

Abstract
Cystic fibrosis (CF), a disease caused by mutations in the CF transmembrane conductance regulator (CFTR) gene, is characterized by chronic bacterial infections and inflammation in the lung. Having previously shown that deletion of CFTR is associated with lower expression of the endogenous anti-inflammatory protein Annexin A1 (AnxA1), we investigated further this possible functional connection using a validated CFTR inhibitor. Treatment of mice with the CFTR inhibitor-172 (CFTR172) augmented the acute peritonitis promoted by zymosan, an effect associated with lower AnxA1 levels in peritoneal cells. Similar results were obtained with another, chemically distinct, CFTR inhibitor. The pro-inflammatory effect of CFTR172 was lost in AnxA1−/−, as well as CFTR−/− mice. Importantly, administration of hrAnxA1 and its peptido-mimetic to CFTR−/− animals or to animals treated with CFTR172 corrected the exaggerated leukocyte migration seen in these animals. In vitro assays with human Polymorphonuclear leukocyte (PMN) demonstrated that CFTR172 reduced cell-associated AnxA1 by promoting release of the protein in microparticles. We propose that the reduced impact of the counterregulatory properties of AnxA1 in CF cells contributes to the inflammatory phenotype characteristic of this disease. Thus, these findings provide an important insight into the mechanism underlying the inflammatory disease associated with CFTR inhibition while, at the same time, providing a novel pharmacological target for controlling the inflammatory phenotype of CF.
Over the past decades our understanding of the underlying causative elements of cystic fibrosis (CF) has significantly improved. We saw a radical shift from innumerable speculations about its origin to a precise definition of causative mutations, specifically those arising in a transmembrane ion channel termed CF transmembrane regulator (CFTR). Clinically childhood mortality has fallen dramatically and the expectancy of CF patients has increased to an average age of more than 30 years.1 However, there is still no cure and no effective control of the exacerbated chronic inflammatory process that leads to the relentless destruction of the lungs and pancreas exist.
Lung disease is associated with the production of a more viscous mucus, resulting from the defect in ion transport across the epithelial cell membrane, and it represents the most life-threatening feature of CF. Several clinical studies indicate that pulmonary neutrophilic inflammation occurs very early in the course of the CF lung disease and often precedes overt signs of colonization or infection. Analysis of bronchoalveolar lavage fluid2 from CF patients of different ages and with a range of disease severity consistently shows a marked rise in neutrophil numbers and elevated amounts of tumor necrosis factor-α, interleukin (IL)−1β and IL-8.3,4,5 The bronchial epithelium is capable of generating all of these inflammatory modulators in large quantities, except for tumor necrosis factor-α, where activated alveolar macrophages and other leukocytes might contribute significantly to the production of this cytokine in CF.6
During the past decade new lines of research have focused on mechanisms of endogenous anti-inflammation and resolution of inflammation.7,8 On an inflammatory challenge, a tightly concerted reaction occurs, leading to the induction of a strong inflammatory response aiming at the deactivation and removal of the initiating insult. In order for this process to prove beneficial to the host, homeostasis needs to be reestablished. In this context, several anti-inflammatory mediators and pathways operate concurrently to assure a strict and timely return to the basal homeostatic state of the inflammatory response. Among these we find the potent anti-inflammatory protein Annexin A1 (AnxA1).9 In resting cells, AnxA1 is predominantly located intracellularly, with a small proportion found on the plasma membrane.10 On cell activation the protein translocates on the cell surface where it encounters its receptor, a specific 7-trans membrane G-protein coupled receptor, termed formyl peptide receptor 2 or lipoxin A4 receptor (FPR2/ALX).11 Acting in a paracrine/autocrine fashion, AnxA1 the triggers signaling pathways that modulate and down-regulate Polymorphonuclear leukocyte (PMN) activation.10,12,13 The actions of AnxA1 are terminated, in a time-dependent manner, by the proteolytic cleavage of the N-terminus, most likely by serine proteases (though, cell specific catabolism is also likely to occur).14
Dysregulation of the AnxA1 system has been associated with exacerbated and prolonged cell activation along with abnormal responses, caused by the lack of “fine tuning” and homeostatic control. Besides modulation of cell activation, other actions recently ascribed to AnxA1 that indicate its multipotent anti-inflammatory properties—and thus might be relevant in the context of chronic inflammation—include: its ability to induced PMN apoptosis15 and their removal by the macrophage.16 In addition, AnxA1 up-regulates the anti-inflammatory cytokine IL-10, which in turn leads to the inhibition of inducible Nitric Oxide Synthase mRNA expression and nitric oxide release.17
Experimental models of CF have relied on the generation of CFTR−/− mouse colonies. One of these colonies revealed a selective absence of the protein AnxA1, a finding associated empirically with exacerbated inflammation (of the gut) and high degree of lethality.18 The same study showed reduced AnxA1 protein levels in human nasal epithelial cells obtained from CF patients. A more recent study reported formation of a complex between AnxA1 and CFTR, encompassing also S100A10 and cytosolic phospholipase A2, This regulatory complex keeps epithelial cells in check; thus, disruption of this complex in vitro leads to elevated tumor necrosis factor-α mediated eicosanoid release.19
The present study sets out with a dual aim: on one hand to establish a model of experimental inflammation linked to CFTR malfunction; on the other hand to extend our knowledge of the potential association between AnxA1 and CFTR to mouse and human neutrophils. This task was fulfilled using the highly selective CFTR inhibitor-172 (CFTR172). Originally identified following a high throughput screening of 50,000 small molecules, CFTR172 is one of six members of the 2-thioxo-4-thiazolidinone class identified as inhibitors. If produces a reversible inhibition of CFTR short-circuit current with a Ki of ∼300 nmol/L.20 Testing this inhibitor in CFTR−/− mice and assessment of the effects of a structurally unrelated CFTR inhibitor, compound GlyH101, reinforced the functional association between CFTR and AnxA1.
Materials and Methods
Model of Zymosan Peritonitis and Treatment with CFTR172
AnxA1−/−,21 backcrossed for more than 10 generations on the C57/Black6 genetic background, and C57/Black6 mice were obtained from B&K Universal (Hull, UK). CFTR−/− mice bred on a C57/Black6 background or their wild-type littermate controls (bred in house) were also used. In all cases mice were 4 to 6 weeks old. Animal work was performed according to UK Home Office regulations (Guidance on the Operation of Animals, Scientific Procedures Act, 1986).
CFTR−/− mice22—and wild-type littermates—were injected with 1 mg of zymosan A (Sigma-Aldrich; Poole, UK) in 0.5 ml PBS as reported,23 following the i.v. injection of 1 μg AnxA1 or vehicle in some of the groups. At the 4-hour time point, animals were euthanized by CO2 exposure, and peritoneal cavities were washed with 3 ml of PBS (Oxoid; Basingstoke, UK) containing 3 mmol/L EDTA (Sigma-Aldrich; Poole, UK) and 25 U/ml heparin. Aliquots of lavage fluid were then stained with Turk’s solution and differential cell counts were performed using a Neubauer hemocytometer and a light microscope (Olympus B061): leukocytes were identified as >95% neutrophils by light microscopy determination. Furthermore, cells from the lavage fluid were stained using an anti-GR1 anti-body (clone: RB6 8C5. eBioscience; Hatfield, UK) to determine total number of GR1 positive (GR1+ve) cells present in the peritoneal cavity. Lavage fluids were then centrifuged at 400 × g for 10 minutes and supernatants stored at −80°C for further analysis.
In the acute CFTR inhibition experiments, several combinations of the CFTR172 inhibitor (Sigma-Aldrich; Poole, UK) and zymosan were tested. Initially, doses of 10 to 250 μg/kg CFTR172 were given 24 hours and 1 hour before the injection of either one of the following three zymosan doses: 0.1, 0.2, and 1 mg per mouse. In parallel groups, CFTR172 was injected only once, 1 hour before the injection of the different doses of zymosan. Dose range for CFTR172 was selected on a previous publication.20 In separate experiments, AnxA1−/−,21 CFTR−/− and C57/Black6 mice were treated with either vehicle or 50 μg/kg CFTR172 24 and 1 hour before treatment with 0.1 mg of zymosan or vehicle. Furthermore, GlyH101 (Calbiochem; Nottingham, UK), another CFTR inhibitor that blocks the channel from its luminal side24 was also tested, at 60 μg/kg dose (equimolar to 50 μg/kg CFTR172) and a treatment regimen −24 and −1 hour, with 0.1 mg zymosan being injected at time 0. In all cases, cells were collected and the cell counts were determined, with the cell supernatants being extracted and stored as outlined above.
To determine the therapeutic benefit of AnxA1 or it peptidomimetic Ac2-26 in controlling the inflammatory processes characterized by CFTR inhibition, mice were treated with 50 μg/kg CFTR172 24 hours and 1 hour before treatment with 0.1 mg of zymosan or vehicle given intraperitoneally; 10 minutes before intraperitoneal treatment mice received a bolus injection of either 1 μg human recombinant (hr)-AnxA1, 50 μg of Ac2-26 or vehicle, all given intravenously (10 ml/kg).
