

Abstract
The development of synthetic process for hollow silica materials is an issue of considerable topical interest. While a number of chemical routes are available and are extensively used, the diameter of hollow silica often large than 50 nm. Here, we report on a facial route to synthesis ultrafine hollow silica nanoparticles (the diameter of ca. 24 nm) with high surface area by using cetyltrimethylammmonium bromide (CTAB) and sodium bis(2-ethylhexyl) sulfosuccinate (AOT) as co-templates and subsequent annealing treatment. When the hollow magnetite nanoparticles were introduced into the reaction, the ultrafine magnetic hollow silica nanoparticles with the diameter of ca. 32 nm were obtained correspondingly. Transmission electron microscopy studies confirm that the nanoparticles are composed of amorphous silica and that the majority of them are hollow.
Keywords: Hollow silica nanoparticles, Fe3O4, Dual-templates, Magnetic nanoparticles
Introduction
Nanoparticles are submicron moieties (diameters ranging from 1 to 100 nm according to the used term, although there are examples of nanoparticles several hundreds of nanometers in size) made of inorganic or organic materials, which have many novel properties compared with the bulk materials [1,2]. The fabrication of uniformly sized hollow nanoparticles (NPs) with controllable size and shape has attracted increasing attentions in many current and emerging areas of nanotechnology. This hollow NPs represent a distinct class of materials that are of interest in the fields of medicine, pharmaceutics, materials science, catalyst and the paint industry [3-7]. Moreover, nanostructured silica materials have attracted many attentions as they process practical applications in the fields of catalysis, sensing, drug delivery and controlled release due to its nontoxic, highly biocompatible, large surface areas and mechanically stable material [8].
Additionally, magnetic NPs are also used in important bio-applications, including magnetic bioseparation and detection of biological entities (cell, protein, nucleic acids, enzyme, bacterials, virus, etc.), targeted drug delivery and biological labels [2]. Owing to its availability, simple synthesis, low-cost and high magnetic responsibility, iron oxide NPs have became a strong candidate, and the application of small iron oxide NPs in in vitro diagnostics has been practiced for nearly half a century [9,10]. However, when naked magnetic NPs are directly exposed to the application system, there are many drawbacks such as easy aggregation, poor stabilization and biodegradation [11]. In order to overcome these limitations, silica seems to be one of the ideal supporting materials since silica matrices embedded with nanomagnets can be easily used to provide functionalities, prevent anisotropic magnetic dipolar attraction in absence of an external magnetic field and enhance the wear and corrosion resistance of the magnetic NPs [12]. From this viewpoint, composite structures based upon nanometer-sized iron oxide and silica also have a lot of potential applications.
As show in previous study, there are two mainly approaches include hard template (inorganic or polymer by layer-by-layer assembly, direct chemical deposition or nanocasting from mesoporous shells) and soft template (emulsion droplets, supramolecular micelles/vesicles, polymer aggregates/vesicles or gas bubbles) methods have been demonstrated to be successful in preparing hollow micro-/nanosized silica spheres [13-19]. It is noteworthy that the size, internal structure and the external morphology of silica NPs have a significant influence on their practical applications. Although quite a lot of preparation methods have been developed up to now, to our best knowledge, there are scarcely reports on synthesis of a particle size of less than 30 nm for hollow silica materials.
Herein, we present an advance in a simple and scalable wet chemical and subsequent annealing treatment synthesis of ultrafine hollow silica NPs (ca. 24 nm) and magnetic hollow silica NPs (ca. 32 nm). The ultrafine hollow silica NPs were synthesized in basic solution using cetyltrimethylammmonium bromide (CTAB) and sodium bis(2-ethylhexyl) sulfosuccinate (AOT) as co-templates and tetraethoxysilane (TEOS) as silica source and then annealed in air. For obtaining the ultrafine magnetic hollow silica NPs, we added hollow magnetite NPs (ca. 100 nm) in the previous process (Fig. 1). We analyse the different aspects of our synthetic approach and discuss the morphology, structure and magnetic properties of these NPs. These systems may be of particular interests for fabricating the hollow nanoshells encapsulating several functionalized materials, such as quantum dots, fluorescent materials and noble metal NPs. Compare with the common single-template method, AOT as a template can be lead to ordered mesoporous materials with controllable shapes, while vesicle templating (CTAB) as well as colloidal templating give rise to hollow nanospheres. The cationic CTA+ surfactant had a strong electrostatic interaction with the functional groups (sulfate and ester) of AOT. The AOT molecules mainly played a role in reducing the surface energy and promoting the formation of helical morphology, as also revealed previously in other systems [20-23].
Figure 1.
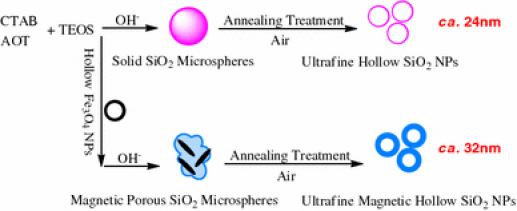
Schematic figure depicting the synthesis process of ultrafine hollow and magnetic hollow SiO2NPs
Experimental Section
Materials
Cetyltrimethylammmonium bromide (CTAB, ultrapure) and sodium bis(2-ethylhexyl) sulfosuccinate (AOT, AR) were purchased from Aladdin Chemical Reagent Co., Ltd; ferrous sulfate (FeSO4·7H2O, AR), sodium hydroxide (NaOH) and potassium hydroxide (KOH, AR) were purchased from Tianjin Kermel Chemical Reagent Co., Ltd; potassium nitrate (KNO3, AR) was purchased from Beijing Hongxing Chemical Reagent Co., Ltd; tetraethoxysilane (TEOS, AR), ethanol (C2H5OH, 95%, AR) andl(+)-glutamic acid (C5H9NO4, BR) were purchased from Sinopharm Chemical Reagent Co., Ltd; and all used as received. The Magnetic Sphere Technology Magnetic Separation Stand (MSS), purchased from Promega (Z5333), was used to separate magnetic particles at washing and selecting steps.
The Synthesis of Hollow Silica NPs
In a typical synthesis, 0.2 g CTAB was dissolve in 96-mL double distilled water, and then dropwise added 0.28 mL NaOH (5 mol·L−1) by pipette, the mixture was stirred at rate of ca. 500 rpm and kept 30 min at room temperature. AOT of 0.1 g was added to the solution and the reaction temperature was raised increasingly to 80 °C, and then dropwise added 1.34 mL TEOS to the mixture by pipette, the resulting mixture was stirred at 80 °C and kept 2 h. A milk white precipitate was observed. After the mixture was cooled to room temperature, the precipitate products washed with water two times and dried at ambient environment (Sample 1, S1). The product was subjected to a serial of isochronal annealing at 550 °C for 5 h in air atmosphere, and the heating rate was 5 °C/min (Sample 3, S3).
The Synthesis of Hollow Magnetite NPs
According to our previous report [20], in a typical synthesis, solution A was prepared by dissolving 2.02 g KNO3 and 0.28 g KOH in 50-mL double distilled water, solution B was prepared by dissolving 0.070 g FeSO4·7H2O in 50-mL double distilled water. Then the two solutions were mixed together under magnetic stirring at a rate of ca. 400 rpm. Two minutes later, solution C (prepared by dissolving 0.18 g glutamic acid (Glu) in 25-mL double distilled water) was added dropwise into the mixed solution. The reaction temperature was raised increasingly to 90 °C and kept 3 h under argon (Ar) atmosphere. Meanwhile, brown solution was observed to change black. After the mixture was cooled to room temperature, the precipitate products were magnetically separated by MSS, washed with ethanol and water two times, respectively, and then redispersed in ethanol.
The Synthesis of Magnetic Hollow Silica NPs
For synthesis of magnetic hollow SiO2NPs, 100 mg hollow magnetite NPs dissolved in 5-mL double distilled water and ultrasonic dispersing 5 min. CTAB of 0.2 g was dissolve in 96-mL double distilled water, and then added the hollow magnetite NPs, the mixture was stirred at rate of ca. 600 rpm and kept 10 min at room temperature, and we obtained the color mixture. Then, 0.28-mL NaOH (5 mol·L−1) by pipette dropwise added into the mixture with the same stirring rate for 30 min. AOT of 0.1 g was added to the solution and the reaction temperature was raised increasingly to 80 °C, and then dropwise added 1.34 mL TEOS to the mixture by pipette, the resulting mixture was stirred at 80 °C and kept 2 h. The gray mixture was observed to change milk white (Sample 2, S2). Likewise, after the mixture was cooled to room temperature, the precipitate products washed with water two times and dried at ambient environment. The product was subjected to a serial of isochronal annealing at 550 °C for 5 h in air atmosphere, and the heating rate was 5°C/min (Sample 4, S4).
Characterization
X-ray Powder Diffraction
Powder X-ray powder diffraction (XRD) patterns of the samples were recorded on a D8 Advance X-ray diffractometer (Germany) using Cu Kα radiation (λ = 0.1542 nm) operated at 40 kV and 40 mA and at a scan rate of 0.05° 2θ s−1.
Transmission Electron Microscopy
Transmission electron microscopy (TEM) and selected-area electron diffraction (SAED) were observed on a JEOL JEM-2010 (HT) transmission electron microscope at an acceleration voltage of 150 kV, and the annealing samples redissolved in water and then dropped on copper grids.
X-ray Photoelectron Spectroscopy
X-ray photoelectron spectroscopy (XPS) measurements were made using a Kratos 800 SIMS. This system uses a focused Mg exciting source for excitation and a spherical section analyzer. The percentages of individual elements detected were determined from the relative composition analysis of the peak areas of the bands. It is based on the relative peak areas and their corresponding sensitivity factors to provide relative compositions.
Nitrogen Adsorption and Desorption
The Brunauer–Emmett–Teller (BET) surface area of the annealing samples was analyzed by nitrogen adsorption in a Micromeritics ASAP 2020 nitrogen adsorption apparatus (USA). All the samples were degassed before the nitrogen adsorption measurement. The BET surface area was determined by a multipoint BET method using the adsorption data in the relative pressure (P/P0) range of 0.05–0.3. A desorption isotherm was used to determine the pore size distribution by the Barret–Joyner–Halender (BJH) method. The nitrogen adsorption volume at the relative pressure (P/P0) of 0.9935 and 0.9957 was used to determine the pore volume and average pore size for annealing samples S3 and S4, respectively.
Superconducting Quantum Interference Device (SQUID) Magnetometry
Magnetic measurements were performed using a Quantum Design MPMS XL-7 SQUID magnetometer. The powder sample was filled in a diamagnetic plastic capsule, and then the packed sample was put in a diamagnetic plastic straw and impacted into a minimal volume for magnetic measurements. Background magnetic measurements were checked for the packing material.
Results and Discussions
The TEM image of as-prepared S1 was shown in Fig. 2a, it can be seen that spherical silica NPs with the range from 100 to 300 nm were obtained. Figure 2b shows the typical TEM micrographs of as-synthesized S2, and the image shows that several acicular magnetite nanocrystals were encapsulated in the silica micelles. To our surprise, hollow nanostructure of magnetite has been transformed into an acicular structure. All particles are needle alike, they have a mean particle length of about 70 nm and a diameter of about 13 nm. In addition, Fig. 2c further displayed the typical TEM images of the hollow Fe3O4NPs. It was found that the Fe3O4NPs had a hollow structure and the overall diameter is around 100 nm, which indicated an oriented aggregation of small Fe3O4NPs. The selected-area electron diffraction (SAED) pattern in the insert of Fig. 2c reveals the polycrystal-like feature of the samples, and their pattern agree well to the structure planes of iron oxide NPs. In our previous report [24], we have confirmed this hollow Fe3O4 NPs are self-aggregates and compose of many ultrafine iron oxide NPs, the earlier mentioned results reveal the hollow Fe3O4 NPs have been dispersed in the reaction.
Figure 2.
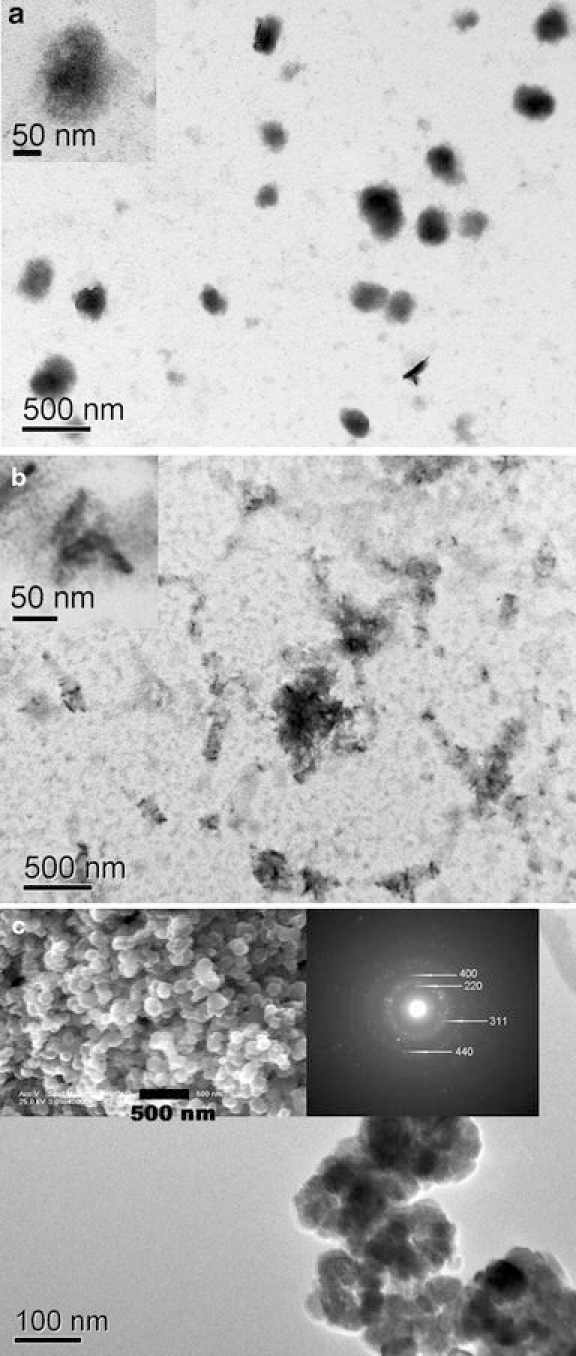
a, b TEM micrographs of samples S1 and S2 (without annealing treatment). The inset is the image of the product with high magnification. c The TEM images and SAED patterns of hollow magnetite NPs
Compare with the samples without annealing treatment, the obvious electron-density differences between the dark edge and pale center of Fig. 3 further confirm the hollow interiors clearly. Figure 3a depicts the typical TEM images of the annealed samples S3 (redissolve in water). From its corresponding size distribution histograms Fig. 3b, it can be seen that the S3 have average size at 23.9 nm and narrow size distribution (The result was statistically analyzed by JEOL SmileView software, analyzing more than 70 resoluble particles). From the insert image at a high magnification, the silica shell appears to be about 4–6 nm thick, which is an important property for applications such as drug delivery or imaging. Figure 3b shows the TEM micrographs of the annealed samples S3 (resolve in water), likewise, from its corresponding size distribution histograms (Fig. 3d), it can be seen that the S4 have average size at 32.0 nm and narrow size distribution. The insert shows that the silica shell appears to be about 8–18 nm thick. Figure 3e shows the magnetic silica NPs at a high magnification and confirmed the iron oxide NPs may be implanted in the shell of S4. Moreover, the corresponding selected-area electron diffraction (SAED) pattern (Fig. 3f) further confirms that the hollow spheres are polycrystalline, and their patterns agree partially to the structure planes of iron oxide NCs. These results suggest that the magnetite NPs have been introduced in the hollow silica NPs and it will cause the increase in particle size and shell thickness correspondingly.
Figure 3.
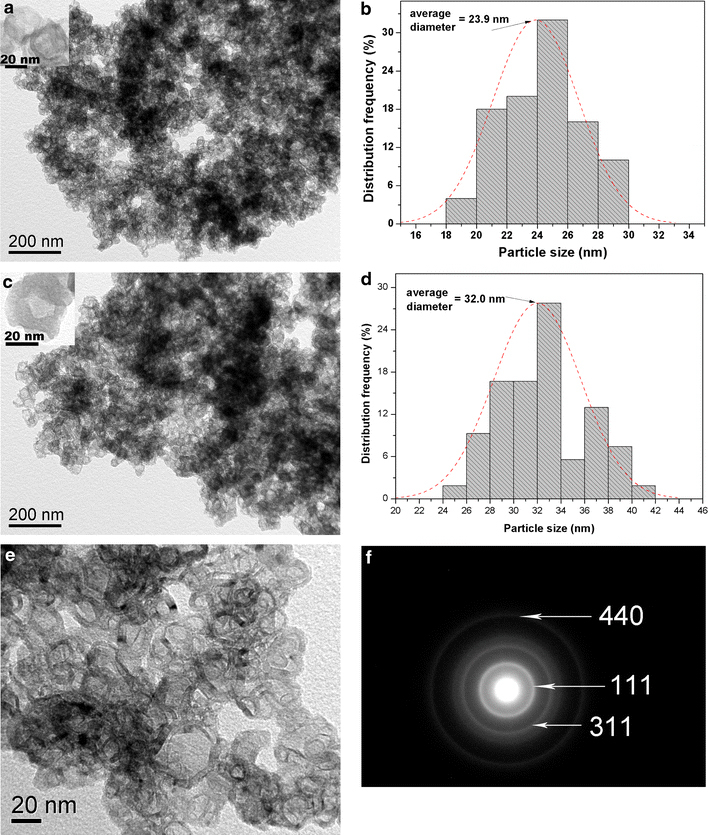
a, c TEM micrographs of annealed samples S3 and S4. The inset is the image of the product with high magnification image. b, d The corresponding size distribution histograms of S3 and S4. e The magnification image of S4 and the in suit SAED pattern (f)
Figure 4a shows the XRD patterns of hollow magnetite NPs and magnetic hollow silica NPs. The (220), (311), (400), (422), (511) and (440) diffraction peaks observed at curve a can be indexed to the cubic spinel structure, and all peaks are in good agreement with the standard Fe3O4 phase (JCPDS card 19-0629). The XRD pattern of S4 showed that the broad diffraction peaks can be assigned to those of amorphous or nanocrystalline silica for 15 < 2θ < 33 (curve b) [25,26]. As expected, the XRD peaks can be assigned to the presence of maghemite were observed with relative intensities, especially the (220) and (311) planes. The peak of planes shifts a litter owing to some gama-Fe2O3 nanoparticles generated by annealing treatment. Maghemite (γ-Fe2O3) can be prepared by oxidation of magnetite (Fe3O4) under air at T = 523 K [24,27]. To identify the composition of the samples, XPS was used to measure the composition and chemical bonding configurations. As shown in Fig. 4b, only one peak (103.9 eV) could be observed in Si 2p spectrum for the S3 and S4, and the result could be assigned to Si 2p for SiO2. Analysis of the XPS spectra revealed the decrease in Si peaks in S4 when the magnetite introduced. Figure 4c shows the XPS spectra for Fe 2p region of S4, two peaks (724.5 and 710.8 eV) could be observed in the spectrum and they could be assigned to Fe 2p3/2 and Fe 2p1/2 for γ-Fe2O3[28].
Figure 4.
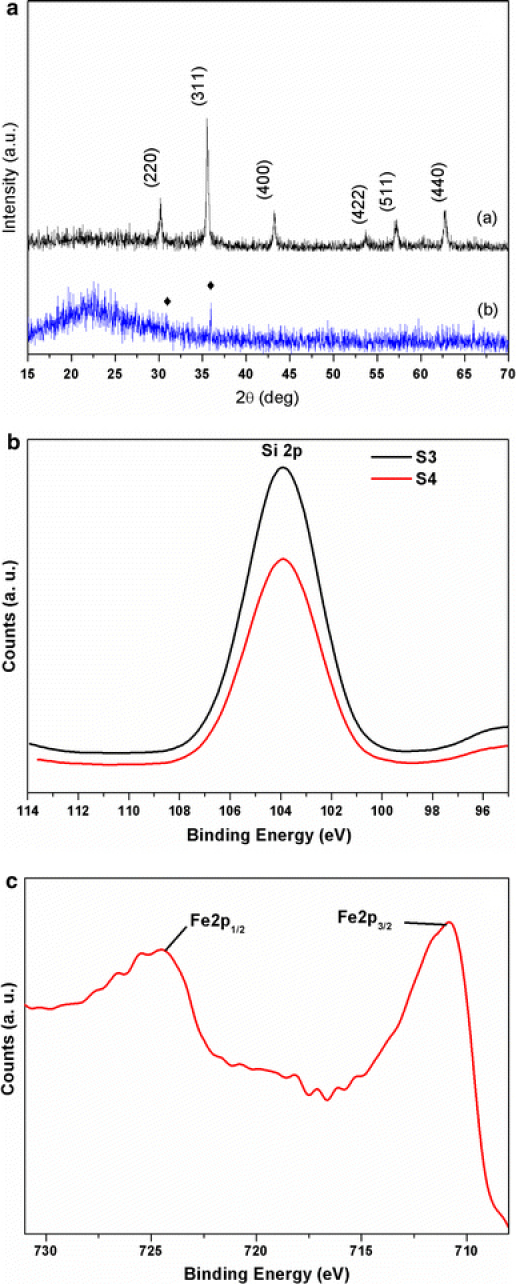
a XRD patterns of hollow Fe3O4NPs and S4. b, c The XPS patterns of Si2p and Fe2p regions, respectively
Nitrogen adsorption/desorption isotherms are measured to determine the specific surface area and porosity of the shell of S3 and S4 at 77 K, and the corresponding results are presented in Fig. 5. The samples S3 and S4 both exhibit a type H3 hysteresis loop according Brunauer–Deming–Deming–Teller (BDDT) classification, indicating the presence of mesopores (2–50 nm) and the pore can be assumed as a cylindrical pore mode [29]. The specific surface area of S3 and S4 according to the Brunauer–Emmett–Teller (BET) method is 213.6 and 417.2 m2/g, respectively. Additionally, it is observed that S4 possess a larger surface area than S3, which is may be derived from the size of nanoparticles composed of iron oxide NPs [30]. The Barret–Joyner–Halender (BJH) adsorption cumulative pore volume of S3 and S4 is 0.61 and 0.83 cm3/g, respectively (between 1.7 and 300 nm width). Moreover, The BJH desorption cumulative pore volume of S3 and S4 is 0.66 and 0.81 cm3/g, respectively (between 1.7 and 300 nm width). Furthermore, the BJH desorption average pore size of S3 and S4 is 7.42 and 5.18 nm, respectively. The results illustrate that some particles may be appeared as open cups due to the loss of organic and surfactant materials during annealing. The mesopores provide pathways by which organic and surfactant can be removed from the interior of the hollow spheres during annealing treatment, without bursting the shells [31].
Figure 5.
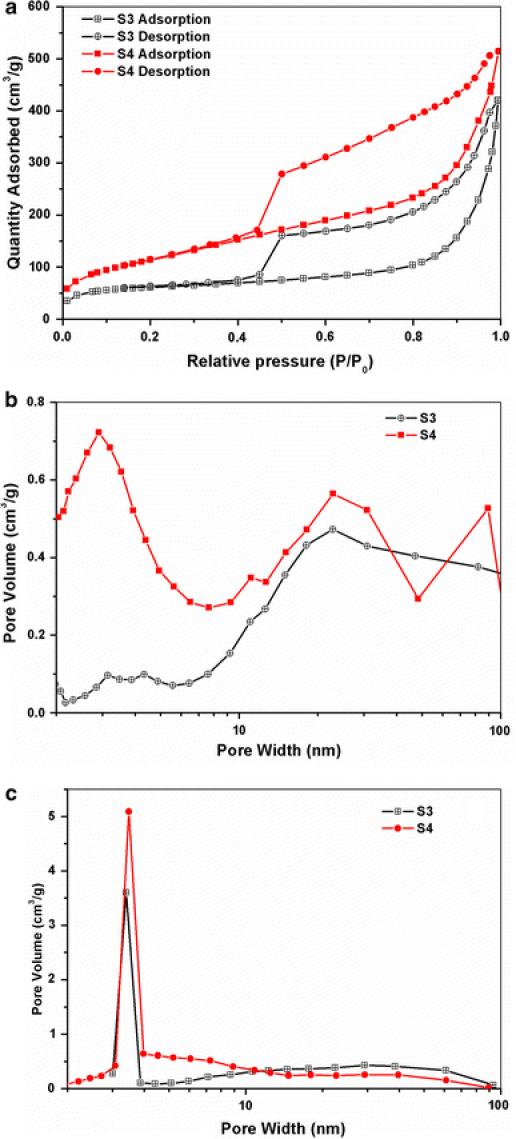
a Nitrogen adsorption–desorption isotherms of S3 and S4. b, c Pore size distribution calculated from adsorption and desorption branch of the isotherms, respectively, using the BJH method
The photographs of annealed samples S3 and S4 on the two parallel glasses are presented in Fig. 6a, it can be seen that the as-obtained S3 powder appears completely white under sunlight illumination, and the as-obtained S4 powder appears completely tan color. Additionally, the result of S4 before and after magnetic separation by an external magnetic field illustrates the facile separation process of the NPs during the experiments. Room-temperature (T = 300 K) magnetization measurement exhibited that there was a slight magnetic hysteresis feature for S4, as shown in Fig. 6b. And the samples with the remnant magnetization (Mr) and coercivity (Hc) being determined to be 0.43 emu/g and 80 Oe, respectively, suggest that the magnetic hollow silica NPs exhibit weak ferromagnetic and soft magnetic behaviors. The results suggest that hollow silica encapsulated the small amount of iron oxide NPs.
Figure 6.
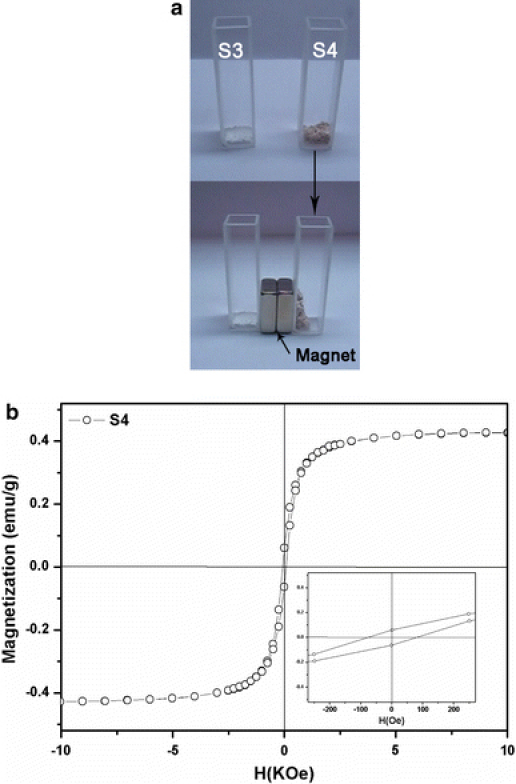
a Photographs of the S3 and S4 before and after magnetic separation by an external magnetic field (this magnet takes from the MSS). b Hysteresis loops of S4
Summary
In conclusion, we have demonstrated a facile, low-cost way to fabricate ultrafine hollow silica NPs and magnetic hollow silica NPs using CTAB and AOT as co-templates. The magnetic hollow silica NPs possesses a larger surface area, and much narrower pore diameter distribution than for the corresponding hollow silica NPs. Moreover, the magnetic hollow silica NPs encapsulated the small amount of iron oxide NPs and can be magnetic separation by an external magnetic field. We expect that our results can provide a solid support for further development of other ultrafine hollow materials, especially for those materials that offer a promising alternative to apply for the target drug delivery, controlled release and the other applications.
Acknowledgments
The author thanks the National Nature Science Foundation of China (No. 10775109), the Specialized Research Fund for the Doctoral Program of Higher Education (No. 20070486069) and the Young Chenguang Project of Wuhan City (No. 200850731371) for financial support. The author also thanks Associate Prof. H. -Y. Zhang of Tsinghua University for assistance with the SQUID measurements.
References
- LaConte L, Nitin N, Bao G. Mater. Today. 2005. p. 32. [DOI]
- Wu W, He QG, Jiang CZ. Nanoscale Res. 2008. p. 397. COI number [1:CAS:528:DC%2BD1cXhsVyhtrvP] [DOI] [PMC free article] [PubMed]
- Yang J, Lee J, Kang J, Lee K, Suh JS, Yoon HG, Huh YM, Haam S. Langmuir. 2008. p. 3417. COI number [1:CAS:528:DC%2BD1cXivV2gtrg%3D] [DOI] [PubMed]
- Cao ZX, Zhang J, Zeng JL, Sun LX, Xu F, Cao Z, Zhang L, Yang DW. Talanta. 2009. p. 943. COI number [1:CAS:528:DC%2BD1cXhsVyrsLvL] [DOI] [PubMed]
- Zhou J, Wu W, Caruntu D, Yu MH, Martin A, Chen JF, O’Connor CJ, Zhou WL. J. Phys. Chem. C. 2007. p. 17473. COI number [1:CAS:528:DC%2BD2sXhtlCkurvE] [DOI]
- Fujiwara M, Shiokawa K, Hayashi K, Morigaki K, Nakahara Y. J. Biomed. Mater. Res. A. 2007. p. 103. COI number [1:CAS:528:DC%2BD2sXjsVSlsbc%3D] [DOI] [PubMed]
- Song CY, Wang CL, Zhu HY, Wu XC, Dong L, Chen Y. Catal. 2008. p. 215. COI number [1:CAS:528:DC%2BD1cXlt1KmsQ%3D%3D] [DOI]
- Feng ZG, Li YS, Niu DC, Li L, Zhao WR, Chen HR, Li L, Gao JH, Ruan ML, Shi JL. Chem. 2008. p. 2629. [DOI] [PubMed]
- Gupta AK, Gupta M. Biomaterials. 2005. p. 3995. COI number [1:CAS:528:DC%2BD2MXisFWr] [DOI] [PubMed]
- Wu W, He QG, Hu R, Huang JK, Chen H. Rare Met. 2007. p. 238. COI number [1:CAS:528:DC%2BD1cXitVWksrY%3D]; Bibcode number [2007AIPC..926..238W]
- Wu W, He QG, Chen H, Tang JX, Nie LB. Nanotechnology. 2007. p. 145609. Bibcode number [2007Nanot..18n5609W] [DOI]
- Shao L, Caruntu D, Chen JF, O’Connor CJ, Zhou WL. J. 2005. p. 10Q908. [DOI]
- Lou XW, Archer LA, Yang ZC. Adv. 2008. p. 3987. COI number [1:CAS:528:DC%2BD1cXhsVartbnK] [DOI]
- Zhang TR, Ge JP, Hu YX, Zhang Q, Aloni S, Yin YD. Angew. 2008. p. 5806. COI number [1:CAS:528:DC%2BD1cXpsVekt7k%3D] [DOI] [PubMed]
- Chen M, Wu LM, Zhou SX, You B. Adv. 2006. p. 801. COI number [1:CAS:528:DC%2BD28XjsV2isro%3D] [DOI]
- Cavaliere-Jaricot S, Darbandi M, Nann T. Chem. 2007. p. 2031 . [DOI] [PubMed]
- Yang J, Lind JU, Trogler WC. Chem. 2008. p. 2875. COI number [1:CAS:528:DC%2BD1cXksl2gur0%3D] [DOI]
- Blas H, Save M, Pasetto P, Boissiere C, Sanchez C, Charleux B. Langmuir. 2008. p. 13132. COI number [1:CAS:528:DC%2BD1cXht1yiur3J] [DOI] [PubMed]
- Zou H, Wu SS, Shen J. Langmuir. 2008. p. 10453. COI number [1:CAS:528:DC%2BD1cXpslOhtbk%3D] [DOI] [PubMed]
- Yang S, Zhou X, Yuan P, Yu M, Xie S, Lu GQ, Yu CZ. Angew. 2007. p. 8579. COI number [1:CAS:528:DC%2BD2sXhtl2rsrnJ] [DOI] [PubMed]
- Che S, Liu Z, Ohsuna T, Sakamoto K, Tersaki O, Tatsumi T. Nature. 2004. p. 281. Bibcode number [2004Natur.429..281C] [DOI] [PubMed]
- Ohsuna T, Liu Z, Che S, Terasaki O. Small. 2005. p. 233. COI number [1:CAS:528:DC%2BD2MXot1Crsw%3D%3D] [DOI] [PubMed]
- Che S, Garcia-Bennett AE, Yokoi T, Sakamoto K, Kunieda H, Terasaki O, Tatsumi T. Nat. 2003. p. 801. COI number [1:CAS:528:DC%2BD3sXptlemt7g%3D]; Bibcode number [2003NatMa...2..801C] [DOI] [PubMed]
- Wu W, Xiao XH, Zhang SF, Li H, Zhou XD, Jiang CZ. Nanoscale Res. 2009. p. 926. COI number [1:CAS:528:DC%2BD1MXoslKisbg%3D]; Bibcode number [2009NRL.....4..926W] [DOI] [PMC free article] [PubMed]
- Grasset F, Labhsetwar N, Li D, Park DC, Saito N, Haneda H, Cador O, Roisnel T, Mornet S, Duguet E, Portier J, Etourneau J. Langmuir. 2002. p. 8209. COI number [1:CAS:528:DC%2BD38Xns1Snur8%3D] [DOI]
- Yoshida H, Kimura K, Inaki Y, Hattori T. Chem. 1997. p. 129.
- Mitov I, Cherkezova-Zheleva Z, Mitrov V. Phys. Status Solidi. 1997. p. 475. COI number [1:CAS:528:DyaK2sXktlGmtrc%3D]; Bibcode number [1997PSSAR.161..475M] [DOI]
- Xu X, Cortie MB. J. Phys. Chem. C. 2007. p. 18135. COI number [1:CAS:528:DC%2BD2sXhtlWqsL%2FE] [DOI]
- Sing KSW, Everett DH, Haul RAW, Moscou L, Pierotti RA, Rouquerol J, Siemieniewska T. Pure Appl. 1985. p. 603. COI number [1:CAS:528:DyaL2MXhvFWrtb4%3D] [DOI]
- Li HQ, Ha CS, Kim I. Langmuir. 2008. p. 10552. COI number [1:CAS:528:DC%2BD1cXhtVCrurrF] [DOI] [PubMed]
- Li WJ, Coppens MO. Chem. 2005. p. 2241. COI number [1:CAS:528:DC%2BD2MXivVeku74%3D] [DOI]