAPDH standard in the present study. Final results for Abca12−/− mouse samples/wild-type mouse samples (%) were determined by 2−(ΔCTAbca12−/− sample − ΔCTcalibrator). We measured mRNA levels five times for each clones. Using similar methods, we quantitatively analyzed these differentiation-specific mRNA expression levels in the primary/subcultured keratinocytes from Abca12−/− and wild-type mice.
Western Blotting
We separated the epidermis from whole skin samples of wild and Abca12−/− mice by 1mol/L NaCl in sterile water at 4°C for 2 hours. For Western blotting, we used epidermal homogenates and proteins extracted from cultured keratinocytes prepared with radioimmunoprecipitation assay (RIPA) buffer comprising 50 mmol/L Tris-HCl, pH7.5, 150 mmol/L NaCl, 1% Nonidet P-40, 0.5% deoxycholate, 0.1% SDS, and Roche protease cocktail tablet (Roche, Basel, Switzerland). Protein concentrations were measured using Micro BCA protein assay kit (Thermo Scientific, Rockford, IL). Protein concentration of the samples for western blotting was from 1 to 2 μg/μl. The 20 μg protein loading per single lane was separated by a 5 to 20% gradient gel SDS-polyacrylamide gel and transferred to polyvinylidene difluoride membranes. Membrane blocking and incubation with antibodies were performed in Tris-buffered saline with 2% nonfat dry milk. Signals were revealed with chemiluminescence reagents and photographed by LAS-1000 mini (Fujifilm Corp, Tokyo, Japan). We also confirmed the loading protein dose by β-actin antibody staining as internal protein control. For analysis of filaggrin solubility and processing, epidermal lysates were prepared with RIPA buffer. Samples of precipitated proteins in the RIPA buffer were solubilized again in 8 mol/L urea before boiling in reducing SDS loading buffer.
Light Microscopy and Immunofluorescence Analysis
For light microscopy, we harvested the newborn pups’ skin and the grafted skin, and fixed them for 24 hours in 10% neutral buffered formalin, dehydrated them in graded ethanol, and embedded them in paraffin. We cut 4-μm sections and stained them with H&E. For immunohistochemistry, the tissue samples were embedded in optimal cutting temperature compound (Sakura Finetechnical Corp., Tokyo, Japan). Frozen tissue sections were cut at a thickness of 5 μm. The sections were blocked with 1% bovine serum albumin (BSA) in PBS for 30 minutes at room temperature, and incubated in primary antibody solution in blocking buffer for 30 minutes at 37°C. Fluorescent labeling was performed with secondary antibodies, followed by propidium iodide (Sigma Chemical Co.) for 5 minutes at room temperature to counterstain nuclei. The stained samples were observed under an Olympus Fluoview confocal laser-scanning microscope (Olympus, Tokyo, Japan).
In Situ Transglutaminase Activity
The procedure for in situ transglutaminase 1 activity assay has been previously described.7,8 In brief, unfixed cryosections of 5 μm were blocked with 100 mmol/L Tris-HCl pH7.4, 1% BSA for 30 minutes, and then incubated with 100 mmol/L Tri-HCl pH7.4, 5 mmol/L CaCl2, 12 μmol/L monodansylcadaverine (Sigma) for 1 hour to detect transglutaminase 1 activity. For negative controls, EDTA was added to the monodansylcadaverine solution to a final concentration of 20 mmol/L. After stopping the transglutaminase 1 reaction with 10 mmol/L EDTA in PBS, sections were incubated with rabbit anti-dansyl antibody (Invitrogen Corp.) in 12% BSA/PBS for 3 hours. Sections were then incubated with fluorescein isothiocyanate-conjugated goat anti-rabbit antibody in 12% BSA/PBS for 30 minutes. Nuclei were counterstained by propidium iodide. The stained samples were observed under an Olympus Fluoview confocal laser-scanning microscope (Olympus).
Immunofluorescence Labeling of Cultured Cells
Immunofluorescence labeling of cultured cells was performed as previously described.5 Briefly, primary/subcultured keratinocytes were fixed in 4% paraformaldehyde for 15 minutes and permeabilized with 0.1% Triton X-100 for 15 minutes at room temperature. Keratinocytes were blocked with 1% BSA in PBS for 30 minutes at room temperature, and incubated in primary antibody solution in blocking buffer for 30 minutes at 37°C and fluorescence labeling was performed with secondary antibodies, followed by propidium iodide for 5 minutes at room temperature to counterstain nuclei. The stained samples were observed under an Olympus Fluoview confocal laser-scanning microscope (Olympus).
Electron Microscopy
Neonatal skin samples and skin grafts were fixed in 5% glutaraldehyde solution, post fixed in 1% OsO4, dehydrated, and embedded in Epon 812 (TAAB Laboratories, Berkshire, UK). All of the samples were ultra-thin sectioned at a thickness of 70 nm, and stained with uranyl acetate and lead citrate. Photographs were taken using a Hitachi H-7100 transmission electron microscope (Hitachi, Tokyo, Japan).
Lipid Analysis
Lipid analysis was performed as previously reported.9 Briefly, lipid analysis was done independently on three Abca12−/− neonates, two wild-type as controls, and two mature Abca12−/− skins 3 months after skin grafting and two mature wild-type skins 3 months after transplantation as control. We separated the epidermis from whole skin specimens of control and Abca12−/− mice by incubation in sterile water at 60°C for 1 minute and homogenized in 0.8 ml of PBS. A total lipid component was extracted from tissue homogenates of epidermis according to conventional methods.9 Lipid analysis in epidermal lysates from neonates and grafted skin was performed by liquid chromatography, electrospray ionization mass spectrometry (LC-ESI-MS) using a HP 1100 liquid chromatography system (Agilent Technologies, Palo Alto, CA).
Measurement of Transepidermal Water Loss
Transepidermal water loss (TEWL) from the skin of neonatal mice and from skin grafted onto SCID mice was measured by evaporimeter (AS-VT100RS: Asahibiomed Corp., Yokohama, Japan), as described previously.5 The AS-VT100RS utilizes the ventilated-chamber method for measuring TEWL. Its hygrometer measures the humidity of incoming air and of outgoing air that has passed over the test area of the skin, and TEWL is calculated from the difference. TEWL measurements were performed on the back of the neonates and the grafted skin onto the back of SCID mice.
Complementary DNA Microarray Analysis for the Gene Expression Profile
Total RNA isolated from primary/subcultured Abca12−/− keratinocytes was extracted as described above. Total RNA concentration was calculated spectrophotometrically, and quality control of RNA was analyzed with an Agilent 2100 Bioanalyzer (Agilent Technologies, Tokyo, Japan). mRNA/cDNA hybrids were generated via T7oligo dT primers, followed by addition of DNA polymerase and ligase (Filgen, Nagoya, Japan) to obtain double-stranded cDNA. The sample tagged with chemiluminescent substrate, Cy3 for the subcultured Abca12−/− keratinocytes, or Cy5 for the primary-cultured Abca12−/− keratinocytes, was hybridized on a microarray chip (Filgen Array mouse 32K, Filgen). We used a mixture of total RNAs from the two cultures for labeling reactions. Fluorescence images for Cy3 and Cy5 dye channels were obtained using a GenePix 4000B scanner (Axon Instruments, CA) and scan data images were analyzed using Microarray Data Analysis Tool version 3.0 software (Filgen).