Semiquantitative and Quantitative Cytokine Level Determinations
The relative abundance of a panel of cytokines was determined using a Mouse Cytokine Array Panel (Panel A: R&D Systems; Abingdon, UK) as outlined in the manufacturers instructions. In brief, membranes were blocked for an hour at room temperature; 200 μl of lavage supernatant were pooled from each group and incubated with the membranes overnight at 4°C following which the membranes were washed and incubated with streptavidin-horseradish peroxidase for 30 minutes at room temperature. Subsequently, each membrane was incubated with an enhanced chemiluminescence detection reagent. The relative abundance of each cytokine kit was ascertained following manufactures instructions, using the internal controls included on each membrane.
Changes in IL-6 levels in the lavage fluids were determined using and IL-6 DuoSet (R&D Systems; Abingdon, UK) following manufacturers instructions. In brief 20 μl of lavage supernatant were used from each sample, and incubated in a 96-well high binding plates (Corning, NY), previously coated with an anti-mouse IL-6 capture antibody. Subsequently plates were washed, blocked for 1 hour before addition of 100-μl lavage fluid samples. Following a 2-hour incubation at room temperature, a washing step was included before a 2-hour incubation with the detecting antibody. Streptavidin-horseradish peroxidase was added for 20 minutes and substrate solution for a further 20 minutes; then, reaction was stopped using the Stop Solution provided and the plate read on a microplate reader (NOVOstar; BMG Labtech; Aylesbury, UK).
Annexin A1 Levels in Human and Mouse Leukocytes
PMN were separated from whole blood as previously described25 according to a protocol approved by the local Research Ethics Committee (P/00/029 ELCHA) and seeded into 12-well plates at a concentration of 2 × 106 cells per ml. Then vehicle or 20 μmol/L CFTR172 inhibitor19 were added for 30 minutes, before stimulation with 10 ng/ml of LPS (E. coli strain 0111:B4; Sigma-Aldrich; Poole, UK) for 4 hours at 37°C in an atmosphere with 5% CO2. Following centrifugation, half of the supernatant was stored at −80°C while the second portion was spun at 100,000 × g for 1 hour to pellet the microparticles26 following which the microparticle-free supernatant was transferred to fresh tubes and stored at −80°C.
For flow cytometric analyses, cell pellets were re-suspended in PBC (a solution containing PBS, 0.15% bovine serum albumin and 1 mmol/L CaCl2; both from Sigma-Aldrich; Poole, UK), whereby the total and cell membrane expression of AnxA1 and FPR2/ALX was determined. Cell membrane immunoreactivity was determined with 5 μg/ml mouse anti-human AnxA1 (clone 1B27), anti-human FPR2/ALX (clone: 6C7-3; kind gift of Dr. Duncan Anderson; Astra Zeneca, UK) or isotype control (IgG1), for 45 minutes on ice. Incubation with a secondary anti-mouse fluorescein isothiocyanate-conjugated antibody (clone:STAR9B. Serotec; Oxford, UK). Total AnxA1 and FPR2/ALX contents were determined in a similar way, though a permeabilization step in 2% paraformaldehyde and 0.03% saponin (30 minutes on ice) was introduced, before incubation with primary and secondary antibodies as above. Flow cytometry was performed with a FACSCalibur (Becton & Dickinson; Oxford, UK), using a photomultiplier tube value of 548 for the FL1 (green) and acquiring ≥10,000 events per sample.
In some cases, AnxA1 expression in mouse peritoneal neutrophils (elicited at 4 hours postzymosan and/or CFTR inhibitors) was also determined using the same permeabilization protocol, and a rabbit anti-AnxA1 antibody (Ab; 1:100: Invitrogen; Calne UK) or control rabbit IgG (Dako; Cambridgeshire, UK). Flow cytometry was performed as above, identifying the predominant (>85%) neutrophil population in the lavage fluids by its scatter characteristics.
Western Blotting Analysis
AnxA1 levels in the human PMN supernatants, in the presence or absence of microparticles, was determined by Western blotting following a previously published protocol.26 In brief, complete or microparticle-free supernatants (50 μl) were boiled with 10 μl of 6× Laemmli buffer for 5 minutes before loading onto a 10% SDS gel. After transfer to polyvinylidene difluoride membrane (Millipore; Watford, UK), and 2 hours blocking using 5% nonfat milk, blots were probed with a mouse anti-human AnxA1 antibody (clone 1B; 0.5 μg/ml) overnight at 4°C before washing and detection with anti-mouse horseradish peroxidase-conjugated antibody (Dako; Cambridgeshire, UK) and enhanced chemiluminescence detection reagent (GE Health care; St. Giles, UK). Subsequently a densitometric analysis was conducted using ImageJ software.28
In some cases, mouse AnxA1 protein in the lavage fluids of the zymosan peritonitis experiments was also determined, whereby 50-μl of cell-free fluids were boiled with 10 μl of 6× Laemlli buffer after which AnxA1 levels were as described above, but using a rabbit anti-AnxA1 Ab (1:5000; InVitrogen; Calne UK) and anti-rabbit horseradish peroxidase-conjugated antibody (Dako; Cambridgeshire, UK) as secondary.
Immunohistochemistry
Cells obtained from the peritoneal lavage were cytospun onto poly-l-lysine coated slides (VWR International; Leicestershire, UK) before incubation with 10% albumin bovine in PBS (PBSA) to block nonspecific binding. A polyclonal rabbit anti-AnxA1 Ab (InVitrogen; Calne UK) was added (final dilution 1/200 in 1% PBSA) and slides incubated overnight at 4°C. As control for the reaction, some sections were incubated with nonimmune rabbit serum (1/200 working dilution. Sigma-Aldrich; Poole, UK) instead of the primary Ab. After repeated washings in 1% PBSA, a goat anti-rabbit IgG (Fc fragment-specific) Ab conjugated to 5 nm colloidal gold (1/100; British BioCell International, Cardiff, UK) was added. Silver enhancing solution (British BioCell International; Cardiff, UK) was used to augment gold particle staining. At the end of the reaction, slides were washed thoroughly in distilled water, counterstained with hematoxylin, and mounted in BIOMOUNT (British BioCell International; Cardiff, UK). Analysis was conducted with a microscope Nikon equipped with a DXM1200 digital camera, using the software LUCIA (Laboratory Universal Computer Image Analysis; Jencons-PLS, London UK).
Data Handing and Statistical Analysis
In vitro experiments with human cells were performed in triplicate using cells from four distinct donors. PMN migration in vivo was conducted using experimental groups of ≥5 mice per group. In all cases data are reported as mean ± SEM. Statistical differences between groups were determined by one-way analysis of variance and, if null hypothesis was rejected, by Student Newman-Keuls test, taking a probability value P < 0.05 as significant.
Results
The CFTR Inhibitor, CFTR172, Exacerbates an Acute Inflammatory Response
The initial aim of these experiments was to develop a pharmacological model suitable to dissect the potential role of AnxA1 in the propagation of the CF condition. Several treatment combinations of CFTR172 (10, 50, and 250 μg/kg) and zymosan (0.1, 0.2, and 1 mg) were tested in conjunction with different treatment regimens using CFTR172 (−24 hours + −1 hour and −1 hour). As expected, zymosan produced a marked influx of Gr1+ve cells into the peritoneal cavity when given at the top dose of 1 mg per mouse (Figure 1A), a response that was unaffected by CFTR172 co-treatment. Thus, we fine tuned the extent of inflammation, administering 0.2 and 0.1 mg zymosan: in either case a significant cell influx was provoked and CFTR172 augmented the number of Gr1+ve cells recovered at the 4-hour time point (Figure 1, B and C). Out of the combinations tested, 50 μg/kg CFTR172 given as pretreatment (−24 hours and −1 hour) and 0.1 mg zymosan gave robust results (twofold increase over zymosan alone) and was chosen for the subsequent experiments. Of note, mice treated with CFTR172 alone also displayed a modest accumulation of Gr1+ve cells above the virtually null values measured in naïve cavities (Figure 1D).
Figure 1.

CFTR172 synergizes with zymosan to produce neutrophil recruitment into the peritoneum. C57/B6 mice were pre-treated i.p. with various doses of CFTR172 given either singularly at −1 hour, or twice at both −24 and −1 hour, as indicated. At time 0, mice were treated with (A) 1 mg, (B) 0.2 mg, or (C) 0.1 mg of zymosan given i.p. in 0.5 ml PBS, with lavage fluids being collected four hours later. Recruited neutrophils were stained with the anti-Gr1 Ab for flow cytometry analysis. D: Various doses of CFTR172 alone were injected i.p. 24 hours and one hour before peritoneal lavage harvesting, and cells processed as above. Data are mean ± SEM of five mice per group. A–C: *P < 0.05, **P < 0.01 vs. zymosan/vehicle group; D: *P < 0.05 vs. naïve group.
This potentiating effect was not restricted to CFTR172. We tested another CFTR inhibitor, structurally unrelated to CFTR172. Administered at the dose of 60 μg/kg, equimolar to 50 μg/kg CFTR172, compound GlyH101 augmented the peritoneal influx of Gr1+ve cells: 20 ± 3.0 × 105/mouse for zymosan alone and 40 ± 3.2 × 105/mouse for zymosan plus GlyH101 (P < 0.01; five mice per group).
AnxA1 Dysregulation in the Inflammatory Response Consequent to CFTR Blockade
Immunohistochemical analysis on cells collected from the peritoneal lavage revealed a significant reduction in cell-associated AnxA1 protein when CFTR172 was administered in conjunction to the low dose zymosan. Figure 2A shows representative images with recruited PMN being highlighted. Using this semiquantitative method cumulative data both for peritoneal neutrophils (Figure 2B, left panel) and macrophages (Figure 2B, right panel) demonstrate that a significant reduction in cell associated AnxA1 immunoreactivity could be detected, in the zymosan + CFTR172 group. Interestingly, in absence of zymosan the CFTR inhibitor appeared to reduced AnxA1 expression in the few PMN present in the peritoneal cavity but not in macrophages (Figure 2B).
Figure 2.

CFTR172 de-regulates the AnxA1 pathway in the mouse peritoneal cells. Mice received CFTR172 given twice at 50 μg/kg −24 and −1 hour, or GlyH101 (60 μg/kg, twice) and then left untreated or injected i.p. with 0.1 mg zymosan at time 0. Cells were collected four hours later. A: Peritoneal cells were stained for AnxA1 content: representative images are shown (arrows indicate neutrophils; arrowheads indicate macrophages). B: Cumulative data of the densitometric analyses as performed for neutrophils and macrophages. C: Cell-free lavage fluids were stained for secreted AnxA1 by Western blotting. D: Cell-associated AnxA1 as determined by flow cytometry in peritoneal neutrophils of mice treated with zymosan alone or together with GlyH101. Panel on the right shows an illustrative histogram with AnxA1 immunostaining as measured in the FL1 channel. In all cases, data are mean ± SEM of five mice per group. B: *P < 0.05 vs. zymosan/vehicle group. C: *P < 0.05 vs. naïve; **P < 0.05 vs. zymosan/vehicle group. D: *P < 0.05 vs. zymosan/vehicle group.
AnxA1 protein rapidly appeared in cell-free peritoneal lavages following treatment with CFTR172 yielding increased levels (Figure 2B), suggesting a possible process of ‘AnxA1 cell leakage’ into the environment when CFTR is inactive (an observation that was also made with the CFTR−/− animals; data not shown). In contrast, the augmented AnxA1 content in the lavage fluids was significantly reduced on animal treatment with CFTR172 (Figure 2C). This suggests that the strong inflammatory reaction produced by the combination zymosan + CFTR172 subverted the normal mechanisms operative in acute inflammation.
Cell-associated AnxA1 levels were reduced also by the other CFTR inhibitor, as quantified by flow cytometry in peritoneal PMN harvested from mice treated with 0.1 mg zymosan alone or together with GlyH101 (60 μg/kg, −24 and −1 hour). Figure 2D reports these data showing an illustrative histogram and the cumulative values out of five mice per group.
To underpin a functional role for AnxA1 in the inflammatory response associated with CFTR inhibition, the active combination was tested in AnxA1−/− mice. Figure 3 reports these data, showing that administration of CFTR172 and zymosan again increased total peritoneal (Figure 3A) and Gr1+ve (Figure 3B) cell recruitment in wild-type mice, but were without effect in AnxA1−/− mice. On treatment with zymosan, AnxA1−/− mice exhibited degree of PMN influx which acted as internal control confirming previous publications.23 Interestingly, the mild inflammatory status induced by administration of CFTR172 in the absence of zymosan was again evident in wild-type mice and even more so in AnxA1−/− mice (Figure 3).
Figure 3.
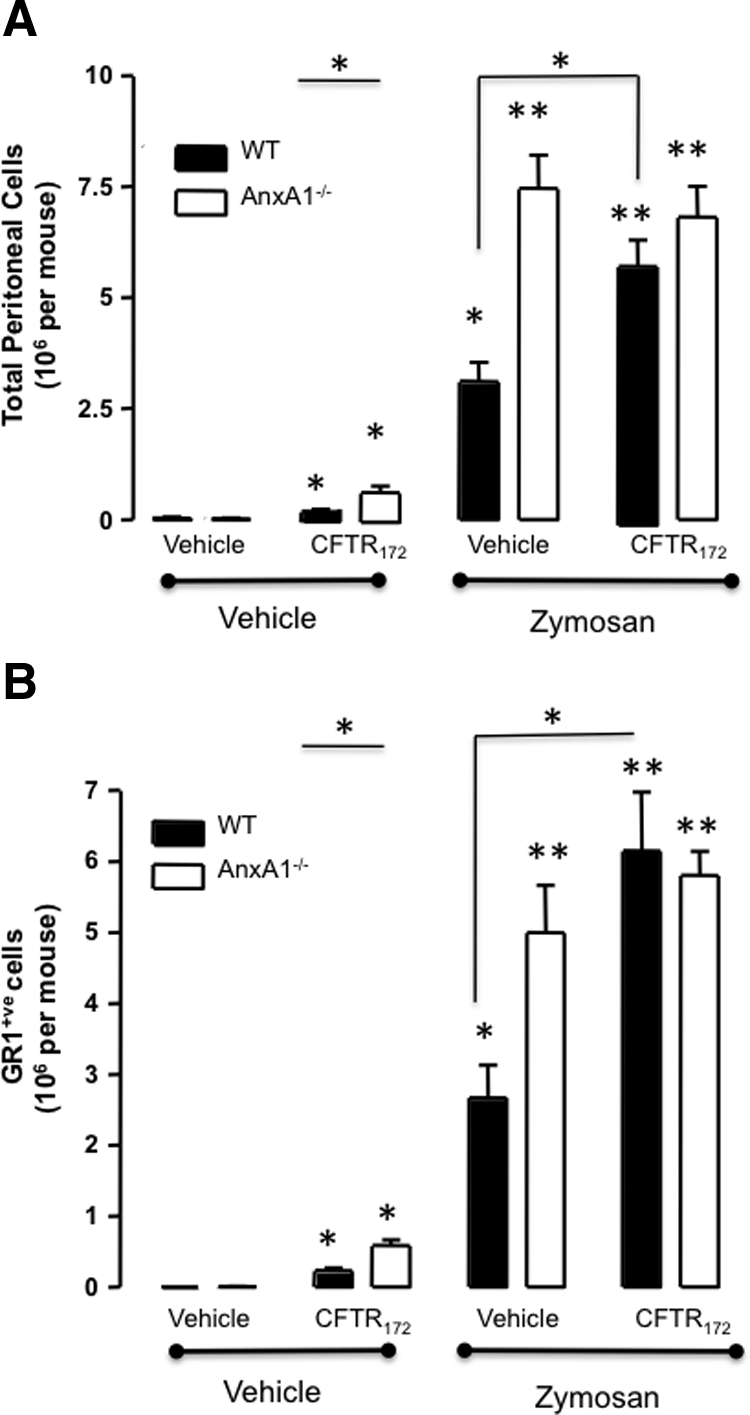
Loss of endogenous AnxA1 is involved in the inflammatory response elicited by CFTR172. AnxA1−/− and wild-type animals were treated with 50 μg/kg CFTR172 24 hours and one hour before the i.p. injection of 0.1 mg of zymosan or vehicle (PBS; 500 μl). Lavage fluids were collected four hours later. A: Total cell migration was assessed by light microscopy. B: Neutrophil migration was quantified after staining with an anti-Gr1 Ab and flow cytometry analysis. Data are mean ± SEM of five mice per group. *P < 0.05, **P < 0.01 vs. vehicle group.
Lack of AnxA1 Modulates Peritoneal Inflammatory Mediators
The analysis of a panel of proteins tested by the array approach revealed that only a few are modulated in these experimental settings. Furthermore, some of these are already increased in AnxA1−/− mice or after CFTR172 treatment (Figure 4). The first differentially modulated marker was soluble intercellular adhesion molecule-1, which appeared elevated in the AnxA1−/− mouse peritoneal cavity even following treatment with the CFTR172 inhibitor alone (Figure 4A). Addition of zymosan cancelled these differences, and a plateau was reached in both wild-type and AnxA1−/− mice. Complement factor 5a was markedly augmented in the AnxA1−/− animals on zymosan injection and this was not further modulated by CFTR172 (Figure 4B). A striking increase in monocyte colony stimulating factor levels was detected in the lavage fluids following CFTR172 administration (Figure 4D). Figure 4C reports the changes in tissue inhibitor of metalloprotease-1, which was singularly regulated by CFTR172 injection, with no major further changes in AnxA1−/− mice.
Figure 4.

Cytokines modified following the pharmacological inhibition of the CFTR in vivo. AnxA1−/− and wild-type animals were treated with 50 μg/kg CFTR172 24 hours and 1 hour before the i.p. injection of 0.1 mg of zymosan or vehicle (PBS; 500 μl). Lavage fluids were collected 4 hours later and assessed for protein array. Panels show (A) soluble intercellular adhesion molecule-1, (B) complement factor 5a, (C) tissue inhibitor of metalloprotease-1, (D) monocyte colony stimulating factor, and (E) IL-6. F: IL-6 levels were quantified by enzyme-linked immunosorbent assay. Data are mean ± SEM of five mice per group. *P < 0.05 vs. respective wild-type group; **P < 0.05 zymosan/vehicle group.
An interesting result was obtained for IL-6, since fluctuation of its levels across the experimental groups mirrored, at least qualitatively, the changes measured for Gr1+ve influx shown in Figure 3. IL-6 was augmented in the peritoneal cavities after zymosan injection and even more so in AnxA1−/− mice (Figure 4E); treatment with CFTR172 increased IL-6 expression in wild-type, but not in AnxA1−/− mice. This result was corroborated by enzyme-linked immunosorbent assay with quantitative differences seen among the experimental groups in line with the changes detected in the array assay (Figure 4F). Interestingly, the IL-6 enzyme-linked immunosorbent assay revealed elevated levels of this cytokine even in cavities treated with the CFTR172 inhibitor alone, an effect that was more striking in AnxA1−/− animals.
Administration of AnxA1 Rescues the Inflammatory Profile of CFTR−/− and CFTR172-Treated Mice
The pro-inflammatory effect of CFTR172 co-injected with low dose zymosan was markedly attenuated when mice where treated with hr-AnxA1 or with peptide Ac2-26 (Figure 5A). The double pretreatment with CFTR172 nearly tripled influx of Gr1+ve cells into the peritoneal cavity over the cell recruitment provoked by zymosan alone (Figure 5A). Treatment of mice with a low dose hrAnxA1 (1 μg corresponding to 27 pmol per mouse) reduced the potentiating effect of CFTR172 by approximately 41%. A similar degree of reduction (55%) was attained when mice were treated with the AnxA1 mimetic, peptide Ac2-26 (50 μg per mouse, equivalent to 323 pmol) (Figure 5A).
Figure 5.
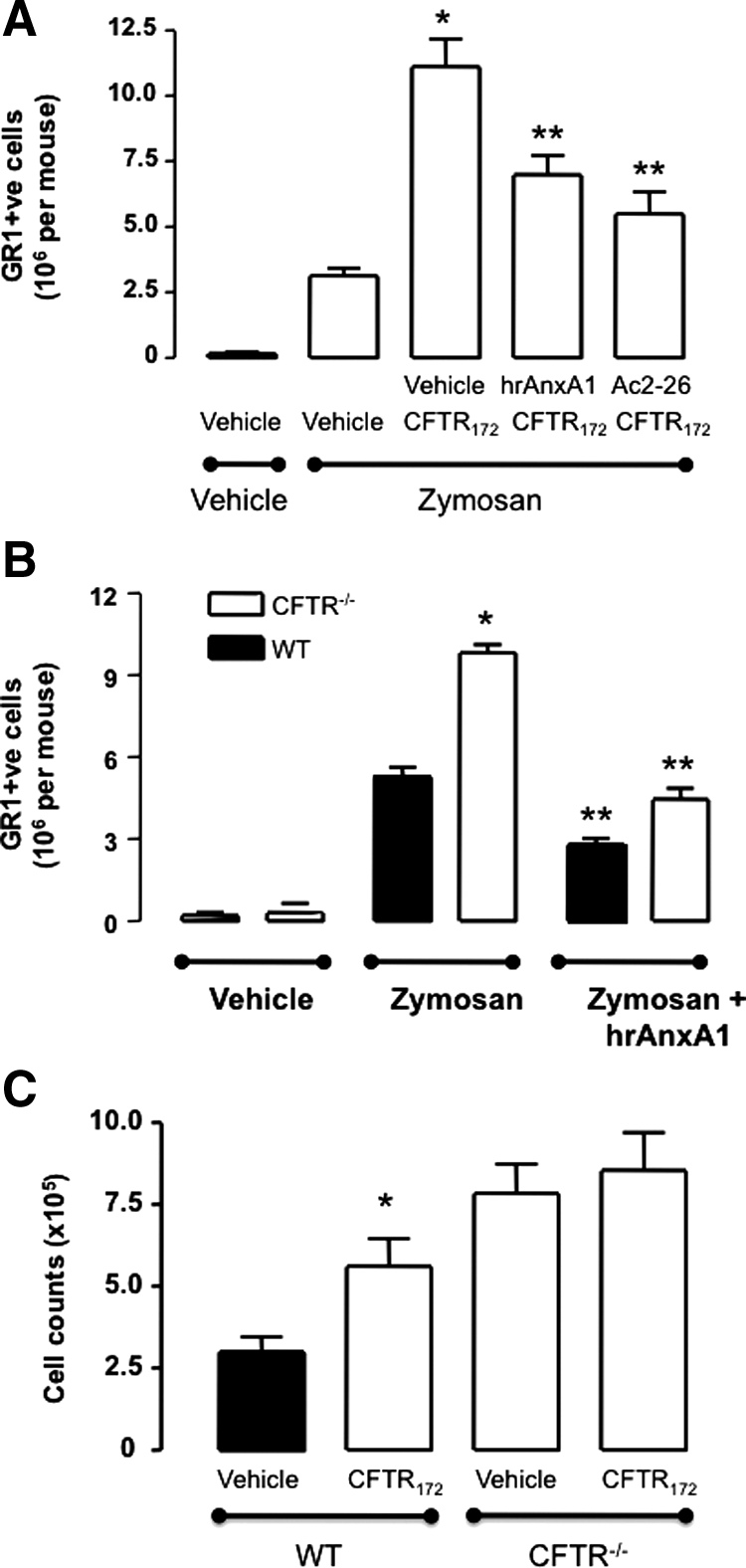
Human recombinant AnxA1 attenuates the inflammatory phenotype associated with CFTR inhibition. A: Wild-type animals were treated with 50 μg/kg CFTR172 24 hours and one hour before the i.p. injection of 0.1 mg of zymosan (PBS; 500 μl). Mice were treated i.v. with saline (200 μl), 1 μg hrAnxA1 or 50 μg peptide Ac2-26. B: Wild-type and CFTR−/− mice received 200 μl i.v. vehicle or hrAnxA1 (1 μg) immediately before the administration of zymosan (1 mg) or vehicle i.p. C: Wild-type and CFTR−/− mice were treated with 50 μg/kg CFTR172 24 hours and 1 hour before the i.p. injection of 0.1 mg of zymosan (PBS; 500 μl). In all sets of experiments peritoneal lavage fluids were collected four hours later, and recruited neutrophils were stained with an anti-Gr1 Ab for flow cytometry analysis. Data are mean ± SEM of five mice per group. A: *P < 0.05 vs. zymosan/vehicle group; **P < 0.05 vs. zymosan/CFTR172 treated group. B: *P < 0.05 vs. zymosan group in wild-type mice; **P < 0.05 vs. zymosan group in CFTR−/− mice. C: *P < 0.05 vs. zymosan/vehicle group.
In this set of experiments the phenotype of CFTR−/− mice was also determined. As expected, these mice had a much higher influx of Gr1+ve cells on zymosan challenge (Figure 5B). Again, this accentuated cell influx was susceptible to the pharmacological effect of hrAnxA1: administered to CFTR−/− at the dose of 1 μg per mouse, which normalized cell influx back to the values observed in wild-type mice (Figure 5B).
The CFTR−/− mouse colony provided strong proof-of-concept to the specificity of CFTR172; in fact, when the combination of zymosan + CFTR172 (50 μg/kg, −24 and −1 hour) was tested in these animals, the potentiating effect of the CFTR inhibitor was no longer detected (Figure 5C).
CFTR172 Deregulates AnxA1 Expression in Human Cells
Finally, we attempted to determine the translational potential for these findings, performing in vitro studies with human primary PMN. This time cells were incubated with CFTR172 alone or together with LPS and the expression of AnxA1 and its receptor FPR2/ALX11,29 was assessed.
Figure 6 reports the cumulative data obtained with freshly isolated PMN. These cells expressed very high levels of intracellular AnxA1, with ∼3% being found on the plasma membrane. CFTR172, added alone to cells, significantly reduced total AnxA1 levels (by approximately 50%); the same reduction was observed with LPS (either alone or with CFTR172) (Figure 6A). Reduction in intracellular AnxA1 levels was associated with an incremental increase in plasma membrane localization of the protein (Figure 6B). The AnxA1 receptor FPR2/ALX followed this pattern only in part: CFTR172 incubation led to a reduction in total levels (Figure 6C) with a marginal increase in the cell surface expression (Figure 6D). LPS augmented FPR2/ALX expression both in total terms (Figure 6C) and on the membrane (Figure 6D).
Figure 6.

The AnxA1 pathway is dysregulated in human PMN following CFTR blockade. Blood-borne PMN were incubated with either vehicle or 20 μmol/L CFTR172 (30 minutes at 37°C). Cells were then stimulated with 10 ng/ml of LPS (four hours at 37°C). Cell pellets were examined for membrane and total detection of AnxA1 (A, C) and FPR2/ALX (B, D) by flow cytometry. Data are mean ± SEM of experiments performed in duplicate with three separate donors. *P < 0.05 vs. vehicle/vehicle group. Insets: representative histograms (all referring to CFTR172 treatment alone: gray histograms, no treatment; open histograms CFTR172).
This reduction in cell-associated AnxA1 was not consequent to reduced gene expression, since real time PCR analysis did not reveal lower gene products for either AnxA1 or FPR2/ALX in extracts of cells treated with CFTR172 alone or together with LPS (data not shown).
The modulation of cell associated AnxA1 in human PMN on incubation with the different treatment protocols (Figure 6) prompted us to test whether the protein could be released via microparticles.26 AnxA1 secretion from the PMN was elevated following the treatment of the cells with CFTR172, although LPS exerted a higher potency on AnxA1 release from PMN than the inhibitor; immunoreactivity in the cell incubation medium is reported in Figure 7A. When microparticles were removed by ultracentrifugation, not much effect was observed on the unstimulated release of AnxA1, whereas the augmenting effect of the three treatment protocols was markedly attenuated (Figure 7B).
Figure 7.
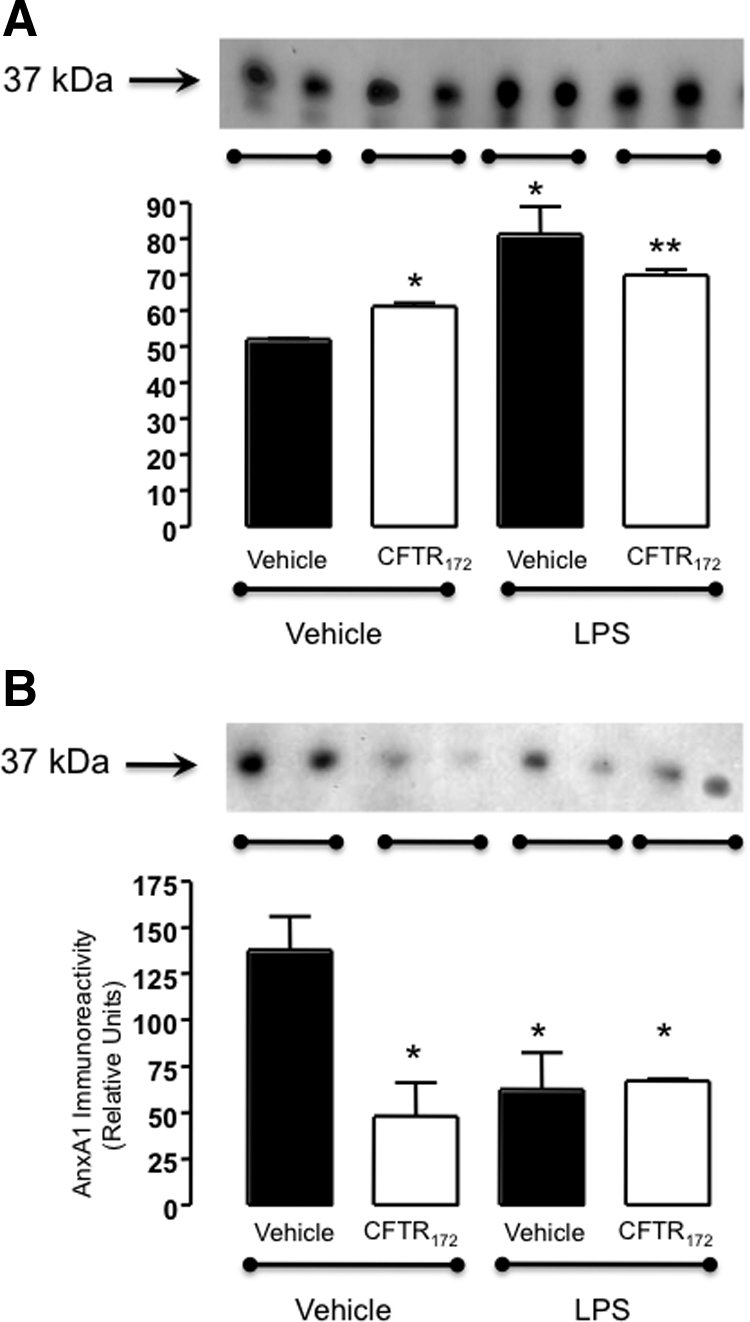
AnxA1 associated with microparticles is released from CFTR172 treated PMN. Supernatants from PMN treated as in Figure 6 were tested for AnxA1 content by Western blotting: (A) Cell-free supernatants; (B) microparticles-depleted supernatants (microparticles removed by ultracentrifugation; see Methods). In each panel, blots show representative duplicates analyzed. The graphs report cumulative data (mean ± SEM) of three independent experiments performed in duplicates. *P < 0.05 vs. vehicle/vehicle group; **P < 0.05 vs. LPS/none group.
Discussion
The data presented here demonstrate unequivocally a functional association between inhibition of CFTR and an acute inflammatory response characterized by neutrophil recruitment and release of pro-inflammatory mediators, including IL-6. We propose that this phenomenon, which is amplified during an ongoing inflammatory process, is caused by the loss of the endogenous anti-inflammatory mediator AnxA1. Finally, we report that acute inhibition of CFTR provokes a partial neutrophil depletion of AnxA1, leading us to propose that it might contribute, at least in part, to the pro-activating and perhaps inflammatory status characteristic of CF.
CF is a monogenic disease where alterations in the sequence of a specific chloride channel, CFTR, lead to chronic inflammation and infection in the lung.30 The disease is controlled with antibiotics given in conjunction, during the acute phase, with anti-inflammatory drugs.31 In this context there is also evidence that some antibiotics could possess anti-inflammatory properties.32 Importantly even in patients receiving combined glucocorticoid plus antibiotic therapy, levels of pro-inflammatory mediators such as IL-6 and IL-8 expressed in CF bronchoalveolar lavage are elevated. Thus suggesting that the predominant cell types responsible for the pathogenesis of this disease are not responsive to such a treatment regimen, hence the reason(s) why the process continues unchecked is still elusive. A recent breakthrough came from analysis of a novel colony of CFTR null mice. A proteomic approach revealed that these animals, which developed a strong and spontaneous pro-inflammatory phenotype leading to lethality, lacked a specific protein, the endogenous anti-inflammatory mediator AnxA1.18
AnxA1 is a glucocorticoid-regulated protein and it is one of the several effectors of endogenous anti-inflammatory system that operates in a cell- and target-specific manner to keep the inflammatory response under check, thereby assuring regulated time- and spatial-dependent responses.10 With respect to PMN and epithelial cell biology, appropriate AnxA1 levels assure a controlled degree of activation on stimulation.25,33,34 Thus, if the protein is absent, these cells would remain activated for longer time, producing higher levels of pro-inflammatory mediators and, in the case of the PMN, displaying a higher migratory response to the site of inflammation.23 Both PMN and epithelial cells possess abundant levels of intracellular AnxA1 and on activation the protein translocates onto the cell surface. Here it is then able to interact with its receptor, which it targets in a paracrine and/or autocrine fashion to assure an appropriate homeostatic degree of activation.10
For all these reasons, the fact that abnormalities in CFTR function lead to a reduction in cellular and tissue levels of this mediator, provides a mechanistic explanation for the augmented inflammation in CF. It should also be noted that the anti-inflammatory and pro-resolving effects of AnxA1 are multiple, since this protein provides exquisite control of neutrophil recruitment by interfering with the process of neutrophil-endothelial cell interaction in the microcirculation.10,23,35 Moreover, AnxA1 increases levels of the anti-inflammatory cytokine IL-1017,36 and promotes apoptosis of recruited cells as well as their removal by phagocytosis.16,37 All these properties are highly relevant to the CF neutrophil.
The dysregulation of the AnxA1 system has been associated with exacerbated and prolonged cell activation and abnormal responses, caused by the lack of “fine tuning” and homeostatic control. The CF neutrophil is characterized by an improper activation,38,39 as well as a reduced apoptosis.40,41 Aberrant cell activation can be controlled by cell incubation with AnxA1 (eg, see34) and AnxA1 null neutrophils display greater chemotaxis and activation (degranulation) compared with wild-type cells stimulated in similar conditions.23 Moreover, AnxA1 can promote PMN apoptosis15 and favors the removal of apoptotic cells by professional phagocytes.16 In addition, AnxA1 up-regulates the anti-inflammatory cytokine IL-10, which in turn leads to the inhibition of inducible Nitric Oxide Synthase mRNA expression and nitric oxide release.17 Interestingly, lower levels of IL-10 have been reported in CF.42,43
Against this background, the present study began by establishing an experimental model where inflammatory responses could be monitored through the administration of a small molecule inhibitor, CFTR172.20 To model the chronic absence of CFTR in the null mice, the inhibitor was administered twice into the mouse peritoneal cavity. It is important to note that this treatment was sufficient to bring into the cavity a small yet significant number of GR1+ve cells. More importantly, when an ongoing inflammatory status was evoked by zymosan, CFTR172 augmented the inflammatory response in a synergistic fashion: this was particularly evident before the maximal cell recruitment was reached, that is, using a low dose of zymosan. The effect of CFTR172 was chiefly associated with endogenous AnxA1 since it was no longer observed in AnxA1 null mice. Of interest, a semiquantitative assessment of AnxA1 protein expression in peritoneal cells revealed a significant reduction in mice pre-treated with CFTR172. Therefore, the model that is emerging is that an inappropriate, possibly untimely, reduction in cell associated AnxA1 would lead to a more pronounced inflammatory response.
These results were validated by two further sets of experiments. First, the potentiating effect of CFTR inhibition on GR1+ve cell trafficking was not restricted to CFTR172 but evident also after treatment of mice with a structurally unrelated inhibitor, compound GlyH101, which blocks the pore of CFTR.24 Secondly, CFTR172 was unable to increase cell recruitment above that promoted by low dose zymosan when administered to CFTR−/− mice. This is a crucial point, since CFTR inhibitors may exert CFTR-independent effects on reactive oxygen species production, mitochondrial failure and activation of the nuclear factor κB signaling pathway,44 all potentially bearing relevance to the cellular response in zymosan peritonitis. However, CFTR172 was not active in CFTR−/− mice strongly indicating a functional link between pharmacological treatment, CFTR and the observed outcome.
Analysis of cell recruitment data were complemented by an unbiased protein array analysis. Interesting observations made here included finding a potential pivotal role for IL-6, which peritoneal levels were augmented by CFTR172 above those produced by zymosan alone; once again, this potentiating effect was no longer observed in AnxA1 null mice. Furthermore, analysis of the lavage fluids obtained from mice treated with CFTR172 alone revealed a slightly increased recruitment of neutrophils in AnxA1 null mice, which dovetails well with the overall augmentation of cell recruitment in this mouse colony. The IL-6 levels observed in the exudates are intriguing since they also marry well with recent clinical observations, where high levels of this cytokine could be measured in CF and proposed to express an altered endothelial status.45
Altogether, we propose that the new model presented here could be valuable to test novel anti-inflammatory therapeutics for the treatment of CF. Results of the experiments conducted with hrAnxA1, as well as with AnxA1-derived peptides,46 indicate that potentiating the AnxA1 pathway could be a viable avenue for controlling CF inflammation. Such an effect could be achieved either using a classical drug approach or by gene therapy. Gene therapy is currently under test in CF47 and soon clinical data will indicate its potential effectiveness. Attempting to ‘restore’ cellular levels of AnxA1 by gene therapy in CF cells is a possibility that derives also from the experiments conducted with primary cells.
As discussed above, the inflammatory phenotype of CF is associated with the hyperactivation of bronchial epithelial cells and neutrophils.39,48,49 Primary epithelial cells taken from CF patients have lower AnxA1 protein expression.18 Here, we complemented data produced with this novel experimental model by focusing our attention to the human PMN. AnxA1 protein is highly abundant in this cell type where it represents between 2 and 4% of total cytosolic proteins.50 In normal conditions, a large pool of cytosolic AnxA1 is contained in gelatinase granules51,52 so that the protein can be promptly externalized on cell adhesion to the endothelium.25 Once on the PMN cell surface, AnxA1 can interact in a paracrine/autocrine fashion with its receptor FPR2/ALX10,29 eliciting inhibitory processes including induction of the detachment process.35,53 We thus reasoned that depleted AnxA1 levels in the CF PMN could contribute to its hyperactivation status.54 To explore this possibility, the second part of the study analyzed the effect of PMN incubation with CFTR172.
Expression of CFTR in the PMN is a contentious issue, with a number of studies providing convincing evidence that it is absent from the PMN cell membrane.55,56 Recently however, evidence has emerged showing CFTR expression in secretory vesicles and phagolysosomes of the PMN,57,58 where it was shown to be important in the bactericidal actions of the latter structures. Such a limited and localized expression could explain the contrasting reports on detection of this protein in the PMN.
We therefore explored the effect of CFTR172 incubation with primary PMN observing a significant depletion of cell-associated AnxA1, ranging from 30 to 60% reduction across blood donors. This effect, shown here for the 20-μM concentrations, was similarly attained with a higher concentration of the inhibitor (50 μmol/L; not shown). It is unclear whether CFTR172 induced mobilization of AnxA1 was secondary to fusion of intracellular granules with the plasma membrane (which seems the physiological path for AnxA1 externalization51) or consequent to ‘leakage’ of cytosolic pool of protein, not associated either with gelatinase or primary granules.51,52 Irrespective of the putative CFTR-dependent mechanisms that translocate AnxA1 onto the plasma membrane, AnxA1 secretion into the medium was by and large occurring via microparticles. We have recently reported this novel mode of externalization, in human PMN, for AnxA1.26 The involvement of microparticles in the mobilization of this protein on cell treatment with CFTR172 was suggested, since supernatant levels of AnxA1 were markedly reduced after ultracentrifugation hence microparticle removal. Figure 8 illustrates a hypothetical model that is emerging when combining data produced with acute CFTR inhibition in mouse and human PMN.
Figure 8.

Hypothetical model for AnxA1 disposition in the PMN on CFTR inhibition. These schemes illustrate the model that could be put forward, based on the experiments presented in this study. In resting PMN, a large proportion (≥50 to 60%) of AnxA1 is contained intracellularly in gelatinase granules, with the remainder primarily found in the cytosol and negligible levels on the cell surface. CFTR is expressed, though this is a contentious issue, in the phagolysosome and/or other vesicles. Acute inhibition (and malfunctioning?) of CFTR is, in a manner yet to be established, associated with ‘leakage’ of the intracellular AnxA1 pool and release of the protein in the supernatant, mainly in microparticles. Importantly, this model still needs to be corroborated with analyses conducted in PMN obtained from cystic fibrosis patients.
Two further points require mention here. Firstly, membrane and supernatant AnxA1 levels—increased after PMN incubation with CFTR172—were mildly augmented in the presence of a leukocyte activator (we used LPS since the presence of toll-like receptor 4 has been described in CF PMN59). Secondly, and more importantly, the mobilization of AnxA1 we monitored here on cell incubation with CFTR172 was not consequent to a cytotoxic effect of the inhibitor (eg, provoking apoptosis) as annexin V staining on these cells was minimal in human PMN incubated for 4 hours at 37°C with or without CFTR172 (both 20 or 50 μmol/L; data not shown). Consistently, apoptotic PMN do not seem to release AnxA1 via the microparticle pathway.37
It is intriguing how a recent study proposed a direct association between AnxA1 and CFTR, admittedly in an epithelial cell line, partly (∼50%) evident in lipid rafts preparations.19 Importantly, cell incubation with CFTR172 disrupted this association yielding an altered distribution of AnxA1. Collectively, these data prompt us to speculate that abnormal function of CFTR leads to a partial depletion of PMN-associated AnxA1 (Figure 8), which might explain, at least in part, the higher degree of activation and extravasation23,25,36,53 observed for this cell type in CF.
In summary, this study presents two distinct, yet related, findings describing on one hand the inflammatory response associated with CFTR inhibition in the mouse and, on the other hand, linking it to a malfunctioning of the endogenous AnxA1 pathway. Moreover, direct evidence for secretion of AnxA1 from human peripheral blood PMN on CFTR inhibition is also shown.
Acknowledgments
We thank Dr. Sabino A. Damazo (University Federal of Mato Grosso, Cuiaba, MT, Brasil) for help with the immunogold labeling experiments and Ms. Delphine Roussel (INSERM, Paris) for her assistance with the in vivo experiments. We acknowledge Prof. Roderick J. Flower’s helpful discussion and help with manuscript revision (Barts and The London School of Medicine, London).
Footnotes
Address reprint requests to Mauro Perretti, Ph.D., The William Harvey Research Institute, Barts and The London School of Medicine, Queen Mary University of London, Charterhouse Square, London, EC1M 6BQ, UK. E-mail: m.perretti@qmul.ac.uk.
Supported by Wellcome Trust (programme grant number 069234/Z/02/Z and 086867/Z/08), the William Harvey Research Foundation, and the French Foundations Vaincre la Mucovicidose’ and ‘Mucoviscidose:ABCF.
References
- Quinton PM. Physiological basis of cystic fibrosis: a historical perspective. Physiol Rev. 1999;79:S3–S22. doi: 10.1152/physrev.1999.79.1.S3. [DOI] [PubMed] [Google Scholar]
- Balough K, McCubbin M, Weinberger M, Smits W, Ahrens R, Fick R. The relationship between infection and inflammation in the early stages of lung disease from cystic fibrosis. Pediatr Pulmonol. 1995;20:63–70. doi: 10.1002/ppul.1950200203. [DOI] [PubMed] [Google Scholar]
- Bonfield TL, Panuska JR, Konstan MW, Hilliard KA, Hilliard JB, Ghnaim H, Berger M. Inflammatory cytokines in cystic fibrosis lungs. Am J Respir Crit Care Med. 1995;152:2111–2118. doi: 10.1164/ajrccm.152.6.8520783. [DOI] [PubMed] [Google Scholar]
- Corvol H, Fitting C, Chadelat K, Jacquot J, Tabary O, Boule M, Cavaillon JM, Clement A. Distinct cytokine production by lung and blood neutrophils from children with cystic fibrosis. Am J Physiol Lung Cell Mol Physiol. 2003;284:L997–L1003. doi: 10.1152/ajplung.00156.2002. [DOI] [PubMed] [Google Scholar]
- Griese M, Kappler M, Gaggar A, Hartl D. Inhibition of airway proteases in cystic fibrosis lung disease. Eur Respir J. 2008;32:783–795. doi: 10.1183/09031936.00146807. [DOI] [PubMed] [Google Scholar]
- Khan TZ, Wagener JS, Bost T, Martinez J, Accurso FJ, Riches DW. Early pulmonary inflammation in infants with cystic fibrosis. Am J Respir Crit Care Med. 1995;151:1075–1082. doi: 10.1164/ajrccm/151.4.1075. [DOI] [PubMed] [Google Scholar]
- Serhan CN, Brain SD, Buckley CD, Gilroy DW, Haslett C, O'Neill LA, Perretti M, Rossi AG, Wallace JL. Resolution of inflammation: state of the art, definitions and terms. FASEB J. 2007;21:325–332. doi: 10.1096/fj.06-7227rev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN, Savill J. Resolution of inflammation: the beginning programs the end. Nat Immunol. 2005;6:1191–1197. doi: 10.1038/ni1276. [DOI] [PubMed] [Google Scholar]
- Gilroy DW, Lawrence T, Perretti M, Rossi AG. Inflammatory resolution: new opportunities for drug discovery. Nat Rev Drug Discov. 2004;3:401–416. doi: 10.1038/nrd1383. [DOI] [PubMed] [Google Scholar]
- Perretti M, D'Acquisto F. Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat Rev Immunol. 2009;9:62–70. doi: 10.1038/nri2470. [DOI] [PubMed] [Google Scholar]
- Perretti M, Chiang N, La M, Fierro IM, Marullo S, Getting SJ, Solito E, Serhan CN. Endogenous lipid- and peptide-derived anti-inflammatory pathways generated with glucocorticoid and aspirin treatment activate the lipoxin A4 receptor. Nat Med. 2002;8:1296–1302. doi: 10.1038/nm786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamal AM, Flower RJ, Perretti M. An overview of the effects of annexin 1 on cells involved in the inflammatory process. Mem Inst Oswaldo Cruz. 2005;100 Suppl 1:39–47. doi: 10.1590/s0074-02762005000900008. [DOI] [PubMed] [Google Scholar]
- Hayhoe RP, Kamal AM, Solito E, Flower RJ, Cooper D, Perretti M. Annexin 1 and its bioactive peptide inhibit neutrophil-endothelium interactions under flow: indication of distinct receptor involvement. Blood. 2006;107:2123–2130. doi: 10.1182/blood-2005-08-3099. [DOI] [PubMed] [Google Scholar]
- Vong L, D'Acquisto F, Pederzoli-Ribeil M, Lavagno L, Flower RJ, Witko-Sarsat V, Perretti M. Annexin 1 cleavage in activated neutrophils: a pivotal role for proteinase 3. J Biol Chem. 2007;282:29998–30004. doi: 10.1074/jbc.M702876200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solito E, Kamal A, Russo-Marie F, Buckingham JC, Marullo S, Perretti M. A novel calcium-dependent proapoptotic effect of annexin 1 on human neutrophils. FASEB J. 2003;17:1544–1546. doi: 10.1096/fj.02-0941fje. [DOI] [PubMed] [Google Scholar]
- Maderna P, Yona S, Perretti M, Godson C. Modulation of phagocytosis of apoptotic neutrophils by supernatant from dexamethasone-treated macrophages and annexin-derived peptide Ac(2-26). J Immunol. 2005;174:3727–3733. doi: 10.4049/jimmunol.174.6.3727. [DOI] [PubMed] [Google Scholar]
- Ferlazzo V, D'Agostino P, Milano S, Caruso R, Feo S, Cillari E, Parente L. Anti-inflammatory effects of annexin-1: stimulation of IL-10 release and inhibition of nitric oxide synthesis. Int Immunopharmacol. 2003;3:1363–1369. doi: 10.1016/S1567-5769(03)00133-4. [DOI] [PubMed] [Google Scholar]
- Bensalem N, Ventura AP, Vallee B, Lipecka J, Tondelier D, Davezac N, Dos Santos A, Perretti M, Fajac A, Sermet-Gaudelus I, Renouil M, Lesure JF, Halgand F, Laprevote O, Edelman A. Down-regulation of the anti-inflammatory protein annexin A1 in cystic fibrosis knock-out mice and patients. Mol Cell Proteomics. 2005;4:1591–1601. doi: 10.1074/mcp.M500019-MCP200. [DOI] [PubMed] [Google Scholar]
- Borot F, Vieu DL, Faure G, Fritsch J, Colas J, Moriceau S, Baudouin-Legros M, Brouillard F, Ayala-Sanmartin J, Touqui L, Chanson M, Edelman A, Ollero M. Eicosanoid release is increased by membrane destabilization and CFTR inhibition in Calu-3 cells. PLoS One. 2009;4:e7116. doi: 10.1371/journal.pone.0007116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T, Thiagarajah JR, Yang H, Sonawane ND, Folli C, Galietta LJ, Verkman AS. Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera toxin-induced intestinal fluid secretion. J Clin Invest. 2002;110:1651–1658. doi: 10.1172/JCI16112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannon R, Croxtall JD, Getting SJ, Roviezzo F, Yona S, Paul-Clark MJ, Gavins FN, Perretti M, Morris JF, Buckingham JC, Flower RJ. Aberrant inflammation and resistance to glucocorticoids in annexin 1−/− mouse. FASEB J. 2003;17:253–255. doi: 10.1096/fj.02-0239fje. [DOI] [PubMed] [Google Scholar]
- Snouwaert JN, Brigman KK, Latour AM, Malouf NN, Boucher RC, Smithies O, Koller BH. An animal model for cystic fibrosis made by gene targeting. Science. 1992;257:1083–1088. doi: 10.1126/science.257.5073.1083. [DOI] [PubMed] [Google Scholar]
- Chatterjee BE, Yona S, Rosignoli G, Young RE, Nourshargh S, Flower RJ, Perretti M. Annexin 1-deficient neutrophils exhibit enhanced transmigration in vivo and increased responsiveness in vitro. J Leukoc Biol. 2005;78:639–646. doi: 10.1189/jlb.0405206. [DOI] [PubMed] [Google Scholar]
- Muanprasat C, Sonawane ND, Salinas D, Taddei A, Galietta LJ, Verkman AS. Discovery of glycine hydrazide pore-occluding CFTR inhibitors: mechanism, structure-activity analysis, and in vivo efficacy. J Gen Physiol. 2004;124:125–137. doi: 10.1085/jgp.200409059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perretti M, Croxtall JD, Wheller SK, Goulding NJ, Hannon R, Flower RJ. Mobilizing lipocortin 1 in adherent human leukocytes downregulates their transmigration. Nat Med. 1996;2:1259–1262. doi: 10.1038/nm1196-1259. [DOI] [PubMed] [Google Scholar]
- Dalli J, Norling LV, Renshaw D, Cooper D, Leung KY, Perretti M. Annexin 1 mediates the rapid anti-inflammatory effects of neutrophil-derived microparticles. Blood. 2008;112:2512–2519. doi: 10.1182/blood-2008-02-140533. [DOI] [PubMed] [Google Scholar]
- Pepinsky RB, Sinclair LK, Dougas I, Liang CM, Lawton P, Browning JL. Monoclonal antibodies to lipocortin-1 as probes for biological function. FEBS Lett. 1990;261:247–252. doi: 10.1016/0014-5793(90)80564-y. [DOI] [PubMed] [Google Scholar]
- Collins TJ. ImageJ for microscopy. Biotechniques. 2007;43:25–30. doi: 10.2144/000112517. [DOI] [PubMed] [Google Scholar]
- Ye RD, Boulay F, Wang JM, Dahlgren C, Gerard C, Parmentier M, Serhan CN, Murphy PM. International Union of Basic and Clinical Pharmacology. LXXIII Nomenclature for the formyl peptide receptor (FPR) family. Pharmacol Rev. 2009;61:119–161. doi: 10.1124/pr.109.001578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elston C, Geddes D. Inflammation in cystic fibrosis–when and why? Friend or foe? Semin Respir Crit Care Med. 2007;28:286–294. doi: 10.1055/s-2007-981649. [DOI] [PubMed] [Google Scholar]
- Fayon M. CF-Emerging therapies: Modulation inflammation. Paediatr Respir Rev. 2006;7 Suppl 1:S170–S174. doi: 10.1016/j.prrv.2006.04.212. [DOI] [PubMed] [Google Scholar]
- Sharma S, Jaffe A, Dixon G. Immunomodulatory effects of macrolide antibiotics in respiratory disease: therapeutic implications for asthma and cystic fibrosis. Paediatr Drugs. 2007;9:107–118. doi: 10.2165/00148581-200709020-00004. [DOI] [PubMed] [Google Scholar]
- Ambrose MP, Hunninghake GW. Corticosteroids increase lipocortin I in alveolar epithelial cells. Am J Respir Cell Mol Biol. 1990;3:349–353. doi: 10.1165/ajrcmb/3.4.349. [DOI] [PubMed] [Google Scholar]
- Zouki C, Ouellet S, Filep JG. The anti-inflammatory peptides, antiflammins, regulate the expression of adhesion molecules on human leukocytes and prevent neutrophil adhesion to endothelial cells. FASEB J. 2000;14:572–580. doi: 10.1096/fasebj.14.3.572. [DOI] [PubMed] [Google Scholar]
- Lim LH, Solito E, Russo-Marie F, Flower RJ, Perretti M. Promoting detachment of neutrophils adherent to murine postcapillary venules to control inflammation: effect of lipocortin 1. Proc Natl Acad Sci USA. 1998;95:14535–14539. doi: 10.1073/pnas.95.24.14535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza DG, Fagundes CT, Amaral FA, Cisalpino D, Sousa LP, Vieira AT, Pinho V, Nicoli JR, Vieira LQ, Fierro IM, Teixeira MM. The required role of endogenously produced lipoxin A4 and annexin-1 for the production of IL-10 and inflammatory hyporesponsiveness in mice. J Immunol. 2007;179:8533–8543. doi: 10.4049/jimmunol.179.12.8533. [DOI] [PubMed] [Google Scholar]
- Scannell M, Flanagan MB, deStefani A, Wynne KJ, Cagney G, Godson C, Maderna P. Annexin-1 and peptide derivatives are released by apoptotic cells and stimulate phagocytosis of apoptotic neutrophils by macrophages. J Immunol. 2007;178:4595–4605. doi: 10.4049/jimmunol.178.7.4595. [DOI] [PubMed] [Google Scholar]
- Witko-Sarsat V, Allen RC, Paulais M, Nguyen AT, Bessou G, Lenoir G, Descamps-Latscha B. Disturbed myeloperoxidase-dependent activity of neutrophils in cystic fibrosis homozygotes and heterozygotes, and its correction by amiloride. J Immunol. 1996;157:2728–2735. [PubMed] [Google Scholar]
- Witko-Sarsat V, Sermet-Gaudelus I, Lenoir G, Descamps-Latscha B. Inflammation and CFTR: might neutrophils be the key in cystic fibrosis? Mediators Inflamm. 1999;8:7–11. doi: 10.1080/09629359990658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriceau S, Kantari C, Mocek J, Davezac N, Gabillet J, Guerrera IC, Brouillard F, Tondelier D, Sermet-Gaudelus I, Danel C, Lenoir G, Daniel S, Edelman A, Witko-Sarsat V. Coronin-1 is associated with neutrophil survival and is cleaved during apoptosis: potential implication in neutrophils from cystic fibrosis patients. J Immunol. 2009;182:7254–7263. doi: 10.4049/jimmunol.0803312. [DOI] [PubMed] [Google Scholar]
- Watt AP, Courtney J, Moore J, Ennis M, Elborn JS. Neutrophil cell death, activation and bacterial infection in cystic fibrosis. Thorax. 2005;60:659–664. doi: 10.1136/thx.2004.038240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonfield TL, Konstan MW, Burfeind P, Panuska JR, Hilliard JB, Berger M. Normal bronchial epithelial cells constitutively produce the anti-inflammatory cytokine interleukin-10, which is downregulated in cystic fibrosis. Am J Respir Cell Mol Biol. 1995;13:257–261. doi: 10.1165/ajrcmb.13.3.7544594. [DOI] [PubMed] [Google Scholar]
- Soltys J, Bonfield T, Chmiel J, Berger M. Functional IL-10 deficiency in the lung of cystic fibrosis (cftr−/−) and IL-10 knockout mice causes increased expression and function of B7 costimulatory molecules on alveolar macrophages. J Immunol. 2002;168:1903–1910. doi: 10.4049/jimmunol.168.4.1903. [DOI] [PubMed] [Google Scholar]
- Kelly M, Trudel S, Brouillard F, Bouillaud F, Colas J, Nguyen-Khoa T, Ollero M, Edelman A, Fritsch J. CFTR inhibitors, CFTRinh-172 and GlyH-101 target mitochondrial functions, independently of chloride channel inhibition. J Pharmacol Exp Ther. 2010;333:60–69. doi: 10.1124/jpet.109.162032. [DOI] [PubMed] [Google Scholar]
- Romano M, Collura M, Lapichino L, Pardo F, Falco A, Chiesa PL, Caimi G, Davi G. Endothelial perturbation in cystic fibrosis. Thromb Haemost. 2001;86:1363–1367. [PubMed] [Google Scholar]
- Perretti M, Dalli J. Exploiting the Annexin A1 pathway for the development of novel anti-inflammatory therapeutics. Br J Pharmacol. 2009;158:936–946. doi: 10.1111/j.1476-5381.2009.00483.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retsch-Bogart GZ. Update on new pulmonary therapies. Curr Opin Pulm Med. 2009;15(6):604–610. doi: 10.1097/MCP.0b013e328331805b. [DOI] [PubMed] [Google Scholar]
- Bartling TR, Drumm ML. Loss of CFTR results in reduction of histone deacetylase 2 in airway epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2009;297:L35–L43. doi: 10.1152/ajplung.90399.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quattrucci S, Napolitano M, Valentini SB, Giuliani A, Bravo E. Neutrophil generation of inflammatory precursors is not modulated by docosahexaenoic acid. Inflamm Res. 2009;58:677–685. doi: 10.1007/s00011-009-0035-5. [DOI] [PubMed] [Google Scholar]
- Rosales JL, Ernst JD. Calcium-dependent neutrophil secretion: characterization and regulation by annexins. J Immunol. 1997;159:6195–6202. [PubMed] [Google Scholar]
- Perretti M, Christian H, Wheller SK, Aiello I, Mugridge KG, Morris JF, Flower RJ, Goulding NJ. Annexin I is stored within gelatinase granules of human neutrophil and mobilized on the cell surface upon adhesion but not phagocytosis. Cell Biol Int. 2000;24:163–174. doi: 10.1006/cbir.1999.0468. [DOI] [PubMed] [Google Scholar]
- Lominadze G, Powell DW, Luerman GC, Link AJ, Ward RA, McLeish KR. Proteomic analysis of human neutrophil granules. Mol Cell Proteomics. 2005;4:1503–1521. doi: 10.1074/mcp.M500143-MCP200. [DOI] [PubMed] [Google Scholar]
- Gavins FN, Kamal AM, D'Amico M, Oliani SM, Perretti M. Formyl-peptide receptor is not involved in the protection afforded by annexin 1 in murine acute myocardial infarct. FASEB J. 2005;19:100–102. doi: 10.1096/fj.04-2178fje. [DOI] [PubMed] [Google Scholar]
- Downey DG, Bell SC, Elborn JS. Neutrophils in cystic fibrosis. Thorax. 2009;64:81–88. doi: 10.1136/thx.2007.082388. [DOI] [PubMed] [Google Scholar]
- Di A, Brown ME, Deriy LV, Li C, Szeto FL, Chen Y, Huang P, Tong J, Naren AP, Bindokas V, Palfrey HC, Nelson DJ. CFTR regulates phagosome acidification in macrophages and alters bactericidal activity. Nat Cell Biol. 2006;8:933–944. doi: 10.1038/ncb1456. [DOI] [PubMed] [Google Scholar]
- Morris MR, Doull IJ, Dewitt S, Hallett MB. Reduced iC3b-mediated phagocytotic capacity of pulmonary neutrophils in cystic fibrosis. Clin Exp Immunol. 2005;142:68–75. doi: 10.1111/j.1365-2249.2005.02893.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Painter RG, Valentine VG, Lanson NA, Jr, Leidal K, Zhang Q, Lombard G, Thompson C, Viswanathan A, Nauseef WM, Wang G. CFTR Expression in human neutrophils and the phagolysosomal chlorination defect in cystic fibrosis. Biochemistry. 2006;45:10260–10269. doi: 10.1021/bi060490t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Painter RG, Bonvillain RW, Valentine VG, Lombard GA, LaPlace SG, Nauseef WM, Wang G. The role of chloride anion and CFTR in killing of pseudomonas aeruginosa by normal and CF neutrophils. J Leukoc Biol. 2008;83:1345–1353. doi: 10.1189/jlb.0907658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koller B, Bals R, Roos D, Korting HC, Griese M, Hartl D. Innate immune receptors on neutrophils and their role in chronic lung disease. Eur J Clin Invest. 2009;39:535–547. doi: 10.1111/j.1365-2362.2009.02145.x. [DOI] [PubMed] [Google Scholar]