Therapeutic Trial with Retinoids on Primary-Cultured Abca12−/− Keratinocytes and Grafted Harlequin Ichthyosis Model Mice Skin
To test the efficacy of a therapeutic trial on primary-cultured Abca12−/− keratinocytes, isotretinoin (purchased by Sigma Chemical Co., St. Louis, MO) was dissolved in dimethyl sulfoxide (DMSO). In addition, etretinate powder (a gift from Chugai Pharmaceuticals, Tokyo, Japan) was dissolved in sterile water.
Primary-cultured keratinocytes were grown in CnT-57 medium (Cellntec Advanced Cell Systems) and then switched into CnT-02 medium (Cellntec Advanced Cell Systems). Twenty-four hours later, the calcium concentration was changed to 1.2 mmol/L in CnT-02 medium with retinoids (10−6mol/L isotretinoin or 10−6mol/L etretinate) dissolved in DMSO or water. The final concentration of DMSO in medium was 0.01%. As control, keratinocytes were cultured in CnT-02 medium with 1.2 mmol/L calcium supplemented with 0.01% DMSO without retinoids. Forty-eight hours later, we extracted protein from keratinocytes.
In a therapeutic trial using grafted HI model mice skin, we dissolved several doses of isotretinoin in soy oil, and single doses (1, 10 mg/kg) of isotretinoin were administered orally into the grafted SCID mice 3 weeks after skin transplantation every day for 10 days.
Statistical Analysis
All statistical analyses were performed using student’s t-tests with sample sizes indicated in the figure legends for each comparison that was made. P values of <0.05 were considered statistically significant.
Results
Abca12−/− Neonatal Mouse Epidermis Exhibited Defective Lipid Distribution, Reduced Expression of Differentiation-Specific Proteins and Profilaggrin/Filaggrin Conversion Defects
As we previously reported,5 Abca12−/− mice (Figure 1B) were typically born with a smaller body size than that of wild-type mice (Figure 1A). Erythematous, rigid skin covered the entire body surface of Abca12−/− neonates. Light microscopy showed a thick, compact cornified layers without the normal basket-weave appearance in the Abca12−/− neonatal skin (Figure 1D), compared with normal neonatal skin (Figure 1C). Electron microscopy of Abca12−/− neonatal skin showed numerous lipid droplets in the granular layer cell cytoplasm (Figure 1F). In wild-type neonatal skin, no lipid droplets were seen although normal keratohyalin granules were observed (Figure 1E). Immunofluorescence staining (Figure 1, G and H) showed Abca12 in wild-type but not abca12−/− mice, with the glucosylceramide/ceramide distribution remarkably sparse at the Abca12−/− neonatal mice granular/cornified layer interface (Figure 1J), compared with the intense labeling in the wild-type neonatal epidermis (Figure 1I). To verify these results in the neonatal Abca12−/− and wild-type skin, we performed lipid analysis using skin samples from both the Abca12−/− and wild-type neonates. Total amounts of epidermal ceramides were significantly reduced in Abca12−/− neonatal mice (Figure 1K). Particularly, amounts/compositions of the CER[EOS], ceramide classes consisting of ester-linked non-hydroxy fatty acids, ω-hydroxy fatty acids and 4-sphinnenines, in Abca12−/− neonatal epidermis were extremely small compared with control mice (see Supplemental Figure S1, A and B at http://ajp.amjpathol.org).
Figure 1.

Abca12−/− neonatal phenotype and lipid trafficking defects. A and B: Gross phenotypes of wild-type and Abca12−/− neonates. C and D: Light microscopy showed a thick compact cornified layer (bracket) without the normal basket-weave appearance in the Abca12−/− mouse skin (D), compared with normal neonatal skin (C) (H&E stain; original magnification, ×40) (Scale bars = 20 μm). E and F: An electron micrograph of the Abca12−/− neonatal skin showed numerous lipid droplets in the cytoplasm of the granular layer cells (F, red arrows). In the wild-type neonatal skin, no lipid droplets were seen and normal keratohyalin granules were observed (E, red arrows) (original magnification, ×3000) (Scale bars = 2 μm). G and H: By immunofluorescence staining, Abca12 expression (Alexa488, green) was detected in the wild-type neonatal mouse skin (G), but not in the Abca12−/− skin (H). I and J: Immunofluorescence staining showed the glucosylceramide/ceramide (GlcCer/Cer) (Alexa 488, green), a major lipid component of lamellar granules and an essential component of the epidermal permeability barrier, to be distributed remarkably sparse in the Abca12−/− neonatal mouse granular/cornified layer interface (J), compared with the intense labeling in the wild-type neonatal epidermis (I). K: In neonatal mice, total ceramides levels were significantly reduced in the epidermis of Abca12−/− mice. (Abca12−/− neonates, n = 3; control neonates, n = 2) (*P < 0.01). GlcCer, glucosylceramide; Cer, ceramide.
Immunofluorescence staining revealed that the keratinocyte differentiation (keratinization)-specific molecules, kallikrein 5 (KLK5), transglutaminase 1 (TGase1), and loricrin, were sparsely distributed in the upper epidermis of neonatal Abca12−/− mice (Figure 2A–J). Immunofluorescence staining for KLK5, a lamellar granule component, was weak in Abca12−/− neonatal mice granular layer (Figure 2B), in contrast to intense labeling in granular and lower cornified layers of wild-type neonatal skin (Figure 2A). In situ TGase1 activity assays with dansyl-cadaverine as a substrate showed neonatal Abca12−/− granular layer keratinocytes exhibited weak TGase1 activity restricted to the cytoplasm (Figure 2D), compared with distinct TGase1 activity with a more peripheral pattern throughout neonatal wild-type granular layer keratinocytes (Figure 2C). Immunofluorescence staining showed loricrin expressed sparsely within neonatal Abca12−/− granular layer cells (Figure 2F), compared with more intense expression in neonatal wild-type granular layer keratinocytes (Figure 2E).
Figure 2.

Reduced epidermal differentiation-associated molecules and defective conversion of profilaggrin to filaggrin in Abca12−/− neonates. A and B: Immunofluorescence staining for kallikrein 5 (KLK5) (Alexa488, green), one of the lamellar granule (LG) contents, was weak in the Abca12−/− neonatal mice granular keratinocyte layers (B), in contrast to its intense labeling in the granular layers and lower cornified layers of wild-type neonatal skin (A). C and D: In situ transglutaminase 1 (TGase1) activity assay (fluorescein isothiocyanate, green) with dansyl-cadaverine showed the granular layer keratinocytes in the Abca12−/− neonates had weak transglutaminase 1 activity only in the cytoplasm (D), compared with distinct transglutaminase 1 activity with the peripheral pattern throughout granular layer keratinocytes in the wild-type neonates (C). Cytoplasmic localization of transglutaminase 1 in Abca12−/− neonates indicated its inability to bind to the cell membrane and to therefore function at its proper place despite the significantly enhanced mRNA expression of transglutaminase 1 (see Figure 2K). E and F: Immunofluorescence staining showed loricrin (Alexa 488, green) expressed sparsely in the Abca12−/− neonatal mice granular layer (F), compared with its intense expression in the granular layer of the wild-type neonatal skin (E). G and H: Both Abca12−/− and wild-type neonatal skin showed intense profilaggrin/filaggrin (proFLG/FLG) (Alexa488, green) expression in the granular layer keratinocytes. However, in Abca12−/− neonatal skin, profilaggrin/filaggrin distribution was also observed throughout the cornified layers. I and J: Desmoglein 1(DSG1) (Alexa 488, green), a cell adhesion molecule unassociated with keratinization, was expressed at the cell periphery in the lower granular and spinous layers of the both Abca12−/− neonatal skin and wild-type mouse skin. (nuclear stain; propidium iodide, red, dotted lines, the skin surface). Original magnification ×40; Scale bars, 20 μm. K: mRNA expression of loricrin, kallikrein 5 (KLK5), involucrin, transglutaminase 1 (TGase1) and filaggrin (FLG) was up-regulated in the Abca12−/− neonatal epidermis. (Abca12−/− neonates, n = 5; wild-type neonates, n = 5, mRNA expression levels of wild-type neonatal epidermis = 1). L: Western blotting of epidermal extracts showed that protein expression of kallikrein 5 (KLK5) and loricrin was lower in the Abca12−/− neonatal epidermis (right) than in the wild-type neonatal epidermis (left). There were no significant differences of desmoglein 1(DSG1), involucrin, β-actin expressions between the Abca12−/− and wild-type epidermis. M: Western blotting with anti-profilaggrin/filaggrin antibody revealed the Abca12−/− neonatal epidermis (right) expressed more profilaggrin/filaggrin protein than wild-type neonatal epidermis (left). High molecular weight smear band corresponding to non-converted profilaggrin peptides were characteristic to the Abca12−/− neonatal epidermis. Western blotting using serial protein dilutions is shown in the supplemental Figure 2 (see Supplemental Figure S2 at http://ajp.amjpathol.org). KLK5, kallikrein 5; LG, lamellar granule; TGase1, transglutaminase 1; CCE, cornified cell envelope; FLG, filaggrin; KHG, keratohyalin granule; DSG1, desmoglein 1; proFLG, profilaggrin; 4FLG, filaggrin tetramer; 3FLG, filaggrin trimer; 2FLG, filaggrin dimer; 1FLG, filaggrin monomer.
KLK5, involucrin, TGase1, loricrin, and filaggrin mRNA expression was up-regulated in neonatal Abca12−/− epidermal keratinocytes (Figure 2K). In contrast, protein expression using epidermal extract, Western blotting demonstrated that loricrin and KLK5 protein expression was reduced in neonatal Abca12−/− epidermal keratinocytes compared with that in neonatal wild-type epidermis (Figure 2L). There were no significant differences in the protein expression of involucrin or control molecules unconnected with the keratinization process, β-actin, and desmoglein1 (DSG1), between Abca12−/− and wild-type neonatal epidermis.
Both Abca12−/− and wild-type neonatal skin demonstrated intense profilaggrin/filaggrin expression within granular layer keratinocytes (Figure 2, G and H). However, in Abca12−/− neonatal skin, profilaggrin/filaggrin distribution was also observed throughout all of the cornified layers (Figure 2H). Western blotting with anti-profilaggrin/filaggrin antibody revealed that neonatal Abca12−/− epidermis exhibited a greater amount of profilaggrin/filaggrin protein than that in the neonatal wild-type epidermis (Figure 2M). In particular, high molecular weight smear bands corresponding to unconverted profilaggrin peptides were characteristic of neonatal Abca12−/− epidermis. Western blotting using the serial dilutions of protein showed that neonatal Abca12−/− epidermis exhibited a filaggrin monomer band, although the ratio of high molecular weight profilaggrin and its derivatives to filaggrin monomer in the Abca12−/− neonatal epidermis was extremely high compared with that of the wild-type neonatal epidermis (see Supplemental Figure S2A at http://ajp.amjpathol.org). The remaining high molecular bands in the neonatal Abca12−/− epidermis indicated defective profilaggrin conversion to filaggrin. Furthermore, we prepared 8 mol/L urea supernatants from precipitated proteins from the Abca12−/− neonatal epidermis in RIPA buffer. Western blotting with 8 mol/L urea supernatants confirmed that a proportion of filaggrin monomer, which is insoluble in the RIPA buffer exists in Abca12−/− neonatal epidermis (see Supplemental Figure S2B at http://ajp.amjpathol.org). In contrast, urea supernatants from precipitated proteins in RIPA buffer of wild-type neonatal epidermis showed only a faint band of filaggrin monomer. This finding indicated that majority of filaggrin monomer in the wild-type neonatal epidermis is soluble in RIPA buffer. These Western blotting results suggested that Abca12−/− neonatal epidermis exhibited not only defective profilaggrin/filaggrin conversion but also alteration of filaggrin monomer solubility. We also performed Western blotting with anti-loricrin antibody using 8 mol/L urea supernatant. Using 8 mol/L urea buffer as well as using RIPA buffer the loricrin band was faint in supernatant samples from the Abca12−/− neonatal epidermis (data not shown). Thus, the solubility of loricrin was unaltered in the Abca12−/− neonatal epidermis and we think that the alteration of solubility in the Abca12−/− neonatal epidermis is specific to filaggrin.
Improved Morphological Abnormalities, Corrected Lipid Distribution, and Restored Expression of Differentiation-Specific Molecules in Abca12−/− Skin Grafts Maintained in Dry Environment
Since Abca12−/− neonates die soon after birth, it was impossible to follow the phenotypic changes in the skin of Abca12−/− mice after birth. Therefore, we grafted their skin onto severe combined immunodeficient (SCID) mice and analyzed its morphological and biochemical alterations in the skin after birth/grafting.
Mature grafted Abca12−/− skin showed hairless keratotic plates at 3 weeks after transplantation onto the backs of SCID mice (Figure 3, A and B). Light microscopic observations revealed that hair follicles and shafts were buried in keratotic plugs in mature Abca12−/− skin (Figure 3, C and D). High power microscopy demonstrated that mature Abca12−/−skin showed discernible keratohyalin granules in the granular layers (Figure 3F) that were completely absent in Abca12−/− neonatal skin (Figure 1, E and F).
Figure 3.

Altered morphology and improvement of ceramide deficiency during maturation of Abca12−/− skin. Gross (A and B) and microscopic (C–L) appearances of wild-type and Abca12−/− skin three weeks after transplantation onto SCID mice. A: Grafted skin of wild-type mice three weeks after transplantation. B: Grafted Abca12−/− skin three weeks after transplantation showed hairless keratotic plates. C and D: Light microscopic observation histology (H&E stain; original magnification ×40; Scale bars = 20 μm). Hair follicles and shafts were buried in keratotic plugs in the mature Abca12−/− skin three weeks after the skin graft (D). E and F: High power views (H&E stain; original magnification ×60; Scale bars = 10 μm). Mature Abca12−/− skin three weeks after transplantation showed discernible keratohyalin granule in the granular layers (F, arrows) that were lacked in Aba12−/− neonatal skin (see Figure 1, D and F). G and H: By immunofluorescence staining, Abca12 expression (Alexa488, green) was detected in the mature wild-type mouse skin (G), but not in the mature Abca12−/− skin (H). I and J: Immunofluorescence staining showed an intense distribution of glucosylceramide/ceramide (GlcCer/Cer) (Alexa 488, green) at the granular/cornified layer interface in mature Abca12−/− epidermis (J, arrows), compared with a sparse distribution in the neonatal Abca12−/− mouse upper epidermis (see Figure 1J). K and L: Ultrastructural observation of the grafted Abca12−/− skins three weeks after transplantation. There were many lipid droplets in the granular layer (K, red arrowheads), however the number of lipid droplets in the cornified layer (L, red arrowheads) was fewer than that of the neonatal Abca12−/− skin (see Figure 1F). Original magnification: ×10000 (K), ×5000 (L); Scale bars: 100 nm (K), 200 nm (L). M: From the lipid analysis of grafted skins, total ceramides levels of mature Abca12−/− epidermis were restored. (Abca12−/− grafted skins, n = 2; control grafted skins, n = 2) (*P < 0.01). GlcCer, glucosylceramide; Cer, ceramide
Immunofluorescence staining showed abca12 staining in wild-type but not abca12−/−mice and intense labeling of glucosylceramides/ceramides at the granular/cornified layer interface in mature grafted Abca12−/− skin (Figure 3, I and J), compared with a sparse distribution in the neonatal Abca12−/− mouse upper epidermis (Figure 1L). Electron microscopy of mature Abca12−/− skin showed many lipid droplets in the granular layer (Figure 3K), although the number of lipid droplets in the cornified layer was reduced when compared with that of neonatal skin (Figure 3L). Using lipid analysis, the amounts of both total ceramides and CER[EOS] were restored in mature Abca12−/− epidermis (Figure 3M, and see Supplemental Figure S1, C and D at http://ajp.amjpathol.org). These results indicate that mature grafted Abca12−/− epidermis was able to obtain a normal ceramide distribution together with a normal composition of ceramides.
Immunolabeling for differentiation-specific molecules confirmed improved keratinization during maturation of the grafted Abca12−/− skin (Figure 4, A–J). Intense KLK5 immunolabeling (Figure 4, A and B), in situ transglutaminase 1 (TGase1) activity (Figure 4, C and D), and loricrin immunostaining (Figure 4, E and F) were distributed throughout the granular layers in mature grafted Abca12−/− skin, compared with a sparse distribution in Abca12−/− neonatal skin (Figure 2, B, D, and F). Increased loricrin and KLK5 immunolabeling intensity was confirmed by Western blot analysis using epidermal extracts from mature grafted Abca12−/− epidermis (Figure 4K).
Figure 4.

Improvement of previously disrupted differentiation-specific protein expression and defective skin barrier function in postnatal Abca12−/− skin. A–J: Immunolabeling for differentiation-specific molecules confirmed improved keratinization during maturation of the grafted Abca12−/− skin. Intense kallikrein 5 (KLK5) immunostaining (A and B, white arrows), in situ transglutaminase 1 (TGase1) activity labeling (C and D) and loricrin immunolabeling (E and F, white arrows) were seen throughout the granular layers in mature grafted Abca12−/− skin, compared with their sparse distribution in the Abca12−/− neonatal skin (see Figure 2, B, D and F). Increased loricrin and KLK5 immunolabeling intensity was confirmed by Western blot analysis using epidermal extracts from mature grafted Abca12−/− skin (K). Mature grafted Abca12−/− skin showed intense profilaggrin/filaggrin labeling in the granular layer (H), similar to the mature grafted wild-type skin (G). Diffuse profilaggrin/filaggrin staining in the entire cornified layers observed in Abca12−/− neonatal skin (see Figure 2H) disappeared in mature grafted Abca12−/− skin. Expression of desmoglein 1 (DSG1), unassociated with keratinization, was not altered in the grafted skin (I and J). Western blotting with anti-profilaggrin/filaggrin antibody revealed that normal conversion of profilaggrin to filaggrin was restored in mature Abca12−/− epidermis (K). Nuclear stain: propidium iodide, red; original magnification ×40; Scale bars = 20 μm. L: The barrier function in the transplanted skin was assessed by measuring transepidermal water loss (TEWL). The Abca12−/− neonates (red bar, left) showed significantly greater TEWL than the wild-type neonates (blue bar, left) showed (*P < 0.001). TEWL levels of mature Abca12−/− epidermis three weeks after the skin graft (red bar, right) were significantly decreased as compared with levels of Abca12−/− neonatal skin (red bar, left) (**P < 0.001), and was similar to levels of mature wild-type skin three weeks after the skin graft (blue bar, right). (Abca12−/− neonates, n = 3; wild-type neonates, n = 3; mature Abca12−/− skin, n = 3; mature wild-type skin, n = 3). KLK5, kallikrein 5; proFLG, profilaggrin; FLG, filaggrin; 4FLG, filaggrin tetramer; 3FLG, filaggrin trimer; 2FLG, filaggrin dimer; 1FLG, filaggrin monomer.
Mature grafted Abca12−/− skin showed intense profilaggrin/filaggrin labeling in the granular layer (Figure 4H), similar to the mature grafted wild-type skin (Figure 4G). The diffuse profilaggrin/filaggrin distribution throughout the cornified layers observed in Abca12−/− neonatal skin (Figure 2H) was not seen in mature grafted Abca12−/− skin (Figure 4H). Western blotting with anti-profilaggrin/filaggrin antibody revealed that the normal conversion of profilaggrin to filaggrin was restored in mature Abca12−/− epidermis (Figure 4K). Epidermal extracts of mature Abca12−/− skin at 3 weeks after transplantation showed low expression of high molecular weight smeared bands and, instead of those, exhibited intense filaggrin monomer bands, compared those with epidermal extracts of Abca12−/− neonatal skin.
Analysis of TEWL as a parameter of skin barrier defects, demonstrated Abca12−/− that neonatal back skin showed significantly greater TEWL than the wild-type neonatal skin (n = 3, P < 0.001) (Figure 4L). However, TEWL levels of mature Abca12−/− skin 3 weeks after the skin graft were significantly decreased as compared with levels of Abca12−/− neonatal skin (n = 3, P < 0.001).
Subcultured Abca12−/− Mouse Keratinocytes Attained Normal Lipid Trafficking in the Cytoplasm, with Restoration of Differentiation-Specific Protein Expression and Intact Profilaggrin/Filaggrin Conversion that Was Defective in Primary-Cultured Abca12−/− Mouse Keratinocytes
To verify the results from grafted skin analysis, we performed a similar analysis using primary versus subcultured Abca12−/− keratinocytes. Immunolabeling with anti-glucosylceramide/ceramide antibody demonstrated a congested glucosylceramide/ceramide pattern in differentiated primary-cultured Abca12−/− mouse keratinocytes after first passage (Figure 5, B and E), compared with an uncongested, diffuse peripheral glucosylceramide/ceramide pattern in differentiated primary-cultured wild-type mouse keratinocytes (Figure 5, A and D). After 10 passages, subcultured Abca12−/− keratinocytes showed a widely distributed, diffuse glucosylceramide/ceramide staining pattern, similar to those of primary-cultured wild-type keratinocytes (Figure 5, C and F). Subcultured wild-type keratinocytes after 10 passages failed to show any alterations in lipid distribution (data not shown). These results indicate that lipid trafficking recovered during 10 passages of subculture in our Abca12−/− keratinocytes.
Figure 5.

Subcultured Abca12−/− mouse keratinocytes exhibited restored intracytoplasmic localization of glucosylceramide/ceramide, protein expression of differentiation-specific molecules and profilaggrin conversion. A–F: Intracytoplasmic localization of glucosylceramide/ceramide in cultured keratinocytes. Immunolabeling demonstrated a congested pattern of glucosylceramide/ceramide (Alexa 488, green) in differentiated primary-cultured Abca12−/− mouse keratinocytes after first passage (B and E), in contrast with the uncongested, diffuse, peripheral pattern in the differentiated keratinocytes of a wild-type mouse (A and D). After 10 passages, subcultured Abca12−/− keratinocytes showed a widely distributed, diffuse glucosylceramide staining pattern (C and F), similar to those of wild-type keratinocytes. Subcultured wild-type keratinocytes after ten passages did not show any alterations in lipid distribution (data not shown). Nuclear stain: propidium iodide, red; original magnification ×60. dotted lines are the cell surface. G: There were no significant differences in loricrin, kallikrein 5 (KLK5), involucrin, transglutaminase 1 (TGase1), and filaggrin (FLG) mRNA expression between primary-cultured Abca12−/− keratinocytes (red bars, left) and primary-cultured wild-type keratinocytes (blue bars, left). Subcultured Abca12−/− keratinocytes (red bars, right) showed higher mRNA levels of loricrin, kallikrein 5, and transglutaminase 1 compared with primary-cultured Abca12−/− keratinocytes (red bars; left) and subcultured wild-type keratinocytes (blue bars, right). No significant difference was observed in involucrin and filaggrin mRNA expression. (primary-cultured Abca12−/− , n = 5; primary-cultured wild-type, n = 5, subcultured Abca12−/− , n = 5; subcultured wild-type, n = 5, mRNA expression levels of Abca12−/− primary cultured keratinocytes = 1). H: Western blotting of differentiated cultured keratinocytes lysates under high Ca2+ condition showed that loricrin and KLK5 expression was lower in Abca12−/− primary-cultured keratinocytes (lane 2) than that of the wild-type primary-cultured keratinocytes (lane 1). Extracts from subcultured Abca12−/− keratinocytes showed restoration of loricrin and KLK5 expression (lane 4, black arrowheads). Western blotting with anti-profilaggrin/filaggrin antibody revealed that the Abca12−/− primary-cultured keratinocytes (lane 2) had reduced expression of mature filaggrin monomer than the wild-type primary-cultured keratinocytes (lane 1). In addition, the lysate from Abca12−/− primary-cultured keratinocytes exhibited plenty of high molecular weight bands, which indicated profilaggrin expression (lane 2). The filaggrin expression pattern was normalized in subcultured Abca12−/− keratinocytes (lane 4). Cell lysates from subcultured Abca12−/− keratinocytes showed an intense filaggrin monomer band that was faint in lysates from Abca12−/− primary first-passage keratinocytes (lane 2 and 4, red arrow). KLK5, kallikrein 5; TGase, transglutaminase; DSG1, desmoglein 1; proFLG, profilaggrin; 4FLG, filaggrin tetramer; 3FLG, filaggrin trimer; 2FLG, filaggrin dimer; 1FLG, filaggrin monomer.
To investigate this altered differentiation state of primary/subcultured Abca12−/− keratinocytes, we performed real-time RT-PCR and immunoblot analysis (Figure 5, G and H). No significant differences were obtained in loricrin, KLK5, involucrin, TGase1 and filaggrin mRNA expression between primary-cultured Abca12−/− and wild-type keratinocytes (Figure 5G). Subcultured Abca12−/− keratinocytes showed higher loricrin, KLK5 and TGase1 mRNA levels compared with primary-cultured Abca12−/− keratinocytes (Figure 5G). Blot extracts from differentiated primary-cultured Abca12−/− keratinocytes showed low expression of loricrin and KLK5 (Figure 5H), as observed in neonatal Abca12−/− mice epidermis (Figure 2L). However, extracts from subcultured Abca12−/− keratinocytes showed restoration of loricrin and KLK5 expression (Figure 5H), as observed in grafted mice epidermis (Figure 4K).
Western blotting with anti-profilaggrin/filaggrin antibody revealed that the filaggrin expression pattern had become normalized in subcultured Abca12−/− keratinocytes (Figure 5H). Cell lysates from10 passage-subcultured Abca12−/− keratinocytes showed an intense filaggrin monomer band that was faint in lysates from Abca12−/− primary first-passage keratinocytes.
Studies of subcultured Abca12−/− keratinocytes demonstrated restored differentiation, at least in part, similar to that observed in mature Abca12−/− skin grafts.
cDNA Microarray Analysis Revealed Up-Regulation of 22 Lipid Metabolic and/or Transport-Related Genes
To investigate the mechanism of restoration of intact keratinocytes differentiation in subcultured Abca12−/− keratinocytes, we analyzed whole gene expression profile of primary/subcultured Abca12−/− keratinocytes maintained under high Ca2+ condition using cDNA microarray methods. We obtained the 35,582 gene expression profile and observed 566 transcripts that were up-regulated more than three-fold in subcultured Abca12−/− keratinocytes compared with primary-cultured Abca12−/− keratinocytes. Among them, we searched for genes related to “lipid metabolism and/or lipid transporter function,” and identified 22 specific transcripts including solute carrier family 22 member 7 (Slc22a7), prostaglandin D2 synthetase (Ptgds), annexin A9 (Anxa9), bactericidal/permeability-increasing protein-like 2 (Bpil2), phosphatidylethanolamine binding protein-2 variant 1 homolog (Pbp2), lipocalin 7 (Tinagl), Gpr119, solute carrier family 5 member 1 (Slc5a1), prostaglandin- endoperoxide synthase 2 (Ptgs2), and solute carrier family 30 member 1 (Slc30a1) (see Supplemental Table S1 at http://ajp.amjpathol.org). Notably, these genes included four ATP-binding transporter family members: Abca17, Abca1a, Abcc5, and Abcb11 that were up-regulated at least three-fold.
Keratinocytes Treated with Retinoids Failed to Exhibit Normal Differentiation and Therapeutic Trials onto Grafted HI Model Mice Failed to Demonstrate Skin Phenotype Recovery
As treatment trials for primary keratinocytes, Western blotting revealed that primary-cultured wild-type keratinocytes expressed a large amount of loricrin peptide and had a decent amount of filaggrin monomer converted from profilaggrin (see Supplemental Figure S3 at http://ajp.amjpathol.org). Both isotretinoin (10−6 mol/L) and etretinate (10−6 mol/L) in the cultured medium for 48 hours lead to remarkable reduction of loricrin protein expression in primary-cultured wild-type keratinocytes, although filaggrin expression patterns were not altered. Neither isotretinoin (10−6 mol/L) nor etretinate (10−6 mol/L) improved the deficient loricrin protein expression or defective profilaggrin conversion in the primary-cultured Abca12−/− keratinocytes under high Ca2+ condition.
As a treatment trial for HI grafted skin, oral administration of various doses of isotretinoin (1 or 10 mg/kg daily for 10 consecutive days) to HI skin graft-recipient SCID mice demonstrated no change in the skin phenotype of grafted HI model mice skin at all (data not shown).
Discussion
Keratinocyte terminal differentiation (keratinization) is remarkably important to skin physiology and essential for epidermal function including barrier formation. Keratinization is a highly regulated process, involving a number of genes and pathways. Until now, abnormalities in keratinocyte differentiation have been reported in HI patients. Morphologically, Buxman et al10 have reported that granular layers were absent or poorly formed in HI patient epidermis. Dale et al reported abnormal lamellar granules and defective profilaggrin conversion both in HI patients skin and their cultured keratinocytes.11 Profilaggrin conversion to filaggrin is a key step during epidermal keratinization. Fleckman et al12 reported that HI keratinocytes in 3 dimensional culture were unable to show the adequate differentiated epithelium and profilaggrin/filaggrin conversion. Recently, Thomas et al reported that the desquamation specific enzyme, kallikrein 5, and cathepsin D, were remarkably reduced in HI model skin using ABCA12 small interfering RNA knockdown keratinocytes.13
In the present study, we have demonstrated that keratinocyte differentiation was severely disrupted in both Abca12−/− HI neonatal model mouse skin and primary-cultured Abca12−/− keratinocytes. The present findings were consistent with previous reports of findings in HI patient’s skin. Our neonatal HI model mice skin showed no apparent keratohyalin granules within any of their granular layer cells, which was consistent with Buxman’s study.10 Furthermore, our immunoblotting results confirmed defective profilaggrin/filaggrin conversion, which was consistent with the studies by Dale et al11 and Fleckman et al.12 Our immunostaining and immunoblotting results showed reduced expression of KLK5 in neonatal Abca12−/−epidermis and primary-cultured Abca12−/− keratinocytes, which was consistent with the study by Thomas et al.13 We therefore believe that our Abca12−/− HI model mouse faithfully reproduces the molecular condition thus far identified in human HI epidermis in addition to mimicking clinical phenotype.5
Zuo et al6 suggested that loss of ABCA12 did not induce a block in the normal processing of profilaggrin to filaggrin in another reported ABCA12 deficient mouse strain, although the solubility of filaggrin was altered. We believe that our data are consistent with Zuo’s study. In the present study, we showed high molecular weight bands in RIPA buffer extracted supernatant from neonatal Abca12−/−epidermis. In study by Zuo et al,6 similar high molecular weight bands were also seen by the Western blotting after detergent buffer supernatant extraction. We also showed an alteration in filaggrin monomer solubility using 8 mol/L urea supernatant, reproducing the findings of Zuo et al.6 The major difference between these two studies is that Zuo et al6 showed no filaggrin monomer band after detergent buffer supernatant extraction in their ABCA12 deficient mouse skin. In contrast, we detected filaggrin monomer band in RIPA supernatant of our Abca12−/−neonatal epidermis. This finding might result from differences in lysis buffer, tissue sample (whole skin or only epidermis), or harvest time (E18.5 or neonate). We conclude that neonatal Abca12−/− epidermis exhibits not only defective profilaggrin/filaggrin conversion but also alterations in filaggrin monomer solubility, as previously suggested by Dale et al,11 Fleckman et al,12 and Zuo et al.6
Based on our results, we propose several new pathogenetic mechanisms for HI (Figure 6). Our results suggest that “an epidermal differentiation defect” is the major pathomechanism in HI. This new pathogenesis model is furthermore able to resolve the conflicting “the barrier insufficiency” theory with the (human) HI clinical phenotype. Our novel “epidermal differentiation” theory can explain HI skin phenotype at birth and even in utero. In our theory, severe hyperkeratosis, characteristic of the HI skin phenotype, was not a compensative hyperkeratosis for barrier insufficiency, but a hyperkeratosis due to defective differentiation and desquamation processes.
Figure 6.
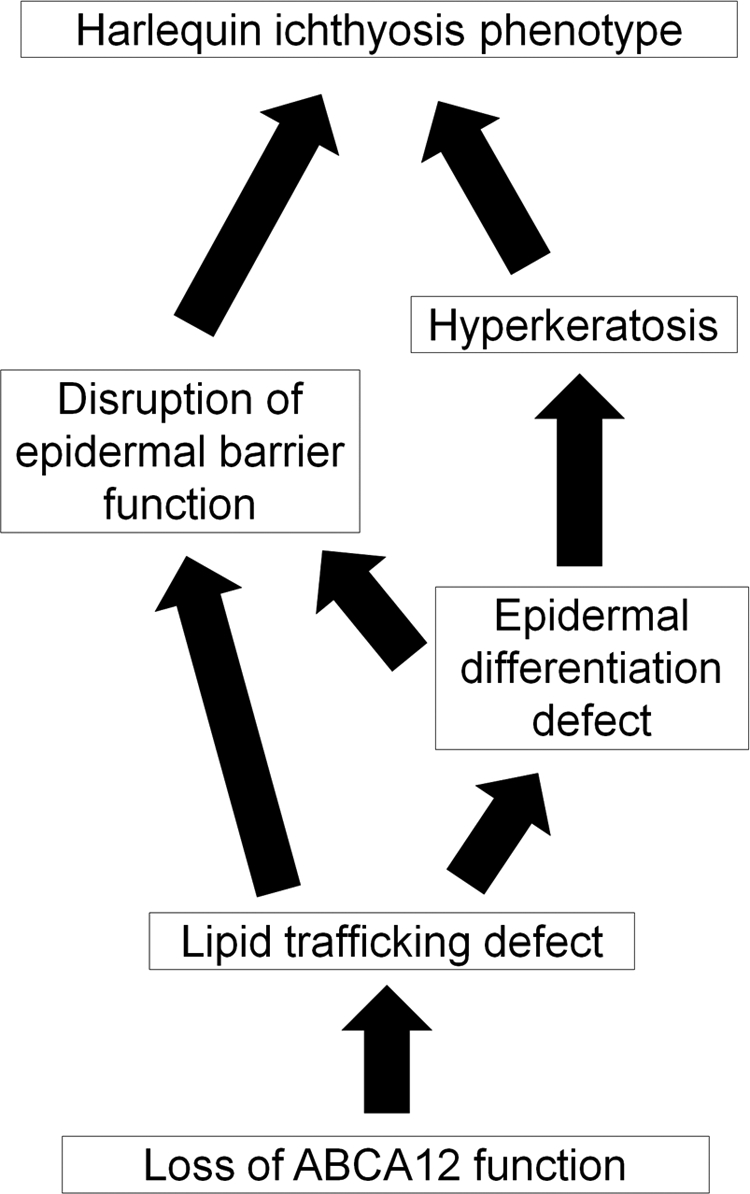
Putative pathomechanism of harlequin ichthyosis. Based on our results, we propose a novel pathogenetic mechanism underlying harlequin ichthyosis (HI). Our results suggested that a lipid trafficking defect due to loss of ABCA12 function leads to not only malformation of the epidermal lipid barrier but also “an epidermal differentiation defect.” In our hypothesis, the severe hyperkeratosis, a characteristic phenotype of HI skin, is not a hyperkeratosis compensating for any barrier insufficiency, but a hyperkeratosis due to both defective differentiation and an abnormal desquamation process. The present scenario explains both the HI skin phenotype at/after birth and even in utero.
Epidermal differentiation defects in our HI model mice suggest that ABCA12 lipid trafficking, which occurs during the early stages of keratinization, is a crucial step for correct keratinocyte terminal differentiation. In normal granular layer keratinocytes, ABCA12 transports lipids and forms lamellar granules.3 Just after the extrusion of lipids from lamellar granules at the granular/cornified layer interface, loricrin accumulates at the cell periphery and is integrated into the insoluble cornified cell envelope by transglutaminases.14 At the final phase of terminal differentiation, profilaggrin undergoes many post-translational modifications, including conversion to functional filaggrin monomers, which aid in the bundling of keratin intermediate filaments and formation of the cornified cell envelope.14,15,16 Serine proteases including KLK5 are involved in the desquamation process after keratinization.17 Our present results suggest that defective lipid trafficking and lamellar granule formation fail to initiate the normal sequence of events of the keratinization process. These facts indicate that ABCA12 lipid trafficking is essential to precisely regulate keratinocyte differentiation.
Interestingly, mature grafted HI model mouse skin and subcultured Abca12−/− keratinocytes showed improvement in the Abca12−/−-related abnormalities observed in neonatal skin and primary-cultured keratinocytes, such as the aberrant ceramide distribution, reduced differentiation-specific protein expression and profilaggrin/filaggrin conversion defects. Similar to our Abca12−/− model mouse skin, improvement in skin manifestations during maturation was observed in several other ichthyotic model mice. Loricrin knockout mice exhibit shiny translucent skin at birth although these mice showed a normal skin phenotype at 4 to 5 days after birth.18 Biochemically, the mice showed increased protein expression of small proline rich proteins, which are also cornified cell envelope components during skin maturation.18 Mature 12R-lipoxygenase knockout mouse skin showed recovery of skin barrier function and restoration of profilaggrin conversion after maturation, which had been lacking in the neonatal mice.19,20 In addition, mature transglutaminase 1 knockout mouse skin also showed skin barrier functional recovery, although exact compensation mechanisms other than hyperkeratosis were not identified.21
To find any clues for the mechanism of compensation in Abca12−/− mice, we conducted cDNA microarray analysis. We could find that several transporters including four ATP-binding cassette (ABC) transporter family genes were up-regulated in subculture compared with primary-cultured Abca12−/− keratinocytes. We consider these up-regulated ABC transporters as prime candidate genes to compensate for the loss of ABCA12 function. These lipid transport/metabolism-related molecules might help in lipid trafficking and/or recovery of epidermal differentiation in Abca12−/− keratinocytes.
The recovery of skin barrier function and self-improvement of epidermal differentiation defects in mature Abca12−/− skin gave us important clues to aid in treatment of HI patients. Infants affected with HI frequently die within the neonatal period, although the survival rate of HI newborns has increased recently with the arrival of neonatal intensive care regimes with retinoid administration.22,23,24,25,26 Our observations in mature Abca12−/− skin further confirmed that the early neonatal period is a critical time defining the prognosis. In this context, neonatal intensive care for HI newborns in the initial neonatal period is important for their survival.
We had already tried systemic retinoid administration to pregnant mice as a fetal therapy, although neither improvement of the skin manifestations nor extension of the survival period was obtained in the Abca12−/− newborns from the treated-mother mice.5 Based on the clinical efficacy of retinoids to HI patients,22,23,24,25,26 we conducted retinoid administration to the primary-cultured Abca12−/− keratinocytes. We could not detect any recovery of differentiation defects in the primary-cultured Abca12−/− keratinocytes treated with retinoids. These results of our therapeutic trials to fetuses in the previous report5 and to primary keratinocytes in this study indicated that retinoids may be ineffective for modifying the epidermal differentiation defects during the fetal period. These facts were consistent with the known effects of retinoids, which enhance proliferation and suppress differentiation of keratinocytes.27,28 Further, we performed a treatment trial on grafted SCID mice using systemic isotretinoin administration. Oral administration of any dose of isotretinoin (either 1 or 10 mg/kg daily, for 10 consecutive days) to the HI-skin-grafted SCID mice failed to improve the skin phenotype of grafted HI skin at all. We failed to obtain any clue to understand the reason why retinoids are only effective for HI patients from our present study.
In conclusion, we have demonstrated that disrupted epidermal keratinocyte differentiation is the pathomechanism involved in HI, and that during maturation, Abca12−/− epidermal keratinocytes regain normal keratinocyte differentiation. This restoration of differentiation is likely to be associated with the skin phenotype improvement observed in HI survivors.
Acknowledgments
We thank Dr. James R. McMillan for proofreading this manuscript.
Footnotes
Address reprint requests to Masashi Akiyama, M.D., Ph.D., or Hiroshi Shimizu, M.D., Ph.D., Department of Dermatology, Hokkaido University Graduate School of Medicine, N15 W7, Kita-ku, Sapporo 060-8638, Japan. E-mail: akiyama@med.hokudai.ac.jp or shimizu@med.hokudai.ac.jp.
Supported in part by a grant-in-aid from the Ministry of Education, Science, Sports, and Culture of Japan (Kiban B 20390304: to M.A.), a grant from Ministry of Health, Labor and Welfare of Japan (Health and Labor Sciences Research grants; Research on Intractable Disease: H21–047 and H22-177: to M.A.), a grant from ONO Medical Research Foundation (T.Y.) and a grant from Kanae Foundation for the promotion of Medical Science (T.Y.).
None of the authors declare any relevant financial relationships.
Supplemental material for this article can be found on http://ajp.amjpathol.org.
References
- Akiyama M. Pathomechanisms of harlequin ichthyosis and ABCA transporters in human diseases. Arch Dermatol. 2006;142:914–918. doi: 10.1001/archderm.142.7.914. [DOI] [PubMed] [Google Scholar]
- Hovnanian A. Harlequin ichthyosis unmasked: a defect of lipid transport. J Clin Invest. 2005;115:1708–1710. doi: 10.1172/JCI25736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama M, Sugiyama-Nakagiri Y, Sakai K, McMillan JR, Goto M, Arita K, Tsuji-Abe Y, Tabata N, Matsuoka K, Sasaki R, Sawamura D, Shimizu H. Mutations in lipid transporter ABCA12 in harlequin ichthyosis and functional recovery by corrective gene transfer. J Clin Invest. 2005;115:1777–1784. doi: 10.1172/JCI24834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelsell DP, Norgett EE, Unsworth H, Teh MT, Cullup T, Mein CA, Dopping-Hepenstal PJ, Dale BA, Tadini G, Fleckman P, Stephens KG, Sybert VP, Mallory SB, North BV, Witt DR, Sprecher E, Taylor AE, Ilchyshyn A, Kennedy CT, Goodyear H, Moss C, Paige D, Harper JI, Young BD, Leigh IM, Eady RA, O'Toole EA. Mutations in ABCA12 underlie the severe congenital skin disease harlequin ichthyosis. Am J Hum Genet. 2005;76:794–803. doi: 10.1086/429844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagi T, Akiyama M, Nishihara H, Sakai K, Nishie W, Tanaka S, Shimizu H. Harlequin ichthyosis model mouse reveals alveolar collapse and severe fetal skin barrier defects. Hum Mol Genet. 2008;17:3075–3083. doi: 10.1093/hmg/ddn204. [DOI] [PubMed] [Google Scholar]
- Zuo Y, Zhuang DZ, Han R, Isaac G, Tobin JJ, McKee M, Welti R, Brissette JL, Fitzgerald ML, Freeman MW. ABCA12 maintains the epidermal lipid permeability barrier by facilitating formation of ceramide linoleic esters. J Biol Chem. 2008;283:36624–36635. doi: 10.1074/jbc.M807377200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama M, Smith LT, Shimizu H. Expression of transglutaminase activity in developing human epidermis. Br J Dermatol. 2000;142:223–225. doi: 10.1046/j.1365-2133.2000.03288.x. [DOI] [PubMed] [Google Scholar]
- Raghunath M, Hennies HC, Velten F, Wiebe V, Steinert PM, Reis A, Traupe H. A novel in situ method for the detection of deficient transglutaminase activity in the skin. Arch Dermatol Res. 1998;290:621–627. doi: 10.1007/s004030050362. [DOI] [PubMed] [Google Scholar]
- Masukawa Y, Narita H, Shimizu E, Kondo N, Sugai Y, Oba T, Homma R, Ishikawa J, Takagi Y, Kitahara T, Takema Y, Kita K. Characterization of overall ceramide species in human stratum corneum. J Lipid Res. 2008;49:1466–1476. doi: 10.1194/jlr.M800014-JLR200. [DOI] [PubMed] [Google Scholar]
- Buxman MM, Goodkin PE, Fahrenbach WH, Dimond RL. Harlequin ichthyosis with epidermal lipid abnormality. Arch Dermatol. 1979;115:189–193. [PubMed] [Google Scholar]
- Dale BA, Holbrook KA, Fleckman P, Kimball JR, Brumbaugh S, Sybert VP. Heterogeneity in harlequin ichthyosis, an inborn error of epidermal keratinization: variable morphology and structural protein expression and a defect in lamellar granules. J Invest Dermatol. 1990;94:6–18. doi: 10.1111/1523-1747.ep12873301. [DOI] [PubMed] [Google Scholar]
- Fleckman P, Hager B, Dale BA. Harlequin ichthyosis keratinocytes in lifted culture differentiate poorly by morphologic and biochemical criteria. J Invest Dermatol. 1997;109:36–38. doi: 10.1111/1523-1747.ep12276450. [DOI] [PubMed] [Google Scholar]
- Thomas AC, Tattersall D, Norgett EE, O'Toole EA, Kelsell DP. Premature terminal differentiation and a reduction in specific proteases associated with loss of ABCA12 in Harlequin ichthyosis. Am J Pathol. 2009;174:970–978. doi: 10.2353/ajpath.2009.080860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bickenbach JR, Greer JM, Bundman DS, Rothnagel JA, Roop DR. Loricrin expression is coordinated with other epidermal proteins and the appearance of lipid lamellar granules in development. J Invest Dermatol. 1995;104:405–410. doi: 10.1111/1523-1747.ep12665896. [DOI] [PubMed] [Google Scholar]
- Candi E, Schmidt R, Melino G. The cornified envelope: a model of cell death in the skin. Nat Rev Mol Cell Biol. 2005;6:328–340. doi: 10.1038/nrm1619. [DOI] [PubMed] [Google Scholar]
- Denecker G, Ovaere P, Vandenabeele P, Declercq W. Caspase-14 reveals its secrets. J Cell Biol. 2008;180:451–458. doi: 10.1083/jcb.200709098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Hoffert U, Wu Z, Schroder JM. Identification of lympho-epithelial Kazal-type inhibitor 2 in human skin as a kallikrein-related peptidase 5-specific protease inhibitor. PLoS One. 2009;4:e4372. doi: 10.1371/journal.pone.0004372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch PJ, de Viragh PA, Scharer E, Bundman D, Longley MA, Bickenbach J, Kawachi Y, Suga Y, Zhou Z, Huber M, Hohl D, Kartasova T, Jarnik M, Steven AC, Roop DR. Lessons from loricrin-deficient mice: compensatory mechanisms maintaining skin barrier function in the absence of a major cornified envelope protein. J Cell Biol. 2000;151:389–400. doi: 10.1083/jcb.151.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epp N, Furstenberger G, Muller K, de Juanes S, Leitges M, Hausser I, Thieme F, Liebisch G, Schmitz G, Krieg P. 12R-lipoxygenase deficiency disrupts epidermal barrier function. J Cell Biol. 2007;177:173–182. doi: 10.1083/jcb.200612116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Juanes S, Epp N, Latzko S, Neumann M, Furstenberger G, Hausser I, Stark HJ, Krieg P. Development of an Ichthyosiform Phenotype in Alox12b-Deficient Mouse Skin Transplants. J Invest Dermatol. 2009;129:1429–1436. doi: 10.1038/jid.2008.410. [DOI] [PubMed] [Google Scholar]
- Kuramoto N, Takizawa T, Takizawa T, Matsuki M, Morioka H, Robinson JM, Yamanishi K. Development of ichthyosiform skin compensates for defective permeability barrier function in mice lacking transglutaminase 1. J Clin Invest. 2002;109:243–250. doi: 10.1172/JCI13563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad RS, Pejaver RK, Hassan A, Dusari S, Wooldridge MA. Management and follow-up of harlequin siblings. Br J Dermatol. 1994;130:650–653. doi: 10.1111/j.1365-2133.1994.tb13114.x. [DOI] [PubMed] [Google Scholar]
- Lawlor F, Peiris S. Harlequin fetus successfully treated with etretinate. Br J Dermatol. 1985;112:585–590. doi: 10.1111/j.1365-2133.1985.tb15268.x. [DOI] [PubMed] [Google Scholar]
- Ward PS, Jones RD. Successful treatment of a harlequin fetus. Arch Dis Child. 1989;64:1309–1311. doi: 10.1136/adc.64.9.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haftek M, Cambazard F, Dhouailly D, Reano A, Simon M, Lachaux A, Serre G, Claudy A, Schmitt D. A longitudinal study of a harlequin infant presenting clinically as non-bullous congenital ichthyosiform erythroderma. Br J Dermatol. 1996;135:448–453. [PubMed] [Google Scholar]
- Singh S, Bhura M, Maheshwari A, Kumar A, Singh CP, Pandey SS. Successful treatment of harlequin ichthyosis with acitretin. Int J Dermatol. 2001;40:472–473. doi: 10.1046/j.1365-4362.2001.01173.x. [DOI] [PubMed] [Google Scholar]
- Eichner R, Kahn M, Capetola RJ, Gendimenico GJ, Mezick JA. Effects of topical retinoids on cytoskeletal proteins: implications for retinoid effects on epidermal differentiation. J Invest Dermatol. 1992;98:154–161. doi: 10.1111/1523-1747.ep12555767. [DOI] [PubMed] [Google Scholar]
- Eichner R. Epidermal effects of retinoids: in vitro studies. J Am Acad Dermatol. 1986;15:789–797. doi: 10.1016/s0190-9622(86)70235-1. [DOI] [PubMed] [Google Scholar